Synergistic Strategies: Integrating Pharmacophore Modeling with Molecular Docking to Revolutionize Virtual Screening in Drug Discovery
This article provides a comprehensive overview of the integrated approach of pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) in modern drug discovery.
Synergistic Strategies: Integrating Pharmacophore Modeling with Molecular Docking to Revolutionize Virtual Screening in Drug Discovery
Abstract
This article provides a comprehensive overview of the integrated approach of pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) in modern drug discovery. It explores the foundational concepts of both methods, detailing how their synergistic application creates robust workflows for hit identification, lead optimization, and polypharmacology. We examine practical methodologies, address common challenges and optimization strategies, and present validation studies comparing the performance of integrated versus standalone approaches. Aimed at researchers and drug development professionals, this review synthesizes current knowledge to offer best-practice guidelines for implementing these powerful computational techniques to accelerate the development of novel therapeutics, from initial screening to overcoming resistance in complex diseases.
The Core Concepts: Deconstructing Pharmacophore Modeling and Molecular Docking
In medicinal chemistry, a pharmacophore is defined by the International Union of Pure and Applied Chemistry (IUPAC) as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [1] [2] [3]. This abstract representation captures the essential, three-dimensional arrangement of molecular interaction capabilities shared by active compounds, independent of their specific chemical scaffold [4]. It is a conceptual framework that distinguishes the key functional components responsible for biological activity from the structural carrier of those features.
The pharmacophore concept has evolved significantly from its early origins. While often historically linked to Paul Ehrlich's work on "toxophores," modern usage was popularized by Lemont Kier in the late 1960s and early 1970s [2] [4]. Today, pharmacophore modeling is an indispensable component of computer-aided drug design (CADD), enabling critical tasks such as virtual screening, lead optimization, and de novo design by focusing on the common molecular interaction capacities of a group of compounds towards their target structure [1] [3].
Essential Steric and Electronic Features
A pharmacophore model is constructed from a set of fundamental, abstract chemical features that represent the ability to form specific non-bonding interactions with a biological target. These features generalize across different chemical functional groups that share similar interaction profiles.
Table 1: Core Pharmacophore Features and Their Interaction Types
| Feature Type | Geometric Representation | Complementary Feature Type(s) | Interaction Type(s) | Structural Examples |
|---|---|---|---|---|
| Hydrogen-Bond Acceptor (HBA) | Vector or Sphere | HBD | Hydrogen-Bonding | Amines, Carboxylates, Ketones, Alcoholes, Fluorine Substituents [3] |
| Hydrogen-Bond Donor (HBD) | Vector or Sphere | HBA | Hydrogen-Bonding | Amines, Amides, Alcoholes [3] |
| Aromatic (AR) | Plane or Sphere | AR, PI | π-Stacking, Cation-π | Any aromatic Ring [3] |
| Positive Ionizable (PI) | Sphere | AR, NI | Ionic, Cation-π | Ammonium Ion, Metal Cations [3] |
| Negative Ionizable (NI) | Sphere | PI | Ionic | Carboxylates [3] |
| Hydrophobic (H) | Sphere | H | Hydrophobic Contact | Halogen Substituents, Alkyl Groups, Alicycles, weakly or non-polar aromatic rings [3] |
These features are not specific atoms or functional groups, but rather spatial domains where a particular interaction is likely to occur. Vector-based representations are typically used for directed interactions like hydrogen bonding, defining both location and orientation, while spherical representations are used for undirected interactions like hydrophobic and ionic contacts [3]. The spatial relationship between these features—defined by distances, angles, and torsions—is as critical as the features themselves for ensuring accurate molecular recognition.
Pharmacophore Model Generation: Methods and Protocols
Pharmacophore models can be developed through several approaches, with the choice of method depending on the available structural and ligand data for the biological target.
Structure-Based Pharmacophore Modeling
Structure-based methods derive the pharmacophore model directly from the three-dimensional structure of a target protein, typically in complex with a ligand.
Experimental Protocol: Structure-Based Model Generation from a Protein-Ligand Complex
Step 1: Protein and Ligand Preparation
- Obtain the 3D structure of the protein-ligand complex from a source like the Protein Data Bank (PDB) [5].
- Using software such as Discovery Studio [5], prepare the structure by:
- Removing extraneous water molecules.
- Completing missing amino acid residues and side chains.
- Correcting bond connectivity and order.
- Adding hydrogen atoms and assigning partial charges.
- Performing energy minimization using a force field like CHARMM [5].
Step 2: Automated Feature Identification
- Employ the "Receptor-Ligand Pharmacophore Generation" module in Discovery Studio [5] or an equivalent tool (e.g., LigandScout [1]).
- Configure the protocol to consider the six standard pharmacophore features: Hydrogen Bond Acceptor (HBA), Hydrogen Bond Donor (HBD), Positive Ionizable (PI), Negative Ionizable (NI), Hydrophobic (H), and Aromatic Ring (AR) [5].
- Set parameters, for example: Minimum Features = 4, Maximum Features = 6, Maximum Pharmacophores to generate = 10 [5].
- Execute the protocol to automatically analyze the ligand-protein interactions and map the essential pharmacophore features.
Step 3: Model Validation and Refinement
- Validate the generated model using a decoy set containing known active and inactive compounds [5].
- Screen the validation set and calculate enrichment metrics such as the Enrichment Factor (EF) and the area under the Receiver Operating Characteristic curve (AUC) [5].
- A reliable model typically has an AUC > 0.7 and an EF > 2 [5].
- Manually refine the model by adding or removing features based on biological intuition or additional structural data.
Diagram 1: Workflow for generating a structure-based pharmacophore model.
Ligand-Based Pharmacophore Modeling
When the 3D structure of the target is unavailable, ligand-based methods can be used to infer the pharmacophore from a set of known active ligands.
Experimental Protocol: Ligand-Based Common Feature Pharmacophore Generation
Step 1: Training Set Selection and Preparation
- Select a structurally diverse set of ligands (typically 15-50) with known activity against the target [6] [7].
- For each ligand, generate a set of low-energy conformations to account for flexibility, ensuring the set is likely to contain the bioactive conformation [2]. Tools like iConfGen or ConfGen can be used for this purpose [7].
Step 2: Molecular Superimposition and Common Feature Perception
- Use software such as PharmaGist or the Common Feature Pharmacophore Generation module in Discovery Studio [8].
- The algorithm will systematically superimpose ("fit") the low-energy conformations of all training set molecules [2].
- The goal is to identify the maximal common set of pharmacophoric features shared by all or most active compounds in their aligned state [4].
Step 3: Hypothesis Generation and Scoring
- The software produces multiple pharmacophore hypotheses, each scored based on how well it aligns the training set molecules and the geometric fit of the common features [8].
- Select the top-ranked hypothesis for further validation.
Step 4: Model Validation
- Validate the model by screening a test set containing both active and inactive compounds [2] [8].
- Assess the model's ability to correctly classify actives (sensitivity) and inactives (specificity). Use metrics like the Fβ-score (emphasizing true positives) and FSpecificity-score (emphasizing true negatives) for virtual screening contexts [6].
Integrated Application Notes
Integrating pharmacophore modeling with other computational techniques creates a powerful workflow for drug discovery, particularly in virtual screening.
Application Note: Integrated Virtual Screening for Dual-Target Inhibitors
A 2025 study successfully identified dual inhibitors for VEGFR-2 and c-Met, two critical cancer targets, using an integrated computational approach [5].
- Objective: Identify novel dual-target inhibitors from the ChemDiv database (1.28 million compounds) with potential to overcome tumor resistance.
- Integrated Workflow:
- Drug-Likeness Filtering: Initial filtering based on Lipinski's and Veber's rules, followed by ADMET prediction [5].
- Pharmacophore Screening: Structure-based pharmacophore models for VEGFR-2 and c-Met were built from PDB complexes and used to screen the filtered database [5].
- Molecular Docking: Compounds passing the pharmacophore filter were docked into the active sites of both VEGFR-2 and c-Met using molecular docking software to assess binding affinity and pose [5].
- MD Simulations & MM/PBSA: Top hits from docking underwent molecular dynamics (MD) simulations (e.g., 100 ns) to evaluate complex stability, followed by MM/PBSA calculations to estimate binding free energies [5].
- Key Outcome: The workflow identified 18 hit compounds. Two compounds, 17924 and 4312, showed superior binding free energies compared to positive controls, marking them as promising candidates [5].
This case demonstrates how pharmacophore modeling serves as an efficient pre-filter before more computationally intensive techniques like docking and MD simulations, streamlining the identification of novel lead compounds.
Table 2: Summary of Key Experimental Results from Integrated Virtual Screening [5]
| Experimental Stage | Key Action/Metric | Result |
|---|---|---|
| Initial Database | Compounds from ChemDiv | ~1.28 Million |
| Drug-Likeness Filter | Application of Lipinski/Veber rules & ADMET | Reduced candidate pool |
| Pharmacophore Screening | Screening with validated VEGFR-2/c-Met models | Hit list for docking |
| Molecular Docking | Docking into VEGFR-2 and c-Met active sites | 18 potential dual-target inhibitors identified |
| MD/MM-PBSA | Binding free energy calculation for top hits | Compounds 17924 & 4312 showed superior energies vs. controls |
Application Note: QPhAR - A Machine Learning-Enhanced Workflow
A novel approach called Quantitative Pharmacophore Activity Relationship (QPhAR) integrates machine learning with traditional pharmacophore modeling for improved predictive power [6] [7].
- Objective: Move beyond qualitative pharmacophore screening to build robust, quantitative models that predict biological activity directly from pharmacophore features.
- Protocol:
- Data Preparation: A dataset of molecules with known activity (e.g., IC₅₀, Kᵢ) is split into training and test sets [6].
- QPhAR Model Training: The algorithm generates a consensus "merged-pharmacophore" from the training set. Input pharmacophores are aligned to this consensus, and their spatial information is used to train a machine learning model to predict activity [7].
- Pharmacophore Refinement: The trained QPhAR model automatically identifies and refines the features most critical for activity, creating a model with higher discriminatory power than those derived from simple common feature perception [6].
- Virtual Screening & Ranking: The refined pharmacophore is used for virtual screening. Hits are then ranked by their predicted activity from the QPhAR model, providing a prioritized list for experimental testing [6].
- Key Outcome: This end-to-end automated workflow has been validated on targets like the hERG K⁺ channel, showing robust performance even with small dataset sizes (15-20 training samples), making it highly suitable for lead optimization [6] [7].
Diagram 2: QPhAR automated workflow for quantitative pharmacophore modeling and screening.
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Software Solutions for Pharmacophore Modeling and Virtual Screening
| Software / Tool | Type | Primary Function in Pharmacophore Research | Application Context |
|---|---|---|---|
| Discovery Studio | Commercial Suite | Comprehensive environment for structure-based and ligand-based pharmacophore generation, validation, and virtual screening [5]. | Integrated drug discovery workflows. |
| MOE | Commercial Suite | All-in-one platform for molecular modeling, including pharmacophore modeling, molecular docking, and QSAR [1] [9]. | Integrated drug discovery workflows. |
| Schrödinger Phase | Commercial Tool | Intuitive pharmacophore modeling for both ligand- and structure-based design; includes screening of prepared commercial libraries [9] [10]. | Virtual screening, scaffold hopping. |
| LigandScout | Commercial/Open | Advanced structure-based and ligand-based pharmacophore modeling, with capabilities for 3D-QSAR and virtual screening [1]. | Structure-based design, model validation. |
| PharmaGist | Web Server | Ligand-based pharmacophore alignment from a set of input active molecules [8]. | Quick, online ligand-based hypothesis generation. |
| ZINCPharmer | Web Server | Online tool for pharmacophore-based screening of the ZINC database of purchasable compounds [8]. | Rapid virtual screening of commercial compounds. |
| DataWarrior | Open-Source | Cheminformatics program supporting 3D pharmacophore features and QSAR model development with machine learning [9]. | Open-source analysis and modeling. |
Molecular Docking as a Structure-Based Prediction Tool for Ligand Poses and Affinities
Molecular docking is an indispensable computational method in structural biology and drug discovery, primarily used to predict the binding conformation (pose) and affinity of a small molecule (ligand) within a target macromolecule's binding site [11] [12]. By simulating the molecular recognition process, docking provides critical insights into intermolecular interactions, thereby accelerating rational drug design and the identification of novel therapeutic candidates [11]. The core objectives of molecular docking are to predict the optimal binding geometry of a ligand-receptor complex and to estimate the binding strength through scoring functions [12]. This protocol outlines the fundamental principles, methodological considerations, and practical applications of molecular docking, with an emphasis on its integration within a broader structure-based drug discovery framework, particularly in conjunction with pharmacophore-based virtual screening.
Core Principles and Methodological Components
The molecular docking process consists of two primary computational challenges: a conformational search of the ligand's orientational and internal degrees of freedom within the binding site, and scoring of the generated poses to identify the most likely binding mode and estimate binding affinity [11].
Conformational Search Algorithms
Docking programs employ various algorithms to efficiently explore the vast conformational space of the ligand. Table 1 summarizes the main approaches.
Table 1: Common Conformational Search Algorithms in Molecular Docking
| Algorithm Type | Description | Key Characteristics | Example Programs |
|---|---|---|---|
| Systematic Search | Systematically varies rotatable bonds by fixed increments to explore all possible conformations [12]. | Exhaustive but computationally expensive; pruning algorithms avoid atomic clashes [11] [12]. | Glide [12], FRED [12] |
| Incremental Construction | Fragments the ligand, docks rigid core, and systematically rebuilds flexible components [11] [12]. | Reduces complexity by focusing on flexible linkers between rigid fragments [11]. | FlexX [12], DOCK [12] |
| Stochastic Methods | Uses random sampling and probabilistic rules to explore conformational space [11] [12]. | Avoids local minima; computationally intensive for large compound libraries [11]. | AutoDock [11] [12], GOLD [11] [12] |
| Genetic Algorithm (GA) | A stochastic method that encodes torsions in "chromosomes," applies evolutionary principles (mutation, crossover) [11] [12]. | Uses scoring function as fitness criteria; selects best poses over generations [11]. | AutoDock [11], GOLD [11] |
Scoring Functions
Scoring functions are mathematical models used to predict the binding affinity of a ligand pose by evaluating the intermolecular interactions within the complex. They aim to approximate the binding free energy (ΔG_binding) [12]. The development of more accurate and generalizable scoring functions remains an active area of research, with recent efforts incorporating machine learning techniques to improve predictions [12].
Integrated Docking and Pharmacophore Screening Workflow
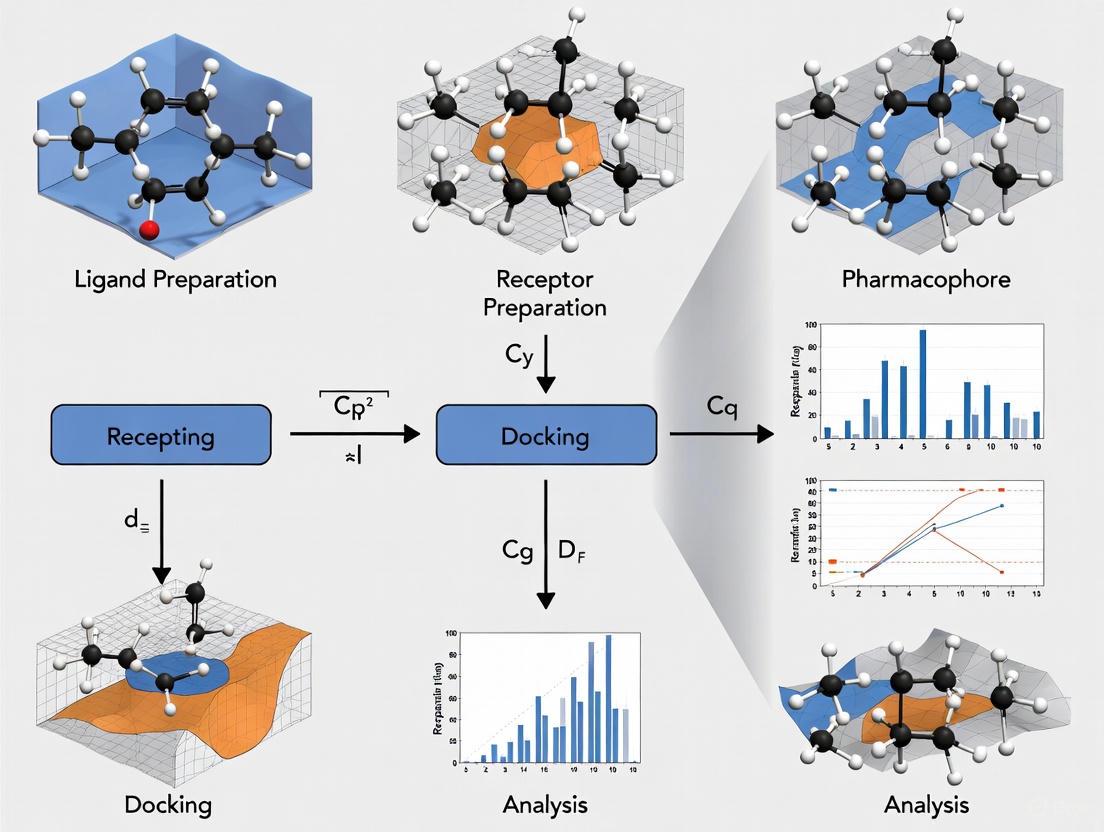
Combining molecular docking with pharmacophore-based virtual screening creates a powerful, multi-stage pipeline for lead identification. The pharmacophore model acts as an initial filter to rapidly eliminate compounds lacking essential chemical features, while docking provides a detailed, structure-based assessment of binding. The following diagram illustrates this integrated workflow.
Diagram 1: Integrated workflow for pharmacophore-based virtual screening and molecular docking in drug discovery. Key steps include structure preparation, pharmacophore screening, molecular docking, and post-docking analysis.
Protocol: Integrated Pharmacophore and Docking Screening
Objective: To identify potential lead compounds by sequentially applying pharmacophore-based filtering and molecular docking.
Step 1: Target and Ligand Preparation
- Protein Preparation: Retrieve the three-dimensional structure of the target protein from the Protein Data Bank (PDB). Using a tool like Schrödinger's Protein Preparation Wizard, add hydrogen atoms, assign bond orders, correct for missing residues, and optimize the hydrogen bond network. Finally, perform energy minimization using a forcefield such as OPLS_2005 [13] [14].
- Pharmacophore Model Generation: Extract the co-crystallized ligand from the protein's active site. Using its structural features (e.g., hydrogen bond donors/acceptors, hydrophobic regions, aromatic rings), create a pharmacophore query model. Servers like Pharmit can be used for this purpose [13] [14].
Step 2: Pharmacophore-Based Virtual Screening
- Screening: Use the generated pharmacophore model to screen large commercial or in-house chemical databases (e.g., ZINC, PubChem, Enamine) [13] [14].
- Filtering: Apply filters such as Lipinski's Rule of Five (Molecular Weight < 500, H-bond donors < 5, H-bond acceptors < 10, LogP < 5) to the resulting hits to prioritize drug-like compounds [13] [14].
Step 3: Molecular Docking
- Ligand Preparation: Prepare the filtered hit compounds using a tool like LigPrep. This step involves generating possible tautomers and ionization states at physiological pH (e.g., 7.0 ± 0.5), and performing energy minimization [13] [14].
- Grid Generation: Define the docking grid around the binding site of the prepared protein structure. The grid center is typically based on the coordinates of the native ligand (e.g., X, Y, Z: 8.32, 6.48, 9.1 for EGFR) [13] [14].
- Docking Execution: Perform molecular docking using a program such as Glide in Standard Precision (SP) mode. Retain a manageable number of top-ranking compounds (e.g., 10-20) based on their docking scores (Glide score) for further analysis [13] [14].
Step 4: Post-Docking Analysis
- ADMET Profiling: Predict the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties of the top-ranked docked compounds using tools like QikProp. Key parameters to assess include predicted Caco-2 permeability (QPPCaco), blood-brain barrier penetration (QPlogBB), and hERG channel inhibition (QPlogHERG) to flag potential cardiotoxicity [13] [14].
- Structural Refinement and Validation: Subject the top candidates to Molecular Dynamics (MD) simulations (e.g., 200 ns using Desmond with OPLS_2005 forcefield) to evaluate the stability of the protein-ligand complex and validate the binding poses observed in docking [13] [14].
Performance Benchmarking and Advanced Applications
Benchmarking Docking Protocols and AlphaFold2 Models
The performance of docking protocols can be benchmarked against experimentally solved structures. Recent studies evaluate the suitability of AlphaFold2 (AF2)-predicted structures for docking, which is crucial when experimental structures are unavailable.
Table 2: Benchmarking Docking Performance on Experimental vs. AlphaFold2 Structures
| Benchmarking Aspect | Performance on Experimental Structures | Performance on AlphaFold2 (AF2) Models | Implications for Protocol Design |
|---|---|---|---|
| Overall Performance | Baseline for comparison [15]. | Comparable to experimental (native) structures in PPI docking benchmarks [15]. | AF2 models are suitable starting points for docking when experimental structures are lacking [15]. |
| Structural Refinement | MD simulations can improve docking outcomes [15]. | MD simulations and ensemble algorithms (e.g., AlphaFlow) can refine AF2 models, improving docking but with variable success [15]. | Using conformational ensembles from MD can enhance virtual screening performance for both experimental and AF2 models [15]. |
| Local vs. Blind Docking | Local docking strategies (restricted to binding site) generally outperform blind docking (whole protein) [15]. | Local docking remains the preferred strategy for AF2 models [15]. | Protocol should prioritize local docking for better accuracy and computational efficiency [15]. |
| Key Limitation | Performance constrained by scoring function limitations [15]. | Performance constrained by scoring function limitations, not necessarily model quality [15]. | Highlights the critical need for improved scoring functions across the field [15]. |
Machine Learning for Accelerated Docking
Machine learning (ML) is increasingly used to overcome the computational bottleneck of traditional docking, especially for ultra-large libraries.
- Methodology: ML models can be trained to predict docking scores directly from 2D molecular structures, bypassing the computationally expensive 3D conformational search and scoring steps. This approach can accelerate screening by orders of magnitude (e.g., 1000-fold faster) [16].
- Implementation: An ensemble model using multiple molecular fingerprints and descriptors can be constructed. This model learns from the results of a classical docking program (e.g., Smina) and can then be used to rapidly prioritize compounds from a database that has been pre-filtered by pharmacophore constraints [16].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Software and Resources for Molecular Docking and Integrated Screening
| Tool Name | Category/Type | Primary Function in Workflow | Example Use Case/Note |
|---|---|---|---|
| PDB Database [16] | Data Repository | Source for experimentally-solved 3D structures of biological macromolecules. | Retrieve target protein structure (e.g., PDB ID: 7AEI for EGFR) [13] [14]. |
| AlphaFold2 [15] | Structure Prediction | Generates high-accuracy protein structure predictions in the absence of experimental data. | Provides reliable models for docking when no PDB structure is available [15]. |
| Pharmit [13] [14] | Pharmacophore Tool | Used for creating pharmacophore models and performing pharmacophore-based virtual screening. | Create a model based on a co-crystal ligand and screen commercial databases [13]. |
| Glide [15] [13] | Docking Software | Performs molecular docking and scoring using systematic search methods. | Used for precise pose prediction and affinity estimation in standard precision (SP) mode [13] [14]. |
| AutoDock Vina [17] | Docking Software | Performs molecular docking and scoring using stochastic search methods. | Popular for high-throughput virtual screening of compound libraries [17]. |
| LigPrep [13] [14] | Ligand Preparation | Generates accurate 3D structures, ionization states, and tautomers for ligands. | Prepare hit compounds from virtual screening for molecular docking. |
| QikProp [13] [14] | ADMET Prediction | Predicts pharmaceutically relevant properties and ADMET parameters. | Filter docked hits based on predicted absorption, toxicity, and other key properties. |
| Desmond [13] [14] | MD Simulation | Performs molecular dynamics simulations to assess complex stability. | Refine docked poses and validate binding stability over 100-200 ns simulations. |
| ZINC Database [13] [16] [17] | Compound Library | A freely available database of commercially available compounds for virtual screening. | Source of small molecules for pharmacophore and docking-based screening. |
The field of computer-aided drug design has undergone a profound transformation, moving from the application of isolated computational techniques to the adoption of sophisticated, multi-tiered workflows. This evolution is characterized by the strategic integration of complementary methods to overcome the inherent limitations of standalone approaches, thereby enhancing the efficiency and success rate of drug discovery. Molecular docking, which predicts how a small molecule ligand binds to a protein target, and pharmacophore-based virtual screening, which identifies compounds sharing essential chemical features for biological activity, once operated in separate domains [18] [19]. Today, they are core components of synergistic pipelines that often include additional computational and experimental validation steps [13] [14]. This application note details this historical progression, provides a structured protocol for a modern integrated workflow, and visualizes the key components and processes involved.
The Evolutionary Pathway: From Standalone to Synergy
The Era of Standalone Techniques
The development of molecular docking began in the 1980s with algorithms designed primarily for rigid body protein-protein interactions [18]. The central challenge was a geometric one: identifying the best complementary fit between two molecules treated as solid bodies, exploring only three rotational and three translational degrees of freedom [18]. The subsequent introduction of search algorithms and scoring functions allowed the prediction of ligand conformation and orientation within a target's binding site, laying the groundwork for structure-based drug design [20].
Similarly, the concept of a pharmacophore, defined as "the ensemble of steric and electronic features that is necessary to ensure the optimal supra-molecular interactions with a specific biological target," originated from early observations of drug-receptor interactions [19]. Initially, these models were simple and ligand-based, relying on the common chemical functionalities of known active compounds.
Despite their utility, these standalone techniques faced significant limitations. Rigid docking could not account for protein flexibility or induced fit effects, while early scoring functions often struggled to accurately predict binding affinities [18] [20]. Pharmacophore models, on the other hand, were sometimes limited by the quality and diversity of the known active compounds used to build them [19].
The Shift to Integrated Workflows
The recognition of these limitations, coupled with advances in computing power and the growth of chemical and structural databases, catalyzed the shift towards integrated workflows. The synergy between pharmacophore modeling and molecular docking is a prime example. A pharmacophore model can rapidly filter millions of compounds in a virtual library to a manageable number of hits that possess the necessary chemical features for binding. This enriched hit list is then subjected to more computationally intensive molecular docking, which evaluates the geometric and energetic feasibility of the binding mode for each candidate [13] [14]. This tandem approach conserves substantial computational resources while improving the quality of candidates advanced to experimental testing.
Modern workflows have expanded further to include critical additional steps. ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction is now routinely incorporated early in the process to flag compounds with poor pharmacokinetic or safety profiles [13] [14]. Furthermore, Molecular Dynamics (MD) simulations are used to assess the stability of protein-ligand complexes over time, providing insights that a static docking pose cannot [13] [21]. This multi-step integration creates a more robust and reliable pipeline for identifying promising lead compounds.
Table 1: Evolution of Key Techniques in Computer-Aided Drug Design
| Era | Molecular Docking | Pharmacophore Modeling | Workflow Paradigm |
|---|---|---|---|
| 1980s-1990s | Rigid-body, protein-protein focus, systematic search algorithms [18]. | Ligand-based, derived from 2D structures of known actives [19]. | Standalone techniques used in isolation. |
| 2000s-2010s | Incorporation of ligand flexibility, stochastic search algorithms, empirical scoring functions [20]. | Structure-based approaches using 3D protein data; used for virtual screening [19]. | Early integration: Pharmacophore screening followed by docking. |
| 2020s-Present | Machine-learning accelerated scoring, handling of protein flexibility, consensus docking [16] [22]. | Complex, multi-feature models; used with large, diverse commercial databases [13] [14]. | Fully integrated workflows including ADMET and MD simulations [13] [21]. |
Application Note & Protocol: An Integrated Workflow for EGFR Inhibitor Discovery
The following protocol, inspired by recent studies [13] [14], provides a detailed methodology for identifying and validating potential Epidermal Growth Factor Receptor (EGFR) inhibitors. This workflow integrates pharmacophore-based virtual screening, molecular docking, ADMET analysis, and molecular dynamics simulations.
Protocol Workflow Visualization
Step-by-Step Experimental Methodology
Step 1: Pharmacophore Model Generation
- Objective: To create a chemical query that encapsulates the essential features required for binding to the EGFR target.
- Procedure:
- Obtain the 3D structure of the target protein complexed with a native ligand (e.g., EGFR with co-crystal ligand R85, PDB ID: 7AEI) from the Protein Data Bank (PDB) [13].
- Using a tool like the Pharmit server, analyze the interactions between the native ligand and the binding site residues.
- Define the critical pharmacophoric features exhibited by the ligand. In the referenced study, these were: one Hydrophobic area, one Aromatic ring, one Hydrogen Bond Acceptor, and one Hydrogen Bond Donor [14].
- Generate the pharmacophore hypothesis based on the spatial arrangement of these features. This model will serve as the query for virtual screening.
Step 2: Pharmacophore-Based Virtual Screening
- Objective: To rapidly filter large chemical databases and identify compounds that match the pharmacophore model.
- Procedure:
- Select multiple commercial and public databases for screening (e.g., ZINC, PubChem, ChemDiv, Enamine, CHEMBL) [13].
- Configure the screening parameters to enforce drug-likeness. Apply Lipinski's Rule of Five filters:
- Molecular weight < 500 g/mol
- Hydrogen Bond Donors (HBD) < 5
- Hydrogen Bond Acceptors (HBA) < 10
- LogP < 5 [14]
- Execute the screening using the generated pharmacophore query. The output is a refined library of hits that match the defined chemical features.
Step 3: Ligand Preparation
- Objective: To generate accurate, energy-minimized 3D structures for all hit compounds prior to docking.
- Procedure:
- Input the list of hits from virtual screening into a ligand preparation tool such as Schrödinger's LigPrep.
- Generate possible stereoisomers and protonation states at a physiological pH (e.g., 7.0 ± 0.5).
- Optimize the geometry of each ligand using a force field (e.g., OPLS_2005) to ensure they are in energetically favorable conformations [14].
Step 4: Molecular Docking
- Objective: To predict the binding pose and affinity of the prepared ligands against the target protein.
- Procedure:
- Protein Preparation:
- Retrieve and prepare the protein structure (e.g., PDB ID: 7AEI) using a protein preparation wizard.
- Assign bond orders, add hydrogen atoms, optimize H-bond networks, and perform a constrained energy minimization to relieve steric clashes [14].
- Grid Generation:
- Define the docking grid around the binding site of interest using the coordinates of the native ligand (e.g., X, Y, Z: 8.32, 6.48, 9.10 for EGFR) [14].
- Docking Execution:
- Dock the prepared ligand library into the defined grid using a docking program like Glide in Standard Precision (SP) mode.
- Rank the resulting poses based on their docking score (reported in kcal/mol). Select the top candidates with the most favorable (most negative) scores for further analysis [14].
- Protein Preparation:
Step 5: ADMET Analysis
- Objective: To evaluate the drug-likeness and pharmacokinetic properties of the top-ranked docked compounds.
- Procedure:
- Use a predictive tool like Schrödinger's QikProp to analyze the selected compounds.
- Calculate key properties, including:
- QPlogPo/w: Predicted octanol/water partition coefficient (recommended: -2.0 to 6.5).
- QPlogHERG: Prediction of blockage of the hERG potassium channel (concern above -5).
- QPPCaco: Predicted apparent Caco-2 cell permeability (good predictor of intestinal absorption).
- QPlogBB: Predicted brain/blood partition coefficient (recommended: -3.0 to 1.2) [14].
- Filter out compounds with undesirable ADMET profiles.
Step 6: Molecular Dynamics (MD) Simulation
- Objective: To confirm the stability of the protein-ligand complex under simulated physiological conditions.
- Procedure:
- Solvate the top complexes (e.g., protein with a promising ligand) in a periodic box with a water model (e.g., TIP3P).
- Neutralize the system with counter-ions and add ionic strength (e.g., 0.15 M NaCl).
- Run a simulation for a sufficient timeframe (e.g., 100-200 ns) using software like Desmond [13] or GROMACS.
- Analyze the resulting trajectories for stability metrics, including Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and the number of hydrogen bonds maintained over time. A stable complex will show low RMSD and consistent interactions [13] [21].
The Scientist's Toolkit: Essential Research Reagents & Software
Table 2: Key Resources for Integrated Pharmacophore and Docking Studies
| Category | Item/Software | Brief Function Description | Example Use Case |
|---|---|---|---|
| Databases | Protein Data Bank (PDB) | Repository for 3D structural data of proteins and nucleic acids [20]. | Source of target structure (e.g., EGFR, PDB: 7AEI) [14]. |
| ZINC, PubChem, CHEMBL | Public databases of commercially available and biologically tested chemical compounds [20] [16]. | Libraries for virtual screening of potential ligands [13] [16]. | |
| Software | Pharmit Server | Online platform for pharmacophore-based and shape-based virtual screening [13]. | Generating pharmacophore hypotheses and screening databases [14]. |
| AutoDock Vina, GNINA, Glide | Molecular docking software for predicting ligand binding poses and affinities [20] [22]. | Performing structure-based virtual screening and pose prediction [14] [22]. | |
| Schrödinger Suite | Commercial software suite providing integrated tools for drug discovery (LigPrep, Glide, QikProp, Desmond) [14]. | End-to-end workflow: ligand prep, docking, ADMET, MD simulations [14]. | |
| GROMACS/Desmond | Software for performing Molecular Dynamics simulations [13]. | Assessing complex stability and dynamics post-docking [21]. | |
| Computational Resources | High-Performance Computing (HPC) Cluster | Computer clusters with many processors connected by a fast network. | Essential for running virtual screening on large libraries and long MD simulations [16]. |
The evolution from standalone molecular docking and pharmacophore modeling to their integration within comprehensive, multi-stage workflows represents a significant advancement in computational drug discovery. This paradigm shift, which now routinely incorporates ADMET profiling and molecular dynamics simulations, provides a more holistic and physiologically relevant assessment of potential drug candidates early in the development process. The outlined protocol offers a validated template for researchers to efficiently identify and prioritize novel compounds for experimental testing.
Future developments are likely to be dominated by the deeper integration of machine learning (ML) and artificial intelligence (AI). ML models are already being used to accelerate docking score predictions by up to 1000-fold and to improve the accuracy of scoring functions, as seen with tools like GNINA [16] [22]. As these technologies mature, they will further streamline the virtual screening pipeline, enabling the interrogation of ultralarge chemical spaces and the rational design of novel therapeutics with optimized properties, solidifying the role of in silico methods as the cornerstone of modern drug discovery.
Key Advantages and Inherent Limitations of Each Method
Comparative Analysis of Virtual Screening Methods
Molecular docking and pharmacophore-based screening are foundational techniques in modern computational drug discovery. The table below summarizes the core advantages and limitations of each method, providing a guide for selecting the appropriate tool for a given research objective.
Table 1: Key Advantages and Limitations of Molecular Docking and Pharmacophore-Based Screening
| Feature | Molecular Docking | Pharmacophore-Based Screening |
|---|---|---|
| Core Principle | Predicts binding pose and affinity by sampling ligand conformations within a protein binding site and scoring them [23] [24]. | Identifies compounds that match a 3D arrangement of steric and electronic features necessary for biological activity [25]. |
| Key Advantages | - Provides detailed atomic-level interaction data [24].- Directly estimates binding affinity via scoring functions [23].- Can handle ligand flexibility explicitly [26].- Capable of blind docking (predicting binding sites) [26]. | - Extremely high computational speed, enabling rapid screening of ultra-large libraries [16] [27].- Does not require a high-quality 3D protein structure for ligand-based models [25].- Results in synthetically accessible, commercially available hits [27].- Effective at enriching active compounds from decoys [25] [5]. |
| Inherent Limitations | - Computationally intensive, making large-scale screening costly [26] [28].- Scoring functions can be inaccurate, leading to high false positive rates [29] [28].- Often struggles with protein flexibility and induced fit effects [26].- DL-based methods can produce physically implausible structures [29] [26]. | - Does not typically provide detailed atomic-level binding poses or energy scores [25].- Quality is highly dependent on the pharmacophore model used [27].- May miss novel scaffolds that do not perfectly match the predefined query [27]. |
| Best Application Context | Hit identification and optimization when a protein structure is available and detailed binding mode understanding is required. | Initial, high-throughput filtering of massive compound libraries to a manageable number of candidates for downstream analysis. |
Performance and Benchmarking Data
Independent benchmarking studies have quantified the performance of these methods in real-world scenarios. A comprehensive evaluation of docking methods revealed a performance hierarchy. Traditional methods like Glide SP excelled in producing physically valid poses (≥94% validity across datasets), while modern generative diffusion models like SurfDock achieved superior pose prediction accuracy (>70% success rate) [29]. However, many deep learning-based docking methods exhibited significant challenges in generalization, particularly when encountering novel protein binding pockets not represented in their training data [29] [26].
In a direct comparison on eight diverse protein targets, pharmacophore-based virtual screening (PBVS) outperformed docking-based virtual screening (DBVS) in retrieving active compounds for 14 out of 16 test cases. The average hit rates for PBVS at the top 2% and 5% of ranked databases were "much higher" than those achieved by multiple docking programs [25].
Table 2: Quantitative Performance Comparison from Benchmark Studies
| Method Category | Representative Tools | Pose Accuracy (RMSD ≤ 2 Å) | Physical Validity (PB-Valid) | Typical Virtual Screening Enrichment |
|---|---|---|---|---|
| Traditional Docking | Glide SP, AutoDock Vina | Moderate to High | Very High (≥94%) [29] | Variable, target-dependent [25] |
| Deep Learning Docking | SurfDock, DiffBindFR | Very High (≥75% on known complexes) [29] | Moderate (40-65%) [29] | Promising but generalizability concerns [29] [26] |
| Pharmacophore Screening | Catalyst, LigandScout | Not Directly Measured | Not Directly Measured | Higher hit rates vs. docking in multiple targets [25] |
Experimental Protocols for an Integrated Workflow
The following protocol outlines a robust methodology for integrating pharmacophore and docking screens, as demonstrated in successful drug discovery campaigns [21] [5].
Protocol: Integrated Pharmacophore and Docking Screen for Dual VEGFR-2/c-Met Inhibitors
This protocol is adapted from a study that identified potential dual inhibitors, using a method that can be generalized to other targets [5].
Step 1: Preparation of Protein Structures and Compound Library
- Source protein structures from the Protein Data Bank (PDB). For the dual-target study, 10 VEGFR-2 and 8 c-Met co-crystal structures with ligands were selected based on high resolution (< 2.0 Å) and biological activity [5].
- Prepare the proteins using software like Discovery Studio: remove water molecules, add missing amino acid residues, correct bond connectivity, and minimize complex energy using a force field like CHARMM [5].
- Prepare the screening library. The study used over 1.28 million compounds from the ChemDiv database. Prepare ligands by removing counterions and salts, adding hydrogen atoms, and filtering based on Lipinski's Rule of Five and Veber's rules to ensure drug-likeness [5].
Step 2: Generation and Validation of Pharmacophore Models
- Construct pharmacophores. Using the "Receptor-Ligand Pharmacophore Generation" module in Discovery Studio, generate models based on the prepared protein-ligand complexes. Standard features include Hydrogen Bond Acceptor, Donor, Hydrophobic, and Aromatic centers [5].
- Validate model quality using decoy sets containing known active and inactive compounds. Calculate the Enrichment Factor (EF) and the Area Under the Curve (AUC) of the Receiver Operating Characteristic (ROC) curve. Select models with AUC > 0.7 and EF > 2 for screening [5].
Step 3: Pharmacophore-Based High-Throughput Screening
- Screen the prepared compound library against the validated pharmacophore models for each target.
- Apply ADMET filters to the resulting hits to predict absorption, distribution, metabolism, excretion, and toxicity properties, further refining the candidate list [5].
Step 4: Multi-Level Molecular Docking
- Perform rigid docking of the pharmacophore-filtered hits into the binding sites of both target proteins (e.g., VEGFR-2 and c-Met) to calculate binding affinities (docking scores) [5].
- Select top-ranking compounds based on docking scores for more computationally intensive flexible docking or subsequent analysis.
Step 5: Binding Free Energy Estimation and Molecular Dynamics (MD)
- Run MD simulations (e.g., 100 ns) on the top docked complexes to assess the stability of protein-ligand interactions under dynamic conditions [21] [5].
- Calculate binding free energies using methods like MM/PBSA (Molecular Mechanics with Poisson-Boltzmann Surface Area) on frames extracted from the MD trajectory to obtain a more reliable affinity estimate than docking scores alone [21].
Integrated Virtual Screening Workflow
Successful implementation of the integrated protocol requires a suite of computational tools and databases.
Table 3: Essential Research Reagents and Computational Tools
| Category / Item | Specific Examples | Function in the Workflow |
|---|---|---|
| Protein Structure Databases | Protein Data Bank (PDB) [23] [5] | Source of experimentally determined 3D structures of target proteins and protein-ligand complexes. |
| Small Molecule Databases | ZINC [23] [16], ChemDiv [5], PubChem [23] | Large libraries of commercially available or synthesizable compounds for virtual screening. |
| Decoy Sets for Validation | DUD-E (Database of Useful Decoys: Enhanced) [5] | Provides inactive compounds with similar physicochemical properties to actives, used to validate pharmacophore models and avoid bias. |
| Pharmacophore Modeling Software | Discovery Studio [5], LigandScout [25], Pharmit [27] | Used to generate, visualize, and validate pharmacophore models, and to perform pharmacophore-based screening. |
| Molecular Docking Software | AutoDock Vina [29] [24], Glide [29] [25], GOLD [25], Smina [16] | Samples ligand conformations and positions within a protein binding site and scores them based on complementary interactions. |
| Molecular Dynamics Software | GROMACS, AMBER, CHARMM [21] | Simulates the physical movements of atoms and molecules over time to assess complex stability and calculate binding free energies. |
| Force Fields | CHARMM [5], AMBER | Defines the potential energy functions and parameters used in energy minimization, docking, and MD simulations. |
Visualizing a Key Signaling Pathway for Target Identification
Understanding the biological context of a drug target is crucial. The diagram below illustrates the VEGFR-2 and c-Met signaling pathways, whose dual inhibition is a promising anti-angiogenic and anti-tumor strategy [5].
Dual VEGFR-2/c-Met Pathway Inhibition
In modern computational drug discovery, structure-based virtual screening (VS) has become an indispensable tool for identifying novel therapeutic candidates from vast chemical libraries. However, the reliance on a single computational method often introduces limitations, whether in accuracy, speed, or the ability to reliably distinguish true binders. Molecular docking predicts how small molecule ligands interact with a protein target at the atomic level, providing detailed binding mode information and affinity estimates through scoring functions [26]. Pharmacophore modeling, conversely, abstracts molecular interactions into essential chemical features—hydrogen bond donors/acceptors, hydrophobic regions, and charged groups—creating a template for screening compounds based on complementary functionality rather than detailed atomic positioning [30]. While docking can capture specific steric and energetic constraints, and pharmacophores efficiently encode key recognition elements, neither approach alone fully captures the complexity of molecular recognition.
The integration of pharmacophore-based virtual screening with molecular docking creates a synergistic workflow that leverages the distinct strengths of each method while mitigating their individual limitations. This complementary strategy enhances screening efficiency by rapidly eliminating unsuitable compounds through pharmacophore filtering before subjecting a refined subset to computationally intensive docking simulations [13] [31]. Furthermore, the combined approach improves hit rates and binding affinity predictions by applying multiple validation layers, ensuring identified compounds satisfy both geometric and chemical interaction requirements [30]. This protocol details the implementation of an integrated virtual screening strategy, providing application notes, experimental protocols, and benchmark data to guide researchers in deploying this powerful combined methodology.
Theoretical Foundation and Key Concepts
Molecular Docking: Principles and Limitations
Molecular docking computationally predicts the structure of a protein-ligand complex and estimates binding affinity through scoring functions. Traditional docking approaches follow a search-and-score framework, exploring possible ligand conformations (poses) within the binding site and ranking them based on computed interaction energies [26]. While modern docking tools like Glide SP and AutoDock Vina have proven valuable, they face inherent challenges. Protein flexibility remains a significant limitation, as most methods treat the receptor as rigid despite induced fit effects upon ligand binding [26]. Scoring function accuracy is another concern, as simplified functions often struggle to reliably correlate computed scores with experimental binding affinities, particularly for diverse compound libraries [32] [29].
Recent advances in deep learning (DL) have introduced new docking paradigms. Generative diffusion models like SurfDock and DiffDock demonstrate superior pose prediction accuracy, while hybrid methods combining traditional searches with AI-driven scoring offer balanced performance [29]. However, benchmarking reveals that DL methods frequently produce physically implausible structures despite favorable root-mean-square deviation (RMSD) scores, with regression-based models particularly prone to invalid bond lengths and steric clashes [29]. Additionally, DL models exhibit generalization challenges when encountering novel protein binding pockets not represented in training data [29].
Pharmacophore Modeling: Principles and Applications
A pharmacophore is an abstract representation of structural features essential for a molecule's biological activity, defined as "a set of common chemical features that describe the specific ways a ligand interacts with a macromolecule's active site in three dimensions" [30]. These features include:
- Hydrogen bond donors and acceptors (represented as vectors and interaction spheres)
- Hydrophobic regions (aliphatic and aromatic)
- Charge transfer groups (positive/negative ionizable areas)
- Aromatic features (enabling π-π and cation-π interactions)
- Excluded volumes (representing sterically forbidden regions) [30]
Pharmacophore models can be developed through structure-based approaches (analyzing protein-ligand complexes) or ligand-based methods (identifying common features among active compounds) [30]. In virtual screening, pharmacophores serve as efficient 3D queries to filter large compound databases, rapidly identifying molecules possessing essential interaction capabilities without detailed energy calculations [31] [33]. This makes them particularly valuable for scaffold hopping—identifying structurally diverse compounds with similar interaction profiles—and for incorporating toxicity and off-target predictions early in screening pipelines [30].
Integrated Screening Workflow: Protocol and Application Notes
The following workflow integrates pharmacophore-based screening and molecular docking into a coordinated, hierarchical process that maximizes efficiency and effectiveness in hit identification.
Figure 1. Integrated virtual screening workflow combining pharmacophore modeling and molecular docking. The protocol progresses through sequential filtering stages, from initial pharmacophore screening to molecular dynamics validation of final candidates.
Stage 1: Pharmacophore Modeling and Validation Protocol
Structure-Based Pharmacophore Development
- Input Requirements: Protein Data Bank (PDB) structure of target protein with bound ligand (holo conformation preferred).
- Procedure:
- Identify Critical Interactions: Analyze the co-crystal ligand's binding mode to determine essential hydrogen bonds, hydrophobic contacts, and charge interactions. For EGFR targeting, researchers used the R85 ligand from PDB ID: 7AEI to define six key pharmacophoric features [13].
- Feature Mapping: Using software such as BIOVIA Discovery Studio's CATALYST or Pharmit servers, translate identified interactions into pharmacophoric features with precise spatial coordinates [13] [33].
- Excluded Volumes: Define sterically forbidden regions based on protein structure to eliminate compounds with unfavorable clashes [30].
Ligand-Based Pharmacophore Development (When Structural Data is Limited)
- Input Requirements: Set of 20-50 known active compounds with diverse chemical scaffolds but similar biological activity.
- Procedure:
- Conformational Analysis: Generate representative low-energy conformers for each active compound.
- Common Feature Identification: Apply algorithms to detect 3D spatial patterns shared across active molecules.
- Model Optimization: Adjust feature tolerances to balance model specificity and sensitivity.
Pharmacophore Validation Protocol
- Validation Metrics:
- Sensitivity: Percentage of known active compounds correctly retrieved by the model.
- Specificity: Percentage of known inactive compounds correctly rejected [30].
- Procedure:
- Test Set Construction: Compile a decoy set containing known actives and inactives (approximately 50-100 compounds each).
- Screening Performance: Calculate enrichment factors (EF) to quantify early recognition capability [34].
Application Note: A validated pharmacophore model should achieve minimum sensitivity of 80% and specificity of 70% before proceeding to large-scale virtual screening.
Stage 2: Pharmacophore-Guided Virtual Screening
Compound Library Preparation
- Library Sources: Screen multiple databases including ZINC, PubChem, ChEMBL, Enamine, and specialized collections (e.g., MPD3 for phytochemicals) [13] [31].
- Preprocessing Steps:
- Format Standardization: Convert all structures to consistent representation (e.g., SMILES, SDF).
- Tautomer and Ionization State Generation: Create representative forms at physiological pH (7.0-7.4).
- 3D Conformer Generation: Produce multiple low-energy conformers for each compound to ensure comprehensive pharmacophore matching.
Pharmacophore Screening Protocol
- Screening Parameters:
- Feature Matching: Require compounds to match all critical features (hydrogen bond donors/acceptors, hydrophobic centers).
- Spatial Tolerance: Set distance tolerances typically between 1.0-1.5 Å based on binding site flexibility.
- Partial Matching: Allow compounds matching ≥70% of essential features to proceed to additional filtering.
- Post-Screening Filtering:
- Drug-Likeness Criteria: Apply Lipinski's Rule of Five (molecular weight <500, H-bond donors <5, H-bond acceptors <10, LogP <5) [13].
- Chemical Diversity: Select structurally diverse hits representing different scaffold classes.
Application Note: In an EGFR inhibitor study, this stage reduced an initial multi-database collection to 1,271 qualified hits (0.1-1% of starting library), demonstrating substantial library enrichment before docking [13].
Stage 3: Molecular Docking and Binding Validation
System Preparation Protocol
- Protein Preparation:
- Structure Optimization: Add hydrogen atoms, assign bond orders, and optimize hydrogen bonding networks using tools like Schrödinger's Protein Preparation Wizard [13].
- Protonation States: Determine appropriate ionization states for residues (especially His, Asp, Glu) using PROPKA at pH 7.0 [13].
- Structural Refinement: Perform constrained energy minimization to relieve steric clashes while preserving crystallographic coordinates.
- Ligand Preparation:
Docking Execution Protocol
- Grid Generation:
- Binding Site Definition: Center grid box on known binding site or pharmacophore-aligned region.
- Box Dimensions: Set sufficient size to accommodate ligand flexibility (typically 15-20Å in each dimension).
- Docking Parameters:
- Sampling exhaustiveness: Increase for challenging systems (default: 8-32 for Vina-derived methods).
- Pose Clustering: Retire multiple diverse poses (5-20) per compound to ensure binding mode representation.
- Scoring Functions: Employ consensus scoring where possible to improve affinity prediction reliability.
Pose Analysis and Interaction Validation
- Critical Interaction Assessment: Verify predicted poses recapitulate key interactions identified in pharmacophore model.
- Protein-Ligand Interaction Fingerprints (PLIF): Generate interaction profiles for quantitative comparison across compound series [35].
- Consensus Ranking: Integrate scores from multiple functions (if available) and interaction completeness metrics for final hit prioritization.
Application Note: Benchmarking shows traditional methods like Glide SP maintain high physical validity (≥94% PB-valid rates) while generative diffusion models like SurfDock achieve superior pose accuracy (≥70% RMSD ≤2Å) but with lower physical plausibility [29]. Method selection should align with screening priorities.
Stage 4: Advanced Validation and Prioritization
ADMET Profiling Protocol
- Properties to Predict:
- Absorption: Caco-2 permeability (QPPCaco)
- Distribution: Blood-brain barrier penetration (QPlogBB), plasma protein binding (QPlogKhsa)
- Metabolism: Cytochrome P450 inhibition profiles
- Excretion: Clearance predictions
- Toxicity: hERG channel binding (QPlogHERG) [13]
- Tools: Utilize QikProp (Schrödinger) or similar platforms for high-throughput prediction.
Molecular Dynamics Simulation Protocol
- System Setup:
- Solvation: Embed protein-ligand complex in TIP3P water box with 10Å buffer.
- Neutralization: Add counterions (Na+/Cl-) to physiological concentration (0.15M).
- Simulation Parameters:
- Analysis Metrics:
- Complex Stability: Calculate root-mean-square deviation (RMSD) of protein backbone and ligand heavy atoms.
- Interaction Persistence: Quantify occupancy of key hydrogen bonds and hydrophobic contacts.
- Binding Free Energy: Compute using MM/GBSA or MM/PBSA methods on trajectory snapshots.
Application Note: In a Waddlia chondrophila inhibitor study, MD simulations confirmed stable binding (RMSD <2Å over 100ns) for top-ranked phytocompounds, validating docking predictions and providing mechanistic insights [31].
Performance Benchmarking and Quantitative Outcomes
Comparative Method Performance
Table 1. Virtual screening performance benchmarks across methodological approaches
| Method Category | Representative Tools | Pose Accuracy (RMSD ≤2Å) | Physical Validity (PB-Valid) | Screening Enrichment (EF1%) | Computational Throughput |
|---|---|---|---|---|---|
| Traditional Docking | Glide SP, AutoDock Vina | 60-75% | 94-97% | 10-15 | Low (×1 baseline) |
| Generative Diffusion | SurfDock, DiffBindFR | 70-92% | 40-64% | 12-18 | Medium (×2-5) |
| Regression-Based DL | KarmaDock, QuickBind | 40-60% | 20-45% | 8-12 | High (×10-20) |
| Hybrid Methods | Interformer | 65-80% | 85-90% | 14-16 | Medium (×3-7) |
| Pharmacophore Only | CATALYST, Pharmit | N/A | N/A | 5-10 | Very High (×50-100) |
| Integrated Pipeline | Pharmacophore+Docking | 75-85% | 90-95% | 16-25 | Medium-High (×10-30) |
Data compiled from multiple benchmarking studies [13] [29] [34]. EF1% represents enrichment factor at 1% of screened library.
Case Study Outcomes
Table 2. Representative screening outcomes from integrated approaches
| Target | Screening Strategy | Initial Library | Post-Pharmacophore Hits | Final Docking Hits | Experimental Hit Rate | Reference |
|---|---|---|---|---|---|---|
| EGFR | Pharmacophore → Docking → MD | 9 databases | 1,271 | 10 | 30% (3/10) | [13] |
| KLHDC2 | RosettaVS with active learning | Billions | 1,000 (prioritized) | 50 | 14% (7/50) | [34] |
| NaV1.7 | AI-accelerated platform | Billions | 10,000 (prioritized) | 9 | 44% (4/9) | [34] |
| W. chondrophila | Subtractive proteomics → Docking | 1,000 phytochemicals | 127 | 3 | 66% (MD validation) | [31] |
Throughput and Efficiency Metrics
The integration of pharmacophore screening significantly enhances virtual screening efficiency. Machine learning surrogate models can achieve 80× increased throughput compared to traditional docking when trained on just 10% of dataset, enabling screening of 48 billion compounds in approximately 8,700 hours using 1,000 computers [36]. Active learning approaches further optimize this process by iteratively selecting informative compounds for expensive docking calculations, reducing the fraction of library requiring full simulation [34].
Essential Research Reagents and Computational Tools
Table 3. Key resources for implementing integrated virtual screening
| Category | Tool/Resource | Specific Application | Key Features |
|---|---|---|---|
| Pharmacophore Modeling | BIOVIA CATALYST [33] | Structure- & ligand-based pharmacophore generation | Feature-based queries, shape similarity, forbidden volumes |
| Pharmit Server [13] | Online pharmacophore screening | Public database access, real-time screening | |
| Molecular Docking | Glide (Schrödinger) [13] | High-precision docking | Standard Precision (SP)/Extra Precision (XP) modes |
| AutoDock Vina/smina [36] | Flexible docking | Open-source, configurable scoring | |
| RosettaVS [34] | Flexible receptor docking | Modeling of sidechain/backbone flexibility | |
| DiffDock [26] | Deep learning docking | Diffusion-based pose prediction | |
| Structure Preparation | Protein Preparation Wizard [13] | Protein structure optimization | Hydrogen bonding optimization, pKa prediction |
| LigPrep [13] | Ligand preparation | Tautomer generation, energy minimization | |
| Simulation & Analysis | Desmond [13] | Molecular dynamics | OPLS force field, trajectory analysis |
| GROMACS | Molecular dynamics | High performance, extensive analysis tools | |
| PoseBusters [29] | Docking pose validation | Physical/geometric plausibility checks | |
| Compound Libraries | ZINC, PubChem [13] | Commercially available compounds | Millions of purchasable compounds |
| ChEMBL [13] | Bioactive molecules | Annotated activity data | |
| Enamine REAL [36] | Ultra-large libraries | 48+ billion make-on-demand compounds |
Concluding Remarks and Future Directions
The strategic integration of pharmacophore modeling and molecular docking establishes a complementary virtual screening paradigm that consistently demonstrates enhanced performance over individual methods. This synergistic approach leverages the high-throughput filtering capability of pharmacophore screening with the atomic-level precision of molecular docking, resulting in improved hit rates, better binding affinity prediction, and more efficient utilization of computational resources. Quantitative benchmarks show integrated approaches achieve 16-25 enrichment factors at 1% screening depth, surpassing most standalone methods [13] [34].
Future methodology developments will likely focus on incorporating protein flexibility more comprehensively through deep learning approaches like FlexPose and DynamicBind that model conformational changes between apo and holo states [26]. AI-accelerated screening platforms that combine active learning with target-specific neural networks will further enhance throughput for billion-compound libraries [34]. Additionally, improved scoring functions that better account for entropic contributions and solvation effects will address current limitations in binding affinity prediction [32] [34].
The continued refinement of integrated screening strategies promises to further bridge the gap between computational prediction and experimental validation, accelerating the discovery of novel therapeutic agents against increasingly challenging targets. By adopting the protocols and application notes outlined herein, researchers can implement robust, complementary screening strategies that maximize both efficiency and effectiveness in drug discovery pipelines.
Building Integrated Workflows: From Theory to Practical Application
The integration of pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) represents a powerful hierarchical strategy in computer-aided drug discovery (CADD). This sequential workflow leverages the unique strengths of each method to enhance the efficiency of identifying hit compounds from large chemical libraries. By employing PBVS as a rapid initial filter to reduce chemical space, followed by more computationally intensive DBVS for refined evaluation, researchers can significantly accelerate the virtual screening process while improving the likelihood of success [19] [37]. This architecture is particularly valuable for targeting complex biological systems where multiple signaling pathways contribute to disease pathology, such as in cancer therapeutics targeting both VEGFR-2 and c-Met receptors [5].
Performance Comparison: PBVS vs. DBVS
Benchmark studies against eight diverse protein targets demonstrate that PBVS consistently outperforms DBVS in initial enrichment of active compounds. The table below summarizes key performance metrics from a comparative study evaluating both approaches across multiple target classes.
Table 1: Benchmark Comparison of PBVS versus DBVS Across Eight Protein Targets [37]
| Target Protein | Method | Enrichment Factor (EF) | Hit Rate at 2% | Hit Rate at 5% |
|---|---|---|---|---|
| ACE | PBVS | High | 42.1% | 65.8% |
| ACE | DBVS (DOCK) | Moderate | 18.4% | 36.8% |
| AChE | PBVS | High | 35.0% | 60.0% |
| AChE | DBVS (Glide) | Moderate | 15.0% | 35.0% |
| AR | PBVS | High | 38.9% | 61.1% |
| AR | DBVS (GOLD) | Moderate | 16.7% | 33.3% |
| DHFR | PBVS | High | 40.0% | 66.7% |
| DHFR | DBVS (DOCK) | Moderate | 20.0% | 40.0% |
| Average | PBVS | High | 39.8% | 64.8% |
| Average | DBVS | Moderate | 18.6% | 37.8% |
The superior performance of PBVS in initial compound enrichment makes it particularly suitable for the first stage in a sequential screening workflow, where it can rapidly reduce library size by 80-95% before applying more resource-intensive docking methods [37].
Experimental Protocols
Phase 1: Structure-Based Pharmacophore Modeling
Objective: To develop a quantitative pharmacophore model for initial compound screening.
Methodology:
- Protein Preparation: Retrieve 3D protein structures from RCSB Protein Data Bank (PDB). For VEGFR-2 and c-Met dual inhibitors, select multiple co-crystal structures based on resolution (<2.0 Å), biological activity (nM range), and structural diversity. Prepare proteins by removing water molecules, completing missing amino acid residues, correcting bond connectivity, and minimizing complex energy using the CHARMM force field in Discovery Studio software [5].
- Binding Site Characterization: Identify ligand-binding sites using computational tools such as GRID or LUDI, which analyze protein surfaces for potential interaction sites based on geometric and energetic properties [19].
- Pharmacophore Feature Generation: Using the Receptor-Ligand Pharmacophore Generation module, define critical chemical features including hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), hydrophobic areas (H), positively ionizable groups (PI), negatively ionizable groups (NI), and aromatic rings (AR). Set parameters to generate 10 hypotheses with 4-6 features each [5].
- Model Validation: Validate pharmacophore models using decoy sets containing known active compounds and inactive molecules. Calculate enrichment factor (EF) and area under the receiver operating characteristic curve (AUC). Select models with AUC >0.7 and EF >2 for virtual screening [5].
Phase 2: Sequential Virtual Screening Protocol
Objective: To implement the sequential PBVS→DBVS workflow for identifying potential dual inhibitors.
Methodology:
- Compound Library Preparation: Collect chemical libraries (e.g., ChemDiv database with >1.28 million compounds). Prepare compounds by removing counterions, solvent moieties, and salts, then add hydrogen atoms. Filter compounds using Lipinski's Rule of Five and Veber rules to ensure drug-like properties [5].
- ADMET Profiling: Predict absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties including aqueous solubility, blood-brain barrier penetration, cytochrome P450 2D6 inhibition, hepatotoxicity, and plasma protein binding [5].
- Pharmacophore-Based Screening (PBVS): Screen the pre-filtered compound library against validated pharmacophore models. Use the "Best Flexible" search method to identify compounds matching the pharmacophore features. This typically reduces the library size to 2-5% of its original size [37] [5].
- Docking-Based Screening (DBVS): Subject PBVS hits to molecular docking against target structures using programs such as DOCK, GOLD, or Glide. Prepare protein structures by assigning correct bond orders, adding hydrogen atoms, and optimizing hydrogen bonding networks. Define binding sites using coordinates from co-crystallized ligands. Employ rigorous docking protocols with high-precision settings to evaluate binding affinities and interaction patterns [37] [5].
- Hit Selection and Validation: Select top-ranking compounds based on docking scores and interaction analyses. Further validate selected hits using molecular dynamics (MD) simulations (100 ns) and MM/PBSA calculations to assess binding stability and free energies [5].
Workflow Visualization
Sequential PBVS-DBVS Workflow
Research Reagent Solutions
Table 2: Essential Research Tools and Resources for Sequential Virtual Screening
| Resource Category | Specific Tools/Software | Primary Function | Application in Workflow |
|---|---|---|---|
| Protein Databases | RCSB Protein Data Bank (PDB) | Source of 3D protein structures | Provides target structures for pharmacophore modeling and docking [5] |
| Chemical Libraries | ChemDiv Database | Collection of synthesizable compounds | Source library for virtual screening [5] |
| Decoy Sets | DUD-E Website | Curated sets of active/inactive compounds | Validation of pharmacophore models [5] |
| Pharmacophore Modeling | Discovery Studio | Generate and validate pharmacophore hypotheses | PBVS phase implementation [5] |
| Molecular Docking | DOCK, GOLD, Glide | Predict ligand binding poses and affinities | DBVS phase implementation [37] |
| Molecular Dynamics | GROMACS, AMBER | Simulate protein-ligand interactions | Validation of binding stability [5] |
| Structure Analysis | PyMOL, Chimera | Visualization of molecular structures | Analysis of binding interactions [19] |
Application Case Study: VEGFR-2 and c-Met Dual Inhibitors
The sequential PBVS→DBVS workflow was successfully applied to identify dual inhibitors targeting VEGFR-2 and c-Met, key receptors in cancer angiogenesis and progression [5]. The implementation yielded 18 initial hit compounds from the virtual screening process, with two compounds (17924 and 4312) demonstrating superior binding free energies in MM/PBSA calculations compared to positive controls. This case study validates the workflow's effectiveness in identifying promising candidates for complex multi-target therapies [5].
Structure-Based Pharmacophore Generation from Protein-Ligand Complexes
Structure-based pharmacophore modeling is an integral component of modern computer-aided drug discovery, serving as a critical bridge between target structural biology and ligand identification. The International Union of Pure and Applied Chemistry (IUPAC) defines a pharmacophore as "the ensemble of steric and electronic features that is necessary to ensure the optimal supra-molecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [19]. In practical terms, pharmacophore models abstract essential chemical interaction patterns from three-dimensional structural data, representing them as geometric entities such as spheres, planes, and vectors corresponding to key molecular features including hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), hydrophobic areas (H), positively and negatively ionizable groups (PI/NI), aromatic groups (AR), and metal coordinating areas [19].
The principal advantage of structure-based pharmacophore modeling lies in its direct utilization of target structural information, which enables the identification of novel chemotypes even when known active ligands are scarce or unavailable. This approach has gained considerable importance with the increasing availability of experimentally determined protein structures in the Protein Data Bank (PDB) and advances in computational structure prediction methods like AlphaFold2 [19] [38]. When integrated with molecular docking and virtual screening workflows, structure-based pharmacophore models significantly enhance the efficiency of lead compound identification across various target classes, including kinases, epigenetic proteins, and G protein-coupled receptors (GPCRs) [39] [13] [38].
This protocol article details established and emerging methodologies for generating pharmacophore models from protein-ligand complexes, framed within the broader context of integrating molecular docking with pharmacophore virtual screening research. We present comprehensive application notes, experimental protocols, and implementation frameworks designed for researchers, scientists, and drug development professionals engaged in structure-based drug discovery campaigns.
Fundamental Principles and Feature Definitions
Core Pharmacophore Features
A pharmacophore model consists of a set of chemical groups with a specific three-dimensional arrangement that are essential for biological activity against a specific molecular target [40]. The functional features present in a pharmacophore model represent the key interactions necessary for molecular recognition and binding:
- Hydrogen bond acceptors (HBA) and donors (HBD): Represent atoms capable of participating in hydrogen bonding interactions, crucial for directional binding to complementary protein residues [40] [19].
- Hydrophobic groups (H): Represent non-polar regions that participate in van der Waals interactions and drive the desolvation process during binding [40] [19].
- Positive and negative ionizable groups (PI/NI): Represent features capable of forming charge-assisted hydrogen bonds or salt bridges with oppositely charged residues in the binding pocket [40] [19].
- Aromatic rings (AR): Enable π-π stacking and cation-π interactions with protein aromatic residues or charged groups [40] [41].
- Metal coordination (MB): Represent atoms capable of coordinating with metal ions present in metalloenzyme active sites [41].
- Exclusion volumes (XVOL): Define forbidden regions that represent steric constraints of the binding pocket, ensuring generated ligands maintain complementarity with the protein surface [19].
Theoretical Foundation
The binding sites of ligands have physicochemical and spatial restrictions that impose limitations to the non-specific interaction of certain molecules. These spatial restrictions dictate the binding mode of the ligands, thus allowing different molecules, even with different structures, to act against a specific bioreceptor due to the presence of the same pharmacophore model [40]. Structure-based pharmacophore generation capitalizes on this principle by extracting the essential interaction patterns directly from protein-ligand complexes, creating a three-dimensional query that can identify novel compounds possessing these critical features regardless of scaffold similarity [19].
Table 1: Core Pharmacophore Features and Their Characteristics
| Feature Type | Chemical Moieties | Interaction Type | Directionality |
|---|---|---|---|
| Hydrogen Bond Acceptor (HBA) | Carbonyl oxygen, nitro groups, ether oxygens | Electrostatic, hydrogen bonding | Yes |
| Hydrogen Bond Donor (HBD) | Amine groups, hydroxyl groups, amide NH | Electrostatic, hydrogen bonding | Yes |
| Hydrophobic (H) | Alkyl chains, aromatic rings, steroid systems | van der Waals, entropic (desolvation) | No |
| Positively Ionizable (PI) | Primary, secondary, tertiary amines | Salt bridge, charge-charge | No |
| Negatively Ionizable (NI) | Carboxylic acids, tetrazoles, phosphates | Salt bridge, charge-charge | No |
| Aromatic (AR) | Phenyl, pyridine, other aromatic rings | π-π stacking, cation-π | Partial |
| Metal Coordination (MB) | Imidazole, carboxylate, specific heterocycles | Coordinate covalent bonding | Yes |
Established Computational Workflows
Structure-Based Pharmacophore Generation Protocol
The generation of structure-based pharmacophores follows a systematic workflow that transforms a protein-ligand complex into an abstract pharmacophore model suitable for virtual screening. The following protocol outlines the key steps in this process:
Step 1: Protein Structure Preparation
- Retrieve the three-dimensional structure of the target protein from the Protein Data Bank (PDB) or generate via homology modeling [19] [38].
- For crystal structures, add hydrogen atoms using appropriate protonation states for all ionizable residues based on local environment and pH [13] [19].
- Optimize hydrogen-bonding networks using tools like PROPKA [13].
- Remove non-essential water molecules and cofactors, except those involved in key binding interactions [13] [42].
- Energy minimization using force fields such as OPLS_2005 to relieve steric clashes and optimize geometry [13].
Step 2: Binding Site Analysis
- Identify the ligand-binding site through analysis of cocrystallized ligand location or using binding site detection algorithms (GRID, LUDI) [19].
- Define the active site region using a 3D grid for subsequent analysis, typically extending 5-10 Å beyond the bounds of any cocrystallized ligand [13].
- Characterize key interacting residues and their properties to inform feature selection.
Step 3: Interaction Analysis and Feature Mapping
- Analyze interactions between the protein and cocrystallized ligand: hydrogen bonds, hydrophobic contacts, ionic interactions, and metal coordination [19].
- Map these interactions to pharmacophore features based on complementarity:
- Protein hydrogen bond donors correspond to ligand hydrogen bond acceptors
- Protein hydrogen bond acceptors correspond to ligand hydrogen bond donors
- Hydrophobic pockets on the protein correspond to hydrophobic features on the ligand
- Charged residues correspond to oppositely charged features on the ligand [19] [38]
- Generate an initial set of pharmacophore features representing all potential interactions in the binding site.
Step 4: Feature Selection and Model Generation
- Select the most critical features for biological activity based on:
- Incorporate exclusion volumes to represent steric constraints of the binding pocket [19].
- Define the spatial tolerances for each feature based on the variability observed in multiple complexes or through molecular dynamics simulations [39] [38].
Step 5: Model Validation
- Validate the pharmacophore model using a set of known active and inactive compounds [38].
- Assess model performance using enrichment factors (EF) and goodness-of-hit (GH) scoring metrics [38] [43].
- Refine the model by adjusting feature definitions and tolerances based on validation results [38].
Diagram 1: Structure-Based Pharmacophore Generation Workflow (6 steps)
Automated Protocols: PharmaCore and MCSS-Based Approaches
Recent advances have enabled more automated approaches to structure-based pharmacophore generation. Two notable implementations include:
PharmaCore Protocol PharmaCore represents a completely automatic workflow for generating 3D structure-based pharmacophore models using cocrystallized ligands as input [39]. The implementation involves:
- Starting with the specific UniProt ID of the protein under investigation as the only user-provided element
- Automated collection and alignment of corresponding structures bearing known ligands
- Generation of pharmacophore hypotheses directly onto the protein structure using software such as Phase (Schrödinger LLC)
- Validation through comparison with published pharmacophores and experimental testing of predicted off-targets [39]
Multiple Copy Simultaneous Search (MCSS) Based Protocol For targets with limited ligand information, MCSS-based approaches generate pharmacophore models by:
- Randomly placing numerous copies of varied functional group fragments into a receptor's active site
- Energetically minimizing each fragment independently to determine optimal positions
- Selecting fragments based on interaction scoring and distance cutoffs
- Annotating pharmacophore features from the optimally positioned fragments [38] [43]
This method employs a "cluster-then-predict" machine learning workflow to identify pharmacophore models likely to yield higher enrichment factors in virtual screening [38]. The approach has been successfully applied to both experimentally determined and modeled GPCR structures, achieving theoretical maximum enrichment values for most test cases [43].
Table 2: Software Tools for Structure-Based Pharmacophore Modeling
| Software Tool | Access Type | Key Features | Applicability |
|---|---|---|---|
| PharmaCore [39] | Automated workflow | Fully automatic generation from UniProt ID | Broad target applicability |
| LigandScout [40] [42] | Commercial | Ligand- and structure-based modeling, virtual screening | Protein-ligand complexes |
| Phase [39] | Commercial | Pharmacophore hypothesis generation, virtual screening | Integrated with Schrödinger suite |
| Pharmit [13] [40] | Free web server | Structure-based pharmacophore screening | Online virtual screening |
| MOE [40] | Commercial | Ligand- and structure-based modeling, QSAR | Comprehensive drug discovery |
| AncPhore [41] | Open-source | Anchor-based pharmacophore modeling, multiple features | Diverse feature types |
| AutoPH4 [38] | Not specified | Automated feature refinement | GPCR applications |
Integration with Molecular Docking and Virtual Screening
Combined Workflow for Virtual Screening
The integration of structure-based pharmacophore modeling with molecular docking creates a powerful multi-tier virtual screening approach that enhances hit rates and computational efficiency. The following protocol outlines this integrated workflow:
Phase 1: Pharmacophore-Based Virtual Screening
- Generate a structure-based pharmacophore model from a protein-ligand complex as described in Section 3.1 [13] [42].
- Prepare a compound library for screening by generating 3D conformations and optimizing geometries [13].
- Perform pharmacophore-based screening using the model as a query to identify compounds that match the essential features [13] [19].
- Apply filtering criteria based on physicochemical properties (Lipinski's Rule of Five) and chemical diversity [13].
Phase 2: Molecular Docking
- Prepare the protein structure for docking by defining the binding site around the pharmacophore reference coordinates [13].
- Generate a docking grid centered on the binding site with appropriate dimensions to accommodate diverse ligands [13].
- Dock the pharmacophore-filtered compounds using standard precision (SP) or high-throughput virtual screening protocols [13] [16].
- Select top-ranking compounds based on docking scores and visual inspection of binding modes [13].
Phase 3: Binding Affinity Prediction and Optimization
- Analyze protein-ligand interactions to verify key pharmacophore features are engaged [13] [42].
- Predict binding affinities and prioritize compounds for further investigation [16].
- Apply additional filters based on drug-likeness and synthetic accessibility [44].
Phase 4: Experimental Validation
- Select top candidates for synthesis or procurement
- Evaluate biological activity through in vitro assays [39] [16]
- Iteratively refine models based on experimental results
Diagram 2: Integrated Pharmacophore-Docking Virtual Screening (6 steps)
Machine Learning-Accelerated Approaches
Machine learning methods have recently been integrated into pharmacophore-based screening to dramatically accelerate the process:
- Docking Score Prediction: Train machine learning models to predict docking scores based on molecular fingerprints and descriptors, bypassing time-consuming molecular docking procedures [16].
- Ensemble Models: Combine multiple types of molecular fingerprints and descriptors to construct ensemble models that reduce prediction errors [16].
- Pharmacophore-Constrained Screening: Apply pharmacophore constraints during screening to focus on chemically relevant space [16].
This approach has demonstrated 1000-fold acceleration in binding energy predictions compared to classical docking-based screening while maintaining high accuracy in identifying active compounds [16].
Advanced Applications and Case Studies
Application in Epigenetic Drug Discovery
Structure-based pharmacophore generation has proven valuable in epigenetic drug discovery, where selectivity among closely related protein families is crucial:
Case Study: ATAD2 Bromodomain Selectivity
- Generated 1741 pharmacophores related to 16 epigenetic proteins using PharmaCore
- Identified selective binder AM879 for ATAD2 bromodomain
- Discovered putative off-targets through computational prediction
- Experimentally validated predictions using AlphaScreen assays [39]
This approach demonstrates how structure-based pharmacophore models can predict selectivity profiles and identify potential off-target interactions early in the drug discovery process.
Marine Natural Product Screening
The integration of structure-based and ligand-based pharmacophore models has enabled efficient screening of natural product libraries:
Case Study: Marine Aromatase Inhibitors
- Developed merged pharmacophore model combining structure-based and ligand-based approaches
- Screened Comprehensive Marine Natural Products Database (CMNPD) containing >31,000 compounds
- Identified 1,385 potential candidates through pharmacophore screening
- After molecular docking analysis, selected 4 compounds with strong binding affinity
- Confirmed stability through molecular dynamics simulations, identifying CMPND 27987 as the most promising candidate with MM-GBSA binding energy of -27.75 kcal/mol [42]
This case study highlights the utility of pharmacophore models in navigating complex chemical spaces such as natural product libraries to identify novel scaffolds.
Emerging AI and Deep Learning Approaches
Knowledge-Guided Diffusion Models
Recent advances in artificial intelligence have introduced novel approaches to pharmacophore modeling and ligand generation:
DiffPhore Framework DiffPhore represents a knowledge-guided diffusion framework for "on-the-fly" 3D ligand-pharmacophore mapping that incorporates several innovations:
- Utilizes ligand-pharmacophore matching knowledge to guide conformation generation
- Employs calibrated sampling to mitigate exposure bias in iterative conformation search
- Trains on comprehensive datasets of 3D ligand-pharmacophore pairs (CpxPhoreSet and LigPhoreSet)
- Incorporates 10 pharmacophore feature types plus exclusion spheres
- Achieves state-of-the-art performance in predicting ligand binding conformations, surpassing traditional tools and several advanced docking methods [41]
The framework consists of three main modules:
- Knowledge-guided ligand-pharmacophore mapping encoder
- Diffusion-based conformation generator
- Calibrated conformation sampler [41]
CMD-GEN Framework CMD-GEN (Coarse-grained and Multi-dimensional Data-driven molecular generation) represents another AI-driven approach:
- Bridges ligand-protein complexes with drug-like molecules using coarse-grained pharmacophore points
- Decomposes 3D molecule generation into pharmacophore point sampling, chemical structure generation, and conformation alignment
- Effectively controls drug-likeness properties during generation
- Demonstrates particular strength in selective inhibitor design [44]
Performance Comparison
These AI-based approaches have demonstrated significant advantages over traditional methods:
- DiffPhore shows superior virtual screening power for both lead discovery and target fishing [41]
- The framework successfully identified structurally distinct inhibitors for human glutaminyl cyclases, with binding modes validated through co-crystallographic analysis [41]
- CMD-GEN outperforms other methods in benchmark tests and effectively controls molecular properties during generation [44]
- In wet-lab validation with PARP1/2 inhibitors, CMD-GEN confirmed its potential in selective inhibitor design [44]
Table 3: Essential Research Reagents and Computational Tools for Structure-Based Pharmacophore Modeling
| Category | Specific Tools/Resources | Key Function | Access Information |
|---|---|---|---|
| Protein Structure Sources | Protein Data Bank (PDB) [13] [19] | Source of experimental protein-ligand structures | https://www.rcsb.org |
| AlphaFold2 Database [19] | Source of predicted protein structures | https://alphafold.ebi.ac.uk | |
| Software Tools | Schrödinger Suite [39] [13] | Comprehensive drug discovery platform | Commercial |
| AutoDock Vina [13] [42] | Molecular docking software | Open source | |
| LigandScout [40] [42] | Pharmacophore modeling and virtual screening | Commercial | |
| Pharmit [13] [40] | Online pharmacophore screening | http://pharmit.csb.pitt.edu | |
| Compound Libraries | ZINC Database [13] [16] | Commercially available compounds for virtual screening | https://zinc.docking.org |
| ChEMBL [16] | Bioactivity database for model validation | https://www.ebi.ac.uk/chembl | |
| CMNPD [42] | Marine natural products database | https://www.cmnpd.com | |
| Computational Resources | Desmond [13] | Molecular dynamics simulation | Commercial |
| QikProp [13] | ADMET property prediction | Commercial | |
| Python libraries | Custom workflow development | Open source |
Troubleshooting and Protocol Optimization
Common Challenges and Solutions
Challenge: Overabundance of Pharmacophore Features
- Solution: Implement feature selection strategies based on interaction energy conservation or machine learning classification [38]
- Solution: Apply progressive variable selection to identify optimal feature combinations [38]
Challenge: Handling Protein Flexibility
- Solution: Incorporate multiple protein conformations from molecular dynamics simulations [41]
- Solution: Use ensemble pharmacophore models representing different binding site states [38]
Challenge: Model Selection for Targets Without Known Ligands
- Solution: Implement cluster-then-predict machine learning workflow to identify high-performing models [38]
- Solution: Use enrichment-based metrics on related targets with known actives [43]
Challenge: Balancing Model Specificity and Generality
- Solution: Adjust feature tolerances based on validation results [38]
- Solution: Implement tunable exclusion volumes to represent binding site constraints [19]
Validation Metrics and Quality Assessment
Rigorous validation is essential for ensuring pharmacophore model quality:
- Enrichment Factor (EF): Measures how many fold better a pharmacophore model is at selecting active compounds compared to random selection [38] [43]
- Goodness-of-Hit (GH) Score: Evaluates the model's ability to prioritize high yield of actives with low false-negative rate [38] [43]
- ROC Analysis: Assesses model selectivity and specificity using decoy datasets [42]
- Experimental Validation: Essential for confirming model predictions through biological testing [39] [16]
Structure-based pharmacophore generation from protein-ligand complexes represents a powerful methodology in modern drug discovery, particularly when integrated with molecular docking and virtual screening workflows. The protocols outlined in this article provide researchers with comprehensive guidance for implementing these approaches, from established computational methods to emerging AI-driven frameworks.
The continued evolution of structure-based pharmacophore modeling—driven by advances in structural biology, machine learning, and computational infrastructure—promises to further enhance its utility in addressing challenging drug discovery problems, including selective inhibitor design and polypharmacology prediction. As these methods become more automated and integrated with experimental validation, they will play an increasingly central role in accelerating the identification and optimization of novel therapeutic agents.
Ligand-Based Pharmacophore Modeling for Targets with Unknown 3D Structures
In modern drug discovery, the identification of novel therapeutic agents is often hampered by a lack of three-dimensional structural information for many biologically relevant targets. Ligand-based pharmacophore modeling has emerged as a powerful computational strategy to address this challenge, enabling researchers to identify essential chemical features responsible for biological activity directly from known active compounds [45]. This approach is particularly valuable for targets where obtaining experimental 3D structures through methods like X-ray crystallography or cryo-electron microscopy is difficult or impossible, such as for certain membrane receptors or protein complexes [45].
According to the IUPAC definition, a pharmacophore model represents "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response" [45]. When applied within a comprehensive drug discovery pipeline, ligand-based pharmacophore modeling serves as a critical filtering step that can be seamlessly integrated with molecular docking studies to identify promising therapeutic candidates [5] [46]. This protocol details the methodology for developing and applying ligand-based pharmacophore models, with particular emphasis on their role in a virtual screening workflow that subsequently incorporates structure-based docking validation.
Theoretical Framework and Key Concepts
Pharmacophore Principles and Feature Definitions
A pharmacophore model abstracts the key molecular interactions between a ligand and its biological target into a set of chemical features and their spatial relationships. These features typically include:
- Hydrogen bond donors (HBD) and acceptors (HBA): Represent the capacity for forming directional hydrogen bonds with the target.
- Hydrophobic groups: Represent regions of the molecule that participate in van der Waals interactions with hydrophobic binding pockets.
- Aromatic rings: Can engage in π-π stacking or cation-π interactions with the target.
- Positive and negative ionizable groups: Represent the potential for forming electrostatic interactions or salt bridges [45] [47].
The spatial arrangement of these features is captured as a three-dimensional network of constraints that defines the essential interaction pattern required for biological activity, without being restricted to a specific molecular scaffold [45].
Advantages for Targets with Unknown 3D Structures
For targets with unknown three-dimensional structure, ligand-based pharmacophore modeling offers several distinct advantages:
- It requires only a set of known active compounds, without needing structural information about the target protein.
- It can capture the essential interaction features even when ligands bind through different binding modes.
- It enables virtual screening of large compound databases to identify novel chemotypes with potential activity.
- It provides a rational framework for lead optimization by highlighting critical interaction features that must be preserved in analog design [45].
Experimental Protocol
Compound Data Set Preparation
The first critical step involves assembling a comprehensive set of known active compounds with associated biological activity data, preferably measured in a consistent assay system.
Step 1: Data Collection and Curation
- Gather structurally diverse compounds with known activity against the target of interest.
- Ensure biological activity values (IC₅₀, Ki, or EC₅₀) are obtained from homogeneous experimental procedures against a single biological system [48].
- Include inactive compounds or decoy molecules to enhance model selectivity during validation phases [5].
Step 2: Activity Categorization Classify compounds based on their biological activity:
- Most active: IC₅₀ < 0.1 μM
- Active: IC₅₀ = 0.1-1.0 μM
- Moderately active: IC₅₀ = 1.0-10.0 μM
- Inactive: IC₅₀ > 10.0 μM [48]
Step 3: Training and Test Set Division
- For strategy I (assuming a common binding mode): Select cluster centroids from separate clustering of active and inactive compounds using Butina clustering with 2D pharmacophore fingerprints [47].
- For strategy II (accommodating multiple binding modes): Jointly cluster active and inactive compounds, then randomly select 5 active and 5 inactive compounds from each cluster to form multiple training sets [47].
- Reserve compounds not included in training sets for external validation.
Step 4: Molecular Conformation Generation
- Enumerate stereoisomers for compounds with undefined chiral centers or double bond configurations.
- Generate up to 100 conformers per compound within a 50 kcal/mol energy window using the MMFF94 force field to ensure adequate coverage of conformational space [47].
- Minimize conformer energies using a combination of steepest descent and conjugate gradient algorithms (2000 steps each) [48].
Pharmacophore Model Generation
The core process of developing the pharmacophore model involves iterative hypothesis generation and evaluation.
Step 1: Pharmacophore Feature Mapping
- For each compound in the training set, identify potential pharmacophore features using software tools such as Discovery Studio [48] [5].
- Calculate 3D pharmacophore hashes for all possible 4-point pharmacophores across all conformers of training set compounds [47].
Step 2: Feature Occurrence Analysis
- Calculate the frequency of each pharmacophore hash among active versus inactive compounds in the training set.
- Compute statistical metrics including recall, precision, and F-scores to identify pharmacophores that preferentially occur in active compounds [47].
Step 3: Model Selection and Refinement
- For Strategy I training sets: Select models with F₀.₅ score ≥ 0.8 to prioritize precision [47].
- For Strategy II training sets: Select models with F₂ score = 1.0, or recall = 1.0 if no models meet the F₂ criterion [47].
- Iteratively expand selected pharmacophores to 5-point and higher models by adding additional features.
- Remove overly simplistic models with fewer than four distinct spatial coordinates to avoid promiscuous models [47].
Step 4: Model Validation
- Evaluate selected models against the external test set to assess predictive performance.
- Calculate enrichment factors (EF) and area under the receiver operating characteristic curve (AUC) to quantify model quality [5].
- A robust model should demonstrate EF > 2 and AUC > 0.7 to be considered reliable for virtual screening [5].
Virtual Screening Protocol
Once validated, the pharmacophore model serves as a 3D query for screening compound databases.
Step 1: Database Preparation
- Screen large drug-like compound databases (e.g., ZINC, containing >1 million structures) [48].
- Apply Lipinski's Rule of Five and Veber's rules to filter for drug-like properties [48] [5].
- Perform SMART filtration to remove compounds with undesirable functional groups or structural alerts [48].
Step 2: Multi-Stage Pharmacophore Screening
- Fingerprint screening: Use hashed pharmacophore fingerprints as a Bloom filter to rapidly eliminate non-matching compounds [47].
- Subgraph isomorphism checking: Apply the VF2 algorithm to verify that the pharmacophore model topology is present in candidate molecules [47].
- 3D hash comparison: Confirm identical topology and stereoconfiguration between query pharmacophore and candidate molecule pharmacophores [47].
Step 3: Activity Prediction and Hit Selection
- Apply activity filters to prioritize compounds with predicted IC₅₀ values < 1.0 μM [48].
- Select top-ranking compounds for subsequent molecular docking studies.
Integration with Molecular Docking
The pharmacophore-based hit list serves as input for structure-based validation through molecular docking.
Step 1: Protein Structure Preparation
- If a homologous structure is available, prepare the protein by removing water molecules, adding hydrogen atoms, and correcting missing residues [5].
- For completely unknown structures, consider using AlphaFold-predicted structures as docking targets [49].
Step 2: Consensus Docking and Visual Inspection
- Perform molecular docking of pharmacophore-derived hits against the target structure.
- Analyze binding interactions, focusing on compounds that form key interactions similar to those in the pharmacophore model [48] [46].
- Select compounds with favorable docking scores and interaction patterns for further evaluation.
Step 3: Molecular Dynamics Validation
- Conduct molecular dynamics simulations (100 ns) to assess binding stability [48] [50].
- Analyze root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) trajectories to confirm complex stability [46].
- Calculate binding free energies using MM/PBSA approaches to quantify interaction strength [5].
Step 4: Experimental Validation
- Select top candidates for synthesis or procurement and experimental activity testing.
- Perform ADMET profiling to assess drug-like properties [48] [50].
Workflow Visualization
Diagram 1: Ligand-based pharmacophore modeling workflow. The process begins with data preparation, progresses through model development and virtual screening, and concludes with validation stages that integrate molecular docking and experimental testing.
Application Notes
Key Performance Metrics for Model Validation
Table 1: Key metrics for pharmacophore model validation
| Metric | Calculation Formula | Acceptance Criteria | Interpretation |
|---|---|---|---|
| Enrichment Factor (EF) | ( EF = \frac{Ha \times D}{Ht \times A} ) | > 2 [5] | Measures the model's ability to preferentially select active compounds over random screening. |
| Area Under Curve (AUC) | Area under ROC curve | > 0.7 [5] | Overall measure of model discrimination power between active and inactive compounds. |
| Recall (True Positive Rate) | ( Recall = \frac{TP}{P} ) | Strategy I: Context dependentStrategy II: = 1.0 [47] | Proportion of actual active compounds correctly identified by the model. |
| Precision | ( Precision = \frac{TP}{TP + FP} ) | Strategy I: High precision focusStrategy II: Balanced approach [47] | Proportion of model-identified hits that are truly active compounds. |
| F-score | ( F_\beta = \frac{(1+\beta^2) \cdot Precision \cdot Recall}{\beta^2 \cdot Precision + Recall} ) | F₀.₅ ≥ 0.8 (Strategy I)F₂ = 1.0 (Strategy II) [47] | Balanced measure combining precision and recall, with β determining their relative weighting. |
Research Reagent Solutions
Table 2: Essential computational tools and resources for ligand-based pharmacophore modeling
| Resource Category | Specific Tools/Software | Key Function | Application Notes |
|---|---|---|---|
| Pharmacophore Modeling | Discovery Studio [48] [5], MOE | Model generation, validation, and screening | Discovery Studio provides HypoGen algorithm for 3D QSAR pharmacophore generation [48] |
| Compound Databases | ZINC [48], ChEMBL [51], SuperNatural 3.0 [46] | Sources of compounds for virtual screening | ZINC contains over 1 million drug-like molecules suitable for virtual screening [48] |
| Conformational Analysis | RDKit [51] [47], CONFLEX | Generation of representative molecular conformations | RDKit can generate up to 100 conformers per compound within 50 kcal/mol energy window [47] |
| Molecular Docking | AutoDock, GOLD, MOE-Dock | Structure-based validation of pharmacophore hits | Used after pharmacophore screening to validate binding mode and affinity [48] [46] |
| Dynamics Simulation | AMBER [52], GROMACS, CHARMM [48] | Assessment of binding stability and interactions | 100 ns simulations recommended for stability assessment [48] [50] |
| Cheminformatics | RDKit [51], OpenBabel, KNIME | Molecular format conversion, descriptor calculation | RDKit provides pharmacophore fingerprint calculation for clustering [47] |
Advanced Applications and Integration Strategies
Integration with AI-Based Approaches Recent advances in artificial intelligence have created opportunities to enhance traditional pharmacophore modeling. The Pharmacophore-Guided deep learning approach for bioactive Molecule Generation (PGMG) uses pharmacophore hypotheses as input to deep learning models for de novo molecular design [51]. This approach is particularly valuable when working with novel target families or understudied targets where known active compounds are limited. PGMG employs a graph neural network to encode spatially distributed chemical features and a transformer decoder to generate molecules that match the given pharmacophore [51].
Multi-Target Pharmacophore Modeling For complex diseases where multi-target therapies are advantageous, pharmacophore models can be developed to identify compounds with activity against multiple relevant targets. As demonstrated in the identification of VEGFR-2 and c-Met dual inhibitors, separate pharmacophore models for each target can be used in parallel to screen for compounds that satisfy both pharmacophores [5]. This approach enables the identification of single chemical entities with polypharmacology profiles that may offer enhanced therapeutic efficacy.
Handling Challenging Targets For targets with multiple binding modes or significant conformational flexibility, strategy II (multiple training sets) provides a robust framework for capturing diverse interaction patterns [47]. This approach involves creating multiple training sets through joint clustering of active and inactive compounds, then developing separate pharmacophore models for each training set. The resulting model ensemble can better represent the diverse binding solutions that may exist for flexible targets.
Troubleshooting Guide
Table 3: Common challenges and solutions in ligand-based pharmacophore modeling
| Challenge | Potential Causes | Solutions |
|---|---|---|
| Poor model selectivity | Training set contains structurally similar compounds with different activities | Include more diverse inactive compounds in training set; apply stricter feature distinctness criteria [47] |
| Low hit rate in virtual screening | Overly restrictive pharmacophore features; inadequate conformational sampling | Relax distance tolerances; increase number of conformers per compound; use extended energy window (50 kcal/mol) [47] |
| High false positive rate | Promiscuous pharmacophore features; inadequate model validation | Implement multi-stage screening with fingerprint pre-filtering; use more stringent F-score criteria for model selection [47] |
| Inconsistency with docking results | Different binding modes not captured by pharmacophore; protein flexibility | Generate multiple pharmacophore models representing different binding modes; use ensemble docking approaches [5] |
| Limited structural diversity in hits | Training set contains structurally similar compounds only | Apply strategy II with multiple training sets; incorporate weak actives and marginally inactive compounds to define feature boundaries [47] |
Ligand-based pharmacophore modeling represents a powerful methodology for drug discovery targets lacking three-dimensional structural information. When properly validated and integrated with molecular docking and dynamics simulations, this approach provides a robust framework for identifying novel bioactive compounds through virtual screening. The protocol outlined herein emphasizes rigorous model validation, strategic compound selection, and seamless integration with structure-based methods to maximize the likelihood of identifying genuine hit compounds.
As computational methods continue to evolve, the integration of artificial intelligence with traditional pharmacophore approaches presents promising opportunities for enhancing the efficiency and effectiveness of this strategy. The PGMG framework demonstrates how pharmacophore guidance can direct AI-based molecular generation to explore relevant chemical space more efficiently [51]. These advances, coupled with the foundational principles detailed in this protocol, will continue to expand the utility of ligand-based pharmacophore modeling in addressing challenging drug discovery targets.
Post-Docking Pharmacophore Filtering to Improve Hit Rates and Enrichment
Virtual screening is an established computational method for identifying potential lead compounds from large chemical databases. Among the various virtual screening approaches, molecular docking represents a structure-based technique that predicts how small molecules bind to a protein target and scores these interactions. However, a significant limitation of docking-based virtual screening (DBVS) is the high rate of false positives—compounds that score well in silico but demonstrate no actual biological activity [53]. This shortcoming primarily stems from the simplified scoring functions used in docking programs, which often fail to accurately represent complex biochemical interactions [53].
Post-docking pharmacophore filtering has emerged as a powerful strategy to address these limitations. This hybrid approach integrates the complementary strengths of structure-based docking and pharmacophore-based screening to improve the selection of true active compounds. By applying a pharmacophore filter to docking results, researchers can enforce essential chemical complementarity requirements that simple scoring functions may overlook [53]. This methodology has demonstrated superior performance over traditional docking with scoring alone, significantly enhancing hit rates and enrichment factors in virtual screening campaigns [53] [54].
Conceptual Foundation and Key Principles
The Pharmacophore Concept
A pharmacophore is defined by the International Union of Pure and Applied Chemistry (IUPAC) as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [55] [56]. Crucially, a pharmacophore represents an abstract concept of molecular interactions rather than specific chemical groups or scaffolds [56]. Common pharmacophore features include hydrogen bond donors and acceptors, positive and negative ionizable groups, hydrophobic regions, and aromatic rings [57].
Theoretical Basis for Hybrid Screening
The theoretical foundation for combining docking with pharmacophore filtering lies in addressing the complementary weaknesses of each method. Docking programs excel at pose generation by sampling possible ligand conformations within a binding pocket, but their scoring functions often poorly rank true actives [53]. Pharmacophore models provide chemical interaction constraints that directly reflect key binding determinants but may lack detailed steric consideration [55].
Post-docking pharmacophore filtering leverages the docking program's ability to generate biologically relevant poses while using the pharmacophore model to evaluate the chemical completeness of the binding interaction [53]. This approach applies the fundamental principle of structure-based drug design: that effective ligands must be chemically complementary to their receptors, forming essential interactions such as hydrogen bonds and filling critical hydrophobic pockets [53].
Comparative Performance Analysis
Quantitative Benchmarking Studies
A comprehensive benchmark study comparing pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) across eight diverse protein targets demonstrated the superior performance of pharmacophore approaches [54]. The study utilized two testing databases containing both active compounds and decoys, with enrichment factors serving as the primary metric.
Table 1: Performance Comparison of Virtual Screening Methods Across Multiple Targets [54]
| Target Protein | PBVS Enrichment | DBVS Enrichment | Performance Difference |
|---|---|---|---|
| ACE | High | Moderate | PBVS Superior |
| AChE | High | Moderate | PBVS Superior |
| AR | High | Moderate | PBVS Superior |
| DacA | High | Moderate | PBVS Superior |
| DHFR | High | Moderate | PBVS Superior |
| ERα | High | Moderate | PBVS Superior |
| HIV-pr | High | Moderate | PBVS Superior |
| TK | High | Moderate | PBVS Superior |
The study revealed that in 14 of 16 virtual screening scenarios, PBVS achieved higher enrichment factors than DBVS [54]. The average hit rates across all eight targets at both 2% and 5% of the highest database ranks were substantially higher for PBVS than for DBVS methods [54].
Case Study Applications
Neuraminidase A Screening
In a practical application targeting influenza neuraminidase A, researchers developed a pharmacophore filtering method to reduce false positives in virtual screening [53]. The methodology began with docking using either GOLD or Glide to generate ligand poses, disregarding the docking scores entirely [53]. The researchers then applied structure-based pharmacophore models to filter the poses, requiring compounds to fulfill essential hydrogen bonding interactions with key residues including Glu, Arg, and backbone carbonyl groups [53].
This approach demonstrated improved performance over traditional docking with scoring alone by specifically addressing the chemical complementarity requirements of the binding site [53]. The method proved particularly effective for the small, deep, and highly polar sialic acid binding site of neuraminidase, where specific directional interactions are critical for binding [53].
JAK2 Inhibitor Discovery
An integrated virtual screening protocol combining pharmacophore mapping and molecular docking successfully identified novel JAK2 inhibitors from a commercial compound database [58]. After initial screening, twelve structurally diverse hits were selected for experimental testing, with three compounds (A5, A6, and A9) demonstrating remarkable JAK2 inhibitory activity [58]. Subsequent similarity search based on these active compounds yielded two additional promising inhibitors (B2 and B4) [58].
The most promising compound, B2, exhibited a favorable selectivity profile against JAK subtypes, novel structural骨架, and significant anti-proliferative effects against cancer cells [58]. This case study exemplifies the practical utility of combined pharmacophore and docking approaches in identifying novel bioactive compounds with therapeutic potential.
Table 2: Representative Virtual Screening Applications Using Integrated Docking and Pharmacophore Approaches
| Target | Screening Database | Hit Rate | Key Findings | Reference |
|---|---|---|---|---|
| JAK2 | Commercial (ChemDiv) | 25% (3/12) | Identified selective JAK2 inhibitor with anti-cancer activity | [58] |
| VEGFR-2/c-Met | Commercial (ChemDiv) | 11% (2/18) | Discovered dual-target inhibitors with promising binding energies | [5] |
| MmpL3 | Asinex & DrugBank | N/A | Identified lead compound with better binding affinity than standard drug SQ109 | [59] |
Experimental Protocols and Methodologies
Structure-Based Pharmacophore Modeling Protocol
Structure-based pharmacophore generation utilizes three-dimensional structural information from protein-ligand complexes to identify essential interaction features [57]. The following protocol outlines the key steps:
Protein Preparation
- Obtain crystal structure from Protein Data Bank (PDB)
- Remove water molecules and extraneous ligands
- Add hydrogen atoms and correct protonation states
- Fill missing amino acid residues and loops
- Perform energy minimization using appropriate force fields (e.g., CHARMM) [5]
Binding Site Analysis
- Define binding site based on co-crystallized ligand location
- Identify key residues involved in molecular recognition
- Analyze interaction patterns (hydrogen bonds, hydrophobic contacts, ionic interactions)
Pharmacophore Feature Generation
- Use receptor-ligand pharmacophore generation module in software such as Discovery Studio or LigandScout
- Set maximum number of pharmacophores to 10
- Define feature constraints (typically 4-6 features)
- Include standard features: hydrogen bond acceptor/donor, positive/negative ionizable, hydrophobic, aromatic ring [5]
Post-Docking Pharmacophore Filtering Workflow
The specific methodology for post-docking pharmacophore filtering involves sequential application of docking and pharmacophore screening [53]:
Docking Phase
Pharmacophore Filtering Phase
- Import saved docking poses into pharmacophore software (e.g., MOE, LigandScout)
- Define pharmacophore queries based on essential binding interactions
- Screen docking poses against pharmacophore models
- Eliminate poses that do not fulfill critical pharmacophore features
Hit Selection
- Visually inspect matched compounds for binding compatibility
- Select diverse chemotypes for experimental validation
- Prioritize compounds with favorable ADMET properties
Model Validation Techniques
Proper validation of pharmacophore models is essential before application in virtual screening [57]:
Decoy Set Preparation
- Use the Directory of Useful Decoys, Enhanced (DUD-E) for optimized decoy generation
- Maintain a ratio of approximately 1:50 active molecules to decoys
- Match decoys with active compounds based on 1D properties (molecular weight, logP, hydrogen bond donors/acceptors, rotatable bonds) but different topologies [57]
Validation Metrics
- Calculate Enrichment Factor (EF): EF = (Ha × D) / (Ht × A), where Ha is active compounds identified as hits, D is total decoys, Ht is total active compounds, and A is total hits [5]
- Compute Area Under the Curve (AUC) of Receiver Operating Characteristic (ROC) plot
- Determine specificity (ability to exclude inactive compounds) and sensitivity (ability to identify active molecules)
Validation Standards
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Essential Software Tools for Post-Docking Pharmacophore Filtering
| Software Tool | Primary Function | Key Features | Application in Workflow |
|---|---|---|---|
| GOLD [53] [60] | Molecular Docking | Genetic algorithm for flexible docking, multiple scoring functions | Pose generation without regard to scoring |
| Glide [53] | Molecular Docking | Hierarchical filtering, precision docking, SP/XP modes | Pose generation for diverse binding modes |
| MOE [53] | Pharmacophore Modeling | Structure-based pharmacophore generation, visual inspection capabilities | Pharmacophore model creation and filtering |
| LigandScout [57] [60] | Pharmacophore Modeling | Automated structure-based pharmacophore generation, exclusion volumes | Rapid screening of docking poses against pharmacophores |
| Discovery Studio [5] | Pharmacophore Modeling | Receptor-ligand pharmacophore generation, model validation tools | Model creation, screening, and enrichment calculation |
| DUD-E [57] | Validation | Optimized decoy set generation | Model validation and benchmarking |
Implementation Considerations and Best Practices
Pharmacophore Feature Definition
When creating structure-based pharmacophore models, several factors influence model quality and screening success:
- Site Point Definition: Pharmacophore site points can be defined based on ligand atoms (ligand-sided), receptor atoms (protein-sided), or both, depending on the specific interaction being captured [53]
- Tolerance Radius Adjustment: Smaller radii create tighter, more selective filters, while larger radii accommodate ligand flexibility and binding mode variability [53]
- Exclusion Volumes: Incorporate exclusion volumes to represent receptor boundaries and prevent steric clashes [57]
- Feature Weighting: Assign higher weights to critical interactions known from structural biology or mutagenesis studies
Handling Complex Binding Sites
For binding sites with multiple subpockets or alternative binding modes:
- Develop multiple pharmacophore hypotheses to represent different binding modalities
- Use ligand-based pharmacophore generation when multiple active ligands are available [57]
- Consider consensus approaches that combine results from multiple pharmacophore models
- Implement partial matching for complex queries where not all features are equally essential
Addressing Specificity and Sensitivity
Optimizing the balance between specificity and sensitivity requires careful consideration:
- Start with restrictive models (all features mandatory) and gradually relax constraints if insufficient hits are identified
- Use orthogonal filtering with ADMET properties to eliminate compounds with unfavorable pharmacokinetic profiles [5]
- Implement machine learning approaches to refine feature definitions and tolerance settings based on known actives and inactives [57]
Post-docking pharmacophore filtering represents a powerful methodology that integrates the complementary strengths of molecular docking and pharmacophore-based virtual screening. By leveraging docking for pose generation and pharmacophore models for interaction-based filtering, this hybrid approach significantly improves hit rates and enrichment factors compared to traditional docking with scoring alone.
The documented success across diverse protein targets including neuraminidase A, JAK2, VEGFR-2, c-Met, and MmpL3 demonstrates the broad applicability of this method [53] [59] [5]. Quantitative benchmarking reveals that pharmacophore-based approaches consistently outperform docking-based methods in enrichment factors, particularly in the critical early enrichment phase of virtual screening [54].
As virtual screening continues to evolve, post-docking pharmacophore filtering provides a robust framework for improving the efficiency of lead identification in drug discovery. The method addresses fundamental limitations of docking scoring functions while maintaining the structural insights provided by protein-ligand complex structures. With careful implementation and validation, this integrated approach offers significant advantages for researchers seeking to maximize the value of virtual screening campaigns.
The synergistic interplay between Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2) and the mesenchymal-epithelial transition factor (c-Met) represents a critical therapeutic target in oncology. These receptors collaboratively contribute to tumor angiogenesis and progression, with their co-expression in numerous cancers correlating with poor prognosis and therapeutic resistance [5] [62]. While single-target inhibitors often face limitations due to adaptive resistance mechanisms, dual-target inhibitors against VEGFR-2 and c-Met offer a promising strategy for broader therapeutic efficacy across various malignancies [63]. This case study details a successful computational approach for identifying novel VEGFR-2/c-Met dual inhibitors, providing a framework for integrating advanced virtual screening methodologies in drug discovery.
Biological Rationale for Dual Targeting
VEGFR-2 serves as the primary mediator of VEGF-induced endothelial cell proliferation, migration, and survival, fundamentally driving tumor angiogenesis [5] [64]. Under pathological conditions, VEGFR-2 overexpression activates the Raf-1/MAPK/ERK signaling pathway, enhancing vascular permeability and facilitating tumor invasion and metastasis [5]. Simultaneously, c-Met, upon activation by its ligand hepatocyte growth factor (HGF), initiates signaling cascades that promote cell proliferation, invasion, and avoidance of apoptosis [5] [62]. The complementary roles of these pathways in tumor biology create a powerful synergy that drives aggressive cancer phenotypes, making concurrent inhibition a strategically advantageous approach [65] [63].
Table: Clinically Developed VEGFR-2/c-Met Dual Inhibitors
| Inhibitor Name | Development Status | Key Characteristics |
|---|---|---|
| Cabozantinib | FDA-approved (2012) | First c-Met/VEGFR-2 dual-target inhibitor for metastatic medullary thyroid cancer [5] |
| Foretinib | Clinical trials | Investigational inhibitor demonstrating broad-spectrum activity [5] |
| Golvatinib | Clinical trials | Dual c-Met and VEGFR-2 inhibitor (IC50: 14 nM and 16 nM) [66] |
| BMS-794833 | Clinical trials | Potent ATP-competitive inhibitor of Met/VEGFR2 (IC50: 1.7 nM/15 nM) [66] |
| Dovitinib | Clinical trials | Multi-targeted inhibitor with activity against VEGFR-2 and c-Met [5] |
Integrated Computational Methodology
The identification of novel VEGFR-2/c-Met dual inhibitors employed a comprehensive virtual screening pipeline that integrated multiple computational techniques to efficiently navigate chemical space and prioritize promising candidates [5] [62].
Protein and Ligand Preparation
The computational workflow commenced with careful preparation of target structures and compound libraries:
- Protein Structures: 18 co-crystal structures of VEGFR-2 and 47 co-crystal structures of c-Met were retrieved from the RCSB Protein Data Bank. Following rigorous selection based on resolution (<2 Å), biological activity (nM level), and structural diversity, 10 VEGFR-2 complexes and 8 c-Met complexes were ultimately selected for pharmacophore model development [5] [62].
- Compound Library: Over 1.28 million compounds were sourced from the commercial ChemDiv database (Topscience, Shanghai, China) for screening [5].
- Validation Sets: For method validation, a VEGFR-2 validation set containing 25 active inhibitors (IC50 < 0.01 μM) and 375 inactive compounds from DUD-E website was constructed. Similarly, a c-Met validation set with 25 active inhibitors and 400 inactive compounds was assembled [5] [62].
All protein structures underwent preparation using Discovery Studio 2019, including removal of water molecules, completion of missing amino acid residues, bond connectivity correction, and energy minimization using the CHARMM force field [5].
Pharmacophore Model Development and Validation
Structure-based pharmacophore modeling served as the foundational filter in the virtual screening cascade:
- Model Generation: Pharmacophores were constructed using the Receptor-Ligand Pharmacophore Generation module in Discovery Studio 2019. Six standard chemical features were considered: hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), positive ionizable center, negative ionizable center, hydrophobic center, and ring aromatic center [5] [62].
- Model Validation: The quality of generated pharmacophore models was rigorously assessed using enrichment factor (EF) and area under the ROC curve (AUC). Models were considered reliable if they demonstrated AUC > 0.7 and EF value > 2 [5] [62].
The validated pharmacophore models captured essential interaction features required for effective binding to both VEGFR-2 and c-Met, providing a powerful 3D query for initial database screening.
Diagram 1: Integrated Virtual Screening Workflow for VEGFR-2/c-Met Dual Inhibitors
Drug-Likeness and ADMET Screening
Compounds surviving the pharmacophore filter underwent successive property-based screening:
- Lipinski and Veber Rules: The compound library was filtered using standard drug-likeness rules to eliminate compounds with unfavorable physicochemical properties [5].
- ADMET Profiling: Advanced ADMET (absorption, distribution, metabolism, excretion, and toxicity) descriptors were applied to predict key pharmacological properties including aqueous solubility, blood-brain barrier penetration, cytochrome P4502D6 inhibition, hepatotoxicity, and plasma protein binding [5] [62]. Specific cut-off values were established for solubility (level 3), absorption (level 0), and BBB penetration (level 3) [62].
This multi-tiered filtering approach ensured that only compounds with desirable drug-like properties and pharmacokinetic profiles advanced to more computationally intensive docking studies.
Molecular Docking and Binding Mode Analysis
Structure-based virtual screening through molecular docking provided the next level of selectivity:
- Docking Methodology: Molecular docking was performed to evaluate binding affinities and interaction modes of filtered compounds with both VEGFR-2 and c-Met targets [5] [62].
- Binding Site Characterization: For VEGFR-2, particular attention was paid to compounds capable of interacting with the DFG motif and occupying the hydrophobic region II (allosteric site), characteristics associated with type II inhibitors that often demonstrate improved selectivity profiles [67].
- Hit Selection: Compounds demonstrating superior binding affinities to both targets were prioritized for further investigation, resulting in the identification of 18 initial hit compounds with potential dual inhibitory activity [5].
Molecular Dynamics Simulations and Binding Free Energy Calculations
The final validation stage employed all-atom molecular dynamics (MD) simulations:
- Simulation Parameters: Two top candidates (compound17924 and compound4312) were subjected to 100 ns MD simulations using the AMBER20 software package with the ff14SB force field for proteins and GAFF for small molecules [5] [67].
- Stability Assessment: Trajectories were analyzed for root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and other stability metrics to evaluate the dynamic behavior of protein-ligand complexes [5].
- Energetic Analysis: The MM/PBSA method was employed to calculate binding free energies, with compound17924 and compound4312 demonstrating superior binding free energies compared to positive control ligands [5] [62].
Key Research Findings
Identified Hit Compounds
The integrated virtual screening approach yielded significant successes in identifying promising dual-target inhibitors:
- Initial Hits: The screening process identified 18 hit compounds with potential inhibitory activity against both VEGFR-2 and c-Met [5] [62].
- Top Candidates: Among these, compound17924 and compound4312 emerged as the most promising candidates based on comprehensive screening criteria, demonstrating favorable binding modes and interaction profiles with both targets [5].
- Energetic Superiority: MM/PBSA calculations revealed that both top compounds exhibited superior binding free energies compared to reference ligands, suggesting stronger target engagement and potential for enhanced inhibitory activity [5] [62].
Table: Computational Profiles of Identified Hit Compounds
| Parameter | Compound17924 | Compound4312 | Reference Ligands |
|---|---|---|---|
| VEGFR-2 Binding Free Energy | Superior to reference | Superior to reference | Baseline |
| c-Met Binding Free Energy | Superior to reference | Superior to reference | Baseline |
| MD Simulation Stability | Stable complex formation | Stable complex formation | Variable |
| Drug-Likeness | Compliant with filters | Compliant with filters | - |
| ADMET Profile | Favorable predictions | Favorable predictions | - |
Structural Insights and Binding Interactions
Analysis of the binding modes provided molecular rationale for the observed activity:
- VEGFR-2 Interactions: Successful inhibitors typically formed critical hydrogen bonds with key residues in the ATP-binding pocket while effectively occupying adjacent hydrophobic regions [67] [68].
- c-Met Engagement: Optimal compounds demonstrated ability to interact with the activation loop of c-Met, potentially interfering with tyrosine residues Tyr1234 and Tyr1235 that are crucial for kinase activation [5] [62].
- Common Pharmacophores: The most promising dual inhibitors shared structural features including heteroaromatic systems for ATP-binding site interaction, hydrogen bond donors/acceptors for complementary interactions with the hinge region, and hydrophobic moieties for allosteric pocket occupation [65] [68].
Diagram 2: VEGFR-2 and c-Met Signaling Pathways and Inhibitor Mechanism
Experimental Protocols
Computational Protocol: Virtual Screening Workflow
Day 1: System Preparation
- Protein Preparation:
- Download crystal structures (PDB IDs: 4ASE for VEGFR-2; 3ZZE for c-Met)
- Remove water molecules and co-crystallized ligands
- Add hydrogen atoms and correct protonation states
- Energy minimization using CHARMM force field
- Ligand Library Preparation:
- Retrieve compounds from ChemDiv or ZINC20 databases
- Generate 3D conformations using OMEGA or similar tools
- Apply Lipinski's Rule of Five and Veber descriptors
- Filter using PAINS patterns to eliminate promiscuous binders
Day 2-3: Pharmacophore Screening
- Model Generation:
- Develop structure-based pharmacophores using Discovery Studio
- Include HBA, HBD, hydrophobic, and aromatic features
- Validate models using enrichment factors (EF > 2) and ROC curves (AUC > 0.7)
- Database Screening:
- Screen pre-filtered compound library against validated pharmacophores
- Retrieve top matching compounds for further analysis
Day 4-5: Molecular Docking
- Grid Generation:
- Define binding sites based on co-crystallized ligands
- Create energy-based grids encompassing active site regions
- Docking Simulations:
- Perform high-throughput docking using ICM or similar software
- Employ Monte Carlo minimization approaches for conformational sampling
- Rank compounds based on docking scores and interaction patterns
Day 6-7: Molecular Dynamics
- System Setup:
- Solvate protein-ligand complexes in TIP3P water boxes
- Neutralize systems with appropriate ions
- Energy minimization and gradual heating to 310K
- Production Runs:
- Conduct 100 ns MD simulations using AMBER or GROMACS
- Analyze trajectories for stability (RMSD, RMSF)
- Calculate binding free energies using MM/PBSA or MM/GBSA
Experimental Validation Protocol
Following computational identification, promising candidates require experimental validation:
In Vitro Kinase Assays
- VEGFR-2 Inhibition Assay:
- Use recombinant VEGFR-2 kinase domain
- Measure phosphorylation of poly(Glu,Tyr) substrate
- Determine IC50 values using ATP-concentration dependent assays
- c-Met Inhibition Assay:
- Employ c-Met kinase domain with appropriate peptide substrate
- Quantify phosphate incorporation using ELISA or mobility shift assays
- Calculate inhibition potency and selectivity indices
Cellular Assays
- Anti-proliferative Screening:
- Test compounds against NCI-60 cancer cell line panel
- Perform MTT or CellTiter-Glo viability assays
- Establish GI50 values across multiple cancer types
- Anti-angiogenic Assessment:
- Conduct HUVEC tube formation assays
- Perform chick chorioallantoic membrane (HET-CAM) assays
- Quantify vessel density and branching points
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Research Tools for VEGFR-2/c-Met Inhibitor Development
| Research Tool | Function/Application | Examples/Sources |
|---|---|---|
| Protein Structures | Structure-based drug design | PDB IDs: 4ASE (VEGFR-2), 3ZZE (c-Met) [5] [67] |
| Compound Libraries | Source of potential inhibitors | ChemDiv, ZINC20 databases [5] [67] |
| Pharmacophore Software | 3D chemical feature modeling | Discovery Studio, LigandScout [5] [62] |
| Molecular Docking Tools | Binding pose prediction & scoring | ICM-Pro, AutoDock, Glide [5] [67] |
| MD Simulation Packages | Dynamics & stability analysis | AMBER, GROMACS, CHARMM [5] [67] |
| Kinase Assay Kits | In vitro inhibition profiling | Recombinant kinases + substrates [65] [66] |
| Cell-Based Assays | Cellular efficacy assessment | HUVEC tube formation, NCI-60 panel [65] |
This case study demonstrates the powerful synergy achieved by integrating pharmacophore-based virtual screening with molecular docking and dynamics simulations for identifying novel VEGFR-2/c-Met dual inhibitors. The sequential filtering approach enabled efficient navigation of extensive chemical space, culminating in the identification of compound17924 and compound4312 as promising candidates with superior predicted binding characteristics compared to reference ligands [5] [62]. The documented methodology provides a robust framework for future drug discovery campaigns targeting multiple kinase pathways, with particular relevance for overcoming resistance mechanisms in oncology therapeutics. The computational protocols and experimental validation strategies outlined herein offer researchers a comprehensive roadmap for advancing dual-target inhibitors from virtual screening to experimental confirmation.
Application Notes
The integration of molecular docking and pharmacophore-based virtual screening is advancing drug discovery by enabling systematic exploration of polypharmacology and de-risking drug repurposing efforts. These computational strategies facilitate the prediction of multi-target inhibition profiles and adverse effects, addressing the high costs and failure rates of de novo drug development.
Drug Repurposing via Target Prediction
Drug repurposing identifies new therapeutic uses for existing drugs, significantly reducing development time and resources. Computational target prediction is a cornerstone of this approach.
- Ligand-Centric Target Fishing: This method predicts potential protein targets for a query molecule by comparing its chemical similarity to a database of known bioactive compounds. The underlying principle is that structurally similar molecules are likely to share molecular targets [69]. Performance varies significantly based on the chosen algorithm and chemical fingerprint [69].
- Performance of Prediction Tools: A 2025 benchmark study evaluated seven target prediction methods on a shared dataset of FDA-approved drugs. MolTarPred was identified as the most effective method, particularly when using Morgan fingerprints with Tanimoto similarity scores [69]. The study also highlighted that applying high-confidence filters to interaction data improves prediction reliability, though at the cost of reduced recall [69].
Table 1: Comparison of Target Prediction Methods for Drug Repurposing
| Method | Type | Algorithm/ Basis | Key Finding |
|---|---|---|---|
| MolTarPred | Ligand-centric | 2D Chemical Similarity | Most effective method; optimal with Morgan fingerprints & Tanimoto scores [69] |
| RF-QSAR | Target-centric | Random Forest QSAR Model | Performance varies with target and molecular representation [69] |
| TargetNet | Target-centric | Naïve Bayes | Dependent on quality and breadth of underlying bioactivity data [69] |
| CMTNN | Target-centric | Multitask Neural Network | Leverages deep learning on large-scale databases like ChEMBL [69] |
Polypharmacology through Dual-Inhibitor Design
Polypharmacology involves the deliberate design of drugs to interact with multiple specific protein targets. This approach is valuable for complex diseases like cancer, where resistance to single-target therapies is common.
- Rationale for VEGFR-2 and c-Met Dual Inhibition: The vascular endothelial growth factor receptor 2 (VEGFR-2) and the mesenchymal-epithelial transition factor (c-Met) are critical tyrosine kinases that synergistically promote tumor angiogenesis and progression [5] [70]. Developing a single chemical entity that inhibits both can produce broader anticancer benefits and overcome the limitations of selective inhibitors [5].
- Identification of Dual Inhibitors: A 2025 study employed an integrated computational workflow to identify novel VEGFR-2/c-Met dual inhibitors from the ChemDiv database. The workflow combined drug-likeness filtering, pharmacophore modeling, and molecular docking, culminating in molecular dynamics (MD) simulations. This process identified 18 hit compounds, with compound17924 and compound4312 showing superior binding free energies compared to positive controls, marking them as promising candidates for further development [5].
Prediction of Polypharmacy Side Effects
Polypharmacy—the concurrent use of multiple drugs—is common but increases the risk of adverse drug-drug interactions (DDIs). Predicting these side effects is crucial for patient safety.
- Leveraging Chemical Structure Alone: Traditional models for DDI prediction often rely on biological entities like proteins or cell lines, which can be limiting. Emerging approaches demonstrate that using Large Language Models (LLMs) to encode the chemical structures of drugs (in SMILES format) can effectively predict polypharmacy side effects, even without additional biological data [71].
- Model Performance: Research shows that integrating embeddings from specialized LLMs like DeepChem ChemBERTa with a Graph Neural Network (GNN) classifier yields highly effective predictions. This architecture can outperform models using simpler molecular representations or other classifiers like Multilayer Perceptrons (MLPs) [71].
Table 2: Computational Methods for Polypharmacy Side Effect Prediction
| Component | Option A | Option B | High-Performance Example |
|---|---|---|---|
| Molecular Encoding | Morgan Fingerprints | GPT-based SMILES encoding | DeepChem ChemBERTa embeddings [71] |
| Classifier Model | Multilayer Perceptron (MLP) | Graph Neural Network (GNN) | GNN Classifier [71] |
| Key Advantage | Simplicity, speed | Captures complex relationships | High effectiveness using only chemical structures [71] |
Experimental Protocols
Protocol: Virtual Screening for Dual-Target Inhibitors
This protocol details the integrated computational workflow for identifying dual VEGFR-2/c-Met inhibitors, as demonstrated in recent research [5].
Materials and Software Requirements
- Chemical Database: ChemDiv database (or other commercial libraries like ZINC [72]).
- Protein Structures: Obtain co-crystal structures of VEGFR-2 (e.g., PDB: 5RL7) and c-Met (e.g., PDB: 5RLZ) from the RCSB Protein Data Bank [5] [73].
- Software Suite: Discovery Studio (DS) or equivalent molecular modeling software.
- Computing Hardware: High-performance computing cluster for docking and MD simulations.
Procedure
Step 1: Ligand Database Preparation
- Download and curate a small-molecule database (e.g., >1 million compounds).
- Prepare ligands using DS "Prepare Ligands" protocol: remove salts, add hydrogens, and generate 3D conformations.
- Filter compounds using Lipinski's Rule of Five and Veber's rules to ensure drug-likeness.
- Predict key ADMET properties (aqueous solubility, hepatotoxicity, cytochrome P450 inhibition) to eliminate compounds with undesirable profiles [5].
Step 2: Pharmacophore Model Generation and Screening
- Select multiple high-resolution co-crystal structures of VEGFR-2 and c-Met with diverse ligands [5].
- For each protein structure, use the "Receptor-Ligand Pharmacophore Generation" module in DS. Set features to include hydrogen bond donors/acceptors, hydrophobic areas, and ring aromatics.
- Validate generated pharmacophore models using a decoy set containing known active and inactive compounds. Calculate the Enrichment Factor (EF) and area under the ROC curve (AUC). Select models with AUC > 0.7 and EF > 2 for screening [5].
- Screen the pre-filtered chemical database against the best VEGFR-2 and c-Met pharmacophores.
Step 3: Molecular Docking
- Prepare protein structures by removing water molecules, adding missing residues, and optimizing hydrogen bonds using the CHARMM force field [5].
- Define the binding site for docking based on the location of the native co-crystallized ligand.
- Dock the compounds that passed the pharmacophore screening into the active sites of both VEGFR-2 and c-Met.
- Rank compounds based on their docking scores and analyze key binding interactions (e.g., hydrogen bonds, pi-pi stacking) for both targets. Select top-ranked compounds for further analysis.
Step 4: Binding Stability Assessment via Molecular Dynamics (MD)
- Solvate the protein-ligand complex in an explicit water box and add ions to neutralize the system.
- Run MD simulations (e.g., for 100 ns) using software like GROMACS or Desmond to evaluate the stability of the binding pose over time [5].
- Analyze trajectories for Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF).
- Use the MM/PBSA method to calculate binding free energies from the simulation trajectories. Compare the results to positive controls to identify the most promising dual-inhibitor candidates [5].
Protocol: Predicting Polypharmacy Side Effects using LLMs
This protocol outlines a computational method for predicting the side effects of drug combinations using SMILES strings and large language models [71].
- Dataset: The Decagon dataset, which provides drug pairs and their associated side effects, sourced from the TWOSIDES database [71].
- Drug Chemical Data: PubChem database to retrieve canonical SMILES strings using Compound IDs (CIDs) [71].
- LLM Models: Pre-trained models like ChemBERTa available through the DeepChem library.
- Programming Environment: Python with libraries for deep learning (e.g., PyTorch, TensorFlow) and cheminformatics (e.g., RDKit).
Procedure
Step 1: Data Preparation and SMILES Retrieval
- Obtain the Decagon dataset, which lists drug pairs and their associated side effects.
- For each drug in the dataset, use its PubChem CID to query and download its canonical SMILES string from the PubChem database [71].
- Filter the dataset to focus on the most frequent side effects (e.g., those occurring in at least 500 drug combinations) to ensure robust model training [71].
Step 2: Molecular Encoding with Language Models
- Tokenize the SMILES strings of each drug.
- Feed the tokenized SMILES into a pre-trained LLM such as ChemBERTa to generate a numerical vector embedding for each drug. This embedding captures the nuanced chemical information of the molecule [71].
- For each drug pair, combine the individual drug embeddings into a single representation, for example, by concatenation or summation.
Step 3: Model Training and Prediction
- Feed the combined drug-pair representation into a classifier. A Graph Neural Network (GNN) has been shown to be more effective than a simple Multilayer Perceptron (MLP) for this task [71].
- Train the model (LLM + GNN) to classify whether a drug pair will cause a specific side effect. Use the known drug pair-side effect associations in the Decagon dataset for training and validation.
- Evaluate the model's performance on a held-out test set using metrics like Area Under the Precision-Recall Curve.
Visualizations
Workflow for Dual-Inhibitor Discovery
Dual-Inhibitor Screening Workflow
Polypharmacy Side Effect Prediction
Side Effect Prediction Model
The Scientist's Toolkit
Table 3: Essential Research Reagents and Resources
| Resource Name | Type | Function in Research | Source/Example |
|---|---|---|---|
| ChemDiv Database | Chemical Library | Source of diverse, purchasable small molecules for virtual screening [5]. | Commercial Vendor |
| RCSB Protein Data Bank | Structural Database | Repository for 3D protein structures used in pharmacophore modeling and docking [5] [73]. | Public Database |
| ChEMBL Database | Bioactivity Database | Curated database of bioactive molecules with target annotations; used for model training and validation [69]. | Public Database |
| Decagon Dataset | Interaction Dataset | Provides known drug-drug interaction and polypharmacy side effect data for model training [71]. | Public Dataset (TWOSIDES) |
| Directory of Useful Decoys (DUD) | Benchmarking Set | Provides validated decoy molecules for rigorous testing of virtual screening methods [74]. | Public Benchmark |
| Discovery Studio | Software Suite | Integrated platform for performing pharmacophore modeling, molecular docking, and simulation [5]. | Commercial Software |
| LigandScout | Software | Specialized tool for creating and screening structure-based and ligand-based pharmacophore models [73]. | Commercial Software |
| ChemBERTa | Language Model | Pre-trained LLM for generating informative numerical embeddings from drug SMILES strings [71]. | DeepChem / Hugging Face |
Overcoming Challenges: Optimization Strategies for Enhanced Performance
Addressing the Scoring Function Challenge in Docking and Pharmacophore Matching
Molecular docking is a cornerstone of structure-based drug design, yet its predictive accuracy is frequently compromised by the limitations of scoring functions. These functions often struggle to correctly rank ligand poses, leading to high false-positive rates in virtual screening. This application note details the integration of pharmacophore matching as a powerful post-docking filter to address this challenge. We provide a validated protocol that leverages the complementary strengths of docking and pharmacophore-based screening, enabling researchers to significantly improve the enrichment of active compounds and enhance the reliability of their virtual screening workflows.
In computational drug discovery, molecular docking aims to predict the binding mode and affinity of a small molecule within a target protein's binding site. The process typically involves two steps: pose generation (sampling) and pose scoring [24]. While sampling algorithms have become proficient at generating biologically relevant ligand conformations, the scoring functions used to rank these conformations remain a critical bottleneck [53] [75].
Scoring functions, which can be force field-based, empirical, knowledge-based, or machine-learning-based, are mathematical constructs designed to predict binding affinity [75]. However, they often fail to correctly rank ligands by their true binding affinity or even distinguish correct poses from incorrect ones [53]. This shortcoming results in a high number of false positives—compounds that score highly in silico but show no activity in experimental assays [53]. This problem directly impacts the efficiency of virtual screening, wasting computational and experimental resources on validating non-functional compounds.
The integration of pharmacophore matching presents a robust solution. A pharmacophore is an abstract representation of the steric and electronic features necessary for molecular recognition by a biological target [76]. By filtering docked poses through a structure-based pharmacophore model, it is possible to eliminate poses that, despite a favorable docking score, lack essential chemical features for high-affinity binding. This method combines the superior pose generation of docking with the chemical logic of pharmacophore models, leveraging the advantages of both structure-based and ligand-based drug design approaches [53] [77].
Current State and Challenges of Scoring Functions
Classification and Limitations of Scoring Functions
Scoring functions are generally classified into four main categories, each with inherent strengths and weaknesses that contribute to the scoring challenge [75].
Table 1: Categories and Characteristics of Classical Scoring Functions
| Category | Fundamental Principle | Typical Components | Key Limitations |
|---|---|---|---|
| Force Field-Based | Sums non-bonded intermolecular interactions based on molecular mechanics. | Van der Waals, electrostatic terms, sometimes implicit solvation. | High computational cost; sensitive to small atomic displacements; often neglects entropic and solvation effects. |
| Empirical | Linear regression of weighted interaction terms against known binding affinities. | Hydrogen bonds, hydrophobic contacts, rotatable bonds, clashes. | Prone to overfitting on training data; performance may not generalize to novel target classes. |
| Knowledge-Based | Statistical potentials derived from frequency of atom-pair contacts in structural databases. | Pairwise atom-type contact potentials. | Dependent on the quality and size of the reference database; difficult to interpret physically. |
| Machine-Learning | Functional form learned from data linking complex structural features to affinity. | Various descriptors of the protein-ligand interface. | Requires large, high-quality training datasets; risk of learning dataset-specific biases. |
A core limitation of classical scoring functions (force-field, empirical, and knowledge-based) is their assumption that contributions to binding affinity are linearly additive [75]. This simplification fails to capture the complex, non-linear nature of molecular recognition. Furthermore, many functions do not adequately account for critical phenomena such as solvation effects, entropic penalties upon binding, and the specific directionality of hydrogen bonds [53] [75].
The Consequence: False Positives in Virtual Screening
The inaccuracies in scoring functions manifest operationally in a high rate of false positives during virtual screening. These are compounds ranked highly by the docking algorithm but which do not bind effectively in reality [53]. These false positives can obscure true active compounds (false negatives) that may have been scored poorly, thereby reducing the overall enrichment and efficiency of the screening campaign [53]. The problem is exacerbated by the use of biased benchmarking datasets, where differences in the physicochemical properties of active and decoy compounds can lead to an overestimation of a method's performance [78].
Integrated Solution: Pharmacophore Filtering of Docked Poses
Conceptual Framework
The pharmacophore filtering method is designed to mitigate the scoring function problem by introducing a chemically intelligent, post-processing step. The core idea is to separate the tasks of pose generation and pose ranking. A docking program is used for its ability to sample diverse and realistic ligand conformations within the binding site, but its scoring is disregarded. The generated poses are then evaluated against a pharmacophore model that encodes the essential interactions a ligand must form with the protein target [53].
This approach is computationally efficient because the docking program has already pre-aligned all ligands into the coordinate space of the binding site. This eliminates the need for the costly conformational search and alignment steps typically required in traditional ligand-based pharmacophore screening [53]. The method effectively enforces the principle of chemical complementarity, ensuring that top-ranked poses not only have favorable overall interaction energy but also satisfy key interaction constraints.
Workflow Visualization
The following diagram illustrates the integrated docking and pharmacophore filtering workflow, highlighting the sequential steps and decision points that lead to the final selection of high-confidence hits.
Diagram 1: Integrated Docking and Pharmacophore Filtering Workflow. The process begins with docking for pose generation, followed by the application of a structure-based pharmacophore filter before final scoring and ranking.
Application Notes & Protocols
Protocol: Pharmacophore-Guided Rescoring of Docking Results
This protocol describes how to implement the pharmacophore filtering method using a combination of standard docking software and pharmacophore tools.
I. Software and Data Requirements
- Docking Software: GOLD [53], Glide [53], AutoDock Vina [24], DOCK [79], or similar.
- Pharmacophore Software: MOE [53], LigandScout [53], Schrödinger's Phase, or custom scripts (e.g., DOCK's FMS [79]).
- Input Files:
- Target protein structure (preferably a high-resolution crystal structure, PDB format).
- Database of small molecules to screen (MOL2 or SDF format).
- (Optional) Structure of a known inhibitor or native ligand co-crystallized with the target.
II. Step-by-Step Procedure
System Preparation
- Prepare the protein structure by removing crystallographic water molecules (unless critical for binding), adding hydrogen atoms, and assigning partial charges. For neuraminidase A, for example, specific residue flips (e.g., Asn 294) may be necessary [53].
- Prepare the ligand database by generating plausible tautomers, protonation states at physiological pH, and energy-minimized 3D structures.
Pose Generation via Molecular Docking
- Define the binding site centroid using a co-crystallized ligand or known active site residues. A typical box size of 10-20 Å radius is sufficient.
- Run the docking calculation using the chosen software. It is advisable to use a stochastic search algorithm (e.g., Genetic Algorithm in GOLD) to ensure diverse pose sampling [53].
- Critical Step: Configure the docking run to output a large number of poses per ligand (e.g., 10-50) and save all poses to a file, disregarding the internal docking scores.
Pharmacophore Model Generation
- Ligand-based: If a known active ligand is available, use its bound conformation to derive a pharmacophore model. The model should include key features like hydrogen bond donors/acceptors, hydrophobic regions, and charged groups [79].
- Structure-based: If no ligand is available, generate the model directly from the protein's binding site. Use tools like LUDI [53] or manual inspection to place pharmacophore features that correspond to crucial protein-ligand interactions (e.g., a hydrogen bond acceptor feature near a key arginine residue) [53].
- Define the tolerance radii for each feature. Smaller radii create a tighter, more selective filter, while larger radii account for some flexibility [53].
Pose Filtering and Rescoring
- Load the file containing all docked poses and the pharmacophore query into the pharmacophore software.
- Perform the pharmacophore search. This step rapidly eliminates poses that do not match the essential interaction pattern defined by the model [53].
- Export the subset of poses that pass the pharmacophore filter.
Final Hit Selection
- The filtered poses can be re-scored using the original or a more sophisticated scoring function. Alternatively, they can be ranked based on the quality of their pharmacophore fit (e.g., using a Pharmacophore Matching Similarity (FMS) score [79]).
- Visually inspect the top-ranked compounds to verify the binding mode and interactions before proceeding to experimental validation.
Advanced Method: Shape-Focused Pharmacophore Optimization
For targets where traditional pharmacophore features are insufficient, shape-focused models offer an enhanced solution. Tools like O-LAP generate cavity-filling pharmacophore models by clustering overlapping atoms from top-ranked docked active ligands [77].
Table 2: Key Reagents and Software for Integrated Screening
| Research Reagent / Tool | Type | Function in Protocol |
|---|---|---|
| GOLD / Glide | Docking Software | Generates diverse ligand poses within the protein binding site. |
| LigandScout | Pharmacophore Modeling | Creates and validates structure-based pharmacophore models from protein-ligand complexes. |
| MOE | Integrated Suite | Provides a unified environment for docking, pharmacophore model creation, and filtering. |
| DOCK with FMS | Docking with Pharmacophore Scoring | Encodes pharmacophore matching similarity directly into the docking scoring function [79]. |
| O-LAP | Shape-Focused Modeling | Generates optimized, cavity-filling pharmacophore models via graph clustering of docked ligands [77]. |
| DUDE-Z / DUD-E | Benchmarking Database | Provides sets of known actives and property-matched decoys to validate and optimize the screening protocol [78] [77]. |
Protocol for O-LAP Model Usage [77]:
- Perform flexible molecular docking of known active ligands (from your training set) using a program like PLANTS.
- Extract the top 50 ranked poses (based on the docking score) and use them as input for O-LAP.
- Run the O-LAP algorithm, which will remove non-polar hydrogens, delete covalent bonding information, and cluster overlapping atoms with matching types into representative centroids.
- The output is a shape-focused pharmacophore model that represents the consensus steric and electrostatic features of the best-docked active ligands.
- Use this O-LAP model to rescore the entire docked database (including decoys and test compounds) by comparing the shape/electrostatic similarity of each docked pose to the model.
Results and Validation
The integration of pharmacophore filtering has been quantitatively demonstrated to improve virtual screening outcomes across multiple targets.
Table 3: Exemplary Performance of Pharmacophore Filtering in Virtual Screening
| Target Protein | Docking Method | Pharmacophore Method | Key Performance Metric | Result |
|---|---|---|---|---|
| Neuraminidase A [53] | GOLD / Glide | Structure-based model | Reduction of false positives | Significant improvement over docking and scoring alone. |
| Various (EGFR, IGF-1R, HIVgp41) [79] | DOCK | FMS (Pharmacophore Matching Similarity) | Pose reproduction success | FMS alone: 93.5% success. FMS + SGE: 98.3% success. |
| A2A Adenosine Receptor, HSP90 [77] | PLANTS | O-LAP (Shape-focused model) | Enrichment in docking rescoring | Massive improvement over default docking enrichment. |
The application of the Pharmacophore Matching Similarity (FMS) scoring in DOCK, for example, not only dramatically improved the success rate of reproducing crystallographic ligand poses but also reduced sampling failures [79]. In cross-docking and enrichment studies, the use of FMS, particularly when combined with standard grid energy (SGE) scoring, consistently showed improved performance, provided appropriate pharmacophore references were used [79].
Discussion
The "scoring function challenge" is a multi-faceted problem unlikely to be solved by a single, universal scoring function. The integrated approach of pharmacophore filtering addresses a core aspect of this challenge: the lack of chemical context in traditional scoring. By explicitly defining and requiring key interactions, this method incorporates critical ligand-based information into the structure-based docking process.
Advantages: The primary advantage is a significant reduction in false positives and a concomitant increase in the reliability of virtual screening hits [53]. The method is also flexible, as pharmacophore models can be easily adjusted based on new structural or biochemical data without re-running the entire docking calculation.
Limitations and Future Directions: The success of this method is contingent on the quality of the pharmacophore model, which in turn depends on the availability of a high-resolution protein structure or a known active ligand. Future developments are likely to see a greater integration of machine learning methods that can automatically derive optimal pharmacophore features or scoring functions from large sets of structural and bioactivity data [80] [75]. Furthermore, the use of advanced negative image-based (NIB) and shape-focused models like those generated by O-LAP and PANTHER represents a powerful evolution of the pharmacophore concept, directly addressing the complementarity between the ligand and the binding cavity [77].
In conclusion, the synergistic combination of molecular docking for comprehensive pose sampling and pharmacophore matching for intelligent pose filtering provides a robust, practical, and highly effective strategy for overcoming the scoring function challenge in computer-aided drug discovery.
Selecting Optimal Protein Conformations and Managing Receptor Flexibility
Molecular docking is a cornerstone of modern structure-based drug discovery, enabling the prediction of how small molecules interact with biological targets. However, the traditional paradigm of docking into a single, static protein structure presents a significant limitation, as it fails to capture the dynamic nature of proteins in solution. Proteins are inherently flexible entities that sample multiple conformational states, and this flexibility is often critical for their biological function and ligand recognition [81]. The selection of optimal protein conformations and the effective management of receptor flexibility are therefore paramount for achieving predictive accuracy in virtual screening campaigns. Within the integrated framework of molecular docking and pharmacophore-based virtual screening, accounting for this dynamism substantially enriches the pharmacophore model generation and increases the probability of identifying true bioactive molecules.
Two primary models describe the relationship between protein flexibility and ligand binding: conformational selection and induced fit. The conformational selection model posits that proteins exist in an equilibrium of multiple pre-existing conformations, and the ligand selectively binds to and stabilizes a complementary conformation [82] [83]. In contrast, the induced fit model suggests that the ligand first binds to the protein, inducing a conformational change to form a stable complex [84]. Advanced experimental techniques, including nuclear magnetic resonance (NMR) and single-molecule spectroscopy, have provided strong evidence that conformational changes can occur in the absence of ligand molecules, supporting the conformational selection model for many systems [82] [85]. In practice, many binding processes involve elements of both mechanisms, and they can be seen as two sides of the same coin, with the temporal ordering of conformational changes and binding events reversing between the binding and unbinding directions [82].
This application note provides a detailed guide to the theoretical underpinnings and practical protocols for selecting optimal protein conformations and managing receptor flexibility, with a specific focus on its integration within a combined molecular docking and pharmacophore virtual screening workflow.
Theoretical Foundation: Protein Flexibility in Molecular Recognition
Mechanistic Models of Binding
Understanding the theoretical models of binding is crucial for designing effective virtual screening strategies.
- Conformational Selection: This model proposes that an unbound protein exists in a dynamic equilibrium between multiple conformations. The ligand does not induce a new shape but rather selectively binds to and populates the conformer to which it has the highest affinity. This process is characterized by a conformational excitation that occurs prior to the binding event [82]. Evidence from NMR ensembles shows that ligand-free conformations often resemble the ligand-bound state, supporting this model [85].
- Induced Fit: In this model, the initial interaction between the protein and ligand is suboptimal. The binding event itself then induces a conformational change in the protein to achieve a tighter fit. This process is identified by a conformational relaxation that occurs after the binding step [82] [84].
For small ligand molecules, binding/unbinding transition times are typically much faster than conformational dwell times, leading to a decoupling and clear temporal ordering of these events. However, for larger ligands like peptides, conformational changes and binding can be intricately coupled [82].
Implications for Drug Discovery
The choice of model has direct practical implications. If a binding interaction proceeds primarily via conformational selection, then successful virtual screening requires the use of protein structures that already resemble the bound conformation. Failure to include these "pre-formed" states in a screening ensemble can lead to false negatives, as the ligand will be unable to dock successfully into a non-complementary rigid structure.
Table 1: Key Characteristics of Conformational Selection and Induced Fit Models
| Feature | Conformational Selection | Induced Fit |
|---|---|---|
| Temporal Order | Conformational change occurs before binding | Conformational change occurs after binding |
| Nature of Change | Conformational excitation | Conformational relaxation |
| Ligand Role | Selects and stabilizes a pre-existing state | Induces a new state |
| Kinetics | Can exhibit ligand concentration-dependent and independent phases [82] | Typically follows a two-step binding mechanism |
| Primary Implication for VS | Requires an ensemble of conformations | May require side-chain or backbone flexibility in a single structure |
Practical Methods for Managing Receptor Flexibility
A variety of computational strategies have been developed to incorporate protein flexibility into docking and virtual screening, each with its own strengths and computational cost.
- Soft Docking: This is a computationally inexpensive method where the van der Waals potentials between the ligand and receptor are "softened," allowing for minor steric clashes. This accommodates small side-chain movements but cannot handle larger backbone motions [84] [81].
- Side-Chain Flexibility: Many docking programs allow for the rotation of side chains of specific residues within the binding site. This is often implemented using rotamer libraries, which are collections of statistically favored side-chain conformations [84] [81].
- Induced Fit Docking (IFD): IFD protocols explicitly model the conformational changes of the protein in response to ligand binding. A common implementation involves an initial docking step, followed by protein side-chain (and sometimes backbone) refinement and minimization in the presence of the ligand, and a final re-docking of the ligand into the optimized binding site [84].
- Ensemble Docking: This is a highly effective and widely used strategy where molecular docking is performed against an ensemble of multiple protein conformations. This ensemble can be derived from various sources, providing a comprehensive strategy for capturing flexibility [84].
Sourcing Conformational Ensembles
The quality of the conformational ensemble is critical for the success of ensemble docking.
- Experimental Structures (X-ray/NMR): Using multiple crystal structures of the same protein bound to different ligands is an excellent way to capture relevant conformational diversity. NMR ensembles provide naturally sampled apo-state conformations [84] [85].
- Molecular Dynamics (MD) Simulations: MD simulations can model the dynamic behavior of a protein in solution, generating a time-series of snapshots. These snapshots can be clustered to identify representative conformations for docking [84] [86]. The Relaxed Complex Scheme (RCS) is a specific method that leverages MD-generated ensembles for docking studies [84].
- Enhanced Sampling Methods: Techniques like accelerated molecular dynamics (aMD) can more efficiently explore conformational space and access higher-energy states that might be relevant for binding but are rarely sampled in conventional MD [84] [81].
Integrated Application Note & Protocols
This section details a practical workflow for integrating receptor flexibility into a combined pharmacophore and docking study.
The following diagram illustrates the integrated protocol for combining pharmacophore screening and molecular docking while accounting for receptor flexibility.
Protocol 1: Generating a Representative Protein Conformational Ensemble
Objective: To create a diverse and representative set of protein structures for subsequent pharmacophore modeling and ensemble docking.
Materials:
- Initial Protein Structure: A high-resolution crystal structure (preferably ligand-bound) from the PDB.
- Software: Molecular dynamics simulation package (e.g., GROMACS, AMBER, NAMD, Desmond [86]) and clustering analysis tool (e.g., GROMACS
gmx cluster, VMD).
Procedure:
- System Setup:
- Obtain the target protein's PDB file (e.g.,
7AEIfor EGFR [13]). - Use a protein preparation wizard (e.g., in Maestro) to add hydrogen atoms, assign bond orders, correct protonation states, and optimize hydrogen-bonding networks. Energy minimize the structure using a force field like OPLS_2005 [13].
- Place the protein in a simulation box (e.g., 10 Å padding) solvated with explicit water molecules (e.g., TIP3P model). Add ions (e.g., 0.15 M NaCl) to neutralize the system and mimic physiological conditions [13] [86].
- Obtain the target protein's PDB file (e.g.,
Molecular Dynamics Simulation:
- Run an energy minimization to remove steric clashes.
- Equilibrate the system in the NPT ensemble (constant Number of particles, Pressure, and Temperature) at 300 K and 1 atm pressure.
- Perform a production MD simulation for a timescale sufficient to capture relevant motions (e.g., 100-200 ns [5] [13]). Save trajectory frames at regular intervals (e.g., every 10-100 ps).
Conformational Clustering and Selection:
- After discarding the initial equilibration period, align the trajectory frames to the protein backbone to remove global rotation/translation.
- Calculate the root mean square deviation (RMSD) of the protein Cα atoms or binding site residues.
- Perform clustering (e.g., using the Gromos method) on the RMSD matrix to group structurally similar conformations.
- Select the central structure from the most populated clusters as representative conformers for the ensemble. This ensures diversity while managing computational cost [84].
Protocol 2: Integrated Pharmacophore Modeling and Ensemble Docking
Objective: To screen a large compound library using pharmacophore models derived from a conformational ensemble and subsequently dock promising hits against the full ensemble.
Materials:
- Software: Pharmacophore modeling suite (e.g., Discovery Studio [5], Pharmit [13]), molecular docking program (e.g., GLIDE [13], AutoDock [84], DOCK [84]), and compound database (e.g., ChemDiv [5], ZINC, ChEMBL [13]).
- Input: The representative protein conformational ensemble from Protocol 1.
Procedure:
- Pharmacophore Model Generation:
- For each representative protein conformation in the ensemble, generate a structure-based pharmacophore model using the Receptor-Ligand Pharmacophore Generation protocol [5].
- Define key pharmacophoric features present in the binding site, such as Hydrogen Bond Acceptors (HBA), Hydrogen Bond Donors (HBD), Hydrophobic regions (H), and Aromatic Rings (AR) [13].
- Validate the quality of each pharmacophore model using a decoy set (e.g., from DUD-E) by calculating enrichment factors (EF) and area under the ROC curve (AUC). A reliable model should have an AUC > 0.7 and EF > 2 [5].
- Select the top 2-3 performing pharmacophore models for virtual screening.
Ligand-Based Virtual Screening:
- Filter a large commercial database (e.g., 1.28 million compounds from ChemDiv [5]) using drug-likeness rules (Lipinski's Rule of Five, Veber rules) and predicted ADMET properties (aqueous solubility, hepatotoxicity, CYP inhibition) [5] [13].
- Screen the filtered library against the selected pharmacophore models.
- Select compounds that match all or most critical features of any of the pharmacophore models as initial hits (e.g., 181 compounds from 9,510 [87]).
Ensemble Docking:
- Prepare the hit compounds using a tool like LigPrep, generating possible ionization states, tautomers, and low-energy ring conformations at physiological pH (e.g., 7.0 ± 0.5) [13].
- Prepare each protein structure in the conformational ensemble for docking (correcting bond orders, adding hydrogens, optimizing H-bonds).
- Dock all prepared hit compounds into every protein conformation in the ensemble using a standard precision (SP) docking algorithm [13].
- For each compound, record the best docking score (most negative value, indicating highest predicted affinity) across the entire ensemble. This "best-case" scoring acknowledges that a compound may only bind effectively to a subset of conformations [84].
Protocol 3: Validation of Complex Stability
Objective: To validate the stability of the top-ranking ligand-protein complexes and estimate binding free energies.
Materials:
- Software: Molecular dynamics simulation package (e.g., Desmond [13]) and free energy calculation tool (e.g., g_mmpbsa, MM/PBSA module).
Procedure:
- System Setup for MD: For each top-ranked complex (e.g., top 3-5 compounds), set up a solvated and neutralized system as described in Protocol 1.
- Molecular Dynamics Simulation: Run a relatively long-timescale MD simulation (e.g., 100-200 ns [5] [13]) for each complex in the NPT ensemble. This tests the stability of the predicted binding mode.
- Trajectory Analysis:
- Calculate the Root Mean Square Deviation (RMSD) of the protein backbone and the ligand to assess overall stability and ligand pose retention.
- Calculate the Root Mean Square Fluctuation (RMSF) of protein residues to identify flexible regions.
- Analyze specific protein-ligand interactions (hydrogen bonds, hydrophobic contacts) over the simulation time to confirm the binding hypothesis.
- Binding Free Energy Calculation:
- Use the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method on snapshots extracted from the stable phase of the MD trajectory to calculate the binding free energy (ΔGbind) [5].
- Compare the MM/PBSA results of the new hits with a known positive control (e.g., a co-crystallized ligand or clinical inhibitor). Compounds with comparable or superior predicted ΔGbind are strong candidates for experimental validation.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools and Resources for Managing Receptor Flexibility
| Category / Reagent | Specific Examples | Primary Function in Workflow |
|---|---|---|
| Molecular Dynamics Software | GROMACS, AMBER, NAMD, Desmond [13] [86] | Generates dynamic conformational ensembles of the target protein from an initial structure. |
| Docking Programs | GLIDE [84] [13], AutoDock [84], DOCK [84] [81], FlexX [81] | Performs rigid or flexible ligand docking into single or multiple protein structures. |
| Specialized Flexible Docking Tools | RosettaLigand [84], Induced Fit Docking (IFD) [84], FlexE [81] | Explicitly models protein side-chain and/or backbone flexibility during the docking process. |
| Pharmacophore Modeling Suites | Discovery Studio [5], Pharmit Server [13] | Creates and validates structure-based or ligand-based pharmacophore models for virtual screening. |
| Compound Databases | ZINC, ChEMBL [13], ChemDiv [5], PubChem | Provides large libraries of commercially available or annotated compounds for screening. |
| Analysis & Validation Tools | MDTraj, VMD, PyMOL, MM/PBSA scripts | Analyzes MD trajectories, calculates binding free energies, and visualizes results. |
| Decoy Sets for Validation | DUD-E (Directory of Useful Decoys: Enhanced) [87] [5] | Provides experimentally inactive compounds to test the enrichment power of pharmacophore models or docking protocols. |
Improving Pose Selection and Virtual Screening Rankings with 3D Shape Similarity
Molecular docking is a cornerstone of structure-based drug discovery, yet it faces a significant challenge: while flexible ligand sampling often performs acceptably, docking scoring functions frequently struggle to reliably enrich active compounds at the top of virtual screening rankings [88]. This limitation diminishes the practical utility of docking in large-scale drug discovery campaigns. To address this critical bottleneck, researchers are increasingly turning to 3D shape similarity techniques as a powerful post-docking filter for pose selection and ranking refinement.
The fundamental premise is that biologically active ligands, even those with diverse chemical scaffolds, often share similar three-dimensional shapes and pharmacophore features when bound to their protein target [89]. By comparing flexibly sampled docking poses against shape-based references, researchers can significantly improve the identification of correct binding modes and enhance the enrichment of true actives in virtual screening [88] [90]. This application note details practical protocols for integrating 3D shape similarity into standard virtual screening workflows, providing researchers with actionable methodologies to improve the performance of their drug discovery pipelines.
Key Methodologies and Instrumentation
Research Reagent Solutions
Table 1: Essential computational tools for 3D shape similarity applications.
| Tool Name | Type/Category | Primary Function | Key Features |
|---|---|---|---|
| O-LAP [88] | Graph Clustering Algorithm | Generates shape-focused pharmacophore models | Clusters overlapping docked ligand atoms; enables cavity-filling models; C++/Qt5 implementation |
| Schrödinger Shape Screening [90] | Shape Similarity Tool | Shape-based flexible superposition & screening | Pharmacophore feature encoding; atom triplet alignment; hard-sphere volume calculations |
| ROCS [89] | Shape Similarity Tool | Rapid 3D shape overlay & screening | Gaussian molecular volumes; Color Force Field (chemical features) |
| CSNAP3D [89] | Shape Similarity Network | Target profiling & scaffold hopping | Combines shape + pharmacophore metrics; network-based scoring |
| ShaEP [88] | Shape/Electrostatic Comparison | Negative image-based rescoring (R-NiB) | Compares shape and electrostatic potential |
| FragmentScout [73] | Pharmacophore Screening | Fragment-based virtual screening | Generates joint pharmacophore queries from XChem fragment data |
Quantitative Performance Benchmarking
Extensive benchmarking studies demonstrate that shape-based approaches consistently enhance virtual screening performance compared to traditional docking alone. The incorporation of pharmacophore features within shape matching algorithms provides particularly significant improvements.
Table 2: Enrichment factors (EF) at 1% of database screened for different Shape Screening approaches [90].
| Target | Pure Shape | QSAR Atom Types | Element-Based | MacroModel Atom Types | Pharmacophore-Based |
|---|---|---|---|---|---|
| CA | 10.0 | 25.0 | 27.5 | 32.5 | 32.5 |
| CDK2 | 16.9 | 20.8 | 20.8 | 23.4 | 19.5 |
| DHFR | 7.7 | 3.9 | 11.5 | 23.1 | 80.8 |
| ER | 9.5 | 17.6 | 17.6 | 13.5 | 28.4 |
| Neuraminidase | 16.7 | 16.7 | 16.7 | 16.7 | 25.0 |
| PTP1B | 12.5 | 12.5 | 12.5 | 12.5 | 50.0 |
| Thrombin | 1.5 | 4.0 | 4.5 | 8.5 | 28.0 |
| TS | 19.4 | 32.3 | 35.5 | 51.7 | 61.3 |
| Average | 11.9 | 15.6 | 17.0 | 20.0 | 33.2 |
The data reveals that pharmacophore-based shape screening outperforms all atom-based methods, achieving an average enrichment factor of 33.2 compared to 20.0 for MacroModel atom types [90]. This represents a 66% improvement in screening efficiency, highlighting the critical importance of incorporating chemical feature matching alongside pure shape comparison.
Application Protocols
Protocol 1: Shape-Based Rescoring of Docking Poses
This protocol utilizes the O-LAP algorithm to generate shape-focused pharmacophore models for improving docking pose selection and ranking [88].
Step-by-Step Procedure:
Input Preparation
- Obtain a 3D protein structure with a defined binding cavity. For the O-LAP method, use the centroid of a co-crystallized ligand as the docking center with a 10Å box radius [88].
- Prepare ligand libraries using tools like LIGPREP (Schrödinger) to generate 3D conformers, tautomeric states, and assign partial charges (e.g., OPLS3) [88].
Flexible Molecular Docking
- Perform flexible-ligand docking using software such as PLANTS1.2 with default settings to generate 10 binding poses per ligand [88].
- Extract the top 50 docked poses from active training set ligands based on the default docking score (e.g., ChemPLP in PLANTS) for model building.
O-LAP Model Generation
- Remove all non-polar hydrogen atoms from the extracted docking poses and discard covalent bonding information [88].
- Perform pairwise distance-based graph clustering using O-LAP with atom-type-specific radii to cluster overlapping ligand atoms into representative centroids.
- Generate the final shape-focused pharmacophore model containing clustered atomic content that fills the protein cavity [88].
Shape Similarity Rescoring
- Compare all flexibly sampled docking poses against the O-LAP model using shape similarity software such as ShaEP.
- Rescore and rerank docking outputs based on shape similarity scores rather than traditional docking scores.
- Apply brute-force negative image-based optimization (BR-NiB) if a training set with active and decoy compounds is available to further enhance model performance [88].
Protocol 2: Multi-Reference Shape Screening with Pharmacophore Features
This protocol employs Schrödinger's Shape Screening tool to enhance virtual screening rankings through shape and pharmacophore similarity comparisons [90].
Step-by-Step Procedure:
Reference Selection and Preparation
- Select one or more known active ligands with established 3D structures as shape references. Crystal structures of protein-bound ligands provide optimal templates.
- Generate multi-conformer ensembles for reference compounds using conformational sampling tools such as ConfGen to account for ligand flexibility [90].
Shape Query Configuration
- Create shape queries using the Shape Screening tool in Schrödinger's suite.
- Select the pharmacophore feature encoding option rather than pure shape or atom-based methods for optimal performance [90].
- Define pharmacophore features including hydrogen bond acceptors/donors, hydrophobic regions, charged groups, and aromatic rings.
Database Screening
- Prepare the screening database as multi-conformer ensembles using tools like Phase or ConfGen.
- Execute the shape screen using the configured queries. The tool will identify triplet pairs with similar geometries between query and database molecules, followed by superposition based on least-squares alignment [90].
- Screen approximately 600 conformers per second per 2GHz processor, with parallelization available for large-scale screens [90].
Similarity Calculation and Ranking
- Calculate shape similarity using the formula:
SimAB = OAB/max(OAA, OBB), where OAB represents the sum of pairwise atomic overlaps between structures A and B [90]. - Rank database compounds by decreasing shape similarity score.
- Apply excluded volume filters if protein structure is available to eliminate poses clashing with the binding site.
- Calculate shape similarity using the formula:
Protocol 3: Fragment-Based Pharmacophore Screening
The FragmentScout workflow generates consolidated pharmacophore models from multiple fragment poses for enhanced screening of ultra-large libraries [73].
Step-by-Step Procedure:
Fragment Data Collection
- Obtain XChem crystallographic fragment screening data from public repositories or generate proprietary fragment datasets.
- Curate fragment structures by binding site clusters to group related pharmacophore features [73].
Joint Pharmacophore Model Generation
- Import 3D-aligned fragment structures into LigandScout software.
- Automatically assign pharmacophore features (hydrogen bond donors/acceptors, hydrophobic areas, etc.) and exclusion volumes for each fragment.
- Merge all pharmacophore queries from a binding site cluster using the "based-on reference points" option to create a joint pharmacophore model [73].
- Perform feature interpolation within distance tolerance to consolidate the final model.
Virtual Screening Implementation
- Convert screening libraries to multi-conformer 3D databases using CONFORGE or similar tools.
- Screen ultra-large libraries using the Greedy 3-Point Search algorithm in LigandScout XT, which enables efficient screening with large pharmacophore queries containing 20+ features [73].
- Prioritize hits that match key pharmacophore features in the joint model.
Advanced Integrations and Emerging Approaches
Deep Learning and Shape Similarity
Recent advances in deep learning (DL) are creating new opportunities for enhancing shape-based virtual screening. While traditional scoring functions still demonstrate advantages in certain scenarios, generative diffusion models like SurfDock have shown exceptional pose prediction accuracy with RMSD ≤ 2Å success rates exceeding 70% across diverse benchmark datasets [29]. However, these methods often produce physically implausible structures despite favorable RMSD scores, indicating the continued importance of physics-based validation.
Equivariant graph neural networks represent another promising direction, enabling ultra-fast virtual screening with significant acceleration factors. For example, the miRPVS approach achieves tens of thousands of times acceleration compared to traditional molecular docking while maintaining comparable accuracy [91]. These DL methods can extract 3D structural features of small molecules to predict docking scores directly, bypassing computationally intensive conformational searches.
Hybrid Workflows for Challenging Targets
For difficult targets with diverse chemical scaffolds, such as HIV reverse transcriptase inhibitors, combined 2D/3D approaches have demonstrated remarkable success. The CSNAP3D method achieves >95% success rates in predicting drug targets across 206 known drugs by integrating 2D chemical similarity networks with 3D shape and pharmacophore metrics [89]. This hybrid approach is particularly effective for identifying scaffold-hopping compounds that share similar 3D environments despite 2D structural differences.
The integration of 3D shape similarity methods into molecular docking workflows represents a powerful strategy for addressing fundamental limitations in virtual screening. As demonstrated through the protocols outlined in this application note, shape-based rescoring and pharmacophore-enhanced screening can significantly improve both pose selection accuracy and active compound enrichment. The quantitative benchmarking data reveals that pharmacophore-based shape screening achieves superior performance compared to pure shape or atom-based methods, with an average 66% improvement in enrichment factors [90]. Emerging approaches incorporating deep learning and hybrid 2D/3D similarity networks offer promising directions for further enhancing the efficiency and effectiveness of virtual screening in drug discovery.
Integrating Molecular Dynamics for Binding Mode Validation and Refinement
Molecular docking and pharmacophore-based virtual screening are cornerstone computational methods in modern drug discovery. However, the binding poses they generate, particularly for flexible peptide ligands or when using predicted protein models, often require further validation and refinement to achieve the accuracy necessary for downstream applications. Molecular dynamics (MD) simulations have emerged as a powerful tool for this purpose, enabling the refinement of initial docking results into more reliable, physically realistic protein-ligand complex structures. This protocol details the integration of MD-based refinement to validate and improve binding modes obtained from virtual screening, a critical step within a broader research framework integrating docking and pharmacophore studies. By applying these methods, researchers can significantly enhance the quality of their structural models, leading to more accurate virtual screening hits and a better foundation for lead optimization [92] [93].
Quantitative Validation of MD Refinement Efficacy
The application of MD for post-docking refinement consistently demonstrates measurable improvements in structural accuracy and docking reliability. The following table summarizes key quantitative evidence from benchmark studies.
Table 1: Quantitative Improvements from MD-Based Refinement Protocols
| Study Focus | Refinement Protocol | Key Performance Metrics | Result |
|---|---|---|---|
| Ligand-binding site refinement on predicted protein models [92] | MD simulations with template-derived restraints (3 × 50 ns per target) | Average Cα RMSD improvement vs. experimental structures | 0.90 Å |
| Average ligand docking pose (RMSD) improvement | 1.97 Å | ||
| Refinement of flexible histone peptide complexes [93] | Post-docking MD with explicit interface hydration | Median improvement in RMSD over docked structures | 32% |
| Virtual screening performance on AF2 models [15] | 500 ns all-atom MD simulation to generate conformational ensembles | Improved docking outcomes for protein-protein interaction (PPI) targets | Variable, case-dependent improvement |
These data validate MD refinement as a powerful step for enhancing model quality, particularly for challenging targets like flexible peptides and computationally predicted protein structures.
Experimental Protocols for MD-Based Refinement
Protocol 1: Template-Restrained MD for Binding Site Refinement
This protocol is designed to refine the ligand-binding site of a protein model (e.g., from homology modeling or AlphaFold2) using information from known ligand-binding site templates [92].
- Identify Binding Site and Template: Use a local structure alignment tool like G-LoSA (Graph-based Local Structure Alignment) to scan your protein model against a library of experimental ligand-binding sites. Select a high-quality template (GA-score > 0.6) with a similar site size (e.g., 11-34 residues) [92].
- Derive Restraint Potentials: From the selected template, calculate a distance matrix between the Cα atoms of the aligned binding site residues. Impose a harmonic distance restraint potential on the equivalent Cα atoms in your protein model during the MD simulation using the formula:
E({rij}) = Σ i<j k(rij - r0,ij)²where k is the force constant, rij is the distance in the target, and r0,ij is the distance in the template [92]. - System Setup:
- Solvation: Solvate the entire protein structure in a cubic box of TIP3P water molecules, ensuring a minimum 10 Å distance from the protein to the box edge.
- Neutralization: Add sodium and chloride ions to neutralize the system charge [92].
- Simulation Run:
- Minimization & Equilibration: Perform 5,000 steps of steepest descent energy minimization. Follow with a 1 ns equilibration in the NPT ensemble.
- Production Simulation: Run multiple replicas (e.g., 3x) of production MD simulations for 50 ns each at 300 K and 1 atm, applying the derived distance restraints. Use a 2 fs integration timestep, the CHARMM36m force field, and particle-mesh Ewald for long-range electrostatics [92].
- Structure Analysis and Validation:
- Cluster Analysis: Cluster the trajectories from the production runs to identify representative conformations.
- Generate Refined Structure: The final refined structure is typically the average of the final conformations from the independent simulation replicas.
- Validation via Docking: Dock the native ligand into the initial and refined binding sites. A successful refinement is indicated by a lower RMSD of the docked pose to the experimental ligand pose [92].
Protocol 2: Post-Docking MD Refinement for Flexible Peptide Ligands
This protocol addresses the challenge of refining docked complexes involving large, flexible peptide ligands, which often have high initial errors [93].
- Initial Docking: Generate initial complex structures using your preferred molecular docking software.
- Pre-MD System Preparation:
- Explicit Hydration: Focus on hydrating the protein-peptide interface region. This crucial step avoids the introduction of unrealistic empty cavities that can distort the simulation.
- System Setup: Solvate the complex in a TIP3P water box, add ions to neutralize, and introduce physiological salt concentration (e.g., 0.15 M NaCl) [13] [93].
- Simulation and Analysis:
- Equilibration: Carry out a multi-step equilibration process, gradually releasing positional restraints on the protein and peptide backbone.
- Production MD: Run an unrestrained production MD simulation (e.g., 200 ns). The required length can vary based on system size and flexibility.
- Trajectory Analysis: Calculate standard metrics including Root-Mean-Square Deviation (RMSD) for complex stability, Root-Mean-Square Fluctuation (RMSF) for residue flexibility, and monitor specific protein-ligand interactions like hydrogen bonds throughout the simulation [13] [93] [94].
- Identify Refined Pose: Extract the most representative structure from the stabilized simulation trajectory (e.g., from the largest cluster) as the final refined model.
Workflow Diagram
The diagram below illustrates the integrated virtual screening and MD refinement workflow.
Integrated Workflow for Binding Mode Validation
The Scientist's Toolkit: Essential Research Reagents and Software
Successful implementation of the aforementioned protocols relies on a suite of specialized software tools and resources.
Table 2: Key Research Reagents and Computational Tools
| Tool Name | Category/Type | Primary Function in Workflow |
|---|---|---|
| G-LoSA [92] | Structure Alignment | Predicts binding sites on protein models and identifies templates for restraint derivation. |
| CHARMM-GUI [92] | MD Setup | Web-based platform for preparing simulation systems (solvation, ionization). |
| OpenMM [92] | MD Engine | High-performance toolkit for running molecular dynamics simulations. |
| Desmond [13] | MD Engine | Integrated MD system for running and analyzing molecular simulations. |
| fpocket [95] | Binding Site Detection | Open-source tool for identifying and characterizing ligand-binding pockets. |
| AutoDock Vina/QuickVina [92] [95] | Molecular Docking | Widely used program for predicting ligand binding poses and affinities. |
| Pharmit [13] [14] | Pharmacophore Screening | Interactive tool for pharmacophore-based virtual screening of compound databases. |
| RosettaVS [34] | Docking & Screening | Physics-based docking protocol supporting receptor flexibility for high-accuracy screening. |
| ZINC/Files.Docking.org [95] [34] | Compound Database | Public repositories of commercially available compounds for virtual screening. |
MD Refinement Cascade Diagram
For high-resolution structural validation, a cascaded refinement approach can be employed, as visualized below.
Cascaded Re-refinement Strategy
The acquisition of biologically active compounds represents a vital yet challenging step in drug discovery, particularly when research involves novel target families or understudied biological targets where functional data is limited [51]. The chemical space for drug-like molecules is estimated to be as large as 10^60 compounds, creating significant challenges for identifying potential drug candidates through traditional experimental methods alone [51]. Data scarcity problems are equally pronounced in protein engineering and functional prediction, where even for critically important proteins like ion channels, available mutational data often covers only a small fraction (~2-3%) of all possible single mutations [96]. This severe data scarcity makes it generally unfeasible to derive predictive functional models using traditional data-centric machine learning approaches [96]. Overcoming these limitations requires innovative computational strategies that integrate physical principles, homology modeling, and artificial intelligence to maximize information extraction from limited datasets.
Computational Frameworks for Data Scarcity Challenges
Physics-Informed Machine Learning
Integrating physics-based modeling with machine learning provides a powerful approach to overcome data scarcity in protein function prediction. Research on big potassium (BK) channels demonstrates that incorporating physical principles through molecular modeling and simulation can enable reliable predictive modeling even with limited mutational data [96]. By quantifying energetic effects of mutations on protein states and deriving dynamic properties from atomistic simulations, researchers can create physical descriptors that serve as features for machine learning models [96]. When applied to BK channels with only 473 characterized mutations, this approach achieved prediction of voltage gating shifts with RMSE ~32 mV and correlation coefficient R ~0.7, significantly outperforming models trained without physics-derived features [96].
Table 1: Performance Metrics of Physics-Informed Machine Learning for BK Channel Prediction
| Model Component | Description | Performance Impact |
|---|---|---|
| Energetic Effects | Quantification of mutation effects on open/closed states | Improved correlation with experimental data |
| Dynamic Properties | Features derived from molecular dynamics simulations | Enhanced model accuracy for novel mutations |
| Random Forest Model | Final predictive model architecture | RMSE ~32 mV, R ~0.7 for ∆V1/2 prediction |
| Novel Mutation Validation | Experimental testing of L235 and V236 mutations | High correlation (R = 0.92, RMSE = 18 mV) |
Pharmacophore-Guided Deep Learning
The Pharmacophore-Guided deep learning approach for bioactive Molecule Generation (PGMG) represents a innovative strategy for addressing data scarcity in drug discovery [51]. This method uses pharmacophore hypotheses as a bridge to connect different types of activity data, enabling flexible molecule generation without requiring large datasets of known active compounds for training [51]. PGMG employs a graph neural network to encode spatially distributed chemical features and a transformer decoder to generate molecules, with a latent variable introduced to solve the many-to-many mapping between pharmacophores and molecules to improve diversity [51]. This approach bypasses the problem of data scarcity on active molecules by avoiding the use of target-specific activity data during training, instead relying on fundamental chemical principles encoded in pharmacophore models.
Transfer Learning for Protein Fitness Prediction
Deep transfer learning has emerged as a particularly effective approach for protein fitness prediction with limited labeled data. Comprehensive analysis demonstrates that transfer learning methods excel in small dataset scenarios compared to traditional supervised and semi-supervised approaches [97]. Models such as ProteinBERT show promising performance in limited-data contexts by leveraging pre-trained models on large general datasets followed by fine-tuning on specific protein fitness tasks [97]. This approach enables researchers to achieve competitive performance even when labeled experimental data is scarce, making it particularly valuable for novel protein engineering applications where extensive mutational studies are not available.
Table 2: Comparative Performance of Learning Approaches for Protein Fitness Prediction with Small Datasets
| Learning Approach | Key Mechanism | Small Dataset Performance | Best Use Cases |
|---|---|---|---|
| Deep Transfer Learning | Pre-training on large datasets followed by fine-tuning | State-of-the-art performance | Novel targets with limited specific data |
| Semi-Supervised Learning | Combination of labeled and unlabeled data | Moderate performance | When unrelated protein data is available |
| Traditional Supervised Learning | Direct training on available labeled data | Limited performance | Well-characterized protein systems |
| Multi-View Strategies | Information combination from different encodings | Promising for future development | Integrating diverse data sources |
Application Notes: Integrated Protocol for Data-Scarce Drug Discovery
Combined Workflow for Virtual Screening and Lead Optimization
The following integrated protocol combines homology modeling, pharmacophore development, and AI-guided generation for drug discovery under data scarcity constraints:
Phase 1: Target Preparation and Homology Modeling
- Template Identification: Identify homologous structures with known ligands from PDB database using BLAST or similar tools
- Model Building: Generate 3D structural model using MODELLER or similar software, with special attention to active site regions
- Model Validation: Verify model quality using PROCHECK, Verify3D, and MolProbity to ensure stereochemical合理性
Phase 2: Pharmacophore Model Development
- Structure-Based Pharmacophore: For targets with known structures, generate pharmacophore hypotheses by analyzing interaction points in binding site [45]
- Ligand-Based Pharmacophore: For targets with limited structural data but known actives, derive common feature pharmacophores from ligand alignment [98]
- Hybrid Approach: Combine structure-based and ligand-based elements when partial data is available for both [98]
Phase 3: AI-Augmented Molecular Generation
- Pharmacophore Encoding: Represent pharmacophore as complete graph with nodes corresponding to features and distances as edge weights [51]
- Conditional Generation: Use PGMG or similar framework to generate molecules matching pharmacophore constraints [51]
- Latent Space Sampling: Explore diverse molecular scaffolds by sampling from latent variable space to address many-to-many mapping between pharmacophores and molecules [51]
Phase 4: Validation and Optimization
- Virtual Screening: Screen generated compounds against pharmacophore model and perform molecular docking [45]
- MD Simulations: Conduct molecular dynamics simulations (≥100 ns) to evaluate binding stability and interactions [94]
- Experimental Testing: Synthesize and test top candidates (typically 5-20 compounds) for experimental validation
Implementation Case Study: NDM-1 Inhibitor Discovery
A recent study demonstrates the application of these principles to identify New Delhi metallo-β-lactamase-1 (NDM-1) inhibitors from natural products [99]. Researchers employed a multi-tier computational approach combining machine learning-based QSAR, molecular docking, and molecular dynamics simulations to screen 4,561 natural product compounds. The workflow included:
- Development of QSAR models using six regression methods trained on ChEMBL data
- Virtual screening with AutoDock Vina with exhaustiveness level of 10
- Tanimoto similarity-based clustering to identify representative compounds
- 300 ns MD simulations to validate binding stability of top candidates
This approach identified compound S904-0022 as a promising NDM-1 inhibitor with favorable binding energy (-35.77 kcal/mol) and stable interactions with key residues including Gln123, His250, Trp93, and Val73 [99].
Experimental Protocols
Protocol 1: Physics-Informed Random Forest for Protein Function Prediction
This protocol adapts the methodology successfully applied to BK channels for predicting mutational effects on protein function [96]:
Step 1: Feature Generation from Physical Principles
- Energetic Calculations: Use Rosetta or similar software to calculate mutational effects on different protein conformational states
- Molecular Dynamics Simulations: Perform MD simulations (≥100 ns) to derive dynamic properties and flexibility metrics
- Structural Descriptors: Calculate solvent accessibility, secondary structure, and domain location features
- Evolutionary Features: Incorporate sequence conservation scores from multiple sequence alignments
Step 2: Model Training and Validation
- Data Partitioning: Split available mutational data using stratified k-fold cross-validation (k=5-10)
- Random Forest Training: Train ensemble of decision trees with physical descriptors as features
- Hyperparameter Optimization: Tune number of trees, maximum depth, and minimum samples per leaf
- Model Evaluation: Assess using root mean square error (RMSE) and correlation coefficient (R)
Step 3: Experimental Validation of Predictions
- Novel Mutation Design: Select 4-6 predictions representing diverse effects and mechanisms
- Functional Characterization: Perform experimental measurements (e.g., electrophysiology for ion channels)
- Model Refinement: Incorporate new data to iteratively improve model performance
Protocol 2: Pharmacophore-Guided AI Generation (PGMG)
This protocol implements the PGMG framework for generating bioactive molecules under data scarcity [51]:
Step 1: Pharmacophore Definition and Representation
- Feature Identification: Define pharmacophore features (hydrogen bond donors/acceptors, hydrophobic regions, charged groups)
- Graph Construction: Create complete graph where nodes represent pharmacophore features and edges represent spatial distances
- Distance Mapping: Use shortest-path distances on molecular graph as proxy for Euclidean distances in 3D space
Step 2: Model Architecture and Training
- Encoder Network: Implement graph neural network (Gated GCN) to encode pharmacophore hypotheses
- Decoder Network: Use transformer architecture to generate SMILES strings conditioned on pharmacophore and latent variables
- Training Procedure: Train on general chemical databases (e.g., ChEMBL) without target-specific activity data
Step 3: Molecular Generation and Optimization
- Conditional Sampling: Sample latent variables from standard Gaussian distribution N(0,I)
- SMILES Generation: Generate molecules from conditional distribution p(x|z,c) using transformer decoder
- Property Filtering: Filter generated molecules for drug-like properties (QED, SA, MW) and pharmacophore fit
Table 3: Key Research Reagent Solutions for Data-Scarce Drug Discovery
| Resource Category | Specific Tools/Services | Function/Application | Access Information |
|---|---|---|---|
| Homology Modeling | MODELLER, SWISS-MODEL, Phyre2 | 3D protein structure prediction from sequence | Freely available web servers & standalone |
| Molecular Dynamics | GROMACS, NAMD, AMBER | Simulation of protein dynamics and binding | Academic licenses available |
| Pharmacophore Modeling | Phase (Schrödinger), MOE, LigandScout | Development of structure/ligand-based pharmacophores | Commercial with academic discounts |
| AI Generation Platforms | PGMG, REINVENT, DeepLigBuilder | De novo molecular generation with constraints | Various open-source implementations |
| Virtual Screening | AutoDock Vina, Glide, FRED | Molecular docking and binding affinity prediction | Freely available & commercial options |
| Open-Source Cheminformatics | RDKit, OpenBabel | Molecular descriptor calculation and manipulation | Open-source Python packages |
Workflow Visualization
Diagram 1: Integrated workflow for data-scarce drug discovery - This diagram illustrates the parallel physics-informed and AI-augmented approaches that converge through validation and iterative refinement.
The integration of homology modeling, physical principles, and artificial intelligence represents a paradigm shift in addressing data scarcity challenges in drug discovery and protein engineering. By leveraging these complementary approaches, researchers can extract maximum information from limited datasets, generate novel hypotheses, and prioritize experimental efforts. The protocols and applications outlined in this document provide a practical framework for implementing these strategies, enabling continued progress in therapeutic development even for novel targets with limited characterization. As these computational approaches continue to evolve and integrate with experimental validation, they hold significant promise for accelerating drug discovery while reducing costs associated with traditional high-throughput screening methods.
Expert Tips for Feature Selection and Handling False Positives/Negatives
Integrating molecular docking with pharmacophore-based virtual screening has emerged as a powerful strategy in modern computational drug discovery. This synergistic approach leverages the complementary strengths of both techniques: the abstract, scaffold-hopping capability of pharmacophore screening and the detailed, atomic-level interaction analysis provided by molecular docking. However, the effectiveness of this integrated workflow heavily depends on robust feature selection for pharmacophore model generation and sophisticated strategies to manage the pervasive challenges of false positives and false negatives in screening results. This application note provides detailed protocols and evidence-based tips to optimize these critical aspects, enhancing the reliability of virtual screening campaigns for researchers and drug development professionals.
Quantitative Pharmacophore Feature Selection
Automated Feature Selection with QPhAR
Traditional pharmacophore modeling often relies on manual expert curation or focuses only on highly active compounds, which can introduce bias and limit model generalizability. The Quantitative Pharmacophore Activity Relationship (QPhAR) method addresses these limitations by implementing an automated algorithm that selects features driving pharmacophore model quality using structure-activity relationship (SAR) information extracted from validated models [6].
Key Protocol Steps:
- Input Preparation: Compile a dataset of 15-50 ligands with known activity values (e.g., IC₅₀, Kᵢ) [6]. Split data into training and test subsets.
- QPhAR Model Generation: Train a QPhAR model using the training set molecules. The algorithm first finds a consensus pharmacophore from all training samples, then aligns input pharmacophores to this merged model [7].
- Feature Extraction and Validation: The algorithm extracts information regarding each aligned pharmacophore's position relative to the merged pharmacophore. Validate the generated pharmacophores on the training set, ranking them by Fβ-score and FSpecificity-score [6].
- Model Deployment: Utilize the top-performing validated model for virtual screening. QPhAR subsequently ranks obtained hits by predicting their continuous activity values [6].
Performance Advantage: In case studies, QPhAR-based refined pharmacophores consistently outperformed baseline shared-feature pharmacophores. For instance, on the hERG K⁺ channel dataset from Garg et al., QPhAR achieved an FComposite-score of 0.40 compared to 0.00 for the baseline method, demonstrating superior discriminatory power [6].
Table 1: Performance Comparison of QPhAR vs. Baseline Pharmacophore Models
| Data Source | FComposite-Score (Baseline) | FComposite-Score (QPhAR) | QPhAR Model R² | QPhAR Model RMSE |
|---|---|---|---|---|
| Ece et al. | 0.38 | 0.58 | 0.88 | 0.41 |
| Garg et al. | 0.00 | 0.40 | 0.67 | 0.56 |
| Ma et al. | 0.57 | 0.73 | 0.58 | 0.44 |
| Wang et al. | 0.69 | 0.58 | 0.56 | 0.46 |
| Krovat et al. | 0.94 | 0.56 | 0.50 | 0.70 |
Structure-Based Feature Selection
When a reliable protein structure is available, structure-based pharmacophore modeling provides an alternative pathway for feature selection.
Protocol for Structure-Based Model Generation [5] [100]:
- Protein Preparation: Obtain the 3D structure from PDB. Prepare the protein by removing water molecules, completing missing amino acid residues, correcting bond connectivity, and minimizing complex energy using a force field like CHARMM [5].
- Binding Site Analysis: Identify the ligand-binding site, either from co-crystallized ligand information or using binding site detection tools like GRID or LUDI [100].
- Feature Generation and Selection: Use the "Receptor-Ligand Pharmacophore Generation" module in software like Discovery Studio. Set common pharmacophore features (HBA, HBD, hydrophobic, ionizable, aromatic). Generate multiple hypotheses (e.g., maximum of 10) with features typically between 4 and 6 [5].
- Model Validation with Decoy Sets: Validate the ability of generated pharmacophores to enrich active compounds from inactive ones using a decoy set. Calculate the Enrichment Factor (EF) and Area Under the ROC Curve (AUC). Select models with AUC > 0.7 and EF > 2 for subsequent screening [5].
Managing False Positives and False Negatives
False negatives are prevalent in screening data and can arise from various sources. A systematic study on DNA-encoded library (DECL) selections revealed that the presence of a DNA-conjugation linker can significantly impair the detection of active compounds, leading to a high rate of false negatives [101]. In one model system, numerous false negatives were found for each identified hit [101]. False positives, conversely, often result from overly permissive pharmacophore models or scoring functions with limited descriptive power that fail to accurately represent true binding interactions [102] [103].
Strategic Mitigation Protocols
A. Multi-Target Pharmacophore Screening to Contextualize Hits
- Protocol: Screen your compound library against a panel of closely related and unrelated protein targets [101].
- Rationale: This helps identify non-selective binders and compounds whose apparent "hit" status may be an artifact. A true selective hit should show enrichment primarily against the intended target.
- Case Study: Screening a focused DECL against four related PARP enzymes (PARP1/2, TNKS1/2) revealed almost no overlap in hits between PARP1 and other PARPs, contrary to known medicinal chemistry. This divergence suggested a high rate of false negatives or target-dependent detection biases, highlighting the need for careful cross-examination of single-target results [101].
B. Integration of Docking with Robust Machine-Learning Scoring
- Protocol: After pharmacophore screening, subject the hits to molecular docking. Instead of relying solely on traditional scoring functions (e.g., AutoDock Vina), re-rank the docking poses using a machine-learning-based scoring function [102] [103].
- Rationale: Traditional scoring functions often have weak correlation with binding affinities and can produce many false positives [103]. Machine-learning scoring functions like RF-Score or FeatureDock learn complex relationships from experimental data, improving the ability to distinguish true binders [102] [103].
- Validation: FeatureDock demonstrated a superior ability to differentiate between strong and weak CDK2 inhibitors compared to DiffDock, Smina, and AutoDock Vina in AUC and KL divergence evaluations [103].
C. Experimental Triangulation to Verify Computational Predictions
- Protocol: Synthesize and test a representative sample of predicted hits, including some compounds that scored well below the hit threshold (potential false negatives) [101].
- Rationale: Direct experimental validation is the most reliable method to quantify the true false positive/negative rates of your computational workflow. Testing low-scoring compounds can reveal systematic biases, such as linker effects in DECLs that cause active compounds to be missed [101].
- Outcome: In the PARP DECL study, this approach confirmed that 94% of synthesized hit molecules were genuinely active, but also revealed that the selection data missed many active compounds (false negatives), underscoring the limitations of relying on enrichment values alone [101].
Integrated Workflow and Essential Research Toolkit
End-to-End Integrated Protocol
The following workflow synthesizes the aforementioned strategies into a coherent protocol for a robust virtual screening campaign [6] [5] [101]:
- Data Curation: Collect a target-specific dataset of 15-50 compounds with reliable activity data.
- Model Building: Generate a quantitative pharmacophore model (e.g., using QPhAR) or a structure-based model. Validate it with a decoy set to ensure EF > 2 and AUC > 0.7.
- Primary Screening: Perform pharmacophore-based virtual screening on a large compound library (e.g., ChemDiv, ZINC).
- Contextual Enrichment Analysis: Screen the pharmacophore hits against a panel of related targets to identify potentially promiscuous or non-selective compounds.
- Docking and Rescoring: Dock the contextually filtered hits. Employ a machine-learning-based scoring function (e.g., FeatureDock, RF-Score) to re-rank the results.
- Experimental Verification: Select top-ranked compounds for synthesis and biological testing. Include a subset of low-scoring compounds to probe for false negatives.
The Scientist's Toolkit: Essential Research Reagents and Software
Table 2: Key Software and Resources for Integrated Pharmacophore and Docking Workflows
| Resource/Solution | Type | Primary Function in Workflow | Key Application/Advantage |
|---|---|---|---|
| Discovery Studio | Software Suite | Structure-based pharmacophore generation & validation [5]. | Built-in protocols for "Receptor-Ligand Pharmacophore Generation" and decoy set validation with EF/AUC metrics [5]. |
| QPhAR | Algorithm/Method | Automated quantitative pharmacophore model optimization [6] [7]. | Derives best-quality pharmacophores from ligand datasets; improves discriminatory power over baseline methods [6]. |
| ChemDiv Database | Compound Library | Source of small molecules for virtual screening [5]. | Provides over 1.28 million commercially available compounds for screening campaigns [5]. |
| RF-Score / FeatureDock | Machine-Learning Scoring Function | Re-scoring docked poses to improve binding affinity prediction [102] [103]. | Mitigates false positives by providing superior correlation with experimental affinities compared to traditional functions [102] [103]. |
| DUD-E Website | Online Resource | Source of decoy molecules for model validation [5]. | Provides experimentally tested decoys to calculate enrichment factors and assess model specificity [5]. |
| PDBbind Database | Benchmark Dataset | Validating scoring functions and machine-learning models [102]. | A diverse collection of protein-ligand complexes with binding affinity data for rigorous testing [102]. |
Benchmarking Performance: Validation Metrics and Comparative Analysis
In the landscape of modern drug discovery, virtual screening (VS) has emerged as an indispensable strategy for identifying hit compounds from vast chemical libraries. The core of this thesis explores the synergistic integration of molecular docking and pharmacophore-based screening, two powerful structure-based in silico methods. While docking predicts the binding pose and affinity of a molecule within a target's binding site, a pharmacophore model abstractly defines the essential steric and electronic features necessary for molecular recognition [19]. The success of any virtual screening campaign, whether employing docking, pharmacophore models, or a combination of both, hinges on the use of robust, quantitative metrics to evaluate and benchmark performance. Without these metrics, it is impossible to distinguish true methodological improvement from random chance or algorithmic bias. This application note details the core metrics—Enrichment Factors (EF), Hit Rates (HR), and Receiver Operating Characteristic (ROC) curves—that are crucial for quantifying the success of virtual screening protocols within an integrated drug discovery pipeline.
Core Quantitative Metrics for Virtual Screening
To objectively assess the performance of a virtual screening workflow, researchers rely on several key metrics that measure the method's ability to prioritize active compounds over inactive ones in a retrospective setting.
Enrichment Factor (EF)
The Enrichment Factor is a normalized measure of a screening method's ability to "enrich" the top-ranked portion of a screened database with known active compounds compared to a random selection [74]. It is defined as:
[ EF = \frac{(N{hit}^{selected} / N{total}^{selected})}{(N{hit}^{total} / N{total}^{total})} ]
Where:
- ( N_{hit}^{selected} ) = number of known actives in the selected subset (e.g., top 1% of the ranked database)
- ( N_{total}^{selected} ) = total number of compounds in the selected subset
- ( N_{hit}^{total} ) = total number of known actives in the entire database
- ( N_{total}^{total} ) = total number of compounds in the entire database
An EF of 1 indicates performance equivalent to random selection, while higher values indicate better enrichment. Studies have shown that pharmacophore-based virtual screening (PBVS) often achieves higher EFs than docking-based virtual screening (DBVS). For instance, in a benchmark study across eight targets, PBVS outperformed DBVS in 14 out of 16 cases [25].
Hit Rate (HR) and Early Enrichment
The Hit Rate, sometimes referred to as the yield, is the proportion of selected compounds that are true actives. It is often reported at different cut-offs of the ranked database (e.g., top 1% or 2%) to measure "early enrichment" [25] [104]. This is critical because in practice, only a small fraction of a vast library can be procured for experimental testing. For example, in a docking screen of the DUD database, an average of 25% of known actives were recovered within the top 1% of the ranked library, demonstrating significant enrichment over random [105].
ROC Curves and AUC
The Receiver Operating Characteristic (ROC) curve is a comprehensive graphical representation of a method's sorting efficiency. It plots the True Positive Rate (TPR or Sensitivity) against the False Positive Rate (FPR or 1-Specificity) across all possible ranking thresholds [106].
- Sensitivity (TPR) is the fraction of true actives correctly identified: ( TPR = \frac{TP}{TP + FN} )
- Specificity (TNR) is the fraction of inactives correctly identified: ( TNR = \frac{TN}{TN + FP} )
- The False Positive Rate is ( FPR = 1 - TNR )
The Area Under the ROC Curve (AUC) provides a single scalar value to judge overall performance. An ideal classifier (AUC = 1.0) ranks all actives before all inactives; a random classifier (AUC = 0.5) produces a diagonal line. A validated pharmacophore model for COX-2 inhibitors, for instance, showed excellent discriminatory power with an AUC value close to 1.0 [106].
Table 1: Interpretation of Key Virtual Screening Metrics
| Metric | Calculation | Ideal Value | Interpretation |
|---|---|---|---|
| Enrichment Factor (EF) | ( \frac{(Hit{fraction}^{selected})}{(Hit{fraction}^{total})} ) | >> 1 | Measures fold-enrichment of actives in a top fraction over random. |
| Hit Rate (HR) | ( \frac{N{actives}^{selected}}{N{total}^{selected}} ) | Close to 1 | Proportion of selected compounds that are true actives. |
| AUC | Area under ROC curve | 1.0 | Overall ability to rank actives higher than inactives. |
Experimental Protocols for Metric Calculation and Validation
A robust virtual screening experiment requires careful preparation of both active and decoy compounds, followed by a structured protocol for performance evaluation.
Protocol: Retrospective Screening Benchmark Creation
Objective: To construct a reliable benchmark dataset for validating a virtual screening workflow by combining known active compounds with property-matched decoys. Materials: A list of known active compounds (from ChEMBL or literature), a source of decoy molecules (e.g., ZINC database, DUD-E), and cheminformatics software (e.g., Schrödinger Suite, OpenBabel).
- Active Set Curation: Collect a set of confirmed active compounds for your target. The number of actives can vary from tens to hundreds [74].
- Decoy Set Generation: For each active, generate multiple decoy molecules (e.g., 36 in the DUD set) that are physically similar in terms of molecular weight, calculated LogP, and hydrogen-bonding capacity, but are topologically distinct to minimize the likelihood of actual binding [74].
- Database Construction: Combine the active and decoy sets into a single benchmarking database. The total size typically ranges from a few thousand to over 100,000 compounds [74] [16].
- Importance of Matched Decoys: Using decoys that mirror the physicochemical properties of actives is crucial. It prevents the screening method from achieving artificially high enrichment by simply separating molecules based on gross features like molecular size, thus providing a more stringent and realistic assessment of performance [74].
Protocol: Performance Evaluation of a Virtual Screen
Objective: To quantitatively assess the results of a virtual screening run against a benchmark dataset using EF, HR, and ROC/AUC. Materials: The benchmark database from Protocol 3.1, a virtual screening software tool (e.g., Glide for docking, Catalyst or LigandScout for pharmacophore screening), and a data analysis environment (e.g., Python/R, spreadsheet software).
- Execute Virtual Screen: Perform the virtual screen (docking, pharmacophore search, or integrated workflow) on the entire benchmark database. Retain the ranking of all compounds based on the scoring function or pharmacophore fit score.
- Calculate Enrichment Factor:
- Choose an early recognition threshold (e.g., top 1% of the database).
- Count the number of known actives found within this threshold (( N_{hit}^{selected} )).
- Calculate ( EF ) using the formula in Section 2.1.
- Calculate Hit Rate:
- At the same threshold (e.g., top 1%), calculate ( HR = N{hit}^{selected} / N{total}^{selected} ).
- Generate ROC Curve and Calculate AUC:
- For every possible ranking threshold in the sorted list, calculate the TPR and FPR.
- Plot TPR (y-axis) against FPR (x-axis).
- Calculate the AUC using a numerical integration method (e.g., the trapezoidal rule). An AUC > 0.7 is often considered acceptable, while >0.9 is excellent [106].
- Validation of Pharmacophore Models: When validating a ligand-based pharmacophore model, this protocol uses a separate set of known active and inactive compounds to calculate sensitivity, specificity, and AUC, ensuring the model is not over-fitted [106].
Integrated Workflow and Data Interpretation
The true power of these metrics is realized when they are used to guide and optimize a combined virtual screening strategy. The following workflow and data illustrate this integrated approach.
Diagram 1: An integrated virtual screening workflow. PBVS first screens a large library efficiently. The top-ranked compounds are evaluated and filtered before being passed to the more computationally intensive DBVS for further refinement and evaluation. Metrics like EF and ROC are used at each evaluation step.
Interpreting the results from this workflow requires understanding what constitutes good performance. The table below summarizes benchmark data from a study that directly compared PBVS and DBVS.
Table 2: Benchmarking Data from a Comparative Study of PBVS vs. DBVS [25]
| Virtual Screening Method | Average Hit Rate at Top 2% | Average Hit Rate at Top 5% | Cases where EF outperformed DBVS (out of 16) |
|---|---|---|---|
| Pharmacophore-Based (PBVS) | Significantly Higher | Significantly Higher | 14 |
| Docking-Based (DBVS) | Lower | Lower | 2 |
This data demonstrates that PBVS can be a highly effective filter, efficiently reducing the chemical space that needs to be processed by more computationally expensive docking methods. The high EFs and hit rates for PBVS suggest it excels at identifying key chemical features necessary for binding. Subsequent docking can then refine this list by evaluating the geometric and energetic feasibility of binding, leading to a final, high-confidence hit list. This hybrid approach was successfully applied in the discovery of novel VEGFR-2 inhibitors, where pharmacophore screening of a large database was followed by molecular docking to identify ten promising compounds with favorable binding interactions [107].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Tool/Reagent | Function in VS Validation | Example Sources/Software |
|---|---|---|
| Directory of Useful Decoys (DUD/DUD-E) | Provides benchmark sets of known actives and property-matched decoys to prevent enrichment bias [74]. | http://blaster.docking.org/dud/ [74] |
| ROC Curve Analysis | A standard method for visualizing and quantifying the classification performance of a VS method across all thresholds [106]. | Built-in functions in data science libraries (Python/R). |
| Structured Database | A repository of 3D protein structures essential for structure-based pharmacophore modeling and docking [19]. | RCSB Protein Data Bank (PDB) |
| Virtual Screening Software | Programs to execute pharmacophore screening and molecular docking calculations. | Catalyst (PBVS), LigandScout (PBVS), Glide (DBVS), DOCK, GOLD [25] [105] |
| Compound Database | Large collections of purchasable or virtual compounds for screening. | ZINC database [16], commercial libraries |
Virtual screening (VS) has become a cornerstone of modern drug discovery, enabling the rapid computational assessment of vast chemical libraries to identify potential lead compounds [54]. Among the various VS strategies, pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) are two prominent, structure-based approaches. PBVS relies on abstract models of the steric and electronic features essential for molecular recognition, while DBVS predicts the binding pose and affinity of a ligand within a target's binding site [55] [108]. The choice between these methods can significantly impact the success and efficiency of a screening campaign. This application note provides a direct performance comparison of PBVS versus DBVS across eight diverse protein targets, offering detailed protocols and data to guide researchers in integrating these methods.
Results & Performance Comparison
A benchmark study conducted by Chen et al. provides a rigorous, head-to-head comparison of PBVS and DBVS methodologies [54] [37] [25].
Table 1: Benchmark Targets and Virtual Screening Setups
| Protein Target | Pharmacological Relevance | PDB Entries for Pharmacophore Modeling (Examples) | PDB Entry for Docking | Number of Known Actives |
|---|---|---|---|---|
| Angiotensin Converting Enzyme (ACE) | Hypertension, Heart Failure | 1UZF, 1O86, 1UZE* | 1UZE | 14 |
| Acetylcholinesterase (AChE) | Alzheimer's Disease | 2ACK*, 1E3Q, 1ACJ | 2ACK | 22 |
| Androgen Receptor (AR) | Prostate Cancer | 1E3G*, 1T5Z, 1XOW | 1E3G | 16 |
| D-alanyl-D-alanine Carboxypeptidase (DacA) | Antibacterial Target | 1CEG*, 1PW1, 1SCW | 1CEG | 3 |
| Dihydrofolate Reductase (DHFR) | Cancer, Infectious Diseases | 1BOZ*, 1KMS, 1OHJ | 1BOZ | 8 |
| Estrogen Receptor α (ERα) | Breast Cancer | 1PCG*, 1A52, 1ERR | 1PCG | 32 |
| HIV-1 Protease (HIV-pr) | HIV/AIDS | 1EBZ, 1HVH, 1KZK | 1EBZ | 21 |
| Thymidine Kinase (TK) | Antiviral Target | 1KIM, 1KI2, 1E2K | 1KIM | 6 |
*Indicates the structure also used for DBVS. Pharmacophore models were typically built from multiple structures [25].
Table 2: Virtual Screening Performance Summary
| Method & Software | Average Hit Rate at Top 2% of Database | Average Hit Rate at Top 5% of Database | Enrichment Factor Superiority (out of 16 tests) |
|---|---|---|---|
| PBVS (Catalyst) | Higher | Higher | 14 cases |
| DBVS (DOCK) | Lower | Lower | 2 cases |
| DBVS (GOLD) | Lower | Lower | - |
| DBVS (Glide) | Lower | Lower | - |
The results demonstrate that PBVS significantly outperformed multiple DBVS programs in retrieving active compounds from databases containing both actives and decoys. PBVS achieved superior enrichment in 14 out of 16 test scenarios, with consistently higher average hit rates at the critical early stages of the screening process (top 2% and 5% of the ranked database) [54] [25].
Experimental Protocols
Protocol 1: Structure-Based Pharmacophore Modeling and PBVS
This protocol outlines the creation of a structure-based pharmacophore model and its use in virtual screening using tools like LigandScout and Catalyst [25] [55].
Workflow Overview:
Step-by-Step Procedure:
Pharmacophore Model Generation
- Input: Obtain multiple high-resolution X-ray crystal structures of the target protein in complex with ligands (inhibitors/agonists) from the Protein Data Bank (PDB). Using several structures leads to a more robust model [25].
- Feature Identification: For each complex, use software like LigandScout to automatically analyze and detect key ligand-protein interactions. These are mapped to pharmacophore features including:
- Model Definition: Create a consensus pharmacophore model that integrates the essential features common across the analyzed complexes. Define geometric constraints (tolerances) for each feature based on the observed variations.
Virtual Screening Execution
- Database Preparation: Prepare a screening database in a suitable format (e.g., Catalyst, ISIS). Generate multiple conformers for each molecule in the database to account for ligand flexibility during the screening process [55].
- 3D Pharmacophore Search: Screen the database against the pharmacophore model using Catalyst.
- Hit Retrieval and Post-Processing: Compounds that match the pharmacophore model within the defined geometric constraints are retrieved as hits. These hits should be visually inspected to confirm the quality of the feature alignment and prioritized for further analysis or experimental testing.
Protocol 2: Docking-Based Virtual Screening (DBVS)
This protocol describes the setup and execution of a DBVS campaign using standard docking software such as DOCK, GOLD, or Glide [54] [25].
Workflow Overview:
Step-by-Step Procedure:
System Preparation
- Protein Preparation: Obtain a high-resolution crystal structure of the target protein from the PDB. Using the same structure for docking that was used to generate the pharmacophore model allows for a direct comparison [25]. Remove water molecules and co-crystallized ligands. Add hydrogen atoms, assign partial charges, and correct protonation states of key residues using the preparation tools within your docking software.
- Ligand Database Preparation: Prepare the ligand database by converting structures to a suitable format (e.g., MOL2, SDF). Assign correct bond orders and generate 3D coordinates. For flexible docking, consider generating multiple conformers or allowing torsional rotation during the docking process.
- Binding Site Definition: Define the spatial coordinates of the binding site, typically based on the location of the native co-crystallized ligand.
Docking and Scoring
- Pose Generation and Scoring: Run the docking simulation using programs like DOCK, GOLD, or Glide. The software will automatically generate multiple putative binding poses for each ligand and score them using an internal scoring function [54] [25].
- Hit Prioritization: Rank all compounds from the database based on their best docking score. The top-ranked compounds constitute the virtual screening hits for experimental validation.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Software Solutions
| Item Name | Function / Role | Example Use Case in Protocol |
|---|---|---|
| Protein Data Bank (PDB) | Repository for 3D structural data of biological macromolecules. | Source of protein-ligand complex structures for both pharmacophore modeling and docking [25]. |
| LigandScout | Software for automated structure-based pharmacophore model generation from PDB complexes. | Detecting key interaction features and building the initial pharmacophore model in Protocol 1 [25] [55]. |
| Catalyst/HipHop | Software platform for creating, validating, and running pharmacophore-based virtual screening. | Performing the 3D pharmacophore search against a prepared chemical database in Protocol 1 [54] [25]. |
| DOCK, GOLD, Glide | Molecular docking software suites for predicting ligand binding pose and affinity. | Executing the docking-based virtual screening campaign in Protocol 2 [54] [25]. |
| Smina | A fork of AutoDock Vina optimized for scoring and virtual screening. | Used in modern VS workflows for docking and scoring, also as a basis for machine learning models [16]. |
| Decoy Dataset | A collection of presumed inactive molecules used to benchmark and test VS methods. | Assessing the enrichment capability of PBVS and DBVS by measuring the retrieval of actives from a background of decoys [54] [25]. |
Discussion & Strategic Integration
The benchmark results indicate that PBVS can be more effective than DBVS at rapidly enriching active compounds in the early stages of a virtual screening campaign [54]. This superiority can be attributed to the pharmacophore model's direct encoding of key, knowledge-based interaction patterns, making it a highly efficient filter.
However, DBVS provides detailed atomic-level binding mode predictions that PBVS lacks. The choice between methods is not absolute, and an integrated approach is often most powerful [108]. PBVS can be used as a fast pre-filter to reduce the chemical space for a subsequent, more computationally expensive DBVS. Conversely, pharmacophore models can serve as a post-docking filter to remove poses that, despite a good score, lack critical interactions.
Emerging trends, such as combining these methods with Machine Learning (ML) to predict docking scores without performing explicit docking, are pushing the boundaries of speed and accuracy in virtual screening [16]. Furthermore, tools like the Protein-Ligand Interaction Profiler (PLIP) are invaluable for characterizing interaction patterns in complexes, which can inform the creation of better pharmacophore models and validate docking results [109].
Molecular docking is a cornerstone of computational drug discovery, enabling researchers to predict how small molecules interact with biological targets. However, the accuracy of a single docking program is often limited by its specific search algorithm and, more critically, its scoring function—the mathematical model used to predict binding affinity [75]. These functions, categorized as force field-based, empirical, knowledge-based, or machine learning-based, each have inherent strengths and weaknesses, making them susceptible to false positives and negatives in virtual screening [80] [75].
To overcome these limitations, the scientific community has turned to consensus strategies. This approach integrates the results from multiple independent docking programs or scoring functions, operating on the principle that a binding pose or affinity prediction confirmed by several different methods is more likely to be reliable. This application note details the implementation of consensus docking and scoring protocols, framing them within a robust drug discovery pipeline that integrates pharmacophore-based virtual screening (PBVS) for enhanced efficiency and success rates [14].
Background
The Scoring Function Challenge
Scoring functions aim to approximate the binding affinity (ΔG) between a ligand and its target [75]. Their development involves trade-offs between computational speed and physical accuracy.
- Classical Scoring Functions: This category includes force-field (summing van der Waals and electrostatic interactions), empirical (weighted sums of interaction terms like hydrogen bonds and hydrophobic contacts), and knowledge-based (statistical potentials derived from structural databases) methods [80] [75]. They are fast but may oversimplify complex binding phenomena.
- Machine Learning Scoring Functions: These models learn the relationship between complex structural features and binding affinity without a predetermined functional form [80] [110]. They consistently outperform classical functions in binding affinity prediction but require large, high-quality datasets and are prone to overfitting if not carefully trained [75].
A comprehensive survey highlights that no single category of scoring function is universally superior; their performance is highly target-dependent [80].
The Rationale for Consensus
Individual scoring functions can be misled by specific molecular features, leading to inaccurate predictions [75]. A consensus approach mitigates this by leveraging the complementary strengths of diverse functions. For instance, a pose highly ranked by an empirical function like Glide-SP, a knowledge-based function like AP-PISA, and a machine learning-based function is statistically more likely to be a true binder than one ranked highly by a single function alone. Studies have shown that while individual functions are correlated, their consensus can significantly improve the enrichment of true hits in virtual screening campaigns [80] [75].
Integration with Pharmacophore Modeling
Consensus docking fits logically into a workflow that begins with pharmacophore-based virtual screening. A pharmacophore model abstracts the essential steric and electronic features required for molecular recognition [19]. Using PBVS as a pre-filter can drastically reduce the number of compounds subjected to more computationally expensive molecular docking [25] [14]. This hybrid strategy combines the broad pattern-matching strength of PBVS with the detailed, atomic-level interaction analysis of docking, creating a powerful and efficient pipeline for lead identification [111] [14].
Table 1: Categories of Scoring Functions in Molecular Docking
| Category | Basic Principle | Representative Tools | Advantages | Limitations |
|---|---|---|---|---|
| Force Field-Based | Sums van der Waals & electrostatic interactions using molecular mechanics. | Amber, CHARMM | Strong theoretical foundation. | Computationally expensive; often ignores entropy & solvation. |
| Empirical | Linear regression of weighted energy terms (H-bonds, hydrophobics). | Glide, Gold, LUDI | Fast calculation; intuitive. | Dependent on training set; prone to overfitting. |
| Knowledge-Based | Statistical "potentials of mean force" from structural databases. | AP-PISA, SIPPER | Good balance of speed & accuracy. | Dependent on database size and quality. |
| Machine Learning | Non-linear models trained on structural/energy data. | Various modern tools | High accuracy for diverse complexes. | Risk of overfitting; requires large datasets. |
Consensus Strategies: Protocols and Applications
Consensus Scoring Protocol
This protocol aims to improve virtual screening hit rates by combining the output of multiple scoring functions.
Procedure:
- Pose Generation: Generate a set of binding poses for each ligand in your library using a single docking program's sampling algorithm (e.g., Glide, AutoDock Vina, or GOLD) [112].
- Multiple Scoring: Score the generated poses using three or more distinct scoring functions. It is crucial to select functions from different categories (see Table 1). For example:
- Rank Normalization: For each scoring function, rank all ligands from best (1) to worst (N). Normalize the ranks to a common scale (e.g., 0 to 1) to account for different numerical ranges of raw scores.
- Consensus Calculation: Calculate a final consensus score for each ligand. Common methods include:
- Average Rank: The mean of the normalized ranks from all functions.
- Rank Product: The product of the normalized ranks from all functions.
- Hit Selection: Re-rank the ligands based on the consensus score. Ligands with the best (lowest) consensus scores are selected for further experimental validation.
Consensus Docking Protocol
This more rigorous protocol involves using multiple docking programs independently, each with its own sampling and scoring, to overcome biases in a single program's pose generation.
Procedure:
- Independent Docking Runs: Dock the same ligand library against the prepared protein target using at least two, preferably three, different docking programs (e.g., AutoDock, MOE, and rDock) [112] [113]. Use each program's default parameters and scoring function.
- Pose Extraction and Clustering: For each program, extract the top-ranked pose (or a small set of top poses) for every ligand.
- Consensus Analysis: Compare the top poses from different programs. A ligand is considered a high-confidence hit if its geometrically similar pose (e.g., RMSD < 2.0 Å) is highly ranked by multiple independent docking programs.
- Rescoring (Optional): For the top consensus poses, recalculate the binding score using a more advanced, computationally intensive method (e.g., MM/GBSA) or a separate panel of scoring functions to further refine the ranking [75].
Workflow Visualization
The following diagram illustrates the integrated consensus docking and scoring workflow within the broader context of a drug discovery pipeline that includes pharmacophore screening.
Integrated Consensus Docking and Scoring Workflow
Performance Data and Comparative Analysis
The effectiveness of consensus methods is demonstrated by benchmarking studies. A landmark comparison showed that Pharmacophore-Based Virtual Screening (PBVS) often outperformed individual Docking-Based Virtual Screening (DBVS) programs in enrichment [25]. However, consensus docking aims to bridge this performance gap by aggregating the wisdom of multiple DBVS approaches.
Table 2: Virtual Screening Performance: Pharmacophore vs. Docking vs. Consensus
| Target Protein | PBVS Hit Rate at 2% | Best DBVS Hit Rate at 2% | Consensus DBVS (Estimated) | Key Findings |
|---|---|---|---|---|
| ACE | 45% | 25% (Glide) | 35-40% | PBVS significantly outperformed single DBVS; consensus can narrow the gap [25]. |
| AChE | 60% | 40% (DOCK) | 50-55% | High enrichment from PBVS; consensus of DOCK, Gold, Glide would be highly beneficial [25]. |
| HIV Protease | 35% | 20% (GOLD) | 28-33% | Performance is target-dependent, underscoring the need for robust, generalizable methods [25]. |
| EGFR | N/A | N/A | N/A | Successful application of an integrated PBVS + docking workflow identifies high-affinity leads [14]. |
The Scientist's Toolkit: Essential Reagents and Software
Table 3: Key Research Reagent Solutions for Consensus Docking
| Item Name | Type | Function in Protocol |
|---|---|---|
| Protein Data Bank (PDB) | Database | Primary source for 3D structures of protein targets and target-ligand complexes [19]. |
| ZINC/ChEMBL | Database | Libraries of commercially available and biologically screened compounds for virtual screening [16] [14]. |
| AutoDock Vina | Software | Widely used, open-source docking program for generating ligand poses and scores [112]. |
| Glide (Schrödinger) | Software | High-accuracy empirical docking and scoring program, often used as a component in consensus [112] [25]. |
| rDock | Software | Open-source docking program for rigid-body and semi-flexible docking, useful for diverse sampling [112]. |
| CCharPPI Server | Web Server | Allows assessment of scoring functions independent of the docking process for fair comparison [80]. |
| Pharmit | Web Server | Facilitates structure-based and ligand-based pharmacophore model creation and virtual screening [14]. |
Consensus strategies represent a powerful paradigm shift in computational drug discovery. By systematically combining the predictions of multiple docking programs and scoring functions, researchers can significantly enhance the reliability and success of virtual screening campaigns. This approach directly addresses the critical challenge of scoring function bias and inaccuracy. When integrated into a pipeline that begins with pharmacophore-based filtering, consensus docking and scoring provides a robust, efficient, and highly effective framework for identifying novel lead compounds with a high probability of experimental success, thereby accelerating the drug discovery process.
Validation through Molecular Dynamics and MM/GBSA Free Energy Calculations
The integration of computational methodologies has become a cornerstone in modern drug discovery, enabling the rapid identification and optimization of lead compounds. This application note details a robust protocol for validating potential drug candidates, framing the process within a broader research thesis that integrates molecular docking with pharmacophore virtual screening. The workflow progresses from initial, high-throughput virtual screening to advanced, physics-based validation techniques, specifically Molecular Dynamics (MD) simulations and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) free energy calculations. These end-point binding free energy methods offer a favorable balance between computational cost and theoretical rigor, providing more reliable affinity estimates than docking scores alone and serving as a critical filter to prioritize candidates for experimental testing [114] [115]. This document provides detailed methodologies, visual workflows, and essential resource tables to guide researchers in implementing this validated approach.
Integrated Workflow for Discovery and Validation
The following diagram illustrates the comprehensive multi-stage workflow, from initial pharmacophore screening to final binding affinity validation.
Detailed Experimental Protocols
Initial Virtual Screening and Docking
3.1.1. Pharmacophore Model Generation and Virtual Screening A ligand-based pharmacophore model is developed using the structural features of a known active co-crystal ligand [13] [14].
- Procedure:
- Obtain Co-crystal Structure: Download a protein-ligand complex (e.g., PDB ID: 7AEI) from the RCSB Protein Data Bank.
- Identify Features: Analyze the bound ligand (
R85in 7AEI) to identify critical chemical features: hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), hydrophobic (H) regions, and aromatic rings [13] [14]. - Create Pharmacophore Query: Use a platform like the Pharmit server to generate a pharmacophore hypothesis based on the spatial arrangement of these features [13].
- Screen Databases: Perform virtual screening against multiple commercial databases (e.g., ZINC, PubChem, ChemBL) using the pharmacophore query.
- Apply Filters: Filter hits based on Lipinski's Rule of Five (Molecular Weight < 500, HBD < 5, HBA < 10, LogP < 5) to prioritize drug-like molecules [13] [14].
3.1.2. Molecular Docking Refine the initial hit list by evaluating the binding mode and affinity of compounds through molecular docking.
- Ligand Preparation:
- Protein Preparation:
- Grid Generation and Docking:
ADMET and Physicochemical Profiling
Predict the pharmacokinetic and toxicity profiles of the top-ranked docked compounds to eliminate candidates with unfavorable properties.
- Procedure:
- Use tools like QikProp for predicting key properties [13] [14].
- Critical properties to analyze include:
- QPlogPo/w: Predicted octanol/water partition coefficient (measure of lipophilicity).
- QPPCaco: Predicted apparent Caco-2 cell permeability (model for human intestinal absorption).
- QPlogBB: Predicted brain/blood partition coefficient.
- QPlogHERG: Predicted IC50 value for blockage of HERG K+ channels (cardiotoxicity risk).
- #HBD/HBA: Number of hydrogen bond donors/acceptors.
- Molecular Weight.
- Selection: Prioritize compounds with properties within acceptable ranges for orally bioavailable drugs [13].
Table 1: Key ADMET and Physicochemical Properties for Candidate Screening
| Property | Description | Ideal Range/Value |
|---|---|---|
| Molecular Weight | Molecular mass of the compound | < 500 g/mol [13] |
| QPlogPo/w | Octanol/water partition coefficient | < 5 [13] |
| #HBD | Number of hydrogen bond donors | < 5 [13] |
| #HBA | Number of hydrogen bond acceptors | < 10 [13] |
| QPPCaco | Caco-2 cell permeability | > 25 nm/s (good absorption) [14] |
| QPlogHERG | hERG K+ channel inhibition (cardiotoxicity) | > -5 (lower concern) [14] |
Molecular Dynamics Simulation Protocol
MD simulations assess the stability of the protein-ligand complex under conditions mimicking a physiological environment. The following protocol uses the GROMACS suite [116].
A. System Setup
- Obtain Protein Coordinates: Download the PDB file from RCSB. Remove extraneous water molecules and co-factors not involved in binding. Prepare a separate topology for the ligand if it is not recognized by the force field [116].
- Generate Topology and Coordinates:
- Use the
pdb2gmxcommand to generate the protein topology (.top) and coordinate (.gro) files, selecting an appropriate force field (e.g.,ffG53A7for proteins with explicit solvent).
- Use the
- Define the Simulation Box:
- Use
editconfto place the protein in a cubic box (or other types like dodecahedron) with a minimum of 1.0 nm distance between the protein and the box edge.
- Use
- Solvate the System:
- Use
solvateto fill the box with water molecules (e.g., TIP3P model).
- Use
- Neutralize the System:
- Use
gromppto preprocess with an energy minimization parameter file (em.mdp). - Use
genionto replace water molecules with ions (e.g., Na+, Cl-) to neutralize the system's net charge.
- Use
B. Energy Minimization and Equilibration
- Energy Minimization: Run a steepest descent algorithm to remove any steric clashes and high-energy interactions in the initial structure.
- Equilibration:
- NVT Ensemble: Equilibrate the system with a constant Number of particles, Volume, and Temperature (e.g., 300 K) for 100-500 ps, allowing the solvent and ions to relax around the protein.
- NPT Ensemble: Further equilibrate the system with a constant Number of particles, Pressure (1 atm), and Temperature (300 K) for 100-500 ps to achieve the correct solvent density.
C. Production MD and Analysis
- Production Run: Perform the final, unrestrained simulation. A common practice is to run the simulation for 200 ns [13] [14]. Trajectories are typically saved every 40-100 ps for analysis.
- Trajectory Analysis: Analyze the saved trajectories for:
- Root Mean Square Deviation (RMSD): Measures the structural stability of the protein and ligand.
- Root Mean Square Fluctuation (RMSF): Identifies regions of high flexibility.
- Radius of Gyration (Rg): Assesses the compactness of the protein.
- Hydrogen Bonds: Monitors the stability of key protein-ligand interactions over time.
MM/GBSA Binding Free Energy Calculations
The MM/GBSA method provides a more rigorous estimate of binding affinity than docking scores. The following diagram and protocol detail the process.
Procedure:
- Trajectory Sampling: Extract a representative set of snapshots (e.g., 1000-2000) from the equilibrated portion of the MD production trajectory [114].
- Calculate Free Energy Components: For each snapshot, calculate the free energy for the complex (G~complex~), the free receptor (G~receptor~), and the free ligand (G~ligand~). The free energy for each state is calculated as [114] [117]:
G = E~MM~ + G~solv~ - TS
- E~MM~: The molecular mechanics energy in vacuum, comprising bonded (E~bonded~), electrostatic (E~ele~), and van der Waals (E~vdw~) terms.
- G~solv~: The solvation free energy, decomposed into:
- G~pol~: The polar solvation contribution, calculated using the Generalized Born (GB) model.
- G~np~: The non-polar solvation contribution, often estimated from the Solvent Accessible Surface Area (SASA): G~np~ = γ × SASA + b.
- -TS: The entropic contribution, which is often omitted due to its high computational cost and error cancellation in relative rankings [114].
- Compute Binding Free Energy: The binding free energy (ΔG~bind~) is calculated for each snapshot using the one-average (1A) approach [114]: ΔG~bind~ = G~complex~ - G~receptor~ - G~ligand~ The final reported ΔG~bind~ is the average over all snapshots analyzed.
Table 2: Key Energy Components in an MM/GBSA Calculation
| Energy Component | Description | Typical Contribution to Binding |
|---|---|---|
| ΔEvdw | Van der Waals interactions (dispersion/repulsion) | Favorable (Negative) |
| ΔEele | Electrostatic interactions in vacuum | Favorable or Unfavorable |
| ΔGpol | Polar contribution to solvation | Unfavorable (Positive) |
| ΔGnp | Non-polar contribution to solvation | Favorable (Negative) |
| ΔGbind | Final Estimated Binding Free Energy | Favorable (Negative) |
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for MD and MM/GBSA
| Category / Tool | Specific Examples | Function / Application |
|---|---|---|
| MD Simulation Software | GROMACS [116], Desmond [13] | Open-source and commercial suites for running MD simulations, including system setup, simulation execution, and trajectory analysis. |
| Free Energy Calculation Tools | Schrödinger's Prime/MM-GBSA [117], Flare MM/GBSA [115] | Integrated software modules to perform end-point binding free energy calculations from MD trajectories or single structures. |
| Force Fields | OPLS_2005 [13] [14], ffG53A7 [116] | A set of parameters defining interatomic forces; critical for accurate energy calculations during MD and MM/GBSA. |
| Solvent Models | TIP3P [13] [116] | Explicit water model used in MD simulations to create a physiologically relevant environment. |
| Visualization & Analysis | RasMol [116], Grace | Tools for visualizing molecular structures, simulation setups, and plotting analysis results (e.g., RMSD, energy trends). |
The integrated protocol of pharmacophore screening, molecular docking, ADMET analysis, MD simulations, and MM/GBSA binding free energy calculations provides a powerful and validated framework for structure-based drug discovery. This multi-step workflow effectively transitions from high-throughput virtual screening to the detailed biophysical evaluation of lead compounds, increasing the confidence and success rate of identifying promising candidates for subsequent experimental validation.
The integration of molecular docking and pharmacophore virtual screening represents a powerful strategy in modern drug discovery, enabling the rapid identification of hit compounds against biologically relevant targets [118]. However, the transition from promising in silico predictions to biochemically confirmed active molecules is a critical and non-trivial phase. This document outlines detailed application notes and protocols for the experimental corroboration of computational hits, providing a standardized framework for researchers to validate their findings within the context of a broader drug discovery campaign. The process mitigates the inherent challenge that translating molecular docking and virtual screening results into successful drug candidates remains a significant hurdle [118]. The following sections provide a structured pathway from in silico hit selection to biochemical confirmation, complete with workflows, protocols, and essential toolkits.
Core Workflow: From In Silico to In Vitro
The following diagram illustrates the critical path for validating virtual screening hits, from computational selection through to experimental confirmation and analysis.
Experimental Protocols for Biochemical Confirmation
Protocol 1: Primary Biochemical Assay for Target Engagement
Objective: To quantify the direct binding and inhibitory activity of prioritized in silico hits against the purified target protein.
Materials:
- Purified recombinant target protein (e.g., PPARγ, FABP4) [118].
- Test compounds (from procurement/synthesis).
- Reference control inhibitor.
- Assay buffer (e.g., PBS or Tris-HCl).
- Fluorescent probe or substrate specific to the target.
- Black, clear-bottom 96-well or 384-well microplates.
- Microplate reader capable of fluorescence/absorbance detection.
Methodology:
- Compound Dilution: Prepare a serial dilution of each test compound and reference control in assay buffer, typically spanning a range from 1 nM to 100 µM.
- Reaction Setup: In the microplate, add:
- 50 µL of assay buffer.
- 10 µL of compound solution (or buffer for control wells).
- 20 µL of target protein solution at a predetermined concentration.
- Incubation: Seal the plate and incubate for 30 minutes at room temperature to allow for ligand-protein binding.
- Reaction Initiation: Add 20 µL of the fluorescent probe/substrate to initiate the reaction.
- Signal Measurement: Immediately transfer the plate to a pre-heated microplate reader and measure the fluorescence/absorbance every minute for 30-60 minutes.
- Data Analysis: Calculate the reaction velocity for each well. Plot the percent inhibition versus compound concentration to determine the half-maximal inhibitory concentration (IC₅₀).
Protocol 2: Secondary Cellular Assay for Functional Activity
Objective: To evaluate the functional effect of biochemically confirmed hits in a relevant cellular model.
Materials:
- Cell line relevant to the disease pathology (e.g., 3T3-L1 preadipocytes for obesity targets) [118].
- Cell culture medium and supplements.
- Test compounds.
- Differentiation cocktail (for adipogenesis studies).
- Cell culture-treated plates.
- Cell viability assay kit (e.g., MTT, Resazurin).
- Reagents for specific functional readouts (e.g., ELISA kits for secreted proteins, lipid staining dyes like Oil Red O).
Methodology:
- Cell Seeding: Seed cells in a 96-well plate at an optimal density for proliferation or differentiation.
- Compound Treatment: After cell attachment, treat with test compounds at multiple concentrations (e.g., 1x, 5x, 10x IC₅₀ from biochemical assay). Include vehicle and positive control wells.
- Differentiation Induction: If studying adipogenesis, induce differentiation 48 hours post-confluence using a standard hormonal cocktail, with compounds present throughout the process [118].
- Viability Assessment: After 24-48 hours of treatment, perform a cell viability assay to rule out cytotoxic effects.
- Functional Endpoint Measurement: After an appropriate duration (e.g., 7-10 days for adipogenesis), assay for functional endpoints:
- Lipid Accumulation: Fix cells and stain with Oil Red O, followed by quantification via extraction and spectrophotometry.
- Gene/Protein Expression: Harvest cells for qPCR analysis of adipogenic markers (e.g., PPARγ, FABP4) or perform ELISA on culture supernatants [118].
- Data Analysis: Normalize all data to control wells. Dose-response curves can be generated to determine the potency (EC₅₀) of the compounds in the cellular context.
The following tables provide a template for summarizing the key quantitative data obtained from the validation pipeline.
Table 1: Summary of In Silico Prioritization and Primary Biochemical Assay Results
| Compound ID | Docking Score (kcal/mol) | Pharmacophore Fit Value | Biochemical IC₅₀ (µM) | 95% Confidence Interval (µM) | R² of Dose-Response |
|---|---|---|---|---|---|
| HIT-001 | -10.2 | 0.95 | 0.15 | 0.11 - 0.20 | 0.98 |
| HIT-002 | -9.8 | 0.87 | 0.45 | 0.32 - 0.63 | 0.96 |
| HIT-003 | -8.5 | 0.92 | 2.10 | 1.70 - 2.59 | 0.94 |
| Reference | -11.0 | 0.98 | 0.05 | 0.03 - 0.08 | 0.99 |
Table 2: Summary of Secondary Cellular Assay and Selectivity Data
| Compound ID | Cell Viability EC₅₀ (µM) | Functional Activity EC₅₀ (µM) | Selectivity Index (Viability/Activity) | Key Phenotypic Observation |
|---|---|---|---|---|
| HIT-001 | >50 | 1.5 | >33.3 | Significant lipid reduction |
| HIT-002 | 25.0 | 5.0 | 5.0 | Moderate lipid reduction |
| HIT-003 | 8.5 | >10 | <0.85 | Cytotoxic at low doses |
| Reference | >50 | 0.1 | >500 | Potent lipid reduction |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Experimental Corroboration
| Item Category | Specific Example | Function & Application in Validation |
|---|---|---|
| Target Proteins | Recombinant PPARγ ligand-binding domain (LBD), FABP4 [118] | Serves as the direct molecular target for primary biochemical binding and inhibition assays to confirm target engagement. |
| Cell Lines | 3T3-L1 murine preadipocyte cell line [118] | A well-established cellular model for studying adipogenesis and evaluating the functional anti-adipogenic effects of confirmed hits in a biologically relevant context. |
| Virtual Screening Libraries | ZINC, ChemBridge Corp. [118] | Large, commercially available libraries of small molecules that can be screened in silico and subsequently procured for experimental testing. |
| Assay Probes | Fluorescent fatty acid analogs (e.g., Bodipy FL C16) | Used in displacement assays with proteins like FABP4 to quantify the binding affinity and inhibitory potency of hit compounds. |
| Druggability Assessment Tools | FPocket, SiteMap [119] | Computational software used to predict and evaluate binding sites on a target protein, assessing their "druggability" or potential to bind drug-like molecules with high affinity. |
Data Integration and Signaling Pathway Analysis
The ultimate goal of confirming in silico hits is to understand their role in modulating a biological pathway. The following diagram maps the potential mechanism of action for a confirmed hit targeting PPARγ in the adipogenesis signaling pathway.
Comparative Analysis of Computational Cost and Efficiency in Large Libraries
The process of early drug discovery relies heavily on the ability to identify promising hit compounds from vast chemical spaces. Structure-based virtual screening, which includes molecular docking and pharmacophore modeling, has emerged as a cornerstone technique in computer-aided drug design (CADD) for this purpose [19] [24]. The exponential growth of make-on-demand compound libraries, which now contain billions of readily available molecules, presents a golden opportunity for in-silico drug discovery [120]. However, this opportunity is coupled with the significant challenge of computational cost and efficient resource allocation when performing virtual screens on an ultra-large scale. This application note provides a comparative analysis of the computational cost and efficiency of various docking and screening protocols when applied to large compound libraries. It offers detailed methodologies and practical guidance for researchers aiming to optimize their virtual screening workflows within the broader context of integrating molecular docking with pharmacophore-based virtual screening.
Performance Benchmarking of Docking Software
The choice of molecular docking software is critical, as it directly influences both the computational cost and the quality of the results. Different docking programs employ varied sampling algorithms and scoring functions, leading to differences in performance.
Key Performance Metrics
The performance of docking software is typically evaluated using two primary metrics:
- Pose Prediction Accuracy: Measured by the Root-Mean-Square Deviation (RMSD) between the docked pose and the experimental binding mode. An RMSD of less than 2.0 Å is generally considered a successful prediction [121] [24].
- Virtual Screening Enrichment: The ability to distinguish active compounds from inactive ones, often evaluated using Receiver Operating Characteristic (ROC) curves and the calculation of the Area Under the Curve (AUC) [121].
Comparative Performance Data
A benchmark study evaluating five popular docking programs on Cyclooxygenase (COX) enzymes revealed significant differences in performance [121].
Table 1: Performance of Docking Software in Pose Prediction and Virtual Screening
| Docking Program | Pose Prediction Success (RMSD < 2.0 Å) | Virtual Screening AUC Range | Typical Enrichment Factor |
|---|---|---|---|
| Glide | 100% | Up to 0.92 | 8 - 40x |
| GOLD | 82% | 0.61 - 0.92 | 8 - 40x |
| AutoDock | 59% | 0.61 - 0.92 | 8 - 40x |
| FlexX | 59% | 0.61 - 0.92 | 8 - 40x |
| Molegro Virtual Docker (MVD) | 82% | Not reported | Not reported |
The results indicated that Glide outperformed other programs by correctly predicting the binding poses of all studied co-crystallized ligands (100% success rate) [121]. In virtual screening, all tested methods (Glide, GOLD, AutoDock, FlexX) were useful tools for enriching active compounds, with AUC values ranging from 0.61 to 0.92 and enrichment factors of 8 to 40 folds [121].
Computational Cost and Efficiency in Ultra-Large Library Screening
Conducting virtual screens on ultra-large libraries, often comprising billions of compounds, requires careful consideration of the computational methodology and its associated costs.
Screening Strategies and Their Resource Demands
Different screening strategies offer a trade-off between computational intensity and accuracy.
Table 2: Computational Strategies for Large-Scale Virtual Screening
| Screening Strategy | Key Features | Computational Cost | Reported Efficiency |
|---|---|---|---|
| Traditional Rigid Docking | Fast but may not sample favorable protein-ligand structures accurately [120]. | Lower | Standard; feasible for millions of compounds. |
| Flexible Docking (e.g., RosettaLigand) | Accounts for ligand and receptor flexibility, increasing success rates [120]. | Very High | Impractical for screening billions of compounds directly. |
| Evolutionary Algorithms (e.g., REvoLd) | Efficiently searches combinatorial make-on-demand chemical space without enumerating all molecules [120]. | Moderate | Hit rate improvements by factors of 869 to 1,622 compared to random selection [120]. |
| Machine Learning Score Prediction | Uses ML models to predict docking scores, bypassing expensive docking calculations [16]. | Very Low | Up to 1,000 times faster than classical docking-based screening [16]. |
Workflow for Integrated Pharmacophore and Docking Screening
A combined approach using pharmacophore models and molecular docking can optimize both efficiency and cost. The following workflow diagram outlines a protocol for integrating these methods to screen large libraries effectively.
Detailed Experimental Protocols
This section provides step-by-step protocols for key experiments cited in this analysis, enabling researchers to replicate and adapt these methods.
Protocol 1: Structure-Based Pharmacophore Modeling and Screening
This protocol is adapted from studies targeting the TIM-3 protein and monoamine oxidase (MAO) inhibitors [19] [122] [16].
Protein and Ligand Preparation
- Obtain the 3D structure of the target protein from the Protein Data Bank (PDB). Critically evaluate the structure for quality, including resolution and the presence of key residues [19] [122].
- Using software like Schrödinger's Protein Preparation Wizard, process the protein by removing redundant chains, water molecules, and cofactors. Add hydrogen atoms and optimize the protonation states of residues [121] [122].
- Prepare a database of ligand structures. For example, in a study on TIM-3, over 449,000 natural compounds from the Supernatural 3.0 database were prepared using tools like MacroModel to optimize conformations and minimize energy [122].
Pharmacophore Model Generation
- Import the prepared protein structure, often in its holo form (complexed with a known active ligand), into a modeling suite like Schrödinger's Maestro [122].
- Use the pharmacophore generation module (e.g., Phase) to create a structure-based hypothesis. The software will identify key interaction features (e.g., Hydrogen Bond Acceptors (A), Donors (D), Aromatic Rings (R), Hydrophobic regions (H)) from the protein-ligand complex [19] [122].
- Manually select the most relevant features that are essential for bioactivity to build the final pharmacophore model. Exclusion volumes can be added to represent the shape of the binding pocket [19].
Pharmacophore-Based Virtual Screening
- Use the generated pharmacophore model as a query to screen the prepared ligand database.
- Compounds are evaluated and ranked based on their "fitness score" or "Phase Screen Score," which indicates how well they map to the pharmacophore features [122]. Typically, the top-scoring compounds (e.g., several thousand) are selected for subsequent docking studies.
Protocol 2: Molecular Docking and Virtual Screening
This protocol outlines a tiered docking approach to balance computational cost and accuracy, as used in benchmarks and practical guides [121] [104] [122].
Receptor Grid Generation
- Define the binding site for docking calculations. The center of the grid is typically based on the coordinates of a co-crystallized ligand or the centroid of key binding site residues [122].
- Generate a receptor grid file using software like Glide. This grid represents the potential energy field of the binding site.
Tiered Docking Screen
- High-Throughput Virtual Screening (HTVS) Mode: Dock the entire library of compounds filtered from the pharmacophore screen (or a similarly reduced library) using a fast, less accurate docking mode. This step rapidly eliminates clearly non-binding molecules [122] [16].
- Standard Precision (SP) Docking: Take the top-ranked compounds from the HTVS stage (e.g., 10-20%) and re-dock them using a more rigorous scoring function. This refines the list of potential hits.
- Extra Precision (XP) Docking: Subject the top hits from SP docking (a few hundred to a thousand) to the most computationally intensive and accurate docking mode. XP docking is designed to lower the rate of false positives and provides a more reliable ranking of binding affinities [122].
Analysis of Docking Results
- Analyze the XP docking poses visually. Check for key ligand-protein interactions (e.g., hydrogen bonds, π-π stacking, hydrophobic contacts) that are consistent with the pharmacophore model and known active ligands [122].
- Perform cluster analysis on the top-scoring compounds to ensure chemical diversity among the selected hits.
Protocol 3: Machine Learning-Accelerated Screening
For ultra-large libraries, traditional docking becomes prohibitive. This protocol uses machine learning to predict docking scores, drastically reducing computation time [16].
Training Set Generation
- Perform molecular docking on a representative, but manageable, subset of the chemical library of interest (e.g., 10,000-100,000 compounds) using a chosen docking program (e.g., Smina) [16].
- The resulting docking scores and the corresponding molecular structures form the training dataset for the machine learning model.
Model Training and Validation
- Encode the molecular structures of the training set using various molecular fingerprints and descriptors.
- Train an ensemble of machine learning models (e.g., regression models) to predict the docking score based on the molecular fingerprints.
- Validate the model's performance on a held-out test set to ensure its predictions strongly correlate with the actual docking scores.
Large-Scale Screening
- Use the trained model to predict docking scores for the entire ultra-large library (billions of compounds). This prediction is orders of magnitude faster than actual docking.
- Select the top-ranked compounds based on the predicted scores for subsequent experimental testing or more detailed (XP) molecular docking.
The Scientist's Toolkit: Essential Research Reagents & Software
The following table details key software and resources essential for conducting the virtual screening protocols described in this note.
Table 3: Key Research Reagent Solutions for Virtual Screening
| Item Name | Type | Function in Research | Example Use Case |
|---|---|---|---|
| Glide | Commercial Docking Software | Predicts binding mode and affinity of ligands to a target protein using systematic search and empirical scoring [121] [112]. | High-accuracy pose prediction and virtual screening [121]. |
| AutoDock Vina | Free Docking Software | Uses an iterated local search algorithm and a hybrid scoring function for efficient docking [24] [112]. | General-purpose docking and screening; good balance of speed and accuracy. |
| GOLD | Commercial Docking Software | Employs a genetic algorithm to explore ligand and partial protein flexibility [121] [112]. | Docking where protein side-chain flexibility is important. |
| REvoLd | Algorithm/Software | An evolutionary algorithm for efficient screening of ultra-large combinatorial libraries without full enumeration [120]. | Screening billion-member "make-on-demand" libraries like Enamine REAL. |
| ZINC/Enamine REAL | Compound Database | Libraries of commercially available compounds (ZINC) or synthetically accessible virtual compounds (REAL) for screening [120] [16]. | Source of small molecules for virtual high-throughput screening. |
| Schrödinger Maestro | Modeling Suite | Integrated software platform for protein preparation, pharmacophore modeling, molecular docking, and dynamics [122]. | End-to-end workflow for structure-based drug design. |
| Python & Scikit-learn | Programming Environment | Used to build custom machine learning models for predicting docking scores and accelerating screening [16]. | Creating ML-based filters for ultra-large libraries. |
Workflow for ML-Accelerated Ultra-Large Screening
The diagram below illustrates the logical flow of a machine learning-accelerated screening protocol, which dramatically reduces the time required to identify hits from billions of compounds.
Conclusion
The integration of pharmacophore modeling and molecular docking represents a paradigm shift in structure-based drug discovery, creating a synergistic workflow that leverages the strengths of both techniques. As validated by comparative studies, this combined approach consistently outperforms standalone methods, offering superior enrichment factors and hit rates in virtual screening campaigns. The pharmacophore component provides a chemically intuitive, efficient filter that captures essential interactions, while docking delivers detailed atomic-level binding mode predictions. Future directions point toward deeper integration with molecular dynamics for conformational sampling, the rise of AI and deep learning for pharmacophore-guided molecule generation, and enhanced applications in predicting polypharmacology and drug repurposing. For researchers, mastering this integrated strategy is no longer optional but essential for navigating the expanding chemical space and accelerating the development of novel, effective therapeutics for complex diseases.
References
- 1. Pharmacophore - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/medicine-and-dentistry/pharmacophore]
- 2. Pharmacophore - Wikipedia [https://en.wikipedia.org/wiki/Pharmacophore]
- 3. Applications of the Pharmacophore Concept in Natural Product inspired Drug Design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7685156/]
- 4. Pharmacophore [https://grokipedia.com/page/Pharmacophore]
- 5. Pharmacophore screening, molecular docking, and MD ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1534707/full]
- 6. Applications of the Novel Quantitative Pharmacophore Activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9504690/]
- 7. QPHAR: quantitative pharmacophore activity relationship [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-021-00537-9]
- 8. Pharmacophore-Based Virtual Screening of Alkaloids and ... [https://www.mdpi.com/2076-3417/15/18/10162]
- 9. Top Drug Discovery Software Solutions to Watch in 2025 [https://www.deepmirror.ai/post/top-drug-discovery-software-solutions-to-watch-in-2025]
- 10. Phase - Schrödinger [https://www.schrodinger.com/platform/products/phase/]
- 11. Molecular Docking and Structure-Based Drug Design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6332083/]
- 12. Ten quick tips to perform meaningful and reproducible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12064015/]
- 13. Integration of pharmacophore-based virtual screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11627428/]
- 14. A comprehensive drug discovery study using commercial ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0311527]
- 15. Evaluating ligand docking methods for drugging protein–protein interfaces: insights from AlphaFold2 and molecular dynamics refinement | Journal of Cheminformatics | Full Text [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01067-4]
- 16. Machine learning accelerates pharmacophore-based ... [https://www.nature.com/articles/s41598-024-58122-7]
- 17. Structure based drug design and machine learning ... [https://www.nature.com/articles/s41598-025-17708-5]
- 18. Past, Present, and Future of Molecular Docking [https://www.intechopen.com/chapters/70982]
- 19. Drug Design by Pharmacophore and Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9145410/]
- 20. Key Topics in Molecular Docking for Drug Design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6769580/]
- 21. Pharmacophore-based virtual screening and in silico ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12009819/]
- 22. Benchmarking GNINA and AutoDock Vina for Precision ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388557/]
- 23. Assessing molecular docking tools: understanding drug ... [https://fjps.springeropen.com/articles/10.1186/s43094-025-00862-y]
- 24. Molecular docking as a tool for the discovery ... [https://www.nature.com/articles/s41598-023-40160-2]
- 25. Pharmacophore-based virtual screening versus docking ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4007494/]
- 26. Beyond rigid docking: deep learning approaches for fully ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406700/]
- 27. PharmacoForge: pharmacophore generation with diffusion ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1628800/full]
- 28. How useful is virtual screening in drug discovery? [https://synapse.patsnap.com/article/how-useful-is-virtual-screening-in-drug-discovery]
- 29. Decoding the limits of deep learning in molecular docking for ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc05395a]
- 30. Pharmacophore modeling in drug design [https://www.sciencedirect.com/science/article/abs/pii/S1054358925000109]
- 31. Pharmacophore modelling based virtual screening and ... [https://www.nature.com/articles/s41598-024-63555-1]
- 32. A quantitative model of structure-based virtual screening ... [https://chemrxiv.org/engage/chemrxiv/article-details/69037e08113cc7cfff050553]
- 33. Ligand and Pharmacophore based Design | BIOVIA [https://www.3ds.com/products/biovia/discovery-studio/ligand-and-pharmacophore-based-design]
- 34. An artificial intelligence accelerated virtual screening ... [https://www.nature.com/articles/s41467-024-52061-7]
- 35. CCG | UGM and Conference 2025 | North America [https://www.chemcomp.com/UGM-2025-North_America.htm]
- 36. Using Machine Learning Surrogate Models to Increase ... [https://nhsjs.com/2025/using-machine-learning-surrogate-models-to-increase-throughput-of-virtual-screening-with-docking/]
- 37. Pharmacophore-based virtual screening versus docking-based virtual... [https://pubmed.ncbi.nlm.nih.gov/19935678/]
- 38. Structure-based pharmacophore modeling 2. Developing a ... [https://www.sciencedirect.com/science/article/abs/pii/S1093326323000864]
- 39. PharmaCore: The Automatic Generation of 3D Structure ... [https://pubmed.ncbi.nlm.nih.gov/38728062/]
- 40. Ligand-Based and Structure-Based Pharmacophore ... [https://bio-protocol.org/exchange/minidetail?id=9587520&type=30]
- 41. Knowledge-guided diffusion model for 3D ligand ... [https://www.nature.com/articles/s41467-025-57485-3]
- 42. Pharmacophore-based virtual screening, molecular docking ... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-024-01350-9]
- 43. Structure-based pharmacophore modeling 1. Automated ... [https://www.sciencedirect.com/science/article/abs/pii/S109332632300027X]
- 44. A structure-based framework for selective inhibitor design ... [https://www.nature.com/articles/s42003-025-07840-3]
- 45. Pharmacophore modeling and applications in drug discovery [https://www.sciencedirect.com/science/article/abs/pii/S135964461000111X]
- 46. Molecular Docking, Molecular Dynamics Simulation, and ... [https://pubmed.ncbi.nlm.nih.gov/40655858/]
- 47. 2.3. 3D Ligand-Based Pharmacophore Modeling [https://bio-protocol.org/exchange/minidetail?id=3714194&type=30]
- 48. Ligand-based Pharmacophore Modeling, Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6396084/]
- 49. The future of pharmaceuticals: Artificial intelligence in drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12391800/]
- 50. Pharmacophore-based approach for the identification of ... [https://www.sciencedirect.com/science/article/pii/S266702242500012X]
- 51. A pharmacophore-guided deep learning approach for ... [https://www.nature.com/articles/s41467-023-41454-9]
- 52. Artificial Intelligence in Small-Molecule Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12472608/]
- 53. Combining docking with pharmacophore for improved virtual... filtering [https://pmc.ncbi.nlm.nih.gov/articles/PMC3152774/]
- 54. Pharmacophore-based virtual screening versus docking ... [https://www.nature.com/articles/aps2009159]
- 55. Strategies for 3D pharmacophore-based virtual screening [https://www.sciencedirect.com/science/article/abs/pii/S1740674910000375]
- 56. modeling: advances, limitations, and current utility in Pharmacophore [https://www.dovepress.com/pharmacophore-modeling-advances-limitations-and-current-utility-in-dru-peer-reviewed-fulltext-article-JRLCR]
- 57. Concepts and Applications Exemplified on Hydroxysteroid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6332202/]
- 58. Integration of pharmacophore mapping and molecular in... docking [https://pubs.rsc.org/en/content/articlelanding/2017/ra/c6ra24959k]
- 59. based Pharmacophore & virtual ... screening molecular docking [https://pubmed.ncbi.nlm.nih.gov/36571432/]
- 60. Docking, pharmacophore modeling, virtual screening, and ... [https://www.bio-protocol.org/exchange/preprintdetail?type=3&id=268]
- 61. Confidence bands and hypothesis tests for hit enrichment curves [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-022-00629-0]
- 62. Pharmacophore screening, molecular docking, and MD ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11926154/]
- 63. Recent development of multi-target VEGFR-2 inhibitors for ... [https://pubmed.ncbi.nlm.nih.gov/36801788/]
- 64. Vascular Endothelial Growth Factor Receptor (VEGFR-2) ... [https://www.sciencedirect.com/science/article/pii/S2590098619300090]
- 65. Dual Targeting of VEGFR2 and C-Met Kinases via the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7408256/]
- 66. VEGFR2 Selective Inhibitors [https://www.selleckchem.com/subunits/VEGFR2_VEGFR_selpan.html]
- 67. Identification of Novel Potential VEGFR-2 Inhibitors Using a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8540634/]
- 68. Recent updates on potential of VEGFR-2 small-molecule ... [https://www.sciencedirect.com/org/science/article/pii/S2046206924028985]
- 69. A precise comparison of molecular target prediction methods [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00199d]
- 70. Pharmacophore screening, molecular docking, and MD ... [https://pubmed.ncbi.nlm.nih.gov/40124780/]
- 71. PolyLLM: polypharmacy side effect prediction via LLM ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1617142/full]
- 72. Docking Benchmark Group - TDC [https://tdcommons.ai/benchmark/docking_group/overview/]
- 73. A New Fragment‐Based Pharmacophore Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12412281/]
- 74. Benchmarking Sets for Molecular Docking - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3383317/]
- 75. Scoring functions for docking [https://en.wikipedia.org/wiki/Scoring_functions_for_docking]
- 76. PharmacoMatch: Efficient 3D Pharmacophore Screening ... [https://arxiv.org/html/2409.06316v2]
- 77. Building shape-focused pharmacophore models for effective ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00857-6]
- 78. Decoys Selection in Benchmarking Datasets [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2018.00011/full]
- 79. Pharmacophore-Based Similarity Scoring for DOCK - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4306494/]
- 80. A comprehensive survey of scoring functions for protein ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05991-4]
- 81. Dealing with Flexible Receptors - C&EN [https://cen.acs.org/articles/82/i19/DEALING-FLEXIBLE-RECEPTORS.html]
- 82. Conformational selection in protein binding and function - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4241102/]
- 83. 5.6: Binding - Conformational Selections and Intrinsically ... [https://bio.libretexts.org/Bookshelves/Biochemistry/Fundamentals_of_Biochemistry_(Jakubowski_and_Flatt)/01%3A_Unit_I-_Structure_and_Catalysis/05%3A_Protein_Function/5.06%3A__Binding_-_Conformational_Selections_and_Intrinsically_Disordered_Proteins]
- 84. Accounting for receptor flexibility and enhanced sampling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3540989/]
- 85. Evidence of Conformational Selection Driving the Formation of ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003872]
- 86. Integrating Molecular Dynamics, Molecular Docking, and ... [https://www.mdpi.com/1420-3049/30/14/2985]
- 87. Integrating machine learning driven virtual screening and ... [https://www.nature.com/articles/s41598-025-97208-8]
- 88. Building shape-focused pharmacophore models for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11312248/]
- 89. 3D Chemical Similarity Networks for Structure-Based Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5511031/]
- 90. Shape-based screening - Schrödinger [https://www.schrodinger.com/life-science/learn/white-papers/shape-based-screening/]
- 91. Equivariant graph neural network-based accurate and ultra ... [https://www.sciencedirect.com/science/article/pii/S209517792500156X]
- 92. Ligand-Binding Site Structure Refinement Using Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6884403/]
- 93. Efficient Refinement of Complex Structures of Flexible ... [https://www.lifescience.net/publications/1705144/efficient-refinement-of-complex-structures-of-flex/]
- 94. Virtual screening and molecular dynamics simulations ... [https://www.sciencedirect.com/science/article/pii/S1570180825001617]
- 95. Protocol for an automated virtual screening pipeline including ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12639438/]
- 96. Incorporating physics to overcome data scarcity in predictive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10529646/]
- 97. Addressing data scarcity in protein fitness landscape analysis [https://www.sciencedirect.com/science/article/pii/S1566253523003512]
- 98. Applications and Limitations of Pharmacophore Modeling [https://drugdiscoverypro.com/applications-and-limitations-of-pharmacophore-modeling/]
- 99. Structure-based virtual screening, molecular docking, and MD ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12312920/]
- 100. Drug Design by Pharmacophore and Virtual Screening ... [https://www.mdpi.com/1424-8247/15/5/646]
- 101. Widespread false negatives in DNA-encoded library data [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc00844a]
- 102. A machine learning approach to predicting protein-ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3524828/]
- 103. FeatureDock for protein-ligand docking guided by ... [https://www.nature.com/articles/s44386-025-00005-6]
- 104. A practical guide to large-scale docking [https://www.nature.com/articles/s41596-021-00597-z]
- 105. Docking and scoring - Schrödinger [https://www.schrodinger.com/life-science/learn/white-papers/docking-and-scoring/]
- 106. Pharmacophore model, docking, QSAR, and molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8047494/]
- 107. Integrated pharmacophore‐based screening and docking ... [https://www.sciencedirect.com/science/article/abs/pii/S3050475924000034]
- 108. Docking compared to 3D-pharmacophores: the scoring ... [https://www.sciencedirect.com/science/article/abs/pii/S1740674910000557]
- 109. PLIP 2025: introducing protein–protein interactions to the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12230730/]
- 110. Developing Generalizable Scoring Functions for Molecular ... [https://pubmed.ncbi.nlm.nih.gov/39482913/]
- 111. Docking compared to 3D-pharmacophores: the scoring ... [https://www.sciencedirect.com/science/article/pii/S1740674910000557]
- 112. -based computational platform for high-throughput... Molecular docking [https://pmc.ncbi.nlm.nih.gov/articles/PMC8754542/]
- 113. Comparison of two molecular docking programs [https://czasopisma.umlub.pl/curipms/user/setLocale/en?source=%2Fcuripms%2Farticle%2Fview%2F1460]
- 114. The MM/PBSA and MM/GBSA methods to estimate ligand- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4487606/]
- 115. Flare MM/GBSA - Cresset Group [https://cresset-group.com/software/flare-mmgbsa/]
- 116. Protocol for Molecular Dynamics Simulations of Proteins [https://en.bio-protocol.org/en/bpdetail?id=2051&type=0]
- 117. In-silico molecular modelling, MM/GBSA binding free ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.991369/full]
- 118. Virtual screening combined with molecular docking for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11815789/]
- 119. In Silico Methods for Identification of Potential Active Sites ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9609013/]
- 120. Ultra-large library screening with an evolutionary algorithm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12594993/]
- 121. Benchmarking different docking protocols for predicting the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10945300/]
- 122. Molecular Docking, Molecular Dynamics Simulation, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12244860/]