Next-Generation Sequencing in Cancer Molecular Profiling: A Comprehensive Guide for Research and Drug Development
Next-generation sequencing (NGS) has fundamentally transformed cancer research and therapeutic development by enabling comprehensive genomic profiling of tumors.
Next-Generation Sequencing in Cancer Molecular Profiling: A Comprehensive Guide for Research and Drug Development
Abstract
Next-generation sequencing (NGS) has fundamentally transformed cancer research and therapeutic development by enabling comprehensive genomic profiling of tumors. This article provides a detailed exploration of NGS technology, from its foundational principles and clinical applications in precision oncology to its crucial role in accelerating drug discovery. It examines key methodological approaches for detecting somatic variants, including single-nucleotide variants (SNVs), insertions/deletions (indels), copy number alterations (CNAs), and structural variants. The content addresses significant implementation challenges such as analytical validation, data interpretation complexities, and reimbursement barriers, while providing practical frameworks for troubleshooting and optimization. Furthermore, it discusses rigorous validation guidelines and comparative effectiveness research essential for clinical translation. Designed for researchers, scientists, and drug development professionals, this resource synthesizes current evidence and best practices to support the effective integration of NGS into cancer research pipelines and precision medicine strategies.
The Genomic Revolution: Understanding NGS Technology and Its Impact on Cancer Biology
Next-generation sequencing (NGS) has fundamentally transformed cancer molecular profiling research, enabling comprehensive genomic characterization that guides diagnostic, prognostic, and therapeutic decisions. This technical guide details the core principles of NGS workflows, from initial library preparation through final data analysis, with specific emphasis on applications in oncology. We provide detailed methodologies for key experiments, quantitative comparisons of current technologies, and standardized bioinformatics approaches tailored to clinical cancer research. The integration of robust NGS methodologies into oncology pipelines has been essential for identifying actionable mutations, tracking clonal evolution, and advancing personalized treatment strategies for cancer patients.
Next-generation sequencing technologies provide massively parallel sequencing capabilities that allow researchers to analyze millions of DNA fragments simultaneously. This high-throughput approach has enabled comprehensive molecular profiling of tumors, revealing the genetic alterations driving oncogenesis, progression, and treatment resistance [1]. The core principle of NGS—massive parallelism—has led to a 96% decrease in sequencing costs per genome since the Human Genome Project, making large-scale cancer genomics studies feasible for research and clinical applications [1].
In cancer research, NGS facilitates a range of applications from targeted panels focusing on known oncogenes and tumor suppressor genes to whole-genome sequencing that reveals complex structural variations and novel drivers. The versatility of NGS platforms allows for analysis of diverse sample types, including challenging formalin-fixed, paraffin-embedded (FFPE) tumor tissues commonly available in pathology archives [2]. As the technology continues to evolve with improved accuracy and throughput, NGS has become an indispensable tool for advancing precision oncology and targeted drug development.
NGS Workflow: From Sample to Insight
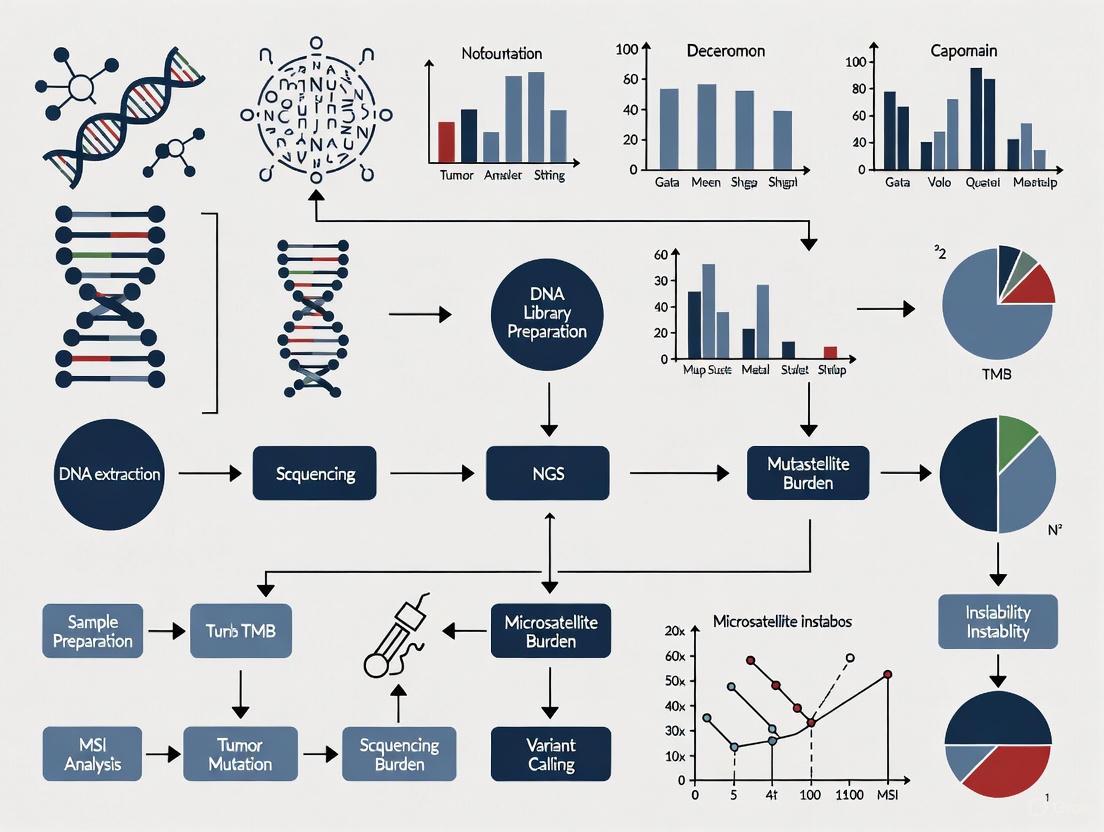
The complete NGS workflow encompasses multiple critical stages, each requiring rigorous quality control to ensure data integrity for downstream cancer research applications. The following diagram illustrates the comprehensive pathway from biological sample to clinical insight in cancer profiling:
Sample Preparation and Quality Control
Proper sample preparation is foundational to successful NGS in cancer research, where starting material is often limited or degraded.
Nucleic Acid Extraction: DNA or RNA is extracted from tumor samples, which can include fresh frozen tissue, FFPE blocks, liquid biopsies, or cell-free DNA [3]. For FFPE samples—common in retrospective cancer studies—specialized extraction kits are required to address formalin-induced cross-linking and fragmentation. The RecoverAll Total Nucleic Acid Isolation Kit is specifically designed for this challenging material [2].
Quality Assessment: Rigorous quality control of extracted nucleic acids is critical. Spectrophotometers (e.g., NanoDrop) assess sample concentration and purity through A260/A280 ratios (~1.8 for DNA, ~2.0 for RNA) [4]. Electrophoresis systems (e.g., Agilent TapeStation or Bioanalyzer) evaluate nucleic acid integrity, particularly important for RNA sequencing where the RNA Integrity Number (RIN) predicts sequencing success [4]. For FFPE-derived DNA, fragment size distribution analysis is essential, as samples with extensive degradation (<200 bp) may not be suitable for certain NGS workflows [2].
Table 1: Essential Research Reagent Solutions for NGS Library Preparation
| Reagent/Category | Specific Examples | Function in NGS Workflow | Cancer Research Application |
|---|---|---|---|
| Nucleic Acid Extraction Kits | GeneJet FFPE DNA Purification Kit, RecoverAll Total Nucleic Acid Isolation Kit | Isolate and purify nucleic acids from challenging sample types | Enable analysis of archival FFPE tumor tissues [2] |
| Library Preparation Kits | KAPA HyperPlus Kit, Illumina AmpliSeq v2 Hotspot Panel | Fragment DNA and attach platform-specific adapters | Prepare sequencing libraries from low-input tumor samples [2] |
| Target Enrichment Systems | SeqCap EZ Target Capture System, AmpliSeq Cancer Panels | Selectively enrich genomic regions of interest | Focus sequencing on known cancer-associated genes [2] [3] |
| Target Enrichment Methods | Hybridization capture, Amplicon-based approaches | Enrich for specific genomic regions | Focus on cancer-relevant genes; hybridization capture allows novel variant discovery [2] [1] |
Library Preparation Methods
Library preparation converts extracted nucleic acids into a format compatible with sequencing platforms through fragmentation, adapter ligation, and optional amplification.
Fragmentation and Adapter Ligation: DNA is fragmented by physical (sonication) or enzymatic methods to optimal sizes (100-800 bp) [3]. Platform-specific adapters containing sequencing primer binding sites are ligated to fragment ends. These adapters often include unique molecular barcodes (indexes) that enable multiplexing—pooling multiple samples in a single sequencing run—significantly reducing per-sample costs [1].
Target Enrichment Strategies: In cancer research, targeted sequencing approaches are commonly employed for their cost-effectiveness and depth of coverage for clinically relevant genes. The two primary enrichment methods are:
- Hybridization Capture: Uses biotinylated probes (e.g., SeqCap EZ system) to pull down target regions from fragmented libraries [2]. This method provides more uniform coverage and better capability to detect novel variants within targeted regions.
- Amplicon-Based: Utilizes PCR primers (e.g., Illumina AmpliSeq) to selectively amplify regions of interest [2]. This approach is highly efficient for small target regions but may miss variants in primer-binding sites.
A recent feasibility study on colorectal cancer FFPE samples demonstrated 94% concordance between these two methods for detecting actionable variants across 15 shared cancer-related genes [2].
Sequencing Platforms and Technologies
The NGS landscape in 2025 offers diverse platforms with distinct characteristics suited to different applications in cancer research. The following diagram compares the core technology approaches of major sequencing platforms:
Table 2: Comparison of Current NGS Platforms (2025)
| Platform | Technology | Read Length | Accuracy | Primary Cancer Applications | Throughput Range |
|---|---|---|---|---|---|
| Illumina | Sequencing by synthesis with reversible terminators [1] | 50-300 bp [5] | High (Q30: 99.9%) [4] | Targeted panels, whole exome, RNA-seq, ChIP-seq [5] | Up to 16 Tb/run (NovaSeq X) [6] |
| Pacific Biosciences (Revio) | Single Molecule Real-Time (SMRT) sequencing with HiFi circular consensus [6] | 10-25 kb | Very High (Q30-Q40: 99.9-99.99%) [6] | Structural variant detection, fusion genes, haplotype phasing [6] | 360 Gb/run [6] |
| Oxford Nanopore (Q20+ Kit14) | Nanopore-based electronic signal detection with duplex reading [6] | 1 kb->2 Mb | High (Simplex: Q20/~99%, Duplex: Q30/>99.9%) [6] | Structural variants, epigenetic modifications, rapid diagnostics [6] | Varies by device (MinION to PromethION) |
Bioinformatics Analysis Pipeline
The bioinformatics pipeline transforms raw sequencing data into interpretable results through a multi-stage process requiring specialized computational tools and reference databases.
NGS Data Formats and Quality Control
Standardized file formats enable interoperability between analytical tools throughout the NGS pipeline. The following diagram illustrates the transformation of data through these formats from sequencing to variant calling:
Primary Analysis (Base Calling): Sequencing instruments generate raw data in platform-specific formats (BCL for Illumina, POD5 for Nanopore, BAM for PacBio) that are converted to FASTQ format [5]. FASTQ files contain nucleotide sequences alongside quality scores for each base, representing the fundamental unit of raw NGS data [7].
Quality Assessment: Tools like FastQC provide comprehensive quality metrics including per-base sequence quality, adapter contamination, and GC content [7]. For cancer samples, special attention should be paid to potential contaminants and sample degradation indicators. The quality score (Q-score) is particularly important, with Q30 (99.9% accuracy) being the standard threshold for high-quality data [4].
Read Trimming and Filtering: Preprocessing tools such as CutAdapt, Trimmomatic, or Nanofilt remove low-quality bases, adapter sequences, and artifacts [4]. This step is crucial for FFPE-derived data where degradation and artifacts are more common.
Secondary Analysis: Alignment and Variant Calling
Read Alignment: Processed reads are aligned to a reference genome (e.g., GRCh38) using aligners like BWA or STAR, generating SAM/BAM files [8]. The alignment process determines the genomic origin of each read, enabling variant identification. For cancer samples, it's recommended to use the hg38 genome build as reference, as it provides more comprehensive coverage of clinically relevant regions compared to older builds [8].
Variant Calling: Specialized algorithms identify differences between the sample and reference genome. The consensus recommendations for clinical NGS bioinformatics pipelines include calling multiple variant types [8]:
- Single nucleotide variants (SNVs) and small insertions/deletitions (indels)
- Copy number variants (CNVs)
- Structural variants (SVs) including insertions, inversions, and translocations
- Short tandem repeats (STRs)
- Loss of heterozygosity (LOH) regions
For cancer applications, both germline (inherited) and somatic (tumor-specific) variants are typically identified, requiring paired tumor-normal analysis when possible.
Tertiary Analysis: Annotation and Interpretation
Variant Annotation: Called variants in VCF format are annotated with biological information using tools that incorporate databases of population frequency (gnomAD), functional prediction (SIFT, PolyPhen), and clinical significance (ClinVar, COSMIC) [1]. For cancer, databases like CIViC and OncoKB provide therapeutic, prognostic, and diagnostic annotations for specific mutations.
Variant Filtering and Prioritization: In cancer research, this critical step identifies clinically actionable variants from background noise and benign polymorphisms. Strategies include:
- Frequency-based filtering against population databases
- Functional impact prediction (missense, truncating, splice-site)
- Pathway analysis and known cancer gene databases
- Hotspot mutation analysis for recurrently altered positions
Recent recommendations emphasize using multiple tools for structural variant calling and in-house datasets for filtering recurrent technical artifacts [8].
Experimental Design and Validation in Cancer Research
Method Validation Protocols
For clinical cancer research, rigorous validation of NGS workflows is essential. The Next-Generation Sequencing Quality Initiative (NGS QI) provides frameworks for validation plans and standard operating procedures [9]. Key validation parameters include:
- Accuracy and Precision: Comparison to orthogonal methods (e.g., Sanger sequencing) and replicate sequencing
- Analytical Sensitivity: Detection of variants at low allele frequencies (critical for heterogeneous tumor samples)
- Specificity: False positive rates across variant types
- Reproducibility: Inter-run and inter-operator consistency
A recent feasibility study implementing NGS in the Chilean public health system demonstrated 80.5% concordance for actionable variants in colorectal cancer samples compared to a validated laboratory, with 98.4% of previously detected variants successfully identified in their implementation [2].
Quality Management and Standards
Implementing a robust quality management system (QMS) is recommended for clinical cancer NGS applications. The NGS QI provides assessment tools and key performance indicators to monitor assay performance over time [9]. Regular monitoring of metrics including coverage uniformity, on-target rates, and variant calling sensitivity ensures consistent performance.
For clinical production, bioinformatics should operate at standards similar to ISO 15189, utilizing off-grid clinical-grade high-performance computing systems, standardized file formats, and strict version control [8]. Reproducibility should be ensured through containerized software environments (Docker, Singularity), and pipelines must be thoroughly documented and tested for accuracy.
The core principles of NGS—from meticulous library preparation through rigorous bioinformatics analysis—provide the foundation for robust cancer molecular profiling research. As sequencing technologies continue to evolve with improvements in accuracy, throughput, and multi-omic capabilities, their integration into oncology research pipelines will further advance our understanding of cancer biology and treatment. The standardized workflows, validation frameworks, and quality control measures outlined in this guide provide a roadmap for implementing NGS in cancer research that generates reliable, reproducible, and clinically actionable genomic insights. Future developments in single-cell sequencing, spatial transcriptomics, and long-read technologies will continue to expand the research and clinical applications of NGS in precision oncology.
The advent of DNA sequencing technologies has fundamentally transformed biological research and clinical diagnostics, with next-generation sequencing (NGS) representing one of the most significant technological breakthroughs since the development of Sanger sequencing in 1977. This paradigm shift is particularly evident in oncology, where the comprehensive genomic profiling enabled by NGS has ushered in a new era of precision oncology. The ability to rapidly and cost-effectively sequence entire cancer genomes allows researchers and clinicians to identify the genetic alterations driving tumorigenesis, thereby facilitating personalized treatment strategies tailored to the specific molecular profile of a patient's cancer [10]. This technical guide provides a comparative analysis of NGS versus traditional sequencing methods, with a specific focus on their application in cancer molecular profiling research for scientists, researchers, and drug development professionals.
The transformative impact of NGS becomes evident when considering the limitations of traditional approaches. Prior to NGS, cancer genetic profiling relied heavily on single-gene assays or small panels that could only detect a limited set of predefined mutations, potentially missing rare or novel genetic alterations that contribute to cancer development and progression [10]. The massively parallel nature of NGS enables the simultaneous analysis of hundreds to thousands of cancer-related genes, providing a comprehensive view of the complex genomic landscape of tumors that was previously unattainable with traditional methods.
Technological Foundations and Comparative Specifications
Fundamental Principles of Sequencing Technologies
Traditional Sanger Sequencing, developed by Frederick Sanger in 1977, operates on the principle of chain-termination with dideoxynucleotides (ddNTPs) [11]. This method involves generating DNA fragments of varying lengths that are terminated at specific bases, which are then separated by capillary electrophoresis to determine the sequence [10]. The key limitation of this technology is its fundamental design—it sequences only one DNA fragment at a time, making it prohibitively slow and expensive for large-scale projects [11]. The Human Genome Project, which relied on Sanger sequencing, took 13 years and cost nearly $3 billion to complete the first human genome sequence [11].
Next-Generation Sequencing employs a fundamentally different approach characterized by massively parallel sequencing. Instead of processing single DNA fragments, NGS platforms simultaneously sequence millions to billions of DNA fragments [11] [10]. The core NGS workflow involves: (1) library preparation through fragmentation of DNA and adapter ligation; (2) cluster generation through amplification to create sequencing features; (3) cyclic sequencing using synthesis with fluorescently-labeled nucleotides; and (4) alignment and data analysis using sophisticated bioinformatics tools [11]. This parallel processing architecture provides NGS with its revolutionary throughput advantage, compressing sequencing timelines from years to hours while dramatically reducing costs [11].
Direct Technical Comparison
Table 1: Technical Comparison of Sanger Sequencing vs. Next-Generation Sequencing
| Feature | Sanger Sequencing | Next-Generation Sequencing |
|---|---|---|
| Throughput | Low - processes one DNA fragment at a time [11] | Extremely high - processes millions to billions of fragments simultaneously [11] |
| Cost per Human Genome | ~$3 billion (Human Genome Project) [11] | Under $1,000, with some services as low as $600 [11] [12] |
| Read Length | Long (500-1000 base pairs) [11] | Shorter (50-600 base pairs for short-read NGS) [11] |
| Primary Applications | Ideal for sequencing single genes or confirming specific variants [10] | Whole-genome sequencing, transcriptomics, epigenetics, metagenomics [10] |
| Data Output | Limited data output [10] | Massive amounts of data (terabases per run) [6] |
| Human Genome Sequencing Time | Years [11] | Hours to days [11] |
| Accuracy | High per-base accuracy (>99.9%) [11] | High overall accuracy achieved through depth of coverage [11] |
Evolution to Third-Generation Sequencing
The sequencing technology landscape continues to evolve with the emergence of third-generation sequencing platforms, which address one of the key limitations of mainstream NGS technologies: short read lengths. Platforms from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) enable the sequencing of much longer DNA fragments—thousands to tens of thousands of bases—without the need for fragmentation [6]. PacBio achieves this through Single Molecule Real-Time (SMRT) sequencing, which observes DNA polymerization in real time within microscopic wells called zero-mode waveguides [6]. Oxford Nanopore employs a fundamentally different approach by measuring changes in electrical current as DNA molecules pass through protein nanopores [6].
These long-read technologies are particularly valuable in cancer research for resolving complex genomic regions that are challenging for short-read NGS, including repetitive elements, structural variants, and gene fusions [11]. While early long-read technologies suffered from higher error rates, significant improvements have been made. PacBio's HiFi reads now achieve over 99.9% accuracy through circular consensus sequencing, while ONT's latest duplex sequencing chemistry exceeds Q30 (>99.9% accuracy) [6]. The convergence of technologies continues, with short-read companies adding long-read capabilities and vice versa, providing researchers with an increasingly sophisticated toolkit for cancer genomics.
Experimental Design and Methodologies for Cancer Profiling
Comprehensive Genomic Profiling Workflow in Cancer Research
The application of NGS in cancer research follows a standardized yet adaptable workflow designed to maximize DNA yield and sequencing quality from often limited and degraded tumor specimens. The BALLETT study (Belgian Approach for Local Laboratory Extensive Tumor Testing), a large-scale multi-center investigation involving 872 patients with advanced cancers, provides an exemplary model of a robust NGS-based cancer profiling protocol [13]. This study demonstrated the feasibility of implementing comprehensive genomic profiling (CGP) across multiple laboratories with a 93% success rate and a median turnaround time of 29 days from inclusion to molecular tumor board report [13].
Key Research Reagent Solutions for NGS in Cancer Studies
Table 2: Essential Research Reagents and Platforms for NGS-Based Cancer Profiling
| Reagent/Platform Category | Specific Examples | Research Function in Cancer Studies |
|---|---|---|
| Commercial CGP Panels | FoundationOne, Tempus, OncoDEEP, MI Profile [14] | Standardized targeted sequencing of cancer-related genes for consistent analysis across studies |
| Library Preparation Kits | Illumina-compatible kits, QIAseq xHYB Long-Read Panels [15] [16] | Fragment DNA and attach adapters for sequencing; specialized kits enable long-read or hybrid capture |
| NGS Platforms | Illumina NovaSeq X, PacBio Revio, Oxford Nanopore [16] [6] | High-throughput sequencing instruments with varying capabilities for short/long-read data |
| Automation Systems | Automated liquid handlers, library prep stations [15] | Increase throughput, reduce human error, and improve reproducibility in sample processing |
| Bioinformatics Tools | DRAGEN platform, various variant callers [17] | Process raw sequencing data, identify mutations, and annotate potential clinical significance |
Analytical Considerations for Cancer NGS
The analytical phase of NGS-based cancer profiling requires specialized approaches to address the unique challenges of tumor genomes. Unlike germline sequencing, cancer sequencing must account for tumor heterogeneity, variable tumor purity, and the distinction between somatic (acquired) and germline (inherited) variants [14]. The BALLETT study implemented a rigorous bioinformatics pipeline that identified not only single nucleotide variants and small insertions/deletions but also copy number variations, gene fusions, and genome-wide biomarkers including tumor mutational burden (TMB), microsatellite instability (MSI), and homologous recombination deficiency (HRD) [13].
Tumor-only sequencing designs, commonly used in cancer research, present specific analytical challenges. The BALLETT study protocol addressed this by considering variants with a variant allele frequency (VAF) greater than 50% as potentially germline in origin and confirming them with validated germline assays when a hereditary cancer syndrome was clinically suspected [13]. Actionable alterations were classified according to established frameworks such as OncoKB, which incorporates FDA approval status, clinical guideline support, and strength of supporting evidence [13]. This meticulous approach to analytical validation ensures that research findings can potentially translate to clinical applications.
Applications in Cancer Molecular Profiling Research
Comprehensive Genomic Characterization in Sarcoma Research
A recent multicenter study investigating advanced soft tissue and bone sarcomas exemplifies the power of NGS in characterizing molecularly complex cancers [14]. This research employed four different commercial NGS kits to analyze 81 patients with metastatic disease, identifying a total of 223 genomic alterations across the cohort, with at least one type of genomic alteration detectable in 90.1% of tumors [14]. The most frequently mutated genes were TP53 (38%), RB1 (22%), and CDKN2A (14%), revealing key insights into the molecular drivers of these rare malignancies [14].
Critically, this study demonstrated that NGS identified actionable mutations in 22.2% of sarcoma patients, rendering them eligible for FDA-approved targeted therapies that would not have been considered based on conventional histopathological diagnosis alone [14]. Additionally, NGS led to a reclassification of diagnosis in four patients, highlighting its utility not only in therapeutic decision-making but also as a powerful diagnostic tool in cases with ambiguous histological features [14]. The functional analysis of genomic alterations revealed potentially targetable changes in key pathways including genomic stability regulation (TP53, MDM2), cell cycle regulation (RB1, CDKN2A/B), and the phosphoinositide-3 kinase pathway (PTEN, PIK3CA) [14].
Large-Scale Implementation Studies
The BALLETT study provides compelling evidence for the feasibility and utility of large-scale NGS implementation in cancer research [13]. In this comprehensive analysis of 756 patients with advanced cancers across 32 different tumor types, actionable genomic markers were identified in 81% of patients—substantially higher than the 21% actionability rate that would have been detected using traditionally reimbursed, small gene panels [13]. The most frequently altered genes in this pan-cancer analysis were TP53 (46%), KRAS (13%), APC (9%), PIK3CA (11%), and TERT (8%) [13].
The study also demonstrated the importance of genome-wide biomarkers detectable only through comprehensive NGS approaches. Tumor mutational burden (TMB-high) was identified in 16% of patients, with particularly high frequencies in lung cancer, melanoma, and urothelial carcinomas [13]. Microsatellite instability (MSI-high) was detected in eight patients, all of whom also exhibited high TMB [13]. Homologous recombination deficiency (HRD) status was analyzed for 100 patients, with 11% showing positive results, including five breast and two ovarian carcinomas [13]. These biomarkers have significant implications for immunotherapy response and targeted treatment approaches.
Emerging Applications: Liquid Biopsies and Resistance Monitoring
Beyond comprehensive tissue profiling, NGS enables several emerging applications that are transforming cancer research. Liquid biopsies, which involve sequencing circulating tumor DNA (ctDNA) from blood samples, provide a non-invasive method for cancer detection, monitoring treatment response, and identifying emerging resistance mechanisms [11]. This approach is particularly valuable for tracking tumor evolution in response to targeted therapies, as cancer cells often develop resistance through additional genetic alterations that can be detected through serial liquid biopsy sampling [11].
The high sensitivity of NGS also facilitates minimal residual disease (MRD) detection, allowing researchers to identify molecular evidence of residual cancer after treatment that would be undetectable by conventional imaging methods [10]. This application has significant implications for understanding cancer recurrence and developing more effective adjuvant therapy strategies. Furthermore, NGS is playing an increasingly important role in immuno-oncology research by enabling comprehensive analysis of tumor-immune interactions, T-cell receptor repertoires, and biomarkers of immunotherapy response such as TMB and MSI [10].
The comparative analysis of NGS versus traditional sequencing methodologies reveals a fundamental technological shift that has transformed cancer molecular profiling research. The massively parallel architecture of NGS provides unprecedented throughput and cost-efficiency, enabling comprehensive genomic characterization that was scientifically and economically unfeasible with Sanger sequencing. This technological advancement has identified actionable genomic targets in the majority of patients with advanced cancers—81% in the BALLETT study compared to just 21% with conventional approaches—highlighting the critical importance of comprehensive genomic profiling in modern oncology research [13].
The applications of NGS in cancer research continue to expand, from diagnostic reclassification and therapeutic targeting to liquid biopsy monitoring and analysis of novel biomarkers such as TMB and HRD. As sequencing technologies continue to evolve, with improvements in long-read sequencing, single-cell analysis, and multi-omic integration, researchers will gain increasingly sophisticated tools to decipher the complex molecular landscape of cancer. For the research community, embracing these technologies and addressing their associated challenges in data analysis, standardization, and implementation will be essential for advancing our understanding of cancer biology and developing more effective, personalized cancer therapies.
Cancer is not a single disease but a complex ecosystem characterized by profound heterogeneity, which represents one of the most significant barriers to effective treatment. Tumor heterogeneity exists at multiple levels—between different patients (inter-tumor heterogeneity) and within individual tumors and patients (intra-tumor heterogeneity) [18]. This variability stems from an evolutionary process where tumors accumulate genetic alterations over time, leading to diverse subpopulations of cancer cells (clones) with distinct molecular profiles [18]. These competing cellular populations exist within a microenvironment comprising various non-cancerous cells, including immune cells, fibroblasts, and vascular endothelial cells, further compounding the complexity [18].
Next-generation sequencing (NGS) has emerged as a transformative technology for deciphering this complexity, enabling comprehensive genomic profiling that reveals the intricate molecular architecture of tumors. Unlike traditional Sanger sequencing, which processes single DNA fragments sequentially, NGS performs massive parallel sequencing, processing millions of fragments simultaneously [10]. This technological leap has significantly reduced the time and cost associated with genomic analysis while providing unprecedented resolution for detecting the genetic alterations that drive cancer progression and therapeutic resistance [10]. The application of NGS in oncology has fundamentally advanced our understanding of tumor biology and is now an essential component of precision medicine approaches aimed at tailoring treatments to the specific molecular characteristics of individual patients' tumors.
Understanding Tumor Heterogeneity: Models and Molecular Mechanisms
Conceptual Models of Tumor Evolution
The development and progression of tumors are governed by two primary, non-exclusive models that explain the emergence of heterogeneity. The clonal evolution model (stochastic model) posits that tumors evolve through a stepwise accumulation of genomic and epigenetic alterations that provide selective advantages to certain cell lineages, leading to their expansion while other populations are depleted [18]. This dynamic process results in continuous tumor remodeling with distinct dimensions of heterogeneity. In contrast, the cancer stem cell (CSC) model (hierarchical model) proposes that tumors are maintained by a subpopulation of cells with stem-like properties that can differentiate into multiple cell types within the tumor [18]. In reality, both models often co-occur, with CSCs frequently representing the cells that acquire critical mutations driving clonal expansion.
Multi-Level Heterogeneity in Cancer
Tumor heterogeneity manifests across multiple molecular dimensions, each contributing to the overall complexity of the disease:
- Genomic heterogeneity: Variations in DNA sequences, including somatic mutations, copy number alterations, and structural rearrangements that differ between tumor regions and individual cells [18].
- Transcriptomic heterogeneity: Differences in gene expression patterns and RNA processing that lead to phenotypic diversity despite identical genetic backgrounds [19].
- Epigenetic heterogeneity: Variable epigenetic modifications that regulate gene expression without altering the underlying DNA sequence, contributing to cellular plasticity and adaptive responses [19].
Table 1: Common Genetic Alterations Across Cancer Types Based on TCGA Data
| Cancer Type | Sample Size | Significantly Altered Genes |
|---|---|---|
| Glioblastoma | 206 | TP53, ERBB2, NF1, PARK2, AKT3, FGFR2, PIK3R1 |
| Lung Adenocarcinoma | 230 | TP53, KRAS, EGFR, STK11, KEAP1, BRAF, MET |
| Breast Cancer | 510 | PIK3CA, TP53, GATA3, CDH1, RB1, MLL3, MAP3K1 |
| Colorectal Cancer | 276 | APC, TP53, KRAS, PIK3CA, FBXW7, SMAD4 |
| Clear Cell Renal Cell Carcinoma | 446 | VHL, PBRM1, BAP1, SETD2, HIF1A |
Data derived from TCGA analysis illustrates the diverse mutational landscapes across different cancer types [18].
NGS Methodologies for Deciphering Tumor Heterogeneity
Core NGS Technology and Workflow
Next-generation sequencing represents a revolutionary advance over traditional sequencing methods, enabling comprehensive genomic analysis with unprecedented speed and accuracy. The fundamental NGS workflow consists of four critical steps:
Sample Preparation and Library Construction: Nucleic acids (DNA or RNA) are extracted from tumor samples and fragmented into appropriately sized pieces (typically around 300 bp). Adapters—synthetic oligonucleotides with specific sequences—are then ligated to these fragments, creating a sequencing library. The library may undergo enrichment steps to isolate specific genomic regions of interest, such as exons or cancer-related genes [10].
Sequencing Reaction: The prepared library is loaded onto a sequencing platform where fragments are amplified and sequenced simultaneously through massive parallel sequencing. The most common technology (Illumina) involves immobilizing library fragments on a flow cell surface, amplifying them to form clusters of identical sequences, and then determining the sequence through cyclic fluorescence detection as fluorescently-labeled nucleotides are incorporated [10].
Data Generation and Primary Analysis: The sequencing instrument detects signals from each cluster in real-time, converting them into raw sequence data (reads) along with quality metrics. The enormous data output—often terabytes per run—requires sophisticated computational infrastructure [10].
Bioinformatic Analysis: Specialized software aligns the generated reads to a reference genome, identifies variations (including single nucleotide variants, insertions/deletions, copy number alterations, and structural variants), and interprets the biological significance of these findings in the context of cancer biology [10].
Comparative Analysis: NGS vs. Traditional Sequencing
Table 2: Comparison of NGS and Sanger Sequencing Technologies
| Feature | Next-Generation Sequencing | Sanger Sequencing |
|---|---|---|
| Cost-effectiveness | Higher for large-scale projects | Lower for small-scale projects |
| Speed | Rapid sequencing | Time-consuming |
| Application | Whole-genome, exome, transcriptome sequencing | Ideal for sequencing single genes |
| Throughput | Multiple sequences simultaneously | Single sequence at a time |
| Data output | Large amount of data | Limited data output |
| Clinical utility | Detects multiple mutation types, structural variants | Identifies specific known mutations |
NGS offers significant advantages in throughput, comprehensiveness, and efficiency for analyzing complex tumor genomes [10].
Advanced NGS Applications for Heterogeneity Analysis
Several sophisticated NGS-based approaches have been developed specifically to address the challenges of tumor heterogeneity:
Single-Cell Sequencing (SCS): This cutting-edge technology enables genomic, transcriptomic, or epigenomic profiling of individual cells, providing the ultimate resolution for analyzing intra-tumor heterogeneity. By classifying tumor cells into distinct subpopulations from multiple spatial regions within a biopsy, SCS allows researchers to trace tumor cell lineages and elucidate mechanisms of therapeutic failure and resistance [18].
Spatial Transcriptomics Integration: Novel computational approaches like Tumoroscope integrate somatic point mutation data from spatial transcriptomics (ST) reads, clone genotypes reconstructed from bulk DNA-seq, and cancer cell counts from H&E-stained images to unravel the clonal composition of each spot within a tumor sample. This enables precise spatial mapping of clones and their mutual relationships [20].
Liquid Biopsies: Analysis of circulating tumor DNA (ctDNA) and circulating tumor cells (CTCs) from blood samples provides a non-invasive method for monitoring tumor heterogeneity and evolution over time, offering insights into therapeutic response and emergence of resistance [19].
Diagram 1: Single-Cell Sequencing Workflow. SCS enables resolution of tumor heterogeneity at the individual cell level [18].
Experimental Design and Protocols for Heterogeneity Studies
Comprehensive Genomic Profiling (CGP) in Multi-Center Studies
Large-scale genomic studies require standardized protocols to ensure reproducible and comparable results across institutions. The Belgian Approach for Local Laboratory Extensive Tumor Testing (BALLETT) study exemplifies a well-designed framework for implementing CGP in clinical decision-making for patients with advanced cancers. This multi-center study enrolled 872 patients from 12 hospitals and established a consortium of nine local NGS laboratories using fully standardized methodology [13].
The study demonstrated a 93% success rate for CGP profiling across diverse tumor types, with a median turnaround time of 29 days from inclusion to molecular tumor board report. The protocol identified actionable genomic markers in 81% of patients—substantially higher than the 21% actionability rate using nationally reimbursed small panels [13]. This highlights the superior capability of CGP for uncovering therapeutic targets in heterogeneous tumors.
Integrated Spatial Genomic Analysis Protocol
The Tumoroscope methodology represents an advanced experimental approach for integrating multiple data types to reconstruct spatial tumor heterogeneity:
Sample Processing: Fresh-frozen tumor tissues are subjected to parallel processing for H&E staining, bulk DNA sequencing, and spatial transcriptomics [20].
Image Analysis: H&E-stained tissue images are analyzed using custom QuPath scripts to identify ST spots within cancer cell-containing regions and estimate cell counts for each spot [20].
Clone Reconstruction: Somatic mutations and allele-specific copy number data from bulk DNA-seq are analyzed using established methods (Vardict, FalconX, and Canopy) to reconstruct cancer clones, their frequencies, and genotypes [20].
Probabilistic Deconvolution: The Tumoroscope model integrates (i) estimated cell counts per spot, (ii) alternate and total read counts for mutations in ST spots, and (iii) clone genotypes and frequencies to infer the proportions of each clone in every spot [20].
Gene Expression Profiling: A regression model uses gene expression data as independent variables and inferred clone proportions as dependent variables to deduce clonal expression profiles [20].
Diagram 2: Tumoroscope Integrated Analysis. This framework combines multiple data types to spatially map tumor clones [20].
Molecular Tumor Board Implementation
Structured interpretation of complex NGS data requires multidisciplinary expertise. Molecular tumor boards (MTBs) comprising oncologists, pathologists, geneticists, molecular biologists, and bioinformaticians provide a critical framework for translating genomic findings into clinical actionable recommendations [21]. Comparative analysis of independent MTBs reveals that while interpretation of single nucleotide variants and clinically validated biomarkers shows high agreement (66% mean overlap coefficient), interpretation of gene expression changes, preclinically validated biomarkers, and combination therapies remains challenging, highlighting areas requiring further standardization [21].
Key Findings and Clinical Implications
Prevalence of Actionable Alterations in Advanced Cancers
Large-scale genomic profiling studies have demonstrated the high frequency of potentially actionable alterations across diverse cancer types. The BALLETT study identified 1,957 pathogenic or likely pathogenic SNVs/indels, 80 pathogenic gene fusions, and 182 amplifications across 276 different genes in 756 patients [13]. The most frequently altered genes included TP53 (46% of patients), KRAS (13%), APC (9%), PIK3CA (11%), and TERT (8%) [13]. Additionally, genome-wide biomarkers with therapeutic implications were common, with 16% of patients exhibiting high tumor mutational burden (TMB-high) and 11% showing homologous recombination deficiency (HRD) in tested cases [13].
Impact on Treatment Selection and Outcomes
The comprehensive assessment of tumor genomics directly influences therapeutic decision-making. In the BALLETT study, the national molecular tumor board recommended biomarker-directed treatments for 69% of patients, with 23% ultimately receiving matched therapies [13]. Real-world evidence confirms that patients receiving treatment following concordant MTB recommendations experience significantly longer overall survival compared to those receiving treatment based on discrepant recommendations or physician's choice alone [21]. The most frequently identified treatment classes include PARP inhibitors, mTOR inhibitors, immunotherapy (immune checkpoint inhibitors), and various receptor tyrosine kinase inhibitors [21].
Table 3: Actionability of Genomic Findings in Advanced Cancers (BALLETT Study)
| Metric | Value | Implication |
|---|---|---|
| CGP success rate | 93% (756/814 patients) | Reliable implementation in clinical setting |
| Patients with ≥1 actionable marker | 81% (616/756 patients) | High potential for treatment personalization |
| Actionability with standard small panels | 21% | 4-fold increase with CGP |
| Patients with multiple actionable alterations | 41% (311/756 patients) | Opportunity for combination therapies |
| Patients receiving MTB-recommended therapy | 23% | Bridge between identification and implementation |
Comprehensive genomic profiling significantly expands therapeutic options for patients with advanced cancers [13].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 4: Essential Research Tools for NGS-Based Heterogeneity Studies
| Category | Specific Tools/Platforms | Function in Heterogeneity Research |
|---|---|---|
| NGS Platforms | Illumina NovaSeq X, Ion Torrent, PacBio Sequel, Oxford Nanopore | High-throughput sequencing with varying read lengths and applications |
| Single-Cell Technologies | 10X Genomics, Fluidigm C1 | Isolation and processing of individual cells for genomic analysis |
| Spatial Omics Technologies | 10X Visium, NanoString GeoMx | Preservation of spatial context in transcriptomic analysis |
| Bioinformatics Tools | DeepVariant, SubcloneSeeker, MethylPurify | Variant calling, clonal decomposition, methylation analysis |
| Data Integration Frameworks | Tumoroscope, Canopy | Probabilistic modeling integrating multiple data types |
| Reference Databases | TCGA, cBioPortal, COSMIC | Contextualization of findings within population-level data |
This toolkit enables comprehensive characterization of tumor heterogeneity at multiple molecular levels [10] [20] [18].
The field of tumor heterogeneity research continues to evolve rapidly, driven by technological innovations and increasing integration of multi-omics approaches. Several promising directions are emerging:
Artificial Intelligence in Genomic Analysis: AI and machine learning algorithms are becoming indispensable for analyzing complex genomic datasets. Tools like Google's DeepVariant utilize deep learning to identify genetic variants with greater accuracy than traditional methods, while AI models analyzing polygenic risk scores help predict disease susceptibility and treatment response [22].
Multi-Omics Integration: Combining genomics with transcriptomics, proteomics, metabolomics, and epigenomics provides a more comprehensive view of biological systems, linking genetic information with molecular function and phenotypic outcomes [22]. This approach is particularly valuable for understanding complex diseases like cancer, where genetics alone does not provide a complete picture.
Liquid Biopsy Applications: The use of circulating tumor DNA (ctDNA) assays offers high specificity and sensitivity for monitoring tumor heterogeneity and detecting minimal residual disease, representing a reliable tool for assessing treatment response [23].
In conclusion, next-generation sequencing has fundamentally transformed our understanding of tumor heterogeneity, revealing the complex genomic architecture that underlies cancer progression and therapeutic resistance. By enabling comprehensive molecular profiling at unprecedented resolution, NGS provides the critical tools necessary to decode this complexity and advance personalized cancer treatment. As sequencing technologies continue to evolve and computational methods become more sophisticated, the integration of NGS into routine oncologic practice promises to further refine our approach to molecularly-driven cancer care, ultimately improving outcomes for patients with diverse malignancies.
The comprehensive molecular characterization of cancer has revealed that the disease is fundamentally driven by acquired genomic aberrations. These alterations span a broad spectrum of types and sizes, ranging from single nucleotide variants (SNVs) to large structural variants (SVs) that can reorganize the genome [24]. Next-generation sequencing (NGS) has revolutionized cancer genomics by enabling researchers to identify these changes in an unbiased, genome-wide fashion, providing unprecedented insights into cancer biology and treatment opportunities [25]. The application of NGS in cancer research has demonstrated that cancer is characterized by a small number of frequently mutated genes and a long tail of infrequent mutations in a large number of genes [25]. This understanding forms the foundation of precision oncology, where molecular profiling guides targeted therapeutic interventions.
The genomic alterations in cancer cells encompass several major categories: single nucleotide variants (SNVs), small insertions and deletions (indels), copy number alterations (CNAs), and structural variants (SVs). Each category contributes differently to oncogenesis, with SVs alone affecting more base pairs in the genome than SNVs and being known drivers of carcinogenesis in at least 30% of cancers [24]. The identification and interpretation of these variants through NGS-based molecular profiling have become crucial components of both cancer research and clinical oncology, enabling informed treatment recommendations based on tumor-specific biomarker status [26].
Methodologies for NGS-Based Variant Detection
Preprocessing and Alignment of Sequencing Data
The initial steps in NGS data analysis are critical for ensuring the quality and reliability of downstream variant calling. NGS platforms generate hundreds of millions of sequence reads per instrument run, which must undergo rigorous quality control procedures. Following each sequencing run, standardized instrument manufacturer-defined pipelines process the signal-based data into sequence reads, including routine quality control on a per-lane or per-region basis to provide metrics of success for each data set [25].
A crucial quality control consideration is read duplication, where the same DNA fragment begets multiple reads or read pairs. This artifact has been attributed to the initial PCR-based library amplification steps and can affect as many as 10% of read pairs. Removal of duplicate reads is advantageous to most downstream analytical approaches since these reads may contain PCR-introduced errors that masquerade as variant nucleotides. The Picard suite provides tools for the de-duplication process that operate on both single-end and paired-end data [25]. In addition to de-duplication, data sets containing reads with insufficient read length, base quality, mapping quality, or paired-end reads having an atypical distribution of insert sizes should be flagged, soft-trimmed, and discarded when necessary to ensure data quality.
For alignment, BWA-MEM is predominantly used prior to SV detection, as it provides secondary alignments to reads mapping to multiple locations rather than placing the reads randomly [24]. The reference genome used also influences alignment performance, with studies adopting GRCh38 (hg38) showing improved alignments and fewer false-positive variants compared to GRCh37 (hg19) [24].
Experimental Design Considerations
Effective detection of somatic variants in cancer requires careful experimental design, particularly regarding sequencing depth and the inclusion of matched normal samples. In practice, a minimum of 20% allele frequency is required for reliable variant detection from tumor-normal pairs, with increasing sequencing depth to 75x-90x for tumor samples improving sensitivity for detecting low-frequency variants [24]. The use of paired tumor-normal samples enables the identification of tumor-unique (somatic) variation by distinguishing variants acquired in tumor cells from those present in the germline or as mosaic variants in healthy cells [24].
Table 1: Key Computational Tools for Detecting Genomic Alterations in Cancer
| Variant Type | Software Tools | Methodology | Key Applications |
|---|---|---|---|
| SNV Detection | SAMtools, SOAPsnp | Bayesian statistical approaches for genotype probabilities | Germline SNP calling |
| Somatic SNV Detection | VarScan, SomaticSniper, SNVmix | Heuristic or probabilistic models comparing tumor-normal pairs | Identification of tumor-specific point mutations |
| Indel Detection | Pindel, GATK Indel Genotyper | Pattern growth approach or heuristic cutoffs | Small insertion/deletion discovery |
| Structural Variant Detection | DELLY, LUMPY, Manta, SvABA, GRIDSS | Combinatorial algorithms integrating multiple read-alignment patterns | Detection of SVs across broad size ranges |
| Copy Number Alteration Detection | EWT, SegSeq, CMDS | Read-depth normalization and change-point analysis | Identification of amplifications and deletions |
Single Nucleotide Variants (SNVs) in Cancer
Biological Significance and Detection Methods
Single nucleotide variants represent the most frequent type of genomic alteration in cancer, arising from errors in DNA replication and repair. These point mutations can have profound functional consequences depending on their genomic context, including activating oncogenes through gain-of-function mutations or inactivating tumor suppressor genes through loss-of-function mutations. Notable examples include recurrent mutations in the KRAS oncogene in pancreatic and colorectal cancers, TP53 tumor suppressor mutations across multiple cancer types, and the IDH1 R132C mutations identified in acute myeloid leukemia (AML) through NGS approaches [25].
The comparison of tumor genomes with their matched constitutional genomes enables the identification of tumor-unique somatic variation in an unbiased, genome-wide fashion. Numerous SNV detection algorithms for NGS data have been developed, with SAMtools and SOAPsnp utilizing Bayesian statistics to compute probabilities of all possible genotypes [25]. However, these tools initially expected a heterozygous variant allele frequency of 50%, which is valid for germline sites but does not hold for somatic sites in most tumors due to normal contamination and/or tumor heterogeneity. This limitation has driven the development of callers designed specifically for somatic mutations, such as SNVmix, which utilizes a probabilistic Binomial mixture model and adjusts to deviation of allelic frequencies using an expectation maximization algorithm [25].
Two specifically developed somatic point mutation discovery algorithms are VarScan and SomaticSniper. VarScan determines overall genome coverage, base quality, and the number of strands observed for each allele, using read counts to infer variant allele frequency and calculating somatic status using Fisher's exact test. This approach makes VarScan well suited for somatic mutation detection in data sets having varying coverage depths, such as from targeted capture [25]. SomaticSniper uses Bayesian theory to calculate the probability of differing genotypes in the tumor and normal samples, reporting a phred-scaled probability that the tumor and normal were identical as the 'somatic' score [25]. These tools have been applied to the analysis of hundreds of tumor and normal pairs for various projects such as The Cancer Genome Atlas and the Pediatric Cancer Genome Project.
Figure 1: Computational Workflow for Somatic SNV Detection in Paired Tumor-Normal Samples
Technical Considerations and Best Practices
Effective SNV detection requires careful consideration of several technical factors. Base quality scores are crucial for distinguishing true variants from sequencing errors, with most pipelines requiring minimum quality scores typically above Q20. Mapping quality is equally important, as ambiguously mapped reads can lead to false-positive variant calls. The optimal minimum mapping quality threshold depends on the read length and complexity of the genomic region, with higher stringency required in repetitive regions [25].
Strand bias represents another critical consideration, as true variants should be supported by reads from both strands. Significant strand bias may indicate mapping artifacts or other technical issues. Additionally, the position of a variant within a read can affect confidence, with variants near read ends typically requiring more stringent filtering due to higher error rates in these regions. For somatic mutation calling, the minimum supporting reads threshold must balance sensitivity and specificity, with many pipelines requiring at least 3-5 supporting reads in the tumor sample and fewer than 1-2 in the normal sample [25].
Table 2: Key Parameters for Somatic SNV Detection
| Parameter | Typical Setting | Purpose | Impact on Results |
|---|---|---|---|
| Minimum Base Quality | Q20-Q30 | Filter sequencing errors | Higher values increase specificity but may reduce sensitivity |
| Minimum Mapping Quality | 20-40 | Filter ambiguous alignments | Reduces false positives in repetitive regions |
| Minimum Supporting Reads (Tumor) | 3-5 | Ensure variant evidence | Higher values reduce false positives but may miss low-frequency variants |
| Maximum Supporting Reads (Normal) | 1-2 | Confirm somatic status | Lower values reduce false positives from germline contamination |
| Minimum Allele Frequency | 5-10% | Filter subclonal variants | Balances detection sensitivity with technical artifacts |
| Strand Bias Filter | p-value > 0.05 | Remove technical artifacts | Eliminates variants supported by only one strand |
Small Insertions and Deletions (Indels)
Detection Challenges and Computational Approaches
Small insertions and deletions (indels) represent another class of common genomic alterations in cancer, with particular importance in microsatellite unstable tumors where defects in DNA mismatch repair lead to elevated rates of indel mutations. While existing alignment tools are generally adequate for mapping reads that contain SNVs, they typically lack the necessary accuracy and sensitivity for reads that overlap indels or structural variants [25]. Most tools by default allow only two mismatches and no gaps in the 'seeded' regions (e.g., the first 28 bp in a read), which prohibits indel-containing reads from aligning to the reference genome correctly.
Paired-end mapping is tremendously helpful in identifying larger indels, when read pair alignment occurs in flanking regions and allows the inference of altered intervening sequences [25]. Specialized tools have been developed to address the challenges of indel detection, with Pindel taking a pattern growth approach borrowed from protein data analysis to detect breakpoints of indels from paired-end reads [25]. While Pindel achieves high specificity, it can suffer from lower sensitivity primarily due to not allowing mismatches during the pattern matching process. SAMtools represents another approach that summarizes short indel information by correcting the effect of flanking tandem repeats, though it tends to produce a large number of indel calls that require additional filtering [25].
Local de novo assembly or multiple alignments around candidate indel sites has proven effective for reducing the number of false-positive indels. This process was used in the analysis of whole-genome data from a basal-like breast cancer and is currently one of the methods utilized in advanced pipelines for indel detection [25]. The GATK Indel Genotyper employs a heuristic cutoff-based approach similar to VarScan, collecting raw statistics such as coverage, numbers of indel-supporting reads, read mapping qualities, and mismatch counts, which are useful for post-filtering of the initial calls [25].
Somatic Indel Identification and Validation
Currently, somatic indel identification is generally achieved by simple subtraction of indels also found present in the normal sample. However, this approach has limitations, particularly for indels with low allele frequency or those occurring in technically challenging genomic regions. A probabilistic model for somatic indel detection represents an unmet need in the field [25]. Such a model would ideally account for the specific error profiles associated with indel detection, including the higher likelihood of alignment errors in repetitive regions and the potential for PCR artifacts to generate false-positive calls.
Validation of putative indel mutations often requires orthogonal methods, such as Sanger sequencing or specialized PCR assays, particularly for indels in homopolymer runs or other low-complexity sequences where alignment uncertainty is high. For clinical applications, careful manual review of aligned reads in visualization tools such as the Integrative Genomics Viewer (IGV) is often necessary to confirm the validity of putative indel calls [25]. The development of more robust statistical frameworks for somatic indel calling remains an active area of research in cancer genomics.
Copy Number Alterations (CNAs) in Cancer
Detection Methods and Normalization Approaches
Copy number alterations, including large amplifications or deletions of chromosomal segments, represent an important class of somatic alteration in cancer with significant functional consequences. Amplifications of oncogenes such as MYC and ERBB2 (HER2) can drive tumor progression, while deletions of tumor suppressor genes like CDKN2A contribute to unchecked cell proliferation. While SNP genotyping data have long been utilized for studying CNAs in cancer, whole-genome sequencing of tumor and matched normal samples enables the identification of CNAs at a scale and precision unmatched by traditional array-based approaches [25].
Accurate inference of copy number from sequence data requires normalization procedures to address certain biases inherent in NGS data. GC content bias arises from mechanistic differences between NGS platforms, while read mapping bias originates from the computational difficulties of assigning relatively short sequences (25-450 bp) to their correct locations in a large, complex reference genome [25]. Approaches have been developed for both GC-based coverage normalization and mapping bias correction. Following these corrections, the unique (non-redundant) read depth can serve as the basis for copy number estimation [25].
Several computational approaches have been developed specifically for CNA detection from NGS data. The EWT algorithm employs a change-point detection method to identify transitions in copy number states, while SegSeq utilizes local change-point analysis and merging to define CNA regions [25]. CMDS focuses specifically on copy number alteration calling in sample populations, enabling the identification of recurrent CNAs across multiple tumors [25]. These methods typically segment the genome into regions of constant copy number, then assign absolute copy number states through comparison with matched normal samples or through ploidy estimation algorithms.
Analytical Considerations for CNA Detection
The accurate detection of CNAs in cancer genomes presents several unique challenges beyond those encountered in germline copy number variation analysis. Tumor samples frequently exhibit aneuploidy, where the entire genome has an abnormal number of chromosomes, complicating the baseline for copy number estimation. Additionally, intratumor heterogeneity can result in multiple subclonal populations with different CNA profiles, making it difficult to determine the true cellular prevalence of any specific alteration [24].
Normal contamination represents another significant challenge, as the presence of non-cancer cells in the tumor sample dilutes the signal from tumor-specific CNAs. This effect can be mitigated through computational methods that estimate purity and ploidy, then adjust the copy number estimates accordingly. Tools such as ASCAT and ABSOLUTE have been developed specifically for this purpose, using allele-specific copy number information to simultaneously estimate tumor purity, ploidy, and absolute copy number states [24].
For targeted sequencing approaches, such as those using gene panels, CNA detection requires specialized methods that compare coverage in target regions to a reference set of normal samples. These approaches are particularly challenging for detecting focal amplifications and deletions, as the limited genomic coverage reduces statistical power. Despite these challenges, CNA detection from targeted sequencing data has proven clinically valuable, particularly for the detection of clinically actionable amplifications in genes such as ERBB2, EGFR, and MET.
Structural Variants (SVs) in Cancer Genomics
Diversity and Detection of Structural Variants
Structural variants encompass a broad range of genomic alterations that affect genome organization, including translocations, inversions, deletions, duplications, and insertions larger than typically defined for indels (often >50 bp). SVs are a major contributor to genomic variation in cancer, affecting more base pairs in the genome than SNVs and having serious phenotypic impact [24]. Some SVs are known to drive carcinogenesis directly, with SVs resulting in gene fusions representing the first recurrent mutations observed in many pediatric cancers [24].
In short-read sequencing data, SVs can be detected based on distinctive patterns in aligned reads. Discordant read-pairs that align with an abnormal distance and/or orientation to the reference genome are particularly suited for detecting large SVs. Split or soft-clipped reads, which are partially mapped reads, can indicate breakpoints with base-pair resolution [24]. The latest generation of SV detection algorithms combines multiple read-alignment patterns to detect SVs across a broad range of types and sizes. DELLY, LUMPY, Manta, SvABA, and GRIDSS employ sophisticated methodologies that achieve high performance in detecting both germline and somatic SVs [24].
Since the optimal detection algorithm differs between SV type and size range, full-spectrum SV detection with high recall and precision currently requires multiple algorithms [24]. The methodology used to combine resulting callsets remains an area of active development, with various tools and in-house pipelines currently in use. Simple integration strategies use reciprocal overlap or breakpoint distance to merge SVs, while more complex solutions combine this with read-evidence integration, local assembly, or machine learning [24]. After overlapping variants are merged, integration of SV callsets from multiple algorithms can either be performed by taking the union or intersection, with the intersection strategy often preferred in cancer research and clinical applications where achieving high precision takes priority over recall [24].
Figure 2: Multi-evidence Approach for Structural Variant Detection in Cancer Genomes
Complex Genomic Rearrangements in Cancer
Recent research has highlighted the prevalence and importance of complex genomic rearrangements (CGRs) in cancer, including phenomena such as chromothripsis (where chromosomes undergo massive shattering and reorganization), chromoplexy (involving interlinked rearrangements across multiple chromosomes), and extrachromosomal DNA (ecDNA) that can amplify oncogenes [27]. In pediatric solid tumors, CGRs have been observed in 47% of tumors, and in the majority of these cases, the CGRs affect cancer driver genes or result in unfavorable chromosomal alterations [27]. The presence of CGRs is associated with more adverse clinical events, highlighting their potential for incorporation into risk stratification or exploitation for targeted treatments [27].
The detection and interpretation of CGRs present unique challenges beyond those encountered with simple SVs. The sheer complexity of these events, with dozens or even hundreds of breakpoints concentrated in localized genomic regions, requires specialized analytical approaches. Tools such as ShatterSeek and ComplexFill have been developed specifically for identifying and characterizing chromothripsis and other complex rearrangement patterns. Additionally, the circular nature of ecDNA molecules necessitates specialized detection approaches, as their rearrangement patterns differ from those of linear chromosomal fragments.
Distinguishing Somatic from Germline SVs
The detection of tumor-specific somatic SVs aims to identify variants that uniquely occur in a patient's tumor cells. Typically, paired tumor-normal samples are used to classify SVs as either germline, mosaic-normal, or tumor-specific variants [24]. This process involves two main steps: the detection of SVs in both samples, followed by differential analysis of the callsets. Somatic SV detection algorithms differ in their approach to identify tumor-specific SVs from paired tumor-normal samples, and as a result can classify the same event differently [24].
DELLY and LUMPY use ad hoc filtering whereby SVs supported by at least one read from the normal sample are removed from the tumor SV callset, which is highly sensitive to contamination [24]. In contrast, Manta uses a probabilistic scoring system for somatic SVs integrating evidence from tumor and normal reads, while SvABA uses both the tumor and normal data during assembly before distinguishing somatic variants [24]. GRIDSS applies extensive rule-based filtering to both single break-ends and breakpoints [24]. Specialized somatic SV detection tools such as Lancet and Varlociraptor account for challenges specific to the identification of tumor-specific SVs, including differences in SV breakpoints and types between tumor and normal samples, the presence of complex rearrangements, and issues inherent to analyzing tumor samples such as contamination, polyploidy, and heterogeneity [24].
Table 3: Computational Tools for Structural Variant Analysis in Cancer
| Tool | Primary Methodology | Variant Types Detected | Somatic Classification Approach |
|---|---|---|---|
| DELLY | Integrated read-pair, split-read, and read-depth | Deletions, duplications, inversions, translocations | Filtering of normal-supported variants |
| LUMPY | Probabilistic framework combining multiple signals | Deletions, duplications, inversions, translocations | Evidence-based somatic scoring |
| Manta | Joint assembly and scoring of tumor-normal pairs | Deletions, duplications, inversions, translocations | Integrated somatic likelihood model |
| SvABA | Assembly-based variant calling | Deletions, insertions, translocations | Joint tumor-normal assembly |
| GRIDSS | Break-end assembly with quality scoring | Deletions, duplications, inversions, translocations | Extensive rule-based filtering |
Table 4: Essential Computational Tools and Databases for Cancer Genomic Analysis
| Resource Category | Specific Tools/Databases | Primary Function | Application in Cancer Genomics |
|---|---|---|---|
| Sequence Alignment | BWA-MEM, Bowtie2 | Map sequencing reads to reference genome | Foundation for all variant detection pipelines |
| Variant Calling | VarScan, SomaticSniper, Strelka | Identify somatic mutations | Detection of SNVs, indels in tumor-normal pairs |
| Structural Variant Detection | DELLY, Manta, GRIDSS | Detect large-scale genomic rearrangements | Identification of SVs including gene fusions |
| Copy Number Analysis | GATK CNV, Sequenza, ASCAT | Infer copy number alterations | Detection of amplifications and deletions |
| Visualization | IGV, Pairoscope | Visual exploration of genomic data | Validation and interpretation of variant calls |
| Annotation | ANNOVAR, VEP, FuncAssociate | Functional consequence prediction | Prioritization of biologically relevant variants |
| Data Integration | cBioPortal, IntOGen | Multi-omics data aggregation | Pathway analysis and cross-cancer comparisons |
The comprehensive characterization of genomic alterations in cancer through NGS technologies has fundamentally transformed our understanding of cancer biology and treatment. The integration of SNV, indel, CNA, and SV analyses provides a more complete picture of the molecular events driving individual tumors, enabling more precise classification and targeted therapeutic approaches. As sequencing technologies continue to evolve, particularly with the increasing adoption of long-read sequencing that can resolve complex genomic regions more effectively, our ability to detect and interpret the full spectrum of cancer-associated variants will continue to improve.
The analytical approaches discussed in this review highlight both the tremendous progress made in computational methods for variant detection and the ongoing challenges in this field. The integration of multiple algorithms and data types remains essential for achieving high sensitivity and specificity across different variant classes. As we move toward increasingly comprehensive genomic profiling in both research and clinical settings, the continued refinement of these methodologies will be crucial for realizing the full potential of precision oncology and for developing more effective, targeted cancer treatments based on the unique molecular alterations present in each patient's tumor.
Precision oncology represents a fundamental shift from histology-based to molecularly-driven cancer treatment. This evolution has been powered by advances in genomic technologies, moving from single-gene tests to comprehensive genomic profiling (CGP). Next-generation sequencing (NGS) serves as the cornerstone of this transformation, enabling simultaneous analysis of hundreds of cancer-associated genes to identify actionable biomarkers for targeted therapy selection [28] [29]. The development of precision oncology was initially constrained by technological limitations, with treatment decisions relying on single-gene tests such as immunohistochemistry (IHC) for hormone receptor status in breast cancer and PCR-based methods for detecting EGFR mutations in lung cancer [29]. The advent of NGS has revolutionized this landscape, making multigene panels and CGP standard tools in clinical oncology and accelerating the development of targeted therapies, especially for rare molecularly-defined cancer subtypes [29].
The Technological Evolution of Molecular Profiling
From Single-Gene Analysis to Comprehensive Genomic Profiling
The initial era of precision oncology relied on single-gene testing methodologies. IHC established the paradigm for biomarker-driven therapy by detecting estrogen and progesterone receptor expression to guide endocrine treatment in breast cancer [29]. Similarly, quantitative HER2 IHC was crucial for identifying patients eligible for trastuzumab therapy [29]. For mutation detection, techniques including Sanger sequencing and PCR-based genotyping were used to screen for somatic EGFR mutations in lung cancer patients to guide treatment with EGFR-selective kinase inhibitors [29].
The limitations of these single-gene approaches became apparent as knowledge of cancer genomics expanded. Testing genes sequentially consumed valuable tissue samples and time, potentially delaying critical treatment decisions [30]. The need for more comprehensive profiling led to the development of multigene NGS panels, which concurrently screen large patient populations for both standard and rare biomarkers, making trials for therapies targeting rare molecular subtypes feasible [29].
Comprehensive Genomic Profiling and the Role of NGS
Comprehensive genomic profiling utilizes NGS to perform detailed genomic analysis of cancers through a single assay, assessing hundreds of genes simultaneously [31] [32]. CGP Interrogates multiple variant types, including single nucleotide variants (SNVs), short insertions and deletions (indels), copy-number variants (CNVs), and gene fusions [13]. Additionally, it can identify genome-wide biomarkers such as tumor mutational burden (TMB), microsatellite instability (MSI), and homologous recombination deficiency (HRD) [13] [28].
The analytical scope of CGP is demonstrated by assays such as the TruSight Oncology Comprehensive (TSO Comprehensive) test, which interrogates over 500 genes from a solid tumor sample [32]. This approach provides a more complete molecular portrait of a patient's cancer compared to single-gene tests or small panels, significantly increasing the likelihood of identifying clinically actionable biomarkers [13] [32].
Complementary Sequencing Approaches
While targeted NGS panels form the current backbone of clinical genomic profiling, complementary sequencing approaches provide additional layers of molecular information:
- Whole-Genome Sequencing (WGS) Interrogates the entire ~3.2 billion base pair human genome, enabling unbiased detection of SNVs, indels, CNVs, structural rearrangements, and mutations in non-coding regions. WGS is considered the gold standard for detecting germline variants associated with hereditary cancer predisposition syndromes and complex structural variants [28].
- Whole-Exome Sequencing (WES) Targets the 1-2% of the genome that encodes proteins, providing high coverage of exonic regions where most actionable alterations reside at a lower cost and complexity than WGS [28].
- Whole-Transcriptome Sequencing (RNA-Seq) Provides dynamic representation of gene expression, enabling identification of oncogenic gene fusions, alternative splicing events, and quantitative transcript levels. RNA-Seq is particularly valuable for detecting clinically actionable fusions that may evade DNA-based sequencing [28].
Clinical Implementation and Impact of CGP
Enhanced Actionable Biomarker Detection
The superior ability of CGP to identify clinically relevant biomarkers is demonstrated by real-world studies. The Belgian BALLETT study, a nationwide multicenter trial, assessed the feasibility of using CGP in clinical decision-making for patients with advanced cancers [13]. This study enrolled 872 patients from 12 Belgian hospitals, with CGP successfully performed in 93% of cases [13].
Table 1: Actionable Biomarker Detection in the BALLETT Study (n=756 patients)
| Metric | CGP with 523-Gene Panel | Standard Small Panels (Estimated) |
|---|---|---|
| Patients with ≥1 actionable marker | 81% (616 patients) | 21% (160 patients) |
| Patients with multiple actionable alterations | 41% (311 patients) | Not reported |
| Patients with both actionable alteration and immunotherapy biomarker | 14% (104 patients) | Not reported |
| Most frequently altered genes | TP53 (46%), KRAS (13%), APC (9%), PIK3CA (11%) | Limited to genes in small panels |
| Immunotherapy biomarkers identified | TMB-high: 16% (124 patients); MSI-high: 8 patients | Limited detection capability |
The BALLETT study also demonstrated the feasibility of decentralized CGP implementation across nine local NGS laboratories using standardized methodology, with a median turnaround time of 29 days from inclusion to molecular tumor board report [13]. This highlights the potential for broader access to CGP when expertise is distributed across multiple centers situated close to clinicians and patients.
Comparison of Sequencing Methodologies in Breast Cancer
The clinical impact of testing breadth is further illustrated in advanced HR+/HER2- breast cancer. A prospective, multicenter study compared single-gene testing using the SiMSen-Seq (SSS) assay for PIK3CA hotspot mutations against broader panel-based sequencing using the AVENIO ctDNA Expanded assay (77 genes) [33].
Table 2: Single-Gene vs. Panel Sequencing in Advanced HR+/HER2- Breast Cancer
| Parameter | SiMSen-Seq (Single-Gene) | AVENIO (77-Gene Panel) |
|---|---|---|
| PIK3CA mutation detection rate | 38.4% | 36.85% |
| Concordance for PIK3CA | Reference | 92.6% overall agreement |
| Additional actionable alterations identified | Limited to PIK3CA | ESR1 (17.5%), other PI3K pathway alterations (40.6%) |
| Ability to interpret negative results | Limited without tumor fraction data | Enhanced with tumor fraction estimation |
| Mutation quantification | Targeted PIK3CA VAF | Comprehensive VAF for all detected variants |
The study demonstrated that while both assays showed high concordance for PIK3CA mutation detection, the broader AVENIO panel identified additional clinically relevant alterations beyond PIK3CA, including ESR1 mutations and other PI3K pathway alterations [33]. This expanded profiling capability is particularly relevant with the development of novel agents targeting these alterations, such as alpelisib for PIK3CA-mutated breast cancer and elacestrant for ESR1-mutated disease [33].
Key Research Reagent Solutions for CGP Implementation
The implementation of CGP in research and clinical settings requires specialized reagents and platforms. The following table outlines essential solutions for comprehensive genomic profiling:
Table 3: Essential Research Reagent Solutions for Comprehensive Genomic Profiling
| Reagent Solution Category | Representative Examples | Primary Function in CGP Workflow |
|---|---|---|
| Comprehensive Genomic Profiling Kits | TruSight Oncology Comprehensive (Illumina) [32] | Simultaneous analysis of 500+ cancer-related genes from DNA and RNA in a single test |
| Targeted NGS Panels | FoundationOneCDx, MSK-IMPACT (505 genes) [29] | Detection of mutations, indels, copy number alterations, and fusions in cancer-associated genes |
| Liquid Biopsy Assays | AVENIO ctDNA Expanded assay (77 genes) [33] | Non-invasive detection of tumor-derived mutations in circulating tumor DNA (ctDNA) |
| Library Preparation Kits | Various platform-specific kits | Preparation of sequencing libraries from tumor DNA/RNA with appropriate adapters and barcodes |
| Hybrid Capture Reagents | Biotinylated probe sets | Enrichment of target genomic regions prior to sequencing |
| Automated Workflow Solutions | Platform-specific automation kits | Streamlined, automated sample-to-report workflow to improve efficiency and reproducibility |
CGP Workflow and Methodological Framework
Implementing CGP requires a standardized workflow from sample acquisition to clinical reporting. The methodology used in the BALLETT study provides a robust framework for CGP implementation [13]:
Detailed Experimental Protocol for CGP
Based on the successful implementation in the BALLETT study and current technological platforms, the core experimental protocol for CGP includes:
Sample Requirements and Quality Control
- Tissue Samples: Formalin-fixed, paraffin-embedded (FFPE) tumor tissue sections with appropriate tumor cellularity (typically >20%)
- Nucleic Acid Extraction: Simultaneous extraction of DNA and RNA using standardized kits with quality assessment via spectrophotometry (NanoDrop) and fluorometry (Qubit)
- Quality Thresholds: DNA integrity number (DIN) >4.0, RNA integrity number (RIN) >6.0 for reliable analysis
Library Preparation and Sequencing
- Library Construction: Fragmentation of DNA and RNA followed by adapter ligation and PCR amplification
- Target Enrichment: Hybridization-based capture using biotinylated probes targeting 500+ cancer-associated genes
- Sequencing Parameters: Minimum coverage of 250-500x for tissue samples, with both DNA and RNA sequencing to detect multiple variant types
Bioinformatic Analysis Pipeline
- Alignment: Mapping of sequencing reads to reference genome (GRCh38) using optimized aligners (BWA, STAR)
- Variant Calling: Multi-algorithm approach for SNVs/indels (MuTect2, VarScan2), CNVs (CONTRA, ADTEx), and fusions (STAR-Fusion, Arriba)
- Annotation: Integration of population databases (gnomAD), cancer-specific databases (COSMIC, TCGA), and clinical knowledgebases (OncoKB)
Current Challenges and Implementation Barriers
Technical and Interpretative Challenges
Despite its demonstrated utility, the widespread implementation of CGP faces several significant challenges:
Tissue Sample Limitations: Insufficient tissue samples remain a critical barrier, with small biopsies and cytological specimens often inadequate for comprehensive testing [30]. In lung cancer, approximately 43% of cases cannot provide all required clinical information due to tissue insufficiency [30].
Data Interpretation Complexity: The analysis of complex genomic data requires specialized bioinformatics expertise and faces challenges including tumor heterogeneity, distinguishing driver from passenger mutations, and interpreting variants of unknown significance [34] [30]. The interpretation of ctDNA results adds additional complexity, requiring classification into categories informative for targeted treatment, non-targeted treatment, or non-informative [30].
Tumor Genomic Heterogeneity: Ongoing mutagenesis and clonal selection lead to evolving mutational profiles, with spatial and temporal heterogeneity potentially limiting the completeness of information from a single biopsy [29].
Access and Economic Considerations
Economic Barriers: The high cost of profiling technologies, including NGS platforms and reagents, creates accessibility challenges, particularly in resource-limited settings [34] [30]. Additional costs associated with specialized personnel (bioinformaticians, molecular biologists) further increase economic barriers.
Access Disparities: Access to NGS-based testing remains uneven, with reported access rates below 40% across Europe [30]. This highlights the need for standardized, cost-effective workflows that can be implemented across diverse healthcare settings.
Future Perspectives in Precision Oncology
Expanding Profiling Technologies
The future evolution of precision oncology will likely involve the implementation of complementary molecular profiling platforms:
Integration of Multi-Omics Approaches: Combining genomic data with transcriptomic, proteomic, and epigenomic analyses will provide more comprehensive insights into tumor biology and therapeutic vulnerabilities [28] [29].
Functional Precision Oncology: Using patient-derived models including organoids and xenografts for ex vivo drug testing may help validate genomic findings and identify effective therapeutic combinations [29].
Long-Read Sequencing Technologies: Emerging sequencing platforms offering long-read capabilities may improve detection of complex structural variants and epigenetic modifications that are challenging for short-read NGS [29].
Clinical Translation and Adoption
The expanding clinical utility of CGP is reflected in the growing proportion of tumors with standard care biomarkers. Analysis of 47,271 solid tumors found that the fraction with standard care biomarkers detectable by tumor NGS increased from 8.9% in 2017 to 31.6% in 2022, reflecting the rapid expansion of targeted therapy options [29]. This trend is expected to continue as more targeted therapies receive regulatory approval.
The tumor profiling market is projected to grow from $11.34 billion in 2024 to $26.56 billion by 2033, driven by rising cancer incidence, technological advances, and increasing adoption of precision oncology approaches [34]. This growth will likely be accompanied by ongoing efforts to address current implementation challenges, particularly regarding data interpretation, standardization, and accessibility.
The evolution from single-gene tests to comprehensive genomic profiling represents a fundamental transformation in cancer assessment and treatment selection. NGS-enabled CGP has dramatically expanded the detection of actionable biomarkers, with studies demonstrating a four-fold increase in actionability compared to traditional small panels [13]. The ongoing refinement of CGP methodologies, combined with growing biomarker-drug pairs and expanding clinical adoption, continues to advance precision oncology. However, realizing the full potential of CGP requires addressing persistent challenges including tissue limitations, data interpretation complexity, and access disparities. Future progress will likely involve the integration of multi-omics approaches, functional validation, and the development of more accessible profiling platforms to further personalize cancer therapy and improve patient outcomes.
From Bench to Bedside: NGS Methodologies and Translational Applications in Oncology
Next-generation sequencing (NGS) has fundamentally transformed molecular oncology research and drug development, enabling comprehensive genomic characterization of tumors. While whole-genome and whole-exome sequencing provide broad discovery power, targeted NGS panels have emerged as the predominant tool for clinical cancer research due to their cost-effectiveness, faster turnaround times, and enhanced sensitivity for detecting low-frequency variants [35] [36]. The strategic design of these panels—balancing gene content, coverage parameters, and technical performance—directly determines their utility in identifying clinically actionable alterations and accelerating therapeutic development.
Targeted panels focus sequencing power on a curated set of genes with established or emerging roles in cancer biology, making them particularly valuable for clinical trial biomarker assessment and diagnostic refinement. The convergence of advanced enrichment technologies, automated library preparation, and sophisticated bioinformatics has enabled researchers to develop panels that detect diverse variant types—including single nucleotide variants (SNVs), insertions/deletions (indels), copy number variations (CNVs), and gene fusions—from minimal input material [37] [38]. This technical guide examines the core principles of strategic NGS panel design, validation methodologies, and implementation frameworks that ensure generated data reliably informs drug development decisions and patient stratification strategies.
Core Design Considerations for Targeted Panels
Defining Panel Content and Genomic Scope
The foundation of effective panel design lies in carefully curating gene content based on intended research applications. Two primary approaches exist: disease-focused panels targeting specific cancer types (e.g., AML panels) and pan-cancer panels covering alterations across multiple malignancies [36] [39]. Research objectives should drive this selection; for instance, therapy selection studies require inclusion of biomarkers with established predictive value for available targeted agents.
Key considerations for content selection:
- Actionable mutations: Include genes with predictive value for targeted therapies (e.g., EGFR, BRAF, ERBB2) [40] [41]
- Prognostic markers: Incorporate variants informing disease outcomes (e.g., TP53, NPM1)
- Diagnostic biomarkers: Cover fusions and mutations essential for classification (e.g., BCR-ABL1, PML-RARA)
- Emerging biomarkers: Include genes under investigation in clinical trials to support novel target discovery
- Germline considerations: Plan for identification of presumed germline findings in cancer predisposition genes [40]
Recent pediatric cancer panel designs exemplify comprehensive content strategy, incorporating 237 genes for solid tumors and 106 fusion partner genes to address the unique genomic landscape of childhood malignancies, which frequently involves structural variants and fusion drivers [38].
Technical Design Parameters and Performance Optimization
The technical architecture of an NGS panel determines its ability to reliably detect genomic alterations. Hybrid capture-based enrichment using biotinylated oligonucleotide probes has become the predominant method for targeted sequencing due to its flexibility and ability to cover large genomic regions, including intronic sequences necessary for fusion detection [36] [37]. This approach demonstrates superior performance for detecting structural variants and copy number changes compared to amplicon-based methods.
Critical technical parameters include:
Table 1: Key Technical Design Parameters for NGS Panels
| Parameter | Considerations | Impact on Performance |
|---|---|---|
| Tiling Strategy | Probe density across target regions (1x, 2x, or advanced tiling) | 2x tiling improves coverage uniformity and accuracy for middle regions [42] |
| Repetitive Region Handling | Automatic masking of highly repetitive sequences | Prevents over-sequencing of uninformative regions and improves variant calling [42] |
| Target Region Definition | Inclusion of promoters, introns, UTRs based on application | Essential for detecting structural variants and non-coding alterations [38] |
| Input Requirements | Minimum DNA input (typically ≥50 ng) | Ensures reliable detection across all targets; lower inputs reduce sensitivity [35] |
Advanced panel designs now incorporate unique molecular identifiers (UMIs) and background noise suppression algorithms to achieve ultra-sensitive variant detection down to 0.01% variant allele frequency (VAF) for minimal residual disease monitoring [43]. This level of sensitivity is particularly valuable for drug development studies assessing early treatment response and emerging resistance mutations.
Validation and Performance Assessment Frameworks
Analytical Validation Protocols
Robust validation is essential to establish panel performance characteristics before implementation in research settings. The Association of Molecular Pathology and College of American Pathologists have established guidelines recommending an error-based approach that identifies potential sources of errors throughout the analytical process and addresses them through test design and quality controls [36].
Comprehensive validation should establish:
- Analytical sensitivity: Detection capability for different variant types at various allele frequencies
- Analytical specificity: Ability to correctly identify wild-type sequences
- Reproducibility: Consistency across replicates, operators, and instruments
- Accuracy: Concordance with orthogonal methods or reference materials
A recent validation of a 61-gene pan-cancer panel demonstrated exceptional performance metrics, achieving 98.23% sensitivity for unique variants and 99.99% specificity at 95% confidence intervals, with both repeatability and reproducibility exceeding 99.99% [35]. Such rigorous validation provides the foundation for reliable data generation in clinical research settings.
Establishing Performance Benchmarks
Performance benchmarks should be established for all critical parameters using well-characterized reference materials and clinical samples. The validation approach should reflect real-world testing conditions, including variety in sample types (FFPE, fresh frozen, liquid biopsy) and tumor content.
Table 2: Representative Performance Metrics for Validated NGS Panels
| Performance Metric | Target Performance | Established Example |
|---|---|---|
| Minimum DNA Input | ≥50 ng | Reliable detection of all 13 mutations in HD701 reference standard [35] |
| Limit of Detection (VAF) | ≤3% for SNVs/Indels | 2.9% VAF established for 61-gene panel; 0.01% for specialized MRD panels [35] [43] |
| Sensitivity | >98% | 98.23% sensitivity for unique variants at 95% CI [35] |
| Specificity | >99.9% | 99.99% specificity for 61-gene panel [35] |
| Coverage Uniformity | >93% | Mean uniformity of 93% achieved in AML panel [39] |
| Concordance with Orthogonal Methods | 100% | 100% concordance for 92 known variants from orthogonal methods [35] |
The following workflow diagram illustrates the key decision points in NGS panel design and validation:
NGS Panel Design Workflow
Implementation in Cancer Research and Drug Development
Clinical Utility and Research Applications
Well-designed NGS panels demonstrate significant impact across multiple research domains, particularly in diagnostic refinement, patient stratification, and therapy selection. A comprehensive study of pediatric cancer patients found that somatic NGS panel testing significantly influenced clinical care in 78.7% of cases, informing diagnosis, prognosis, and treatment planning [38]. Similarly, in adult cancers, comprehensive genomic profiling identified actionable alterations in 41.6% of advanced cancer patients, though only 3.6% ultimately received genomically matched therapy, highlighting both the potential and challenges in translating genomic findings to treatment [40].
Key applications in drug development:
- Clinical trial stratification: Enrichment of trial populations based on molecular alterations
- Biomarker discovery: Identification of novel predictive biomarkers for targeted therapies
- Response monitoring: Detection of emerging resistance mutations during treatment
- Mechanistic studies: Elucidation of drug mechanisms through comprehensive genomic profiling
The development of specialized panels for minimal residual disease (MRD) monitoring represents a particularly advanced application, with newer panels achieving sensitivity of 0.01% VAF for detecting residual leukemia cells after treatment [43]. This capability provides a powerful tool for assessing treatment efficacy in clinical trials and understanding the dynamics of tumor evolution under therapeutic pressure.
Turnaround Time and Workflow Integration
Turnaround time (TAT) represents a critical practical consideration in panel design, particularly for time-sensitive clinical research applications. While external laboratory testing typically requires approximately 3 weeks, optimized in-house panels can reduce TAT to 4 days through streamlined workflows and automated library preparation [35] [38]. This acceleration enables more rapid integration of genomic findings into research decisions and patient management.
Integration strategies for research workflows:
- Automation compatibility: Designing panels compatible with automated liquid handling systems
- Streamlined bioinformatics: Implementing user-friendly analysis pipelines with pre-configured settings
- Multi-omic approaches: Combining DNA and RNA sequencing from the same sample
- Longitudinal analysis: Incorporating tools for tracking genomic evolution over time
The implementation of automated hybridization capture steps using robotic systems has demonstrated significant benefits, enabling batch processing of 96 samples with resulting improvements in efficiency and cost-effectiveness for hematological malignancy testing [37].
Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for NGS Panel Development
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| Target Enrichment | Hybrid capture probes (SureSelect, Cell3 Target) | Sequence-specific capture of genomic regions of interest [38] [37] |
| Library Preparation | SureSelectQXT, Archer Universal RNA Reagent Kit | Fragmentation, adapter ligation, and amplification for sequencing [38] |
| Automation Systems | Hamilton robot programs | Automated hybridization capture for batch processing [37] |
| Reference Materials | HD701, NA12878, commercial FFPE controls | Assay validation, quality control, and performance monitoring [35] [36] |
| Bioinformatics Tools | Sophia DDM, DeepVariant, Archer analysis | Variant calling, annotation, and interpretation [35] [22] |
Emerging Trends and Future Directions
The landscape of NGS panel design continues to evolve, driven by technological innovations and expanding clinical applications. Several emerging trends are particularly noteworthy:
AI-Enhanced Bioinformatics: Artificial intelligence and machine learning algorithms are increasingly being integrated into variant calling pipelines, with tools like Google's DeepVariant demonstrating superior accuracy compared to traditional methods [22]. These approaches are particularly valuable for interpreting complex variants and filtering artifacts in challenging sample types.
Liquid Biopsy Applications: The development of specialized panels for circulating tumor DNA (ctDNA) analysis enables non-invasive tumor genotyping and monitoring. Custom panels can achieve detection limits of 0.125% for known variants and 0.3% for novel variants in liquid biopsy applications, supporting applications in therapy response monitoring and resistance detection [37].
Multi-omic Integration: Combined DNA and RNA sequencing panels provide comprehensive molecular profiling from limited specimen material. The relationship between different molecular profiling approaches and their applications can be visualized as follows:
Multi-Omic Integration Approach
The ongoing convergence of advanced genomics, CRISPR-based functional screening, and AI-driven analytics promises to further refine panel design and application, enabling more personalized and effective cancer therapeutic strategies [41]. As these technologies mature, strategic NGS panel design will remain essential for translating genomic insights into meaningful advances in cancer research and drug development.
Next-generation sequencing (NGS) has become a cornerstone of precision oncology, enabling comprehensive genomic profiling of tumors to guide diagnosis, prognosis, and treatment selection [10] [44]. The fidelity of this profiling critically depends on the initial library preparation step, where two principal methods—hybrid capture and amplicon-based enrichment—dominate the landscape [45] [46]. This guide provides an in-depth technical comparison of these two approaches, framing their capabilities, methodologies, and optimal applications within modern cancer molecular profiling research.
Core Principles and Workflows
The fundamental difference between these methods lies in how they isolate genomic regions of interest prior to sequencing.
Amplicon-Based Target Enrichment
Amplicon sequencing (Amplicon-Seq) uses a "library-then-capture" approach centered on polymerase chain reaction (PCR) [45]. It employs a multitude of primers to directly amplify the specific genomic targets from a sample. A key limitation is its requirement for precise primer matching, particularly at the 3' end, as even minor sequence mismatches can lead to amplification failure or significant bias [45]. This method is celebrated for its rapid, streamlined workflow, minimal input DNA requirements, and high on-target rates, making it ideal for focused panels [47] [48] [49].
Hybrid Capture-Based Target Enrichment
Hybrid capture employs a "capture-then-library" or "library-then-capture" strategy [45] [46]. In the widely adopted solution-based method, sheared genomic DNA is first converted into a sequencing library with adapter ligation. Subsequently, biotinylated oligonucleotide probes are hybridized with the library in solution, specifically binding to the target regions. These probe-target complexes are then captured and purified using streptavidin-coated magnetic beads before being amplified and sequenced [46]. A major advantage is its higher mismatch tolerance, allowing probes to bind successfully to target regions with only ~70-75% sequence similarity, which is particularly beneficial for analyzing genetically diverse samples or when using a reference genome from a related species [45].
The following diagram illustrates the distinct workflows for each method.
Performance Comparison in Cancer Genomics
The choice between amplicon and hybrid capture methods has profound implications for the success of cancer genomic studies. The table below summarizes key performance metrics and characteristics critical for research and clinical applications.
Table 1: Performance and Characteristic Comparison of NGS Library Prep Methods
| Aspect | Amplicon-Based Sequencing | Hybrid Capture-Based Sequencing |
|---|---|---|
| Mismatch Tolerance | Low; requires perfect primer match, especially at 3' end [45] | High; allows ~70-75% sequence similarity [45] |
| Typical Input DNA | Low (e.g., 20 ng) [50] [49] | High; requires more due to fragmentation losses [45] [49] |
| Workflow Simplicity | High; fewer steps, faster (e.g., 3 hours for some panels) [47] [49] | Low; more steps, time-consuming (can be 1-2 days) [45] [47] |
| On-Target Rate | Naturally high (>96%) [49] | Lower than amplicon; requires optimization [47] |
| Coverage Uniformity | Can be lower with high PCR background [49] | High; superior uniformity [47] [49] |
| Variant Detection | Excellent for SNVs/Indels [50] [45] | Excellent for SNVs, Indels, CNVs, and Fusions [50] [46] |
| TMB/MSI/HRD | Possible with specific panels [50] | Reliable for complex biomarkers (TMB, MSI, HRD) [50] [51] |
| Panel Scalability | Flexible, but traditionally limited in multiplex scale [49] | Virtually unlimited; suitable for exome-scale panels [47] |
| Cost per Sample | Generally lower [47] | Higher [47] |
The table above highlights trade-offs. Amplicon-based methods, such as the Oncomine Comprehensive Assay Plus (OCA Plus), demonstrate high proficiency in detecting simple biomarkers like single nucleotide variants (SNVs), insertions/deletions (indels), and fusions, with reported concordance rates of 94.8% and 94.2%, respectively, against orthogonal methods [50]. However, its performance for complex biomarkers like tumor mutational burden (TMB) and microsatellite instability (MSI) was lower, with concordance of 81.3% and 80.8%, respectively [50]. Its requirement for perfect primer matching can be a limitation in analyzing samples with unknown or highly variable sequences [45].
Hybrid capture excels in applications requiring comprehensive genomic analysis. Its ability to reliably detect a wide range of alterations, including copy number variations (CNVs) and structural variants (SVs), and complex biomarkers like TMB and MSI makes it a robust choice for large-scale profiling [50] [46] [51]. Its higher tolerance for sequence mismatches also makes it more suitable for phylogenetics or when a complete reference genome for the target species is unavailable [45].
Method Selection Guide for Research Applications
Choosing the appropriate method is a strategic decision that depends on research goals, sample characteristics, and available resources.
Table 2: Method Selection Guide for Oncology Research Applications
| Application | Recommended Method | Rationale |
|---|---|---|
| Hotspot/Gene Signature Validation | Amplicon-Seq | High sensitivity, cost-effective for small targets [48] |
| CRISPR QC/Genome Editing | Amplicon-Seq | Precise validation of on-/off-target edits [48] [49] |
| Large Panels/Exome Sequencing | Hybrid Capture | Superior uniformity and scalability for large regions [47] [49] |
| Complex Biomarker Analysis (TMB) | Hybrid Capture | Broader, more unbiased genomic context improves accuracy [50] [51] |
| Phylogenetic/Evolutionary Studies | Hybrid Capture | Higher mismatch tolerance allows use of related reference genomes [45] |
| Analysis of Degraded/FFPE DNA | Amplicon-Seq | Works well with low input and fragmented DNA [49] |
| Liquid Biopsy (cfDNA) | Hybrid Capture | More effective for short, fragmented cfDNA; suitable for methylation capture [45] |
Experimental Protocols for Cancer Research
This section details standard protocols for implementing each method, as utilized in recent cancer genomics studies.
Protocol: Amplicon-Based Sequencing (e.g., Oncomine Comprehensive Assay Plus)
This protocol is adapted from a 2025 multicenter evaluation of the OCA Plus panel for profiling solid tumors [50].
- Step 1: Nucleic Acid Isolation. Extract DNA and RNA from Formalin-Fixed Paraffin-Embedded (FFPE) tumor samples using a validated kit (e.g., QIAamp DNA FFPE Tissue Kit). Assess quality and quantity, requiring a minimum of 20 ng of DNA and RNA. A tumor cell content of at least 10% should be confirmed by a pathologist [50] [51].
- Step 2: Library Preparation. Treat DNA with uracil DNA glycosylase (UDG) to remove deaminated cytosines that cause C>T artifacts. Synthesize cDNA from RNA. Prepare libraries manually according to the OCA Plus protocol. The panel covers 501 genes for DNA analysis and 49 driver genes for RNA-based fusion detection [50].
- Step 3: Templating and Sequencing. Template the libraries using an Ion Chef System. Sequence the prepared libraries on an Ion GeneStudio S5 Plus System using Ion 550 chips [50].
- Step 4: Data Analysis. Use Torrent Suite and Ion Reporter software for base calling, alignment (to GRCh37/hg19), and variant annotation. Apply specific filter chains for variant calling and complex biomarker calculation (e.g., TMB, MSI) [50].
Protocol: Hybrid Capture-Based Sequencing (e.g., SNUBH Pan-Cancer Panel)
This protocol is derived from a 2024 real-world clinical study implementing an NGS cancer panel [51].
- Step 1: Sample Preparation and DNA Extraction. Perform manual microdissection on FFPE samples to enrich tumor regions. Extract genomic DNA using a kit (e.g., QIAamp DNA FFPE Tissue Kit). Quantify DNA using a fluorometer (e.g., Qubit) and assess purity via spectrophotometry (A260/A280 ratio of 1.7-2.2). A minimum of 20 ng of DNA is required [51].
- Step 2: Library Preparation and Target Enrichment. Shear DNA enzymatically or mechanically. Ligate sequencer-specific adapters with sample barcodes. For target enrichment, use a solution-based hybrid capture method (e.g., Agilent SureSelectXT Target Enrichment Kit). Hybridize the library with a pool of biotinylated probes targeting the panel's genes (e.g., 544 genes). Capture the probe-bound targets using streptavidin-coated magnetic beads, followed by washing and amplification [51].
- Step 3: Sequencing. Sequence the final enriched library on a platform such as the Illumina NextSeq 550Dx. The average mean depth of coverage for the cohort in the cited study was 677.8x, with a minimum coverage threshold [51].
- Step 4: Bioinformatic Analysis. Align reads to the reference genome (hg19). Use tools like Mutect2 for SNV/indel detection (with a VAF threshold ≥2%), CNVkit for CNV analysis, and LUMPY for gene fusions. Calculate MSI and TMB using dedicated algorithms [51].
Essential Research Reagents and Materials
Successful implementation of NGS library preparation methods relies on a suite of specialized reagents and tools.
Table 3: Research Reagent Solutions for NGS Library Preparation
| Item | Function | Example Products/Brands |
|---|---|---|
| Nucleic Acid Extraction Kit | Isolates high-quality DNA/RNA from complex biological samples (e.g., FFPE tissue). | QIAamp DNA FFPE Tissue Kit (Qiagen) [51] |
| Target-Specific Primer Panels | Multiplexed primers for amplifying regions of interest in amplicon-based methods. | Oncomine Comprehensive Assay Plus (Thermo Fisher) [50], CleanPlex Panels (Paragon Genomics) [49] |
| Biotinylated Probe Panels | Probes for hybridizing to and capturing target regions in hybrid capture methods. | Agilent SureSelectXT (Agilent Technologies) [51] |
| Streptavidin Magnetic Beads | Bind to biotinylated probe-target complexes for purification and separation. | Component of SureSelectXT and other hybrid capture kits [46] [51] |
| Library Preparation Master Mix | Contains enzymes and buffers for PCR, adapter ligation, and other enzymatic steps. | Ion Torrent NGS Reverse Transcription Kit (Thermo Fisher) [50] |
| UDG Enzyme | Treats DNA to remove deaminated cytosines, reducing sequencing artifacts in FFPE samples. | Uracil DNA Glycosylase (Thermo Fisher) [50] |
| NGS Platform | Instrumentation for performing massively parallel sequencing. | Ion GeneStudio S5 Plus (Thermo Fisher), Illumina NextSeq 550Dx [50] [51] |
In cancer molecular profiling research, the decision between hybrid capture and amplicon-based library preparation is not a matter of superiority but of strategic alignment with the study's objectives. Amplicon-based methods offer a fast, sensitive, and cost-efficient solution for focused panels and high-throughput screening of known targets. In contrast, hybrid capture provides a robust, comprehensive, and flexible platform for discovery-oriented research, large genomic regions, and complex biomarker analysis like TMB. Advances in multiplex PCR are blurring the lines, with newer amplicon technologies overcoming traditional limitations in panel size and uniformity [49]. As NGS continues to propel precision oncology forward, the informed selection and ongoing refinement of these library preparation approaches will remain fundamental to unlocking the molecular secrets of cancer.
Comprehensive Genomic Profiling (CGP) represents a transformative approach in cancer research and clinical oncology, enabling the simultaneous detection of hundreds of biomarkers across multiple genomic variant classes through next-generation sequencing (NGS) technologies. Unlike traditional single-gene tests or limited panels, CGP provides nucleotide-level resolution for identifying single nucleotide variants (SNVs), insertions/deletions (indels), copy number variants (CNVs), gene fusions, and splice variants, while also quantifying genomic signatures such as tumor mutational burden (TMB) and microsatellite instability (MSI) [52]. This comprehensive approach maximizes the potential for discovering clinically actionable alterations that drive precision oncology initiatives, facilitating both therapeutic development and personalized treatment strategies for cancer patients across diverse malignancy types.
The transition from sequential single-gene testing to multiplexed CGP represents a paradigm shift in cancer molecular profiling. Where traditional approaches required multiple separate tests that consumed precious tissue samples and extended turnaround times, CGP consolidates biomarker detection into a single multiplex assay [52]. This consolidation is particularly valuable in advanced cancers where biopsy material is limited, as it preserves samples while providing a more complete genomic landscape of each patient's tumor. For research scientists and drug development professionals, this comprehensive data generation enables the discovery of novel biomarkers, identification of resistance mechanisms, and development of targeted therapies for patient subpopulations defined by specific genomic characteristics rather than solely by tumor histology.
Technical Foundations of Comprehensive Genomic Profiling
Core Principles and Methodological Framework
CGP operates on the fundamental principle of massively parallel sequencing, a hallmark of NGS technologies that enables the simultaneous analysis of millions of DNA fragments [44]. This represents a significant advancement over first-generation Sanger sequencing, which processes individual DNA fragments sequentially with limited throughput and higher costs for large-scale analyses [10]. The methodological framework of CGP encompasses four critical stages: (1) sample preparation and library construction, (2) target enrichment, (3) sequencing, and (4) bioinformatic analysis and interpretation [10]. Each stage requires rigorous optimization and quality control to ensure the accuracy and reproducibility of results, particularly when implemented in clinical research settings where findings may inform therapeutic development decisions.
The library preparation phase begins with nucleic acid extraction from tumor specimens, typically formalin-fixed paraffin-embedded (FFPE) tissue or less frequently from liquid biopsy sources. The quality and quantity of extracted DNA are critically assessed, with minimum thresholds of 20 ng DNA and A260/A280 ratios between 1.7-2.2 representing typical quality standards [51]. Following extraction, genomic DNA undergoes fragmentation—through physical, enzymatic, or chemical methods—to appropriate sizes (approximately 300 bp), after which adapter sequences (synthetic oligonucleotides with platform-specific sequences) are ligated to fragment ends [10]. These adapters facilitate both the amplification of fragment libraries and their attachment to sequencing platforms. For targeted CGP approaches, an enrichment step isolates coding sequences or specific genomic regions of interest, typically accomplished through polymerase chain reaction (PCR) amplification with specific primers or hybridization with exon-specific probes [10].
Comparative Analysis of Genomic Profiling Approaches
Table 1: Comparison of Genomic Profiling Technologies in Cancer Research
| Feature | Single-Gene Tests | Targeted Panels | Comprehensive Genomic Profiling (CGP) | Whole Exome Sequencing |
|---|---|---|---|---|
| Number of Biomarkers | Single biomarker | Dozens of specific genes | Hundreds of genes | ~20,000 genes |
| Variant Types Detected | Limited to specific variant types | Limited to panel-specific variants | SNVs, indels, CNVs, fusions, TMB, MSI | Primarily SNVs and indels |
| Tissue Consumption | High (with iterative testing) | Moderate | Low (single test) | Low to moderate |
| Novel Discovery Potential | None | Low | High | Highest |
| Clinical Actionability | Limited to single gene | Variable | High | Limited by interpretative challenges |
| Cost-Effectiveness | Costly for multiple genes | Moderate for targeted approach | High value for breadth | Lower for clinical applications |
| Turnaround Time | Rapid for single test | Moderate | Single test replaces multiple | Lengthy |
| Coverage Depth | Very high | High | High with uniform coverage | Variable |
As illustrated in Table 1, CGP occupies a strategic position between limited targeted panels and extensive whole exome sequencing, offering an optimal balance between comprehensive genomic coverage and clinically actionable results. While single-gene tests remain useful for validating specific alterations, their iterative application consumes significant tissue resources and may delay research conclusions [52]. Targeted panels offer improved efficiency but typically cover only specific genomic regions rather than entire gene sequences, potentially missing important alterations in non-targeted regions [52]. Whole exome sequencing provides exhaustive coverage but often with inadequate depth for detecting lower-frequency variants and presents significant interpretive challenges for clinical translation [52].
Experimental Protocols and Workflow Specifications
Laboratory Implementation Framework
The implementation of CGP in research settings requires standardized protocols to ensure reproducible and reliable results. The SNUBH Pan-Cancer version 2.0 protocol exemplifies a robust workflow for CGP implementation [51]. This protocol begins with manual microdissection of FFPE tumor specimens to select representative tumor areas with sufficient cellularity (typically >20% tumor content). DNA extraction employs specialized kits designed for FFPE material (e.g., QIAamp DNA FFPE Tissue kit), with quality assessment through fluorometric quantification (Qubit dsDNA HS Assay) and purity verification (NanoDrop Spectrophotometer) [51].
Following quality control, library preparation utilizes hybrid capture-based target enrichment (e.g., Agilent SureSelectXT Target Enrichment Kit) with panels covering hundreds of cancer-relevant genes. The prepared libraries undergo quantitative and qualitative assessment through Bioanalyzer systems, with size thresholds of 250-400 bp and minimum concentration requirements (typically ≥2 nM) [51]. Sequencing occurs on established platforms such as Illumina NextSeq 550Dx, with a minimum depth of coverage (typically >500x) and >80% of targets achieving 100x coverage as quality thresholds. This rigorous approach ensures consistent performance across research samples and enables reliable detection of somatic variants present at low variant allele frequencies.
Figure 1: CGP Experimental Workflow illustrating the three major phases of comprehensive genomic profiling, from sample preparation through final interpretation.
Bioinformatic Analysis Pipeline
The computational analysis of CGP data requires a sophisticated bioinformatic pipeline to transform raw sequencing data into clinically interpretable results. The foundational step involves alignment of sequencing reads to a reference genome (typically hg19/GRCh37) using optimized aligners such as BWA-MEM or Bowtie2 [44] [51]. Subsequent variant calling employs specialized tools: Mutect2 for SNVs and small indels, CNVkit for copy number variations, and LUMPY for structural variants including gene fusions [51]. Variant annotation utilizes resources like SnpEff combined with comprehensive cancer databases to characterize the functional impact of identified alterations.
For genomic signature analysis, CGP pipelines incorporate specialized algorithms: MSI status determination using mSINGs or similar tools, and TMB calculation as the number of eligible mutations per megabase of sequenced genome [51]. Critical to research applications is the implementation of rigorous filtering criteria, including minimum depth thresholds (typically ≥200x), variant allele frequency cutoffs (commonly ≥2-5%), and population frequency filtering (excluding variants with >1% frequency in population databases) [51]. The final variant classification follows established frameworks such as the Association for Molecular Pathology guidelines, which categorize alterations into four tiers based on clinical significance: Tier I (variants of strong clinical significance), Tier II (variants of potential clinical significance), Tier III (variants of unknown significance), and Tier IV (benign or likely benign variants) [51].
Key Research Reagents and Solutions for CGP Implementation
Table 2: Essential Research Reagents and Platforms for CGP Workflows
| Category | Specific Products/Platforms | Research Application | Technical Considerations |
|---|---|---|---|
| Nucleic Acid Extraction | QIAamp DNA FFPE Tissue Kit | Isolation of high-quality DNA from challenging FFPE specimens | Optimized for fragmented, cross-linked DNA from archival tissues |
| Target Enrichment | Agilent SureSelectXT | Hybrid capture-based enrichment of target regions | Customizable target content; effective for large gene panels |
| Library Preparation | Illumina TruSight Oncology Comprehensive | Integrated DNA and RNA library preparation | Standardized workflow for simultaneous genomic and transcriptomic profiling |
| Sequencing Platforms | Illumina NextSeq 550Dx, NovaSeq | High-throughput sequencing | Balance between read length, depth, and cost for large panels |
| Variant Callers | Mutect2, CNVkit, LUMPY | Detection of SNVs, CNVs, and structural variants | Algorithm selection depends on variant type and signal-to-noise ratio |
| Variant Annotation | SnpEff, OncoKB, ClinVar | Functional and clinical interpretation of variants | Integration of multiple databases improves classification accuracy |
| MSI Detection | mSINGs, MSIsensor | Assessment of microsatellite instability | Requires established thresholds for MSI-H classification |
| TMB Calculation | Custom algorithms | Quantification of tumor mutational burden | Dependent on panel size and filtering criteria for accurate estimation |
The selection of appropriate research reagents and bioinformatic tools dramatically impacts the quality and interpretability of CGP data. As shown in Table 2, each component of the workflow requires careful consideration of technical specifications and compatibility with research objectives. For drug development professionals, standardized reagents and platforms facilitate reproducibility across studies and enable meta-analyses of genomic data from multiple research cohorts. The integration of annotation resources with clinical trial databases further enhances the ability to identify potential therapeutic targets and stratify patient populations for clinical trial enrollment based on molecular profiles.
Clinical Research Applications and Validation Studies
Research Validation Across Malignancies
CGP has demonstrated significant utility across diverse cancer types in research settings, revealing previously uncharacterized molecular heterogeneity and identifying potential therapeutic targets. In advanced soft tissue and bone sarcomas—a group of malignancies with limited treatment options and complex genomic landscapes—CGP identified actionable mutations in 22.2% of patients, making them eligible for FDA-approved targeted therapies [14]. The most frequent alterations occurred in TP53 (38%), RB1 (22%), and CDKN2A (14%) genes, highlighting key pathways involved in sarcoma pathogenesis [14]. Importantly, CGP facilitated reclassification of diagnosis in four patients, demonstrating its value in refining pathological classification beyond conventional histomorphology [14].
In non-small cell lung cancer (NSCLC), real-world evidence from 3,884 patients demonstrated that CGP identified one or more actionable biomarkers in 32% of cases, compared to only 14% with single-gene testing [53]. This enhanced detection rate translated into improved clinical outcomes, with CGP-tested patients showing significantly longer median overall survival (15.7 months versus 7 months) and higher rates of matched targeted therapy utilization [53]. For research applications, these findings validate CGP as a powerful tool for patient stratification in clinical trials and for identifying novel biomarker-therapy associations across diverse cancer types.
Biomarker Discovery and Therapeutic Matching
Table 3: Actionable Alterations Identified Through CGP in Research Studies
| Cancer Type | Most Frequently Altered Genes | Actionable Alteration Rate | Common Therapeutic Implications |
|---|---|---|---|
| Advanced Sarcomas [14] | TP53 (38%), RB1 (22%), CDKN2A (14%) | 22.2% | CDK4/6 inhibitors, PARP inhibitors, MDM2 antagonists |
| Non-Small Cell Lung Cancer [51] | KRAS (10.7%), EGFR (2.7%), BRAF (1.7%) | 26.0% (Tier I variants) | EGFR inhibitors, KRAS G12C inhibitors, BRAF/MEK inhibitors |
| Multiple Solid Tumors [52] | Varies by histology | 30-50% across studies | Histology-agnostic therapies for MSI-H, NTRK fusions, high TMB |
| Rare/Refractory Cancers [52] | Diverse molecular profiles | 43.4% | Off-label targeted therapies, clinical trial enrollment |
The comprehensive nature of CGP enables detection of rare but clinically significant alterations that might be missed by limited testing approaches. In colorectal cancer research, CGP has characterized somatic mutations in both canonical genes (TP53, APC) and previously unreported genetic alterations, expanding understanding of molecular carcinogenesis pathways [54]. Similarly, in gallbladder cancer, CGP has identified targetable alterations in BRCA1/2, EGFR, and ERBB2 genes, revealing potential therapeutic opportunities for a malignancy with historically limited treatment options [54]. The ability to simultaneously assess multiple biomarker classes positions CGP as an essential discovery tool for identifying novel therapeutic targets and resistance mechanisms across the cancer spectrum.
For drug development professionals, CGP facilitates the identification of patient populations most likely to respond to investigational therapies, particularly in basket trials that enroll patients based on molecular alterations rather than tumor histology. The detection of genomic signatures such as TMB and MSI status further enables immunotherapy development, as these biomarkers predict response to immune checkpoint inhibitors across multiple cancer types [52]. As targeted therapy options expand, CGP provides the necessary comprehensive molecular profiling to match patients with appropriate therapeutic strategies based on the unique molecular characteristics of their tumors.
Integration with Emerging Research Technologies
The research utility of CGP continues to expand through integration with complementary genomic technologies and computational approaches. Single-cell sequencing methodologies provide unprecedented resolution of intratumoral heterogeneity and tumor microenvironment interactions, revealing cellular subpopulations with distinct molecular features and potential differential treatment responses [44] [54]. Spatial transcriptomics technologies further enhance this capability by preserving topological information, enabling researchers to map genomic alterations within specific tumor regions and their relationship to the tumor-immune interface [44].
Liquid biopsy approaches using circulating tumor DNA (ctDNA) represent another transformative application, enabling non-invasive genomic profiling through CGP when tissue samples are unavailable or insufficient [53]. Research studies have demonstrated that combining tissue and liquid biopsy profiling identifies more patients with actionable mutations than either method alone. In NSCLC, 65.7% of actionable mutations were detected by both methods, while 29% were identified exclusively through tissue profiling and 5.5% were detected only in liquid biopsy despite successful tissue analysis [53]. This complementary approach provides a more comprehensive assessment of tumor genomics, potentially capturing heterogeneity across different metastatic sites.
Artificial intelligence and machine learning algorithms are increasingly being applied to CGP data to enhance pattern recognition, biomarker discovery, and outcome prediction. These computational approaches can identify complex relationships between multiple genomic alterations and treatment responses, potentially revealing novel predictive biomarkers that would be difficult to detect through conventional statistical methods [44] [54]. As these technologies mature, their integration with CGP workflows will further advance precision oncology research by enabling more sophisticated analysis of complex genomic datasets and accelerating the translation of molecular insights into therapeutic strategies.
Comprehensive Genomic Profiling represents a fundamental advancement in cancer research methodologies, consolidating the detection of hundreds of biomarkers into streamlined workflows that generate actionable insights for therapeutic development. Through its ability to simultaneously assess multiple variant classes and genomic signatures across extensive gene sets, CGP provides researchers with a powerful tool for understanding cancer biology, identifying novel therapeutic targets, and stratifying patient populations for clinical trial enrollment. The continued evolution of CGP technologies—including integration with single-cell analysis, liquid biopsy, and artificial intelligence—promises to further expand its research applications and enhance our understanding of cancer genomics. For drug development professionals and translational researchers, CGP has become an indispensable component of the precision oncology toolkit, driving the development of more effective, biomarker-driven therapeutic strategies across diverse cancer types.
Next-generation sequencing (NGS) has emerged as a transformative technology in oncology drug discovery, providing unprecedented capabilities for comprehensive genomic analysis. By enabling rapid, cost-effective, and high-throughput sequencing of DNA and RNA, NGS technologies have fundamentally reshaped approaches to target identification and validation in precision oncology. The technology's capacity to process millions of DNA fragments simultaneously has significantly reduced the time and cost associated with genomic sequencing, making large-scale studies feasible for research and clinical applications [10] [55]. This technological revolution has positioned NGS as a cornerstone in the shift toward molecularly-driven cancer care, allowing researchers to identify disease-associated genetic variants with unprecedented efficiency and scale.
The integration of NGS into oncology research has been accelerated by dramatic reductions in sequencing costs – from approximately $100 million per genome in 2001 to under $1,000 today [56] – coupled with continuous improvements in sequencing speed and accuracy. These advancements have enabled researchers to move beyond single-gene assays to comprehensive genomic profiling that captures the full complexity of tumor genomics. In the context of drug discovery, NGS provides critical insights into cancer mechanisms, therapeutic targeting opportunities, and biomarkers for patient stratification, ultimately accelerating the development of targeted therapies and personalized treatment strategies [10] [55].
NGS Technologies and Methodologies
Core Sequencing Technologies and Platforms
Next-generation sequencing encompasses several distinct technology platforms, each with unique methodologies for template preparation, sequencing, and imaging. The fundamental principle unifying all NGS platforms is the massive parallel sequencing of spatially separated, immobilized DNA templates, enabling millions of simultaneous sequencing reactions [57]. The four major technological approaches currently dominating the field include: (1) Complementary metal-oxide semiconductor (CMOS) technology used by Ion Torrent Personal Genome Machine, which employs ion-sensitive field-effect transistors to detect hydrogen ions released during DNA polymerization; (2) Single-molecule real-time (SMRT) sequencing utilized by Pacific Biosciences, enabling real-time observation of DNA synthesis; (3) Incorporation of fluorescently labeled reversible terminators (FLRT) implemented in Illumina platforms; and (4) Combination of emulsion PCR and pyrosequencing used by Roche/454 systems [57].
Each technology offers distinct advantages in read length, accuracy, throughput, and cost parameters, making them suitable for different applications in drug discovery. Illumina platforms typically provide read lengths of 50-300 bp with 98% accuracy, while Pacific Biosciences SMRT sequencing offers much longer read lengths (up to 14,000 bp) while maintaining high accuracy [57]. The selection of an appropriate sequencing platform depends on the specific research objectives, with considerations including the need for detection of structural variants, requirement for quantitative analysis, and balance between throughput and read length.
Template Preparation Methods
The initial stage of any NGS workflow involves template preparation, which fundamentally determines data quality and applicability for different research questions. Three well-established approaches for template creation are utilized across platforms:
Clonally amplified templates rely on PCR-based amplification (emulsion PCR or bridge PCR) to generate sufficient signal for detection. This method requires sample concentrations of less than 20 ng/μL and is susceptible to amplification bias in AT-rich and GC-rich regions [57].
Single-molecule templates are prepared and immobilized on solid surfaces without amplification, reducing sequencing error rates and avoiding amplification bias. This approach requires minimal preparation materials (<1 μg) and can accommodate larger DNA molecules, facilitating longer read lengths [57].
Circle templates represent a recently developed library preparation method that dramatically reduces error rates through rolling circle replication. This approach is particularly suitable for cancer profiling, diploid and rare-variant calling, microbial diversity, immunogenetics, and environmental sampling [57].
For quantitative NGS analyses such as transcriptome or gene expression profiling, single-molecule templates are recommended to avoid sequence amplification bias. For qualitative analyses including methylation or mutational analysis, amplified templates are preferred to capture complete genomic sequences without arbitrary sequence loss [57].
Comparison with Traditional Sequencing Methods
NGS technologies offer significant advantages over traditional Sanger sequencing, which sequences DNA fragments individually through chain-termination with dideoxynucleotides (ddNTPs) followed by capillary electrophoresis [10]. The critical differences between these approaches are summarized in the table below:
Table 1: Comparison of Next-Generation Sequencing and Sanger Sequencing
| Feature | Next-Generation Sequencing | Sanger Sequencing |
|---|---|---|
| Cost-effectiveness | Higher for large-scale projects | Lower for small-scale projects |
| Speed | Rapid sequencing | Time-consuming |
| Application | Whole-genome sequencing, targeted sequencing | Ideal for sequencing single genes |
| Throughput | Multiple sequences simultaneously | Single sequence at a time |
| Data output | Large amount of data | Limited data output |
| Clinical utility | Detects mutations, structural variants | Identifies specific mutations |
The massive parallelism of NGS enables comprehensive genome, transcriptome, and epigenome analyses that are essential for personalized medicine approaches in oncology, solidifying its role as a cornerstone of modern genomic research and clinical diagnostics [10].
NGS in Target Identification
Genetic Variant Discovery and Association Studies
NGS technologies have revolutionized target identification in oncology by enabling systematic discovery of disease-associated genetic variants through large-scale genomic studies. Population-wide sequencing studies leverage electronic health records and NGS data to identify associations between genetic mutations and specific cancer phenotypes, streamlining the discovery of disease-causing variants [56] [55]. These approaches facilitate the identification of novel therapeutic targets by comparing genomic sequences from healthy and tumor tissues to pinpoint somatic mutations, structural variations, and copy number alterations driving oncogenesis [10].
The application of NGS in target identification extends beyond simple variant discovery to functional annotation and pathway analysis. By integrating genomic data with transcriptomic and epigenomic information, researchers can identify not only mutated genes but also dysregulated pathways and networks that represent potential therapeutic intervention points. For example, in osteoarthritis research, NGS identified ADAMTS-4 as a therapeutic target, enabling the development of inhibitors to slow cartilage degradation – a breakthrough beyond symptom management [56]. Similar approaches in cancer research have identified numerous targetable mutations in genes such as EGFR, BRAF, and ALK, leading to development of effective targeted therapies.
Loss-of-Function Mutation Analysis
A powerful application of NGS in target validation involves the analysis of naturally occurring loss-of-function (LoF) mutations in human populations. By identifying individuals with LoF mutations in genes encoding potential drug targets and correlating these genetic variants with phenotypic outcomes, researchers can confirm target relevance and predict potential therapeutic effects and safety concerns [56] [55]. This approach provides human genetic evidence to prioritize molecular targets, potentially de-risking drug development programs by indicating both efficacy and safety profiles before significant investment in compound development.
The integration of LoF mutation analysis with NGS-based phenotypic studies enables a comprehensive understanding of target biology, including potential compensatory mechanisms and unintended consequences of target inhibition. For instance, population studies of individuals with LoF mutations in PCSK9 revealed both reduced LDL cholesterol levels and decreased cardiovascular risk without apparent adverse effects, providing strong genetic validation for PCSK9 inhibition as a therapeutic strategy for cholesterol management [56]. Similar approaches in oncology help identify targets whose inhibition is likely to yield therapeutic benefits with acceptable safety margins.
DNA-Encoded Chemical Libraries
NGS technologies have enabled innovative approaches to early drug discovery through DNA-encoded chemical libraries (DELs). This technology combines combinatorial chemical synthesis with DNA tagging to create vast libraries of small molecules that can be screened against protein targets of interest. Following incubation with a target protein, bound molecules are identified through NGS of their DNA tags, dramatically accelerating the identification of lead compounds [56].
DEL technology leverages the massive sequencing capacity of NGS platforms to screen libraries containing billions of compounds in a single experiment, significantly increasing throughput compared to traditional high-throughput screening methods. The application of NGS in DEL screening has transformed early drug discovery by enabling more efficient exploration of chemical space and identification of novel chemical starting points for drug development programs against targets identified through genomic approaches.
NGS Target Identification Workflow
NGS in Target Validation
Functional Genomics Approaches
NGS enables comprehensive functional genomics studies that are critical for target validation in oncology drug discovery. By integrating CRISPR-based screens with NGS readouts, researchers can systematically evaluate the functional consequences of gene knockouts or perturbations across the genome in cancer models. These approaches identify genes essential for cancer cell survival or growth, providing strong validation for potential therapeutic targets [55]. The combination of CRISPR screens with NGS analysis allows for genome-wide functional assessment, prioritizing targets based on their essentiality in specific cancer contexts.
Single-cell RNA sequencing (scRNA-seq) represents another powerful NGS application for target validation, enabling characterization of gene expression patterns at individual cell resolution within heterogeneous tumor samples. This technology provides insights into cellular subpopulations, tumor microenvironment interactions, and transcriptional networks dysregulated in cancer [55]. For example, automated scRNA-seq library preparation systems can process up to 96 samples concurrently while cutting hands-on time by nearly 7.5 hours compared to manual methods, with studies showing high reproducibility (gene expression correlations of R = 0.971 between automated and manual methods) [58].
Patient-Derived Models and Organoids
The integration of NGS with advanced disease models such as patient-derived organoids has created powerful platforms for target validation and drug repurposing, particularly for rare cancers [55]. NGS allows for comprehensive molecular characterization of these models, ensuring their genetic fidelity to original tumors and enabling monitoring of genetic stability during culture. Corning's specialized organoid culture products, when combined with NGS analysis, provide valuable insights into molecular characteristics of organoids, helping researchers understand disease mechanisms and validate potential therapeutic targets [55].
The application of NGS in patient-derived model systems enables functional validation of targets in contexts that more closely resemble human tumors than traditional cell lines. By sequencing DNA or RNA from organoids before and after genetic manipulation or drug treatment, researchers can assess target engagement, mechanism of action, and resistance mechanisms. Furthermore, NGS can monitor quality and stability of organoids over time by assessing changes in gene expression or genetic alterations, ensuring reliability and reproducibility of these models for target validation studies [55].
Biomarker Discovery and Companion Diagnostic Development
NGS plays a crucial role in biomarker discovery for patient stratification and development of companion diagnostics, which are essential components of targeted therapy development. Comprehensive genomic profiling through NGS identifies genetic signatures that predict drug response, resistance mechanisms, or adverse effects, enabling development of biomarkers for clinical trial enrichment and eventual companion diagnostics [56] [26]. In 2024, the FDA further expanded approvals of NGS-based tests to be used in conjunction with immunotherapy treatments for oncology, indicating rapid growth in this area [59].
The validation of biomarkers through NGS requires careful experimental design and analytical validation to ensure clinical utility. For metastatic breast cancer (mBC), clinical guidelines recommend comprehensive germline and somatic profiling to identify candidates for targeted therapies against alterations in genes such as BRCA1/2, PIK3CA, AKT1, PTEN, ESR1, NTRK, and others [26]. NGS-based molecular profiling allows clinicians to identify cancer genomic alterations, enabling informed treatment recommendations based on tumor-specific biomarker status [26]. Several studies have demonstrated clear benefits of this approach, with mBC patients who received NGS testing and appropriate targeted therapy showing prolonged progression-free survival compared to patients who did not receive NGS testing [26].
Table 2: Key NGS Applications in Target Identification and Validation
| Application | Methodology | Output | Impact on Drug Discovery |
|---|---|---|---|
| Genetic variant discovery | Population sequencing, association studies | Disease-associated variants, novel targets | Identifies new therapeutic targets based on human genetic evidence |
| Loss-of-function analysis | Natural variant correlation with phenotypes | Target validation, safety prediction | Confirms target relevance and predicts therapeutic outcomes |
| DNA-encoded libraries | NGS screening of tagged compound libraries | Lead compounds | Accelerates identification of chemical starting points |
| Functional genomics | CRISPR screens with NGS readout | Essential genes, synthetic lethal interactions | Prioritizes targets based on functional essentiality |
| Patient-derived organoids | NGS characterization of model systems | Preclinical validation | Provides physiologically relevant models for target validation |
| Biomarker discovery | Comprehensive genomic profiling | Predictive biomarkers, companion diagnostics | Enables patient stratification and personalized therapy |
Research Reagent Solutions
The successful implementation of NGS in drug discovery requires specialized reagents and consumables optimized for specific workflow steps. The following table details essential research reagent solutions and their applications in NGS-based target identification and validation:
Table 3: Essential Research Reagent Solutions for NGS Workflows
| Reagent Category | Specific Examples | Function in NGS Workflow | Application in Target ID/Validation |
|---|---|---|---|
| Library preparation kits | Illumina TruSight Oncology 500, Pillar Biosciences assays | Fragment DNA, add adapters, amplify libraries | Target enrichment, biomarker detection |
| Target enrichment systems | Twist Bioscience targeted enrichment | Capture specific genomic regions of interest | Focused sequencing of cancer-related genes |
| Automation reagents | Beckman Coulter liquid handling reagents | Enable automated library preparation | High-throughput screening, reproducibility |
| Single-cell reagents | 10x Genomics single-cell assay kits | Barcoding and library prep for single cells | Tumor heterogeneity analysis, microenvironment |
| Specialized enzymes | Watchmaker Genomics custom enzymes | Optimized polymerases, ligases for NGS | Improve library complexity, reduce bias |
| Quality control reagents | Corning clean-up kits, QC standards | Assess library quality, remove contaminants | Ensure sequencing data reliability |
| Organoid culture reagents | Corning specialized surfaces and media | Support 3D growth of patient-derived models | Preclinical target validation in relevant systems |
These specialized reagents form the foundation of robust NGS workflows in drug discovery, enabling researchers to generate high-quality genomic data for target identification and validation. The selection of appropriate reagents depends on specific research objectives, sample types, and sequencing platforms. Strategic partnerships between reagent manufacturers and automation companies have been instrumental in developing integrated solutions that streamline NGS workflows, reduce costs, and improve reproducibility [58]. For example, the partnership between Beckman Coulter Life Sciences and Pillar Biosciences has enabled development of single-tube, one-day workflows for solid tumor, liquid biopsy, and haematology assays, facilitating rapid processing of patient samples with on-target rates exceeding 90% and 100% base coverage [58].
Data Analysis Frameworks
Primary Data Processing and Quality Control
NGS data analysis begins with primary processing and quality control to ensure data integrity before biological interpretation. The initial cleaning phase involves removing low-quality sequences, adapters, and contaminants from raw sequencing data [60]. Quality assessment typically utilizes Phred scores, which indicate the probability of incorrect base calls, with a score of 30 representing 99.9% accuracy (one error per 1,000 bases) [60]. Tools like FastQC provide comprehensive quality metrics through visualizations that help researchers identify potential issues including sequence quality degradation, adapter contamination, or overrepresented sequences.
Data exploration follows quality control, employing techniques such as principal component analysis (PCA) to reduce data dimensionality and identify patterns, outliers, and sample relationships [60]. PCA transforms high-dimensional NGS data into two-dimensional space defined by principal components that capture the greatest variance in the dataset, enabling visualization of sample clustering based on biological or technical factors. This step is crucial for understanding data structure, identifying batch effects, and ensuring that experimental groups are comparable before downstream analysis.
Advanced Analytical Approaches
After initial processing, NGS data undergoes application-specific analysis tailored to research objectives. For whole genome sequencing, common analyses include variant calling (identification of single nucleotide polymorphisms, insertions/deletions), structural variant detection, and microsatellite marker analysis [60]. RNA sequencing data typically involves differential gene expression analysis, pathway enrichment, and co-expression network construction to identify dysregulated genes and pathways in cancer samples. Epigenomic analyses focus on identifying differentially methylated regions, histone modification patterns, and chromatin accessibility changes.
Advanced analytical approaches increasingly leverage machine learning and artificial intelligence to extract biological insights from complex NGS datasets. AI-driven tools facilitate variant calling, functional annotation, and predictive modeling of variant effects on protein function and disease phenotypes [55]. Cloud-based platforms enable scalable and collaborative NGS data analysis, providing the computational resources necessary for processing large genomic datasets [59]. These platforms often integrate multiple algorithms and visualization tools, allowing researchers to interactively explore NGS datasets and interpret complex genomic features relevant to target identification and validation.
NGS Data Analysis Pipeline
Emerging Trends and Future Perspectives
Technological Innovations
The NGS landscape continues to evolve rapidly, with several technological innovations poised to further transform target identification and validation in oncology drug discovery. Long-read sequencing technologies from Pacific Biosciences and Oxford Nanopore are improving resolution of complex structural variants and repetitive genomic regions that were previously challenging to characterize [55]. Single-cell sequencing approaches provide unprecedented insights into cellular heterogeneity within tumors, revealing rare cell populations and tumor microenvironment interactions that may represent therapeutic opportunities or resistance mechanisms [55].
Spatial transcriptomics represents another cutting-edge innovation, enabling researchers to visualize gene expression patterns within tissue architecture context, preserving crucial spatial information lost in conventional single-cell approaches [55]. Liquid biopsy sequencing continues to advance, allowing non-invasive monitoring of tumor dynamics through detection of circulating tumor DNA in blood samples [59]. These technological advancements are complemented by improvements in real-time sequencing, epigenomic profiling, and high-throughput functional genomics, collectively expanding the toolbox available for target discovery and validation.
Automation and Integration
Strategic partnerships between sequencing platform manufacturers, automation companies, and reagent suppliers are driving increased automation and integration of NGS workflows [58]. These collaborations develop streamlined solutions that reduce manual intervention, improve reproducibility, and broaden access to cutting-edge genomic technologies. For example, automation of Illumina's TruSight Oncology 500 assay has compressed extended workflows into a three-day process powered by continuously operating robotic systems, reducing hands-on time from approximately 23 hours to just six hours per run while improving data quality [58].
The integration of complementary technologies within automated NGS workflows is particularly valuable for drug discovery applications requiring high throughput and reproducibility. Partnerships between companies like Beckman Coulter Life Sciences and Watchmaker Genomics focus on developing automated library preparation systems with enhanced sequencing performance [58]. Similarly, collaboration with 10x Genomics integrates automated library preparation with advanced single-cell capabilities, enabling processing of up to 96 samples concurrently while significantly reducing hands-on time [58]. These integrated, automated solutions make NGS technologies more accessible to smaller laboratories and institutions in resource-constrained settings, democratizing access to cutting-edge genomic tools for target discovery.
Challenges and Implementation Barriers
Despite the tremendous potential of NGS in drug discovery, several challenges remain for widespread implementation. Data management represents a significant hurdle, as NGS generates terabytes of sequencing data requiring robust bioinformatics infrastructure and expertise [56]. The integration of NGS-based biomarkers into regulatory frameworks for drug approval requires standardization and validation to ensure consistency across platforms and laboratories [56]. Ethical considerations surrounding genetic privacy and data sharing also need addressing for broader NGS implementation [10].
A multi-stakeholder survey revealed that inconsistent payer coverage, high out-of-pocket costs for patients, and challenges in managing reimbursement processes can lead to suboptimal utilization of NGS in clinical practice [26]. Interestingly, 33% of payers surveyed were not aware of current somatic biomarker testing recommendations from NCCN guidelines, highlighting the need for broader education on NGS clinical utility [26]. These implementation barriers underscore the importance of ongoing stakeholder education, development of clear clinical guidelines, and establishment of coverage policies that support appropriate use of NGS in oncology drug discovery and development.
Table 4: NGS Market Analysis and Growth Projections
| Market Segment | 2024 Revenue Share | Projected CAGR | Key Growth Drivers |
|---|---|---|---|
| Product Type (Consumables) | 48.5% | - | Recurring demand for reagents, kits, and cartridges |
| Technology (Targeted Sequencing) | 39.6% | - | Cost-effective, high-precision analysis for biomarker discovery |
| Application (Target Identification) | 37.2% | - | Critical role in early-stage discovery of genetic drivers |
| End-User (Pharma & Biotech) | 46.2% | - | Investment in high-throughput screening and target validation |
| Workflow (Sequencing) | 41.5% | - | Central role in generating genomic data |
| Overall NGS in Drug Discovery Market | - | 18.3% (2025-2034) | Precision medicine expansion, chronic disease burden, AI integration |
The paradigm of oncology drug development has shifted fundamentally with the advent of precision medicine, moving from population-based approaches to biomarker-driven strategies that enable personalized treatment selection. Biomarker-driven clinical trials represent a sophisticated methodological framework that uses molecular characteristics to guide patient stratification, treatment assignment, and outcome assessment. This approach is particularly crucial in oncology, where tumor heterogeneity necessitates precise targeting of molecular alterations to achieve therapeutic efficacy. The integration of comprehensive genomic profiling, particularly through next-generation sequencing (NGS), has become the cornerstone of this transformation, providing the analytical foundation for identifying actionable biomarkers across diverse cancer types.
The role of NGS in cancer molecular profiling research extends beyond simple mutation detection to encompass comprehensive genomic characterization that informs clinical trial design. As the tumor profiling market demonstrates substantial growth—projected to reach $26.56 billion by 2033 with a CAGR of 9.92%—the infrastructure supporting biomarker-driven trials continues to expand technologically and methodologically [34]. This growth reflects both the rising incidence of cancer globally and the increasing reliance on precision oncology approaches that customize treatments according to a tumor's specific genetic composition [34]. The convergence of advanced genomic technologies with innovative clinical trial designs has created new opportunities for enhancing drug development efficiency and success rates.
Core Designs for Biomarker-Driven Clinical Trials
Biomarker-driven clinical trials employ several distinct methodological frameworks, each with specific applications, advantages, and limitations. Understanding these designs is essential for researchers and drug development professionals seeking to optimize trial strategies for targeted therapies.
Biomarker-Stratified Design
The biomarker-stratified design represents the most comprehensive approach for evaluating biomarker utility in clinical trials. In this design, all patients are enrolled and randomized regardless of biomarker status, but randomization is stratified by biomarker status to ensure balance across treatment arms [61]. The primary analysis focuses on testing treatment effects within each biomarker-defined subgroup, allowing for direct comparison of therapeutic efficacy across different molecular profiles.
This design provides unbiased estimates of benefit-to-risk ratios across biomarker-defined subgroups and the overall population [61]. A key advantage is its ability to assess whether a biomarker is useful for selecting the optimal treatment for individual patients. For example, the NCCTG-0723 (MARVEL) trial for second-line advanced non-small cell lung cancer (NSCLC) employed this design to evaluate whether EGFR FISH status could guide treatment selection between erlotinib and pemetrexed [61]. The biological hypothesis postulated that EGFR FISH-positive patients would derive greater benefit from erlotinib, while FISH-negative patients might benefit more from pemetrexed.
From a methodological perspective, stratified randomization ensures that all patients will have tissue available for biomarker assessment, though this is not strictly necessary for validity if complete biomarker ascertainment can be guaranteed [62]. When biomarker status is not evaluated upfront, careful planning is required to anticipate unavailable biomarker measurements and ensure adequate sample sizes in relevant subgroups [61]. The INTEREST trial experience highlights this consideration, where only 374 of 1466 randomized patients had tissue available for biomarker evaluation, substantially limiting the assessment of the biomarker question [61].
Enrichment Design
The enrichment design restricts patient enrollment to those with specific biomarker values, typically biomarker-positive patients [61] [63]. This approach is appropriate when compelling preliminary evidence suggests that treatment benefit is likely confined to a particular biomarker-defined subgroup, or when equipoise exists only for patients with specific molecular characteristics.
Enrichment designs offer significant advantages in efficiency for signal detection, particularly for targeted therapies with strong mechanistic rationale linked to a biomarker [63]. The CALGB-10603 trial exemplifies this approach, restricting eligibility to acute myeloid leukemia patients with FLT3 mutations and randomly assigning them to standard treatment with or without the FLT3 kinase inhibitor midostaurin [61]. Patients without the FLT3 mutation were excluded from the study entirely.
While enrichment designs can accelerate drug development for biomarker-defined populations, they carry the limitation of potentially narrowing regulatory labels and provide no information about treatment effects in biomarker-negative patients [63]. Successful implementation requires robust assay validation and careful upfront planning regarding biomarker prevalence and companion diagnostic requirements [63].
Biomarker-Strategy Design
The biomarker-strategy design compares a biomarker-guided treatment approach against a non-guided control strategy. In its simplest form, patients are randomized to either a control arm that receives standard treatment or an experimental arm where treatment is selected based on biomarker status [61]. This design evaluates the clinical utility of the biomarker itself rather than focusing solely on treatment efficacy.
An example of this approach is the ERCC1 trial in NSCLC, where patients in the control arm received cisplatin+docetaxel, while those in the biomarker-strategy arm were switched to gemcitabine+docetaxel if classified as cisplatin-resistant based on ERCC1 expression [61]. More complex variations can guide decisions among three or more treatments, as demonstrated by the Tumor Chemosensitivity Assay Ovarian Cancer study, which used a luminescence assay to select from 12 chemotherapy regimens in the biomarker-strategy arm [61].
Table 1: Comparison of Biomarker-Driven Clinical Trial Designs
| Design Type | Key Features | Best Use Cases | Advantages | Limitations |
|---|---|---|---|---|
| Biomarker-Stratified | All patients randomized; stratification by biomarker status; analysis by subgroup | When biomarker utility is uncertain; requires broad population assessment | Provides unbiased treatment effect estimates across subgroups; maximizes randomization benefits | Requires larger sample sizes; more complex analysis |
| Enrichment | Enrollment restricted to biomarker-positive patients | Strong preliminary evidence of efficacy limited to biomarker-positive subgroup | Efficient signal detection; smaller sample sizes; faster completion | No information on biomarker-negative patients; may narrow regulatory label |
| Biomarker-Strategy | Compares biomarker-guided vs. non-guided treatment strategies | Evaluating clinical utility of biomarker itself | Tests overall value of biomarker-based decision making | Complex interpretation; requires larger sample size than enrichment designs |
All-Comers and Basket Trial Designs
Beyond the three primary designs, additional approaches have emerged to address specific challenges in precision oncology. The all-comers design enrolls both biomarker-positive and negative patients without stratification, assessing biomarker effects retrospectively through subgroup analysis [63]. This approach is typically used for earlier phase trials where biomarker effects are not yet well understood, though it risks diluting overall results if the drug is only effective in a specific biomarker-defined subgroup [63].
Tumor-agnostic biomarker-driven basket trials represent another innovative approach, where patients with biomarker-positive tumors across different cancer types are enrolled into separate study arms [63]. These trials leverage Bayesian methods to share information across cohorts, enhancing statistical efficiency when appropriate. This design offers high operational efficiency through a single protocol addressing multiple candidate indications and naturally accommodates adaptive elements that allow individual arms to be expanded or discontinued based on early efficacy signals [63].
The Role of Next-Generation Sequencing in Biomarker Discovery and Validation
Next-generation sequencing has revolutionized biomarker discovery and validation by enabling comprehensive genomic profiling that informs clinical trial design and patient stratification. The technical sophistication of NGS platforms and analytical pipelines provides the foundation for reliable biomarker identification in modern oncology trials.
NGS Data Analysis Workflow
The NGS data analysis workflow comprises three core stages: primary, secondary, and tertiary analysis [64]. Primary analysis assesses raw sequencing data for quality and converts binary base call files into FASTQ format, which contains nucleotide sequences and quality scores [64]. Key quality metrics assessed during this stage include sequencing yield, error rate, Phred quality scores (with Q>30 representing <0.1% base call error), percentage of sequences aligned, cluster density, and phasing/prephasing percentages [64].
Secondary analysis converts data into biological results through read cleanup, sequence alignment, and mutation calling [64]. Read cleanup involves trimming adapters, removing low-quality reads, and deduplication using unique molecular identifiers (UMIs) to correct for PCR and sequencing errors [64]. Sequence alignment maps reads to reference genomes using tools like BWA and Bowtie 2, producing Binary Alignment Map (BAM) files that facilitate visualization of read pileups and mismatches [64]. For RNA sequencing, additional steps include correction of sequence bias, quantitation of RNA types, and determination of strandedness [64].
Tertiary analysis generates biological interpretations and clinical recommendations by connecting genomic features to biological knowledge [5]. This stage identifies genetic mutations of interest, interprets their functional significance, and provides actionable insights for clinical decision-making, including recommendations for targeted therapies based on identified biomarkers [64].
NGS Data Analysis Workflow: This diagram illustrates the three-stage process of NGS data analysis, from raw data processing to clinical interpretation.
Diagnostic Accuracy of NGS in Clinical Applications
The diagnostic accuracy of NGS-based genomic profiling has been rigorously evaluated across cancer types, establishing its reliability for clinical decision-making. In advanced NSCLC, blood-based circulating tumor DNA (ctDNA) analysis has emerged as a viable alternative to tissue biopsy, particularly when tumor tissue is inadequate or unavailable [65] [66]. The tumor fraction (TF) in ctDNA significantly impacts diagnostic accuracy, with TF>1% demonstrating 100% positive percent agreement (PPA) for actionable mutations compared to tissue-based testing [65] [66]. In contrast, the ctDNA TF low group showed substantially lower PPA (47.5%) for actionable mutations [65].
The correlation between blood-based tumor mutational burden (bTMB) and tissue-based TMB (tTMB) also varies by tumor fraction, with correlation coefficients of 0.13 for ctDNA TF low versus 0.71 for ctDNA TF high groups [65]. Similarly, PPA for bTMB was 31.3% for ctDNA TF low and 92.3% for ctDNA TF high, with negative percent agreement (NPA) at 100% and 85.6%, respectively [65]. These findings support the use of blood-based TP-NGS for detecting clinically actionable mutations when ctDNA tumor fraction is sufficiently high.
In advanced soft tissue and bone sarcomas, NGS-based genomic profiling has demonstrated significant clinical utility despite the rarity and heterogeneity of these tumors. A retrospective multicenter analysis of 81 patients identified 223 genomic alterations, with detectable alterations in 90.1% of patients [14]. The most common alteration types were copy number amplifications (26.9%) and deletions (24.7%), with TP53 (38%), RB1 (22%), and CDKN2A (14%) representing the most frequently mutated genes [14]. Actionable mutations were identified in 22.2% of patients, making them eligible for FDA-approved targeted therapies [14].
Table 2: NGS Diagnostic Performance in Advanced NSCLC
| Parameter | ctDNA TF High (TF>1%) | ctDNA TF Low | Clinical Implications |
|---|---|---|---|
| Positive Percent Agreement (PPA) for Actionable Mutations | 100% | 47.5% | Blood-based NGS reliable when TF sufficient |
| bTMB vs tTMB Correlation | 0.71 | 0.13 | Strong correlation only in high TF context |
| PPA for bTMB | 92.3% | 31.3% | Accurate TMB assessment requires adequate TF |
| Negative Percent Agreement (NPA) for bTMB | 85.6% | 100% | High specificity across TF levels |
NGS Data Formats and Computational Considerations
Effective NGS data management requires understanding specialized file formats optimized for massive genomic datasets. The FASTQ format serves as the universal standard for raw sequence data, containing nucleotide sequences and per-base quality scores [5]. For alignment data, the Sequence Alignment/Map (SAM) format provides human-readable alignment information, while its binary equivalent (BAM) offers compressed, computationally efficient storage [5]. The CRAM format extends compression further using reference-based algorithms, reducing file sizes by 30-60% compared to BAM [5].
Critical handling considerations include verifying file integrity after transfers, maintaining consistent coordinate systems (0-based vs 1-based indexing), preserving metadata throughout analysis pipelines, and using appropriate compression levels balancing file size and access speed [5]. These computational aspects form the infrastructure supporting robust biomarker identification and validation in clinical trials.
Essential Research Reagents and Platforms for Biomarker-Driven Research
The execution of biomarker-driven clinical trials requires specialized reagents, platforms, and analytical tools that ensure reliable biomarker assessment and interpretation.
Table 3: Essential Research Reagent Solutions for Biomarker-Driven Trials
| Reagent/Platform | Function | Application in Biomarker Trials |
|---|---|---|
| FoundationOneCDx | Comprehensive genomic profiling | Tissue-based targeted NGS for solid tumors |
| FoundationOneLiquid CDx | Liquid biopsy genomic profiling | Blood-based ctDNA analysis for actionable mutations |
| Tempus xT assay | NGS-based genomic profiling | Comprehensive tumor sequencing across cancer types |
| OncoDEEP | Genomic analysis platform | Detection of genomic alterations in tumor samples |
| MI Profile | Molecular profiling platform | Multi-analyte biomarker assessment |
| Unique Molecular Identifiers (UMIs) | Molecular barcoding | Error correction in NGS; accurate variant calling |
| PhiX Control | Sequencing quality control | Monitoring sequencing accuracy and error rates |
| BWA/Bowtie 2 | Sequence alignment tools | Mapping sequencing reads to reference genomes |
| SAMtools | Alignment processing | Manipulation and analysis of BAM/SAM files |
| Integrative Genomic Viewer (IGV) | Visualization tool | Visual exploration of genomic data and alterations |
Methodological Protocols for Key Experiments
Protocol for NGS-Based Biomarker Discovery in Sarcoma
The multicenter analysis of advanced soft tissue and bone sarcomas provides a representative protocol for NGS-based biomarker discovery [14]. This study employed a retrospective, cross-sectional design including 81 adult patients with STS (n=61) or bone sarcoma (n=20), excluding gastrointestinal stromal tumors (GIST) and Kaposi sarcoma due to their distinct molecular characteristics [14].
Sample processing involved either biopsy or surgical resection followed by standardized pathological examination by sarcoma-specialized pathologists [14]. Comprehensive molecular profiling was performed using four different NGS kits (FoundationOne, Tempus, OncoDEEP, and MI Profile) in CLIA-regulated laboratories [14]. Genomic alterations including insertions/deletions, copy number variations, and structural rearrangements were recorded, along with tumor mutation burden (TMB) and microsatellite instability (MSI) status when available [14].
Bioinformatic analysis included functional assessment of genomic alterations across key pathways: genomic stability regulation (TP53, MDM2), cell cycle regulation (RB1, CDKN2A/B, CDK4), DNA repair (RAD genes), phosphoinositide-3 kinase (PI3K) pathway (PTEN, PIK3CA, mTOR, RICTOR), and receptor tyrosine kinase pathway (ALK, FGFR) [14]. Actionable alterations were determined based on clinical annotations from NGS platforms and reclassified according to OncoKB criteria, incorporating FDA approval status, clinical guideline support, and strength of supporting evidence [14].
Protocol for Evaluating NGS Diagnostic Accuracy in NSCLC
The evaluation of NGS diagnostic accuracy in advanced NSCLC followed a rigorous methodological framework [65] [66]. Participants were enrolled in a Precision Oncology Program, with TP-NGS conducted using both FoundationOneCDx (tissue) and FoundationOneLiquid CDx (blood) assays [65]. The study included an unpaired cohort (n=340) and a paired cohort (n=221) for direct comparison between tissue and liquid biopsy approaches [65].
Statistical analysis focused on sensitivity metrics, particularly positive percent agreement (PPA) for actionable mutations between tissue and blood-based testing stratified by ctDNA tumor fraction [65]. Correlation between blood-based TMB (bTMB) and tissue-based TMB (tTMB) was assessed using correlation coefficients, with PPA and NPA calculated for bTMB classification [65]. Actionable mutations were identified, with particular attention to common EGFR mutations representing 65.8% of actionable alterations in the paired cohort [65].
Biomarker-Driven Trial Design Framework: This decision tree illustrates the strategic selection process for appropriate clinical trial designs based on biomarker knowledge and trial objectives.
Biomarker-driven clinical trials represent the methodological cornerstone of precision oncology, enabling more targeted patient stratification and enhanced trial success rates. The integration of NGS-based genomic profiling has transformed the landscape of cancer clinical research, providing comprehensive molecular characterization that informs trial design and patient selection. As the field evolves, several emerging trends are shaping the future of biomarker-driven trials.
The growing adoption of liquid biopsy approaches for biomarker assessment addresses critical challenges in tissue acquisition, particularly in advanced cancers where tumor tissue may be inadequate or inaccessible [65] [66]. The demonstrated diagnostic accuracy of blood-based NGS, especially in patients with high ctDNA tumor fraction, supports its integration into clinical trial workflows for longitudinal monitoring and resistance mechanism detection [65]. Additionally, innovative computational approaches, including large language models (LLMs), show promise for enhancing biomarker-based trial matching by extracting and structuring genomic biomarkers from unstructured clinical trial descriptions [67].
Future directions in biomarker-driven trials will likely involve more sophisticated adaptive designs that incorporate real-time biomarker data, increased utilization of multi-analyte biomarker panels, and greater integration of artificial intelligence for biomarker discovery and validation. As regulatory frameworks evolve to accommodate these complexities, the continued refinement of biomarker-driven trial methodologies will accelerate the development of personalized cancer therapies and improve outcomes for patients across diverse cancer types.
The therapeutic landscape of oncology has been transformed by precision medicine, creating an urgent need for diagnostic tools that can dynamically track a patient's response to therapy. While traditional imaging techniques like computed tomography (CT) remain the gold standard for monitoring tumor size, they lack the sensitivity to detect molecular changes and minimal residual disease (MRD) at a microscopic level [68]. Tissue biopsies, though informative, are invasive, impractical for serial monitoring, and fail to capture the full spatial and temporal heterogeneity of tumors [69] [68]. In this context, liquid biopsy has emerged as a pivotal, minimally invasive modality for cancer surveillance.
Liquid biopsy involves the analysis of circulating tumor-derived components in biofluids, most commonly blood. Circulating tumor DNA (ctDNA), a fraction of cell-free DNA (cfDNA) shed into the bloodstream by apoptotic or necrotic tumor cells, has become one of the most promising biomarkers [68] [70]. Its short half-life, estimated between 16 minutes and several hours, allows it to provide a real-time snapshot of tumor burden and clonal evolution [68]. The integration of Next-Generation Sequencing (NGS) technologies has been fundamental to unlocking the potential of ctDNA analysis, enabling comprehensive genomic profiling from a simple blood draw and solidifying its role within modern cancer molecular profiling research [71] [72].
Technical Foundations of ctDNA Biology and Analysis
Origin and Characteristics of ctDNA
Circulating tumor DNA is released into the bloodstream through various mechanisms, primarily apoptosis and necrosis of tumor cells [68]. These small DNA fragments carry the same genetic alterations found in the parent tumor, including point mutations, copy number variations (CNVs), insertions, deletions (indels), and epigenetic modifications [69] [72]. The quantity of ctDNA in circulation correlates with disease burden, ranging from less than 0.1% of total cfDNA in early-stage cancers to over 90% in advanced metastatic disease [68]. Beyond genetic sequence, other characteristics like fragmentation patterns and end motifs can differentiate ctDNA from normal cfDNA, adding another layer of diagnostic information [68].
Essential Research Reagent Solutions
Successfully isolating and analyzing ctDNA requires a suite of specialized reagents and tools. The following table details key components of a researcher's toolkit.
Table 1: Essential Research Reagents and Materials for ctDNA Analysis
| Research Reagent/Material | Function and Importance in ctDNA Workflow |
|---|---|
| Cell-Free DNA Blood Collection Tubes | Stabilizes nucleated blood cells to prevent genomic DNA contamination during sample transport and storage, preserving the integrity of plasma cfDNA [70]. |
| DNA Extraction Kits (for plasma) | Designed to efficiently recover short, fragmented cfDNA molecules from plasma with high purity and yield, which is critical for downstream applications [68]. |
| Unique Molecular Identifiers (UMIs) | Short nucleotide barcodes ligated to individual DNA molecules prior to PCR amplification. UMIs enable bioinformatic correction of PCR errors and sequencing artifacts, ensuring high-fidelity variant detection [68]. |
| PCR Master Mixes (for dPCR/ddPCR) | Optimized reagent formulations for precise and sensitive amplification of target sequences in digital PCR platforms, allowing for absolute quantification of mutant allele copies [70]. |
| Hybridization Capture Probes | Biotinylated oligonucleotide probes designed to enrich for specific genomic regions of interest (e.g., a cancer gene panel) from a sequencing library before NGS [68]. |
| NGS Library Preparation Kits | Reagent sets for converting isolated cfDNA into a sequencing-ready library, including steps for end-repair, adapter ligation, and PCR amplification [68] [72]. |
Methodologies for ctDNA Monitoring in Treatment Response
Core Analytical Technologies
Two primary technological approaches are employed for the detection and quantification of ctDNA in the context of treatment monitoring: PCR-based methods and NGS-based methods.
Digital PCR (dPCR) and Droplet Digital PCR (ddPCR): These methods partition a sample into thousands of individual reactions. ddPCR provides absolute quantification of target molecules without the need for standard curves, offers a faster turnaround time, and is highly effective for tracking known mutations in a background of wild-type DNA [70]. Its simplicity and cost-effectiveness make it ideal for repetitive monitoring of specific variants in clinical trials and routine care [70].
Next-Generation Sequencing (NGS): NGS offers a more comprehensive view of the tumor genome. Techniques like whole-exome sequencing (WES) and whole-genome sequencing (WGS) can identify novel alterations, while targeted approaches such as CAPP-Seq and TEC-Seq allow for ultra-deep sequencing of specific gene panels to detect low-frequency variants [68]. A key advantage of NGS is its ability to analyze multiple biomarkers simultaneously from a single sample, including mutations, CNVs, and indels [73] [72]. To overcome the high error rates of standard NGS, advanced error-correction methods like SaferSeqS and Duplex Sequencing are used, which significantly improve detection sensitivity and specificity [68].
Key Experimental Protocols for Treatment Monitoring
A standard experimental workflow for ctDNA-based treatment response monitoring involves several critical stages, from blood collection to data interpretation.
Table 2: Key Methodological Steps in ctDNA Treatment Response Monitoring
| Protocol Step | Detailed Methodology & Considerations |
|---|---|
| 1. Blood Collection & Processing | Collect blood in cell-stabilizing tubes (e.g., Streck, PAXgene). Process within 4-6 hours via a two-step centrifugation protocol (e.g., 1,600 x g for 10 min, then 16,000 x g for 10 min) to obtain platelet-poor plasma [70]. |
| 2. cfDNA Extraction | Extract cfDNA from plasma using commercial silica-membrane or magnetic bead-based kits. Quantify yield using fluorescent assays (e.g., Qubit) and assess fragment size distribution (e.g., Bioanalyzer) [68]. |
| 3. Assay Selection & Setup | For known targets: Use ddPCR with probe assays specific to the mutation(s) of interest. For discovery/panels: Use targeted NGS with a panel of cancer-associated genes. Incorporate UMIs during library prep for error correction [68] [70]. |
| 4. Data Analysis & Quantification | ddPCR: Use Poisson statistics to calculate the concentration of mutant and wild-type alleles from positive/negative droplet counts. NGS: Generate UMI consensus sequences; variant calls are made based on a predefined variant allele frequency (VAF) threshold (e.g., >0.1%) after error correction [68]. |
| 5. Calculating Molecular Response | Track ctDNA dynamics over time. Key metrics include: • ctDNA clearance: Conversion from detectable to undetectable ctDNA levels. • % change from baseline: Calculate the reduction in mutant allele concentration or VAF after treatment initiation [68] [70]. |
Diagram 1: ctDNA analysis workflow.
Clinical Applications and Quantitative Data Interpretation
Monitoring Treatment Efficacy and Detecting Resistance
The most immediate clinical application of ctDNA is the real-time assessment of treatment efficacy. A decrease in ctDNA levels, often termed "molecular response," can precede tumor shrinkage observed on imaging. For instance, in the ctMoniTR project, a pooled analysis of eight clinical studies in advanced NSCLC showed that patients whose ctDNA levels dropped to undetectable within 10 weeks of starting tyrosine kinase inhibitor (TKI) therapy had significantly better overall survival and progression-free survival [70]. Similarly, Foundation Medicine's research demonstrated that serial monitoring of ctDNA tumor fraction (the proportion of ctDNA in total cfDNA) is strongly associated with clinical benefit from immunotherapy and targeted therapy across lung, breast, and other solid tumors [74].
Furthermore, the high sensitivity of NGS allows for the early detection of acquired resistance mechanisms. For example, the emergence of mutations in genes like ESR1 in breast cancer or KRAS in colorectal cancer during treatment can inform clinicians about the need to modify therapeutic strategies long before clinical progression is evident [68].
Detecting Minimal Residual Disease (MRD)
Following curative-intent surgery or radiotherapy, the detection of MRD is a powerful predictor of relapse. ctDNA analysis can identify MRD with higher sensitivity than imaging. The clearance of ctDNA post-treatment is associated with a significantly reduced risk of recurrence, while its persistence or subsequent reappearance indicates residual disease or imminent relapse, sometimes months before radiographic evidence [69] [68] [75]. This capability allows for patient risk stratification and the potential for early therapeutic intervention.
Quantitative Data from Clinical Studies
Robust quantitative data from recent studies underscores the clinical validity of ctDNA monitoring.
Table 3: Key Quantitative Findings from Recent ctDNA Monitoring Studies
| Study / Trial (Cancer Type) | Intervention | Key ctDNA Metric & Findings | Correlated Clinical Outcome |
|---|---|---|---|
| ctMoniTR Project (aNSCLC) [70] | Tyrosine Kinase Inhibitors (TKIs) | ctDNA clearance to undetectable levels within 10 weeks. | Improved Overall Survival and Progression-Free Survival. |
| LUNG-MAP Study (NSCLC) [74] | Various (Real-World) | Elevated baseline ctDNA tumor fraction (≥1%). | Associated with worse Overall Survival, despite improved mutation detection. |
| NIMBUS Trial (Breast Cancer) [74] | Dual Immune Checkpoint Blockade | Changes in ctDNA tumor fraction during treatment. | Strong association with clinical benefit. |
| MyPathway (Pan-Tumor) [74] | Immune Checkpoint Inhibitors | Serial monitoring of ctDNA tumor fraction. | Correlation with treatment response. |
Current Challenges and Future Directions
Despite its promise, the clinical adoption of ctDNA faces several hurdles. Lack of technical standardization across different platforms and laboratories affects reproducibility [76] [70]. Pre-analytical variables, such as blood collection timing and tube type, need strict protocols [70]. Biologically, non-malignant sources of mutations like clonal hematopoiesis of indeterminate potential (CHIP) can lead to false-positive results [70]. Furthermore, achieving sufficient analytical sensitivity remains a challenge in early-stage cancers and low-shedding tumors [68].
Future progress hinges on collaborative efforts to harmonize assays, validate clinical utility in large prospective trials, and integrate ctDNA data with other modalities like imaging and tissue biopsy. Emerging techniques, including the analysis of fragmentomics and methylation patterns in cfDNA, are poised to further enhance the sensitivity and specificity of liquid biopsies [68] [70]. As these challenges are addressed, ctDNA analysis is poised to become an indispensable tool in precision oncology, accelerating drug development and enabling more personalized, dynamic cancer care.
Next-generation sequencing (NGS) has revolutionized immuno-oncology by providing comprehensive tools for profiling key biomarkers that predict response to immune checkpoint inhibitors (ICIs). This technical guide details the integrated analysis of tumor mutational burden (TMB), microsatellite instability (MSI), and the tumor microenvironment (TME) using NGS-based approaches. These biomarkers have gained FDA approval as tissue-agnostic predictors for immunotherapy response, enabling precision oncology across diverse cancer types. The convergence of these biomarkers provides a more robust framework for patient stratification than any single marker alone, yet presents significant technical and standardization challenges that researchers must navigate for successful clinical implementation. This whitepaper examines the technical specifications, experimental protocols, and analytical frameworks required for optimal assessment of these critical immuno-oncology biomarkers within the broader context of cancer molecular profiling research.
Biomarker Foundations and Clinical Significance
Biomarker Definitions and Interrelationships
Tumor Mutational Burden (TMB) is defined as the total number of somatic mutations per megabase (mut/Mb) of sequenced genome region, representing a quantitative measure of mutational load in tumor tissue. TMB functions as a proxy for neoantigen load, with higher mutation rates increasing the probability of generating immunogenic neoantigens recognizable by T-cells [77] [78]. The resulting increased immunogenicity enhances potential response to ICIs across multiple cancer types [78].
Microsatellite Instability (MSI) status indicates a deficiency in the DNA mismatch repair (MMR) system, leading to accumulation of insertion/deletion mutations at short, repetitive DNA sequences (microsatellites). MSI-high (MSI-H) tumors exhibit markedly elevated mutation rates and represent a distinct molecular subtype with characteristic clinical and pathological features [77] [79].
The Tumor Microenvironment (TME) comprises the complex ecosystem surrounding tumors, including immune cells, stromal cells, blood vessels, and signaling molecules. The functional state of the TME critically determines anti-tumor immune responses and ultimately influences immunotherapy efficacy [80] [81].
These biomarkers exhibit significant biological and clinical interdependencies. MSI-H tumors frequently demonstrate high TMB, with studies showing approximately 78% of TMB-H/MSI-H neoplasms harbor genetic/epigenetic alterations in MMR genes [79]. Research indicates MSI-H colorectal cancers (CRCs) display more abundant immune cell infiltration, higher expression of immune-related genes, and greater immunogenicity compared to microsatellite stable (MSS) counterparts [81]. This inflammatory TME contributes to their enhanced sensitivity to ICIs.
Clinical Validation and Predictive Power
Robust clinical evidence supports TMB and MSI as predictive biomarkers for immunotherapy response. A real-world retrospective study of 157 patients with TMB-H (≥20 mut/Mb) and/or MSI-H solid tumors demonstrated significantly improved outcomes with immunotherapy compared to chemotherapy [77]. The objective response rate (ORR) for immunotherapy was 55.9% versus 34.4% for chemotherapy, with median progression-free survival (PFS) of 24.2 months versus 6.75 months, respectively (P = 0.042) [77]. The PFS ratio (PFS2/PFS1) favoring immunotherapy over chemotherapy was 4.7, highlighting the substantial clinical benefit [77].
Similarly, MSI-H status predicts dramatic responses to ICIs. The KEYNOTE-016 study demonstrated a 62% objective response rate in pre-treated MSI-H CRC patients, with most responses being durable [81]. This led to the first tissue-agnostic FDA approval for pembrolizumab in 2017 for MSI-H solid tumors [77].
Emerging evidence suggests these biomarkers may have prognostic significance beyond predictive value for immunotherapy. A 2024 study of 102 MSS metastatic colon cancer patients found high TMB (>10 mut/Mb) was associated with significantly longer median overall survival compared to low TMB (70.0 vs. 45.0 months, HR: 0.45; P = 0.0396), identifying TMB as an independent prognostic factor [82].
Table 1: Clinical Outcomes by Biomarker Status
| Biomarker Profile | Cancer Type | Treatment | Objective Response Rate | Median PFS | Study |
|---|---|---|---|---|---|
| TMB-H (≥20 mut/Mb) | 27 solid tumor types | Immunotherapy | 55.9% | 24.2 months | [77] |
| TMB-H (≥20 mut/Mb) | 27 solid tumor types | Chemotherapy | 34.4% | 6.75 months | [77] |
| MSI-H | Colorectal Cancer | Immunotherapy | 62% | Not reached | [81] |
| MSS/TMB-H (>10 mut/Mb) | Metastatic Colon Cancer | Standard Treatments | N/A | 70.0 months (OS) | [82] |
| MSS/TMB-L (≤10 mut/Mb) | Metastatic Colon Cancer | Standard Treatments | N/A | 45.0 months (OS) | [82] |
Technical Specifications for NGS-Based Biomarker Assessment
NGS Panel Requirements for TMB Analysis
Targeted NGS panels provide a cost-effective alternative to whole exome sequencing (WES) or whole genome sequencing (WGS) for TMB assessment, but require careful consideration of technical specifications [78]:
- Panel Size: For accurate TMB estimation, panels should cover at least 1.0-1.5 Mb of genomic territory, with optimal performance in the 1.5-3.0 Mb range. Larger panels reduce confidence intervals and improve extrapolation accuracy [78].
- Gene Content: Panels should focus on cancer driver genes (approximately 375 known genes) but must balance this with the need for neutral regions without positive selection to avoid TMB overestimation [78].
- Variant Types: TMB calculation typically includes only somatic, non-synonymous coding mutations (missense, nonsense, indels). However, the ability to detect synonymous mutations can improve calculation accuracy by reducing sampling noise [78].
- Bioinformatic Considerations: Robust pipelines must implement filters for germline variants (using population databases and, ideally, matched normal samples), sequencing artifacts, and driver mutations that may inflate TMB estimates [78].
MSI Detection by NGS
NGS-based MSI detection offers advantages over traditional PCR-based methods by simultaneously assessing hundreds to thousands of microsatellite loci, potentially increasing sensitivity [79]. Key methodological considerations include:
- Loci Selection: Panels should include a sufficient number of mononucleotide and dinucleotide repeats representative of genome-wide microsatellite instability.
- Bioinformatic Algorithms: Specialized tools compare length distributions at microsatellite loci between tumor and normal samples, with MSI-H typically defined as instability at ≥30% of loci [79].
- Concordance with IHC: Studies demonstrate high concordance (98%) between NGS-based MSI calling and deficient MMR protein expression by immunohistochemistry [79].
Diagram 1: NGS biomarker assessment workflow
TME Profiling via NGS
NGS enables comprehensive TME characterization through multiple approaches [80]:
- Immune Cell Deconvolution: Computational algorithms (e.g., CIBERSORT) use gene expression data to infer relative abundances of 22 immune cell types within the TME [81].
- T-Cell Receptor (TCR) Repertoire Sequencing: Assessment of T-cell diversity and clonality provides insights into antigen-specific immune responses.
- Gene Expression Profiling: Immune-related gene signatures can classify TME into immunologically "hot" or "cold" phenotypes.
- Multimodal Integration: Combining DNA-based markers (TMB, MSI) with RNA-based immune signatures provides the most comprehensive TME assessment.
Table 2: Technical Specifications for NGS-Based Biomarker Assessment
| Parameter | TMB Analysis | MSI Detection | TME Profiling |
|---|---|---|---|
| Recommended Panel Size | 1.5-3.0 Mb | Gene-agnostic (focused on microsatellite regions) | Varies by approach |
| Primary Data Type | Somatic mutations | Microsatellite length alterations | Gene expression, TCR sequences |
| Key Bioinformatic Methods | Mutation calling, germline filtering | MSI calling algorithms, reference comparison | Deconvolution algorithms, clustering |
| Quality Metrics | Coverage uniformity, sequencing depth | Loci coverage, background signal | RNA quality, library complexity |
| Common Thresholds | TMB-H: ≥10 mut/Mb (variable by cancer type) | MSI-H: ≥30% unstable loci | Immune-rich vs. immune-poor |
| Standardization Status | Evolving (QuIP guidelines) | Established | Emerging |
Experimental Protocols and Methodologies
Sample Preparation and Quality Control
Optimal sample preparation is critical for reliable NGS-based biomarker assessment:
- Tissue Processing: Formalin-fixed, paraffin-embedded (FFPE) tissues represent the most common specimen source. Ensure fixation time of 6-48 hours in 10% neutral buffered formalin to prevent DNA degradation [79] [80].
- DNA Extraction: Use specialized kits for FFPE-derived DNA (e.g., MagCore Genomic DNA FFPE One-Step Kit). Assess DNA quality via fluorometry and fragment analyzer, with recommended DNA integrity numbers (DIN) >4.0 for reliable TMB assessment [82].
- Tumor Content: Pathological review should confirm minimum tumor content of 20-30% to ensure accurate mutation calling and avoid false-negative results.
- Library Preparation: Employ hybrid capture-based methods for target enrichment. Input 50-200ng of DNA, with fragmentation optimized for FFPE-derived DNA. Incorporate unique molecular identifiers (UMIs) to reduce sequencing artifacts and improve mutation detection sensitivity [82] [78].
Sequencing and Data Generation
- Sequencing Platform: Utilize Illumina-based platforms (NovaSeq 6000) for high-throughput sequencing. Achieve minimum coverage of 150-200x in tumor samples, with >100x uniformity across targeted regions [82].
- Control Samples: Include both positive and negative control samples in each sequencing run. Process without-template controls to identify contamination and reference standards with known TMB/MSI status for quality assurance.
- Data Output: Generate paired-end reads (2x75bp or 2x100bp) sufficient to cover the entire target region with minimum 100x coverage. FastQ files serve as the primary data output for downstream analysis [82].
Bioinformatic Analysis Pipelines
TMB Calculation Protocol:
- Alignment: Map sequencing reads to reference genome (GRCh37/38) using optimized aligners (BWA-MEM).
- Variant Calling: Identify somatic mutations using paired tumor-normal analysis when possible. For tumor-only workflows, implement rigorous germline filtering using population frequency databases (gnomAD) and computational predictions.
- Mutation Filtering: Exclude known driver mutations, synonymous variants, and variants in genomic regions with poor mapping quality. Retain only coding, non-synonymous mutations for TMB calculation.
- TMB Calculation: Divide the count of eligible mutations by the size of the coding target territory in megabases. Report as mutations per megabase (mut/Mb) [78].
MSI Analysis Protocol:
- Microsatellite Loci Analysis: Evaluate sequencing data at hundreds to thousands of microsatellite loci included in the panel.
- Instability Assessment: Compare length distribution at each locus between tumor and normal reference. Apply statistical models to identify significant shifts indicative of instability.
- MSI Scoring: Calculate percentage of unstable loci. Classify as MSI-H (≥30% unstable loci), MSI-L (10-29% unstable), or MSS (<10% unstable) [79].
TME Deconvolution Protocol:
- Gene Expression Quantification: Generate normalized counts or FPKM values for all genes.
- Cellular Decomposition: Apply computational methods (CIBERSORT, EPIC, MCP-counter) to estimate immune cell abundances from bulk RNA-seq data.
- Immune Signature Analysis: Calculate scores for specific immune populations (T-cells, macrophages) and functional states (cytotoxicity, exhaustion) [80] [81].
Diagram 2: Biomarker interrelationships in immunotherapy response
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for NGS-Based Immuno-Oncology
| Reagent/Platform | Primary Function | Key Features | Representative Examples |
|---|---|---|---|
| Targeted NGS Panels | Comprehensive genomic profiling | 1.5-3.0 Mb size; 500+ cancer-related genes; TMB & MSI analysis | FoundationOne CDx, Tempus xT, TruSight Oncology 500 |
| Hybrid Capture Reagents | Target enrichment for NGS | Optimized for FFPE DNA; unique molecular identifiers; dual-indexing | Illumina TruSeq, IDT xGen, Twist Target Enrichment |
| FFPE DNA Extraction Kits | Nucleic acid isolation from archival tissues | Designed for cross-linked DNA; deparaffinization steps; integrity assessment | MagCore Genomic DNA FFPE Kit, QIAamp DNA FFPE Tissue Kit |
| Immune Deconvolution Software | TME characterization from RNA-seq | Estimates 20+ immune cell types; validated signatures; user-friendly interfaces | CIBERSORT, EPIC, MCP-counter |
| Variant Calling Pipelines | Somatic mutation detection | Germline filtering; artifact removal; TMB calculation | Sentieon, GATK, VarScan |
| MSI Analysis Algorithms | Microsatellite instability detection | Hundreds of loci; tumor-normal comparison; statistical scoring | MSIsensor, mSINGS, NGS-specific algorithms |
Implementation Challenges and Standardization Efforts
Analytical Validation and Quality Assurance
Implementing robust NGS assays for TMB, MSI, and TME assessment requires rigorous validation:
- Reproducibility: Demonstrate high inter-run and inter-laboratory concordance for TMB measurement (R² > 0.95 across the analytical range).
- Accuracy: Establish agreement with orthogonal methods (WES for TMB, PCR for MSI, IHC/FACS for immune cells).
- Precision: Determine repeatability and reproducibility with ≤20% coefficient of variation for TMB values.
- Limit of Detection: Establish minimum DNA input, tumor content, and sequencing depth required for reliable biomarker assessment [78].
Clinical Implementation Barriers
Multiple studies identify significant challenges in translating NGS-based biomarkers to routine clinical practice:
- Turnaround Time: Real-world data indicates median turnaround time of 73 days from specimen collection to final NGS report, potentially delaying treatment decisions [77].
- Cost and Reimbursement: Inconsistent payer coverage and high out-of-pocket costs limit access, with 33% of payers unaware of current biomarker testing recommendations in guidelines [26].
- Interpretation Complexity: Evolving thresholds for TMB-H classification (10 vs. 20 mut/Mb) and tissue-specific differences create confusion in clinical application [77] [78].
- Sample Quality: Suboptimal DNA from FFPE tissues or low tumor cellularity can lead to test failures or inaccurate results [26].
Standardization Initiatives
Multiple organizations are addressing standardization needs:
- Quality Assurance Initiative Pathology (QuIP): Provides guidelines for TMB assessment, recommending reporting in mut/Mb and using panels ≥1 Mb [78].
- Friends of Cancer Research: Leads multi-stakeholder efforts to harmonize TMB measurement across platforms and establish universal thresholds.
- ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT): Creates framework for classifying genomic alterations based on evidence level for targeted therapies [26].
Future Directions and Research Applications
The integration of TMB, MSI, and TME profiling represents the future of immunotherapy biomarker development. Emerging applications include:
- Dynamic Monitoring: Serial liquid biopsy-based assessment of TMB and MSI during treatment to monitor evolution of resistance mechanisms [26].
- Multimodal Integration: Combining genomic (TMB, MSI), transcriptomic (immune signatures), and proteomic (PD-L1) data for composite biomarker scores with enhanced predictive power.
- Spatial Analysis: Advanced technologies enabling spatial profiling of the TME to understand geographic relationships between tumor cells and immune populations.
- Machine Learning Approaches: Development of integrated algorithms that simultaneously consider multiple biomarker inputs to predict immunotherapy outcomes with higher accuracy.
For researchers and drug development professionals, comprehensive biomarker assessment now represents a fundamental component of immuno-oncology development programs. The systematic integration of TMB, MSI, and TME profiling using robust NGS methodologies provides critical insights into mechanisms of response and resistance, enabling more effective patient stratification and novel therapeutic combinations. As standardization improves and analytical frameworks mature, these biomarkers will increasingly guide both clinical practice and drug development strategies across the spectrum of cancer types.
Navigating Implementation Challenges: Optimization Strategies for Robust NGS Workflows
The integration of next-generation sequencing (NGS) into oncology represents a paradigm shift in cancer care, enabling comprehensive molecular profiling to guide targeted therapy and personalized treatment decisions. Despite its transformative potential and endorsement by major clinical guidelines, the widespread adoption of NGS-based tumor profiling in clinical and research settings faces significant economic and systemic challenges. The barriers of inconsistent reimbursement, complex payer coverage policies, and questions regarding cost-effectiveness create substantial impediments to realizing the full potential of precision oncology. This whitepaper provides a technical analysis of these critical barriers, synthesizing recent quantitative evidence to inform researchers, scientists, and drug development professionals working to advance NGS integration in cancer research and clinical practice. Within the broader thesis on NGS in cancer molecular profiling research, understanding these implementation challenges is crucial for developing strategies that ensure equitable access and sustainable adoption of genomic technologies.
Quantitative Analysis of NGS Implementation Barriers
Recent multi-stakeholder surveys provide compelling quantitative evidence regarding the primary barriers impeding NGS implementation in oncology. The data reveal consistent concerns across healthcare providers, payers, and patients that directly impact research protocols and clinical translation.
Table 1: Perceived Barriers to NGS Adoption Among Healthcare Providers [26] [83] [84]
| Barrier Category | Specific Challenge | Percentage Reporting | Stakeholder Group |
|---|---|---|---|
| Reimbursement & Coverage | Inconsistent payer coverage | 87.5% | Physicians (Oncologists, Surgeons, Pathologists) |
| High out-of-pocket costs for patients | 87.5% | Physicians | |
| Prior authorization requirements | 72.0% | Physicians | |
| Paperwork/administrative duties | 67.5% | Physicians | |
| Knowledge & Guidelines | Lack of knowledge of NGS testing methodologies | 81.0% | Physicians |
| Lack of clear clinical guidelines | 74.0% | Payers | |
| Lack of internal expertise on NGS | 39.0% | Payers | |
| Evidence Base | Lack of clinical utility evidence | 80.0% | Physicians |
| Lack of internal consensus on which NGS tests to cover | 45.0% | Payers |
Table 2: Payer Perspectives on NGS Coverage Challenges [26]
| Payer Barrier | Percentage Ranking as Top 3 Barrier | Implications for Research |
|---|---|---|
| Unaware of current NCCN guidelines for biomarker testing | 33% | Highlights need for improved guideline dissemination |
| Lack of clear clinical guidelines | 74% | Underscores importance of robust clinical validity data in study design |
| Lack of internal consensus on which NGS tests to cover | 45% | Suggests value of standardized test evaluation frameworks |
| Absence of internal expertise on NGS | 39% | Indicates need for educational initiatives targeting payers |
The data demonstrates that reimbursement challenges constitute the most frequently cited barrier, affecting 87.5% of physicians surveyed [83] [84]. Specifically, prior authorization requirements (72.0%) and administrative burdens (67.5%) create significant operational friction that can delay testing and impact research protocols [83]. Importantly, a knowledge gap persists among payers, with 33% unaware of current National Comprehensive Cancer Network (NCCN) biomarker testing recommendations and 74% citing unclear guidelines as a primary concern [26]. This disconnect between guideline development and payer implementation highlights a critical opportunity for researcher-payer engagement to facilitate appropriate coverage policies.
Methodological Frameworks for NGS Barrier Research
Multi-Stakeholder Survey Methodology
Understanding NGS implementation barriers requires robust methodological approaches that capture perspectives across the healthcare ecosystem. Recent studies have employed comprehensive survey methodologies to quantify these challenges [26].
Study Population and Recruitment: A 2025 multi-stakeholder analysis recruited 367 participants across the United States, including medical oncologists (n=109), nurses and physician assistants (n=50), lab directors and pathologists (n=40), payers (n=31), and metastatic breast cancer patients (n=137). Recruitment utilized multiple channels including market research vendors and an internally developed database of healthcare experts to minimize selection bias. Stratified quotas ensured diversity across geographic regions, practice types, and institutional settings [26].
Survey Design and Validation: The survey instrument was developed through a series of 60-minute double-blinded phone-based interviews with representative stakeholders. Beta-testing ensured question clarity, appropriate answer options, neutrally framed questions, and optimal survey flow. To reduce social desirability bias, the survey was designed to be fully anonymous, encouraging candid responses about implementation barriers [26].
Data Collection Parameters: Quantitative online surveys captured data on testing rates, reimbursement challenges, coverage policies, perceived barriers, and demographic information. Practice characteristics included patient volume, testing rates, geographic region, and practice type (private, academic, community-based). Payer characteristics included plan type (commercial vs. CMS) and plan size [26].
Cost-Effectiveness Analysis Framework
Economic evaluations of NGS testing employ sophisticated methodologies to assess value across different testing strategies and clinical scenarios. A 2024 systematic review of 29 cost-effectiveness studies established three primary methodological approaches for evaluating NGS economics [85].
Direct Testing Cost Comparison: This approach compares the direct expenses of NGS-based testing versus sequential single-gene testing. Analysis includes reagent costs, equipment, and personnel requirements. Studies consistently demonstrate that targeted NGS panels (2-52 genes) become cost-effective when 4+ genes require analysis, with larger panels (hundreds of genes) generally less cost-effective under this narrow framework [85].
Holistic Testing Cost Analysis: This expanded methodology incorporates indirect costs including turnaround time, healthcare personnel requirements, number of hospital visits, and overall hospital expenditures. Holistic analyses consistently show NGS advantages through reduced administrative burden, faster time to treatment decisions, and optimized resource utilization compared to sequential testing approaches [85].
Long-Term Outcomes Assessment: This comprehensive approach evaluates incremental cost-effectiveness ratios (ICERs) considering long-term patient outcomes, quality-adjusted life years (QALYs), and total healthcare costs. While targeted therapies identified through NGS often exceed conventional cost-effectiveness thresholds, these analyses capture valuable patient benefits including improved survival and treatment matching [85].
Table 3: NGS Cost-Effectiveness Methodologies and Findings [85]
| Analysis Methodology | Key Parameters Measured | Primary Findings | Research Implications |
|---|---|---|---|
| Direct Testing Cost Comparison | Reagent costs, equipment, personnel time | Targeted NGS cost-effective when 4+ genes tested; larger panels less cost-effective | Supports targeted panel selection based on clinical context |
| Holistic Testing Cost Analysis | Turnaround time, staff requirements, hospital visits, institutional costs | NGS reduces administrative burden and resource utilization | Captures systemic efficiencies beyond direct test costs |
| Long-Term Outcomes Assessment | ICERs, QALYs, overall survival, total healthcare costs | Targeted therapies often exceed cost thresholds but provide significant patient benefit | Supports value-based pricing frameworks for targeted therapies |
Cost-Effectiveness Evidence for NGS in Oncology
The economic value proposition of NGS represents a critical evidence gap for many healthcare systems and payers. Recent systematic analyses provide compelling data regarding the cost-effectiveness of NGS across various clinical contexts and testing methodologies.
A comprehensive 2024 systematic review of 29 cost-effectiveness studies across 12 countries and 6 oncology indications revealed that targeted panel testing (2-52 genes) consistently demonstrated cost savings compared to conventional single-gene testing when four or more genes required analysis [85]. This finding held across multiple healthcare systems and economic contexts, supporting the economic viability of focused NGS panels in biomarker-driven oncology.
When holistic testing costs were incorporated into economic analyses, NGS demonstrated significant advantages through reduced turnaround time, decreased healthcare personnel requirements, fewer hospital visits, and lower overall institutional costs [85]. This expanded economic perspective captures the systemic efficiencies enabled by comprehensive genomic profiling compared to sequential single-gene testing approaches.
Evidence from the ATLAS study in advanced NSCLC demonstrates the clinical value proposition of NGS, with centralized NGS testing increasing the detection of druggable mutations from 7.9% to 25.9% compared to local pathology assessments [86]. This enhanced detection capability directly impacts therapeutic decision-making, with 34.5% of patients having molecular alterations matching available clinical trials in their country [86].
Comparative cost analyses reveal important distinctions between testing methodologies. Multiplatform profiling approaches that integrate NGS with other molecular analyses demonstrated significantly lower treatment costs per cycle (£995) compared to NGS-only guided treatments (£2,795) [87]. This cost differential reflects the tendency of NGS-only approaches to guide more expensive targeted therapies, while multiplatform profiling more frequently identifies conventional chemotherapies with comparable efficacy [87].
NGS Testing Workflow and Technical Requirements
The complete NGS testing workflow encompasses multiple technical stages from sample acquisition through clinical reporting, each with specific resource requirements and quality control checkpoints.
The NGS testing workflow involves three distinct phases, each with critical quality control checkpoints. The pre-analytical phase encompasses sample acquisition through nucleic acid extraction, requiring stringent quality assessment to ensure adequate material for sequencing. The analytical phase includes library preparation and actual sequencing, with platform-specific quality metrics ensuring data integrity. The post-analytical phase involves complex bioinformatic analysis and clinical interpretation, ultimately generating actionable reports for therapeutic decision-making [9] [14].
Essential Research Reagents and Platforms
Implementation of NGS in research and clinical settings requires specific technical resources and platform solutions. The following table details essential research reagents and their functions in the NGS workflow.
Table 4: Essential Research Reagent Solutions for NGS Implementation [9] [14] [17]
| Reagent Category | Specific Examples | Technical Function | Implementation Considerations |
|---|---|---|---|
| Nucleic Acid Extraction Kits | QIAGEN DNA/RNA kits, Thermo Fisher Scientific extraction reagents | Isolation of high-quality DNA/RNA from diverse sample types | Yield optimization for limited samples, FFPE-compatible protocols |
| Library Preparation Kits | Illumina Nextera, Thermo Fisher Scientific Ion AmpliSeq, Oncomine Focus Assay | Fragmentation, adapter ligation, target enrichment | Input DNA requirements, target capture efficiency, hands-on time |
| Sequencing Chemistry | Illumina SBS, Ion Torrent semiconductor, Oxford Nanopore R10 | Nucleotide incorporation detection, signal capture | Read length, error profiles, throughput requirements |
| Quality Control Assays | Bioanalyzer, TapeStation, Qubit assays, qPCR | Quantitation, fragment size distribution, integrity assessment | Sensitivity thresholds, sample consumption, throughput |
| Bioinformatics Tools | Illumina DRAGEN, Google DeepVariant, GATK, custom pipelines | Base calling, alignment, variant annotation, interpretation | Computational requirements, validation complexity, reporting capabilities |
Payer Coverage Policy Analysis
Understanding payer perspectives is crucial for addressing coverage barriers impeding NGS adoption. Recent survey data reveals that 33% of payers lack awareness of current NCCN biomarker testing recommendations, creating a fundamental knowledge gap that directly impacts coverage policies [26]. This awareness deficit contributes to the 74% of payers who identify unclear clinical guidelines as a primary barrier to NGS coverage [26].
The coverage decision-making process is further complicated by internal structural challenges within payer organizations. Approximately 45% of payers report lacking internal consensus on which NGS tests to cover, while 39% cite insufficient internal expertise on NGS technology as a significant barrier [26]. These findings indicate substantial opportunities for researcher-payer collaboration to develop evidence frameworks that support consistent coverage policies.
Coverage limitations manifest most significantly in prior authorization requirements, which 72% of physicians identify as a major reimbursement challenge [83]. The administrative burden associated with these requirements creates operational friction, with 67.5% of physicians citing paperwork and administrative duties as significant barriers to NGS utilization [83]. These procedural hurdles can delay testing and subsequent treatment initiation, potentially impacting patient outcomes and clinical trial enrollment.
The NGS coverage decision framework illustrates the multifactorial nature of payer policy determination. Clinical guideline recommendations, evidence quality, economic considerations, and internal payer capabilities collectively influence coverage policies. Specific considerations include appropriate testing timing, biomarker targets, specimen types, clinical utility evidence, cost-effectiveness metrics, and internal administrative capacity [26] [83] [85].
The integration of NGS into cancer molecular profiling research faces significant yet addressable barriers related to reimbursement, payer coverage, and cost-effectiveness. Quantitative evidence demonstrates that reimbursement challenges affect 87.5% of physicians, primarily through prior authorization requirements (72.0%) and administrative burdens (67.5%) [83]. Simultaneously, payer coverage decisions are hampered by knowledge gaps, with 33% of payers unaware of current NCCN guidelines and 74% citing unclear guidelines as a primary barrier [26].
Economic analyses reveal that targeted NGS panels demonstrate cost-effectiveness when four or more genes require testing, particularly when holistic costs including turnaround time and resource utilization are considered [85]. The systematic evaluation of these barriers through robust methodological frameworks provides researchers, scientists, and drug development professionals with evidence-based strategies to advance NGS integration in oncology. Addressing these implementation challenges requires multidisciplinary collaboration across researchers, clinicians, payers, and policymakers to establish sustainable economic models that support personalized cancer care through comprehensive molecular profiling.
Next-Generation Sequencing (NGS) has fundamentally transformed oncology research and drug development by enabling comprehensive genomic profiling of tumors. The successful application of this powerful technology, however, is critically dependent on a factor that precedes the actual sequencing: sample quality and tumor purity. Specimens with inadequate tumor content or degraded nucleic acids can generate misleading or uninterpretable results, compromising research validity and potentially derailing drug development pipelines. Within the context of molecular profiling research, the pre-analytical phase—encompassing specimen selection, collection, processing, and pathological review—constitutes a foundational determinant of data reliability. This technical guide establishes best practices for researchers and drug development professionals seeking to optimize specimen quality for NGS applications, thereby ensuring the generation of robust, reproducible, and clinically translatable genomic data.
Specimen Types for Molecular Analysis: Advantages and Limitations
The choice of specimen type is a primary consideration in research design, with each offering distinct advantages and presenting specific challenges for genomic analysis.
Formalin-Fixed Paraffin-Embedded (FFPE) Tissue
FFPE specimens represent the most widely available resource for cancer genomics research. Their widespread use in pathology departments makes them invaluable for retrospective studies. However, the formalin fixation process introduces DNA cross-linking and fragmentation, which can adversely affect sequencing library preparation and downstream analysis [88]. To mitigate these effects, fixation time should be carefully controlled; for core biopsies, a fixation time of 6–24 hours is optimal, while prolonged fixation (exceeding one week) results in severe DNA degradation that may preclude reliable mutation detection [88]. Prior to nucleic acid extraction, a dedicated section must be stained with Hematoxylin and Eosin (H&E) and subjected to pathological review to determine tumor cell percentage and assess tissue viability.
Fresh Frozen Tissue
Fresh frozen tissue, typically snap-frozen in liquid nitrogen within 30 minutes of resection, provides the highest quality DNA and RNA for NGS applications [88]. This preservation method avoids the cross-linking artifacts inherent to formalin fixation, making it the gold standard for genome, transcriptome, and epigenome studies. For research involving complex assays such as whole genome sequencing or spatial transcriptomics, fresh frozen material is preferable. Intra-operative sampling requires macroscopic dissection followed by microscopic confirmation of tumor cell content from an adjacent section to ensure specimen adequacy.
Liquid Biopsy and Circulating Tumor Cells (CTCs)
Liquid biopsy, which analyzes circulating tumor DNA (ctDNA) or circulating tumor cells (CTCs) from peripheral blood, offers a minimally invasive means of sampling tumor DNA. This approach enables real-time monitoring of tumor evolution and treatment response, which is particularly valuable for assessing tumor heterogeneity and tracking the emergence of resistance mechanisms [89] [90]. However, the analytical challenge is significant, as the tumor-derived nucleic acids can represent a very small fraction (0.01%–93%) of the total cell-free DNA [88]. For plasma preparation, 6–10 mL of whole blood should be collected into EDTA tubes, processed within 6 hours to separate plasma, and centrifuged to remove cellular debris before cfDNA extraction and storage at -80°C [88]. CTCs are exceptionally rare in circulation (approximately 1 per 10^6–10^7 mononuclear cells) [89], necessitating sophisticated enrichment technologies like the CellSearch system (immunomagnetic enrichment for EpCAM) or microfluidic CTC-Chip platforms prior to molecular characterization [89].
Cytological Samples
Cellular specimens such as pleural or ascitic fluids represent a valuable source of tumor material when tissue biopsies are unattainable. The processing of these samples involves cytocentrifugation to create a cell pellet, which can be used for smear preparation, cell block formation, or direct nucleic acid extraction. Given the typically low tumor cellularity of these specimens, their effective use in NGS requires highly sensitive detection methods capable of identifying mutations present in a small fraction of the analyzed cells [88].
Table 1: Comparative Analysis of Specimen Types for NGS in Cancer Research
| Specimen Type | Optimal DNA Quality | Primary Advantages | Key Limitations | Ideal NGS Applications |
|---|---|---|---|---|
| FFPE Tissue | Moderate to Poor (Fragmented) | Widely available, enables retrospective studies, linked to clinical pathology | DNA/RNA cross-linking and degradation, variable quality | Targeted sequencing, mutation profiling in validated cohorts |
| Fresh Frozen Tissue | High (Intact Nucleic Acids) | Gold standard for nucleic acid integrity, no cross-linking artifacts | Requires specialized infrastructure, not routinely collected | Whole genome/exome sequencing, transcriptomics, multi-omics |
| Liquid Biopsy (cfDNA/CTCs) | Variable (ctDNA is fragmented) | Minimally invasive, enables serial monitoring, captures heterogeneity | Low tumor DNA fraction in total cfDNA, requires ultra-sensitive assays | Therapy resistance monitoring, MRD detection, tracking clonal evolution |
| Cytological Samples | Variable | Alternative when tissue is unavailable, minimally invasive | Often low tumor cellularity, limited material | Targeted sequencing when tissue is unavailable |
Tumor Purity Assessment and Pathologist Review
Macrodisection and Microdissection Protocols
The accurate determination of tumor purity is a critical pre-analytical step. The process begins with a pathologist reviewing an H&E-stained section adjacent to the area destined for DNA/RNA extraction. Tumor purity is quantified as the percentage of viable tumor cells relative to all nucleated cells in the sample, including stromal cells, lymphocytes, and other non-neoplastic elements [88]. For specimens with heterogeneous cellularity, macrodisection techniques are employed to manually isolate tumor-rich regions from the surrounding non-malignant tissue. In cases of extensive stromal infiltration or for the analysis of specific tumor subpopulations, laser capture microdissection (LCM) provides a precise method for isolating pure cell populations under microscopic visualization. This technique is particularly valuable for research aimed at understanding the genomic features of specific histological patterns or the tumor-stroma interface.
Tumor Purity Thresholds for NGS Platforms
The required tumor purity varies significantly depending on the sensitivity of the NGS platform employed. Traditional Sanger sequencing, with a detection sensitivity of approximately 15-20%, necessitates a high tumor cell content (generally ≥50%) to avoid false-negative results [88] [90]. In contrast, modern NGS panels, especially those utilizing unique molecular identifiers (UMIs) and deep sequencing (>500x coverage), can reliably detect variants at allele frequencies as low as 1% with a minimum tumor purity of 5-10% [91]. For whole genome or exome sequencing of heterogeneous tumors, a higher purity (≥30%) is recommended to ensure adequate power for the detection of subclonal mutations and accurate copy number alteration calling.
Table 2: Technical Specifications and Tumor Purity Requirements for Genomic Technologies
| Technology | Principle | Detection Sensitivity | Minimum Tumor Purity | Sample Input Requirements |
|---|---|---|---|---|
| Sanger Sequencing | Dideoxy chain termination | ~15-20% | ≥50% | High-quality DNA (50-100 ng) |
| NGS (Panel) | Massive parallel sequencing | ~1-5% (with deep coverage) | 5-10% | DNA from FFPE/frozen (10-50 ng) |
| NGS (WES/WGS) | Massive parallel sequencing | ~5-10% (for subclones) | ≥30% | High-quality DNA (50-100 ng for WES, >100 ng for WGS) |
| Digital PCR | Absolute nucleic acid quantification | ~0.1-1% | 1-5% | DNA (10-20 ng) |
| ARMS-PCR | Allele-specific amplification | ~0.1-1% | 1-5% | DNA (10-20 ng) |
Experimental Workflows for Specimen Processing and Analysis
Standardized Workflow for FFPE Tissue Processing
A rigorous, standardized protocol is essential for ensuring consistent and reliable results from FFPE specimens. The workflow begins with sectioning, where 5-10 consecutive tissue sections of 5-10 μm thickness are prepared. The first and last sections are H&E-stained for pathological assessment of tumor content and necrosis. If the tumor percentage meets the required threshold, the intervening unstained sections are used for nucleic acid extraction. Following deparaffinization with xylene and ethanol washes, proteinase K digestion is performed to release nucleic acids from the protein cross-links. DNA is then purified using column-based or magnetic bead-based methods. The quality control of extracted DNA involves spectrophotometric (A260/A280) or fluorometric quantification, followed by fragment size analysis (e.g., Bioanalyzer) to assess the degree of fragmentation, which is a key predictor of NGS success.
Integrated Protocol for CTC Enrichment and Molecular Characterization
The analysis of CTCs involves a two-step process: enrichment followed by detection/characterization. For enrichment, the CellSearch system employs immunomagnetic beads coated with anti-EpCAM antibodies to positively select epithelial cells from whole blood [89]. The enriched cell fraction is then identified as CTCs based on positive staining for cytokeratins (CK8/18/19), negative staining for the leukocyte marker CD45, and positive nuclear staining with DAPI [89]. For downstream molecular analysis, the AdnaTest system combines immunomagnetic enrichment (using a cocktail of antibodies against tumor-associated antigens) with subsequent RT-PCR analysis of tumor-specific transcripts [89]. Emerging microfluidic platforms like the CTC-Chip and its successor, the HB-Chip, use antibody-coated microposts in a microfluidic chamber to enhance the efficiency of CTC capture from whole blood, improving yield for molecular studies [89].
NGS Workflow from Specimen to Data Interpretation
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of NGS-based cancer profiling requires access to specialized reagents and platforms. The following toolkit outlines critical components for research in this field:
RosetteSepTM / OncoQuick: These kits employ density gradient centrifugation for CTC enrichment. RosetteSep uses antibody-mediated cross-linking to deplete hematopoietic cells, while OncoQuick uses a porous barrier during centrifugation to separate CTCs from other blood components [89].
CellSearch System: This FDA-cleared platform provides standardized CTC enumeration. It uses EpCAM-coated magnetic beads for positive selection, followed by immunofluorescent staining (CK8/18/19+, CD45-, DAPI+) for definitive CTC identification [89].
CTC-Chip/HB-Chip: These microfluidic devices contain microposts (CTC-Chip) or herringbone structures (HB-Chip) coated with anti-EpCAM antibodies. They optimize cell-antibody contact by controlling blood flow dynamics, significantly improving CTC capture efficiency over previous methods [89].
QIAGEN, Roche, Illumina NGS Panels: Commercially available targeted sequencing panels (e.g., Qiagen GeneRead, Illumina TruSight, Roche AVENIO) contain pre-designed primers and probes for amplifying cancer-related genes. These panels are optimized for performance with FFPE-derived DNA and typically require 10-50 ng of input material [91].
PolyPhen-2 (Polymorphism Phenotyping v2): This computational algorithm predicts the potential functional impact of amino acid substitutions on protein structure and function. It generates a score from 0.0 (benign) to 1.0 (probably damaging), aiding in the prioritization of missense mutations identified through NGS [91].
In the era of precision oncology, the reliability of NGS-based molecular profiling is inextricably linked to the quality of the input biological specimens. Adherence to standardized protocols for specimen selection, processing, and pathological review is not merely a procedural formality but a fundamental scientific requirement. By implementing the best practices outlined in this guide—rigorous assessment of tumor purity, appropriate selection of specimen type matched to research objectives, and utilization of optimized experimental protocols—researchers and drug developers can significantly enhance the validity and translational potential of their genomic findings. As NGS technologies continue to evolve toward single-cell and multi-omic analyses, the principles of quality specimen management will remain the cornerstone of robust cancer research and therapeutic innovation.
Next-generation sequencing (NGS) has fundamentally transformed oncology research, enabling comprehensive genomic profiling that identifies driver mutations, biomarkers, and novel therapeutic targets across diverse cancer types [90]. This paradigm shift toward precision oncology, however, comes with a significant computational challenge: the massive volume and complexity of data generated by modern sequencing technologies. Bioinformatics pipeline optimization has therefore become a critical discipline for researchers seeking to derive meaningful biological insights from NGS data while managing associated computational burdens.
The scale of this data challenge is substantial. While analyzing a single sample may cost less than $1, processing millions of data points can result in monthly data processing expenses reaching tens or even hundreds of thousands of dollars [92]. Furthermore, inefficient semi-manual workflows requiring full-time employees to manage unstable pipelines compound these expenses while slowing research progress. Within clinical oncology, these inefficiencies directly impact patient care by delaying the identification of actionable mutations for targeted therapies [26] [14].
This technical guide examines comprehensive strategies for optimizing bioinformatics workflows within the context of cancer molecular profiling research. By addressing computational bottlenecks, implementing efficient data management practices, and establishing reproducible analytical frameworks, researchers can significantly enhance the reliability and throughput of their NGS-based oncology studies.
The Computational Landscape of NGS in Oncology
NGS technologies generate unprecedented amounts of data through massively parallel sequencing, simultaneously analyzing millions of DNA fragments to provide a comprehensive genomic landscape of tumors [90]. The data intensity of these approaches stems from both the volume of sequencing reads and the complexity of subsequent analytical processes required for variant identification, annotation, and interpretation.
In cancer genomics, this challenge is particularly pronounced due to several factors:
- Tumor Heterogeneity: Analyzing complex karyotypes with numerous genomic alterations requires deeper sequencing and sophisticated variant calling algorithms [14].
- Multi-Sample Comparisons: Matched tumor-normal analyses and longitudinal monitoring increase data processing requirements.
- Comprehensive Profiling: Large gene panels, whole exome sequencing (WES), and whole genome sequencing (WGS) generate substantially more data than targeted approaches [93].
The bioinformatics pipelines that process this data typically follow a structured lifecycle encompassing four critical stages: pipeline development, deployment across environments, automation and data management, and results interpretation [92]. Each stage presents distinct optimization opportunities for managing computational complexity.
Table 1: NGS Technology Comparison in Oncology Applications
| Technology | Typical Read Length | Error Profile | Optimal Oncology Applications | Data Output per Run |
|---|---|---|---|---|
| Illumina (Short-read) | 75-300 bp | Low (0.1-0.6%) | Variant detection, gene expression, targeted panels | High (GB to TB range) |
| Oxford Nanopore (Long-read) | 100-100,000+ bp | Higher (~5%) but improving | Structural variant detection, fusion genes, epigenetics | Medium to High |
| PacBio (Long-read) | 10,000-100,000 bp | Low with HiFi mode | Complex rearrangement resolution, haplotype phasing | Medium to High |
Strategic Framework for Pipeline Optimization
Effective optimization requires a systematic approach that balances computational efficiency with analytical accuracy. Research indicates that optimization efforts typically require at least two months to complete but can yield time and cost savings ranging from 30% to 75% [92]. A phased implementation strategy focusing on three critical components delivers the most consistent results.
Stage 1: Analysis Tools and Algorithms
The foundation of any efficient bioinformatics pipeline is the selection and optimization of analytical tools. This initial stage requires identifying performance bottlenecks and either implementing improved tools or developing custom solutions when none exist.
Error Correction Benchmarking: In cancer research, accurate variant detection is paramount, particularly for identifying low-frequency subclonal populations. A comprehensive benchmarking study of computational error-correction methods revealed that performance varies substantially across different types of datasets, with no single method performing best on all examined data [94]. The evaluation used gain, precision, and sensitivity metrics to assess tools including Coral, Bless, Fiona, Pollux, BFC, Lighter, Musket, Racer, RECKONER, and SGA.
Table 2: Performance Metrics of Select Error Correction Tools for WGS Data
| Tool | Algorithm Type | Precision Range | Sensitivity Range | Optimal Use Case |
|---|---|---|---|---|
| BFC | k-mer based | 0.85-0.95 | 0.75-0.90 | General purpose WGS |
| Fiona | k-mer based | 0.80-0.92 | 0.82-0.95 | Homogeneous genomes |
| Lighter | k-mer based | 0.88-0.96 | 0.78-0.88 | Memory-efficient processing |
| Musket | k-mer based | 0.82-0.94 | 0.80-0.92 | Multithreaded environments |
Tool Selection Criteria: When optimizing analysis components, researchers should prioritize tools that demonstrate high precision in their specific cancer genomics context, as false positives in variant calling can lead to erroneous conclusions about therapeutic targets. The k-mer size parameter significantly influences correction accuracy, with increased k-mer size typically offering improved performance, though this relationship varies across tools [94].
Stage 2: Workflow Orchestration
As pipeline complexity grows, workflow orchestrators become essential for managing computational resources and execution dependencies. Technologies such as Nextflow, Snakemake, and Cromwell provide robust frameworks for defining and executing complex pipelines across diverse computing environments.
The implementation of a dynamic resource allocation system helps prioritize operations based on dataset size, preventing over-provisioning and reducing computational costs [92]. In one large-scale implementation, Genomics England transitioned to Nextflow-based pipelines to process 300,000 whole-genome sequencing samples for the UK's Genomic Medicine Service, demonstrating the scalability of optimized workflow orchestration [92].
Stage 3: Execution Environment Optimization
The execution environment must be carefully configured to match pipeline requirements, particularly for cloud-based workflows where misconfigurations can lead to substantial unnecessary expenses [92]. Key considerations include:
- Storage Architecture: Implementing tiered storage solutions that balance access speed against cost
- Compute Resource Allocation: Right-sizing instances based on specific pipeline requirements
- Containerization: Using Docker or Singularity for environment consistency
- Data Transfer Optimization: Minimizing movement of large datasets between storage and compute resources
Cloud-based systems contribute to flexible NGS research by enabling remote data access, scalability, and efficient data management. They help address NGS data challenges by providing scalable storage solutions, high-performance computing resources, and efficient data-sharing capabilities [95].
Experimental Protocols and Methodologies
High-Fidelity Sequencing with Unique Molecular Identifiers
For detecting low-frequency variants in heterogeneous cancer samples, UMI-based approaches significantly enhance accuracy by mitigating sequencing errors.
Protocol: UMI-Based Error Correction [94]
- Library Preparation: Attach UMIs to each molecule during library preparation prior to amplification
- Sequencing: Cluster UMIs and sequence using standard NGS protocols
- Consensus Building: Group reads by their UMI tags and generate consensus sequences
- Variant Calling: Apply standard variant callers to the consensus reads
Validation: In a benchmarking study, this approach generated error-free reads for T-cell receptor repertoire and intra-host viral population datasets, providing a gold standard for evaluating computational error-correction methods [94].
Automated Library Preparation Protocol
Automation technologies can maximize the precision and efficiency of NGS workflows, particularly for high-throughput oncology applications [95] [96].
Protocol: Automated Library Preparation Using ExpressPlex Technology [96]
- Sample Processing: Normalize input DNA (extracted plasmid or PCR products)
- Automated Liquid Handling: Transfer samples to 384-well plates using systems such as SPT Labtech's firefly or Tecan Fluent
- Library Preparation: Execute the ExpressPlex workflow (90-minute protocol)
- Quality Control: Quantify libraries using fluorometric methods or qPCR
- Pooling and Sequencing: Normalize and pool libraries for sequencing
Performance Metrics: This automated approach enabled preparation of 1,536 libraries in 24 hours by a single user, with highly consistent results and balanced read counts without manual normalization [96].
Essential Research Reagent Solutions
Table 3: Key Research Reagents for Optimized NGS Workflows
| Reagent Category | Specific Examples | Function in Workflow | Optimization Benefit |
|---|---|---|---|
| Nucleic Acid Extraction & Repair | SureSeq FFPE DNA Repair Mix [97] | Removes cross-links and damage artifacts from FFPE samples | Enables use of challenging clinical specimens; reduces false positives |
| Library Preparation | ExpressPlex Library Prep Kit [96] | Streamlined library construction in 90 minutes | Reduces hands-on time; compatible with automation |
| Target Enrichment | SureSeq Targeted Cancer Panels [97] | Hybridization-based capture of cancer-related genes | Better uniformity than amplicon approaches; fewer false positives |
| Quality Control | AMPure XP Beads [97] | Size selection and purification | Maintains fragment size distribution; removes contaminants |
| Unique Identifiers | UMIs and UDIs [97] | Molecular barcoding for error correction | Differentiates true variants from artifacts; enables multiplexing |
Implementation Case Study: Genomics England
A exemplary implementation of bioinformatics pipeline optimization at scale is Genomics England's transition to Nextflow-based pipelines for processing 300,000 whole-genome sequencing samples [92]. This project demonstrates several key optimization principles:
Migration Strategy: The implementation utilized agile methodologies with bi-weekly sprints and continuous stakeholder feedback to ensure a smooth transition while maintaining high-quality outputs through rigorous testing frameworks.
Technical Architecture: The solution leveraged Nextflow and the Seqera Platform to replace an internal workflow engine, creating a more scalable and maintainable system capable of supporting the UK's Genomic Medicine Service.
Operational Framework: The project successfully balanced innovation with operational reliability within a conservative healthcare environment, prioritizing patient benefits while enabling future scalability.
Optimizing bioinformatics pipelines for NGS data analysis is no longer optional but essential for advancing cancer research and precision oncology. The systematic approach outlined in this guide—addressing analysis tools, workflow orchestration, and execution environments—provides a framework for managing the data volume and computational complexity inherent in modern oncology genomics.
As NGS technologies continue to evolve and their clinical applications expand, the principles of pipeline optimization will become increasingly critical for drug development professionals and translational researchers. By implementing these strategies, research organizations can significantly enhance their analytical capabilities, reduce operational costs, and accelerate the translation of genomic discoveries into improved cancer treatments.
The integration of artificial intelligence with optimized bioinformatics workflows represents the next frontier in NGS data analysis, promising further enhancements in variant interpretation, pattern recognition, and predictive modeling for oncology applications [93] [90]. As these technologies mature, the foundational optimization principles outlined here will ensure that computational infrastructure remains an enabler rather than a bottleneck in cancer research.
The integration of Next-Generation Sequencing (NGS) into cancer molecular profiling has fundamentally transformed oncology research and clinical practice. This technology enables comprehensive genomic characterization of tumors, facilitating personalized treatment strategies and targeted therapeutic interventions [10]. However, the complexity of NGS workflows—spanning wet-lab procedures, sophisticated instrumentation, and advanced bioinformatics—introduces multiple potential failure points that can compromise data reliability and patient safety [9] [98]. In this context, robust Quality Management Systems (QMS) have become indispensable for ensuring the analytical validity, reproducibility, and regulatory compliance of NGS-based oncogenomics research.
A QMS provides a structured framework of coordinated activities to direct and control organizations regarding quality, investigating the entire laboratory system rather than isolated components [99]. For cancer researchers utilizing NGS, implementing a rigorous QMS is not merely an administrative exercise but a fundamental scientific necessity to ensure that genomic findings driving therapeutic decisions are technically sound and clinically actionable. The Centers for Disease Control and Prevention (CDC) and the Association of Public Health Laboratories (APHL) have championed this cause through the Next-Generation Sequencing Quality Initiative (NGS QI), which develops tools and resources to help laboratories build effective quality systems [9].
QMS Frameworks and Standards for NGS in Oncology
Core Quality System Essentials (QSEs)
The Clinical and Laboratory Standards Institute (CLSI) outlines 12 Quality System Essentials (QSEs) that form the foundational architecture of a comprehensive QMS for NGS operations [99]. These QSEs address critical aspects ranging from document control and equipment management to personnel competency and process improvement. The CDC and APHL have crosswalked their NGS QI documents with regulatory, accreditation, and professional bodies including the FDA, Centers for Medicare and Medicaid Services, and College of American Pathologists to ensure current and compliant guidance on these QSEs [9].
For oncology applications, specific QSEs demand particular attention. Personnel management requires specialized training and competency assessment for bioinformaticians and laboratory technologists handling cancer specimens [9]. Equipment management ensures the proper qualification, calibration, and maintenance of NGS platforms generating data for treatment decisions. Process management establishes standardized procedures for critical steps from nucleic acid extraction through variant interpretation, while information management safeguards the integrity and confidentiality of sensitive genomic data [100].
Regulatory and Accreditation Landscape
Multiple regulatory frameworks and accreditation standards govern NGS applications in cancer research and diagnostics, with significant overlap and occasional divergence in requirements (Table 1). The College of American Pathologists (CAP) provides comprehensive QC metrics for clinical diagnostics with emphasis on pre-analytical, analytical, and post-analytical validation [100]. The Clinical Laboratory Improvement Amendments (CLIA) establish standards for sample quality, test validation, and proficiency testing in U.S. clinical laboratories [98]. Internationally, the European Medicines Agency (EMA) offers technical guidance on NGS validation for clinical trials and pharmaceutical development, while the In Vitro Diagnostic Regulation (IVDR) establishes a robust regulatory framework for diagnostic devices in the European Union [100].
Table 1: Key Organizations and Their Quality Focus Areas in NGS
| Organization | Primary Focus Areas | Relevance to Cancer Research |
|---|---|---|
| CAP (College of American Pathologists) | Comprehensive QC metrics for clinical diagnostics; pre-analytical, analytical, and post-analytical validation [100] | Accreditation for clinical cancer genomics laboratories |
| CLIA (Clinical Laboratory Improvement Amendments) | Standards for sample quality, test validation, proficiency testing [100] [98] | Regulatory compliance for patient testing |
| ACMG (American College of Medical Genetics and Genomics) | Technical standards for clinical NGS, variant classification, reporting [100] | Interpretation and reporting of hereditary cancer variants |
| FDA (Food and Drug Administration) | Analytical validation, bioinformatics pipelines, clinical application of NGS-based diagnostics [100] | Regulatory approval for NGS-based companion diagnostics |
| EMA (European Medicines Agency) | NGS validation for clinical trials and pharmaceutical development [100] | European translational cancer research |
| GA4GH (Global Alliance for Genomics and Health) | Data sharing, privacy, interoperability standards [100] | Multi-institutional cancer genomics collaborations |
Implementing QMS for NGS Workflows in Cancer Research
The Total Testing Process: A Phase-Based Approach
Implementing effective quality control across the entire NGS workflow requires a phase-based approach that addresses unique challenges at each step. The Association of Public Health Laboratories categorizes this process into pre-analytical, analytical, and post-analytical phases, each with distinct quality considerations [100].
The pre-analytical phase encompasses sample collection, nucleic acid extraction, and library preparation—steps particularly challenging with cancer specimens that may be derived from formalin-fixed paraffin-embedded (FFPE) tissue with associated DNA fragmentation and cross-linking [51]. Quality indicators at this stage include DNA/RNA integrity, quantification measurements, and library quality metrics. The analytical phase involves the actual sequencing process on platform-specific instruments, with quality parameters including cluster density, error rates, and percentage of bases above quality thresholds (e.g., Q30) [100]. The post-analytical phase covers bioinformatics analysis, variant interpretation, and reporting, requiring rigorous validation of pipelines and classification frameworks based on organizations such as the Association for Molecular Pathology, which categorizes variants into tiers of clinical significance for cancer [51].
Validation Requirements for Clinical Oncology Applications
For NGS tests used in cancer treatment decisions, validation represents a cornerstone of the QMS. The New York State Department of Health provides extensively referenced guidelines for somatic genetic variant detection that establish key performance indicators [98]. These include:
- Accuracy: Recommended minimum of 50 samples composed of different material types
- Precision: Recommended minimum of three positive samples for each variant type
- Analytical sensitivity and specificity: Determination of positive and negative percentage agreement compared to a gold standard
- Robustness: Assessment of likelihood of assay success under variable conditions [98]
In oncology applications, validation must address the specific variant types relevant to cancer genomics, including single nucleotide variants (SNVs), insertions/deletions (indels), copy number variations (CNVs), gene fusions, and complex structural variants. The SNUBH Pan-Cancer study demonstrated a practical approach to validation, implementing thresholds such as variant allele frequency (VAF) ≥ 2% for SNVs/INDELs and average CN ≥ 5 for copy number gains, while using established tools like Mutect2 and CNVkit for variant detection [51].
Quality Control Metrics and Monitoring
Essential QC Parameters Across Workflow Stages
Systematic quality monitoring requires tracking specific, quantifiable metrics throughout the NGS workflow. Different regulatory and professional organizations emphasize varying parameters, though several core metrics receive universal attention (Table 2). The NGS Quality Initiative provides tools for identifying and monitoring Key Performance Indicators (KPIs) that serve as early warning systems for workflow deterioration [9].
Table 2: Quality Control Parameters Emphasized by Various Organizations
| QC Parameter | CAP | CLIA | EuroGentest | NIST/GIAB | ACMG | AMP | RCPA | ACGS |
|---|---|---|---|---|---|---|---|---|
| Sample Quality | X | X | X | X | X | X | X | X |
| DNA/RNA Integrity | X | X | X | X | X | X | X | X |
| Library QC | X | X | X | X | X | X | X | |
| Depth of Coverage | X | X | X | X | X | X | X | X |
| Base Quality (e.g., Q30) | X | X | X | X | X | X | ||
| Reads Mapped | X | |||||||
| GC Bias | X | X |
Based on data from [100]
For cancer panel sequencing, the SNUBH study implemented comprehensive QC checks including DNA concentration quantification with Qubit dsDNA HS Assay, purity assessment with NanoDrop Spectrophotometer (A260/A280 ratio between 1.7-2.2), library size and quantity determination with Agilent 2100 Bioanalyzer system (250-400 bp target), and minimum coverage thresholds (at least 80% of bases at 100× coverage, with average mean depth of 677.8×) [51]. This multi-parameter approach ensures detection of low-frequency variants critical in cancer heterogeneity studies.
Quality Tools and Documentation
The NGS QI has developed numerous freely available tools to support quality implementation, with the most widely used documents including the QMS Assessment Tool, Identifying and Monitoring NGS Key Performance Indicators SOP, NGS Method Validation Plan, and the NGS Method Validation SOP [9]. These resources provide templates for standardizing quality documentation, which typically follows a three-tier hierarchy: policies (high-level principles), standard operating procedures (detailed step-by-step instructions), and records (documentation of activities performed) [98].
Technical Notes (TN) serve as preventive quality assurance methods, functioning as inspection records that accompany samples through the entire workflow. These documents ensure comprehensive quality management documentation and ultimately serve as quality certificates attesting to proper procedural adherence [98].
Advanced Technologies and Methodologies
Emerging Sequencing Platforms and Their Quality Implications
The NGS technology landscape continues to evolve rapidly, with significant implications for quality management. Oxford Nanopore Technologies has introduced duplex sequencing capable of Q30 (>99.9%) accuracy, enabling applications previously challenging for nanopore technology, such as low-frequency variant detection in cancer [6]. Pacific Biosciences offers HiFi reads that combine long-read sequencing (10-25 kilobases) with high-fidelity (Q30-Q40 accuracy) through circular consensus sequencing [6]. These technological advancements expand the scope of detectable cancer genomic alterations but necessitate revalidation of established workflows.
The convergence of sequencing modalities creates new opportunities and challenges for cancer researchers. Short-read companies are adding long-read or synthetic-long-read capabilities, while long-read companies have launched short-read platforms [6]. This technological diversification enables more comprehensive cancer genome characterization but requires quality systems that can accommodate multiple platforms and integrated data analysis approaches.
Experimental Protocols for Oncology Applications
Robust NGS protocols for cancer research incorporate quality checkpoints at each technical stage. The following methodology from the SNUBH Pan-Cancer study exemplifies a validated approach:
Nucleic Acid Extraction and QC:
- Manual microdissection of representative tumor areas with sufficient tumor cellularity
- DNA extraction using QIAamp DNA FFPE Tissue kit (Qiagen)
- DNA quantification with Qubit dsDNA HS Assay kit on Qubit 3.0 Fluorometer
- Purity assessment with NanoDrop Spectrophotometer (A260/A280 ratio 1.7-2.2)
- Minimum input: 20 ng DNA [51]
Library Preparation and Target Enrichment:
- Hybrid capture method using Agilent SureSelectXT Target Enrichment Kit
- Library size and quantity assessment with Agilent 2100 Bioanalyzer system and Agilent High Sensitivity DNA Kit
- Size target: 250-400 bp
- Concentration threshold: 2 nM/μL [51]
Sequencing and Data Analysis:
- Platform: NextSeq 550Dx (Illumina)
- Alignment reference: hg19
- Variant calling: Mutect2 for SNVs/indels, CNVkit for copy number variations, LUMPY for gene fusions
- Minimum thresholds: VAF ≥ 2% for SNVs/INDELs, read counts ≥ 3 for fusions
- MSI detection: mSINGs
- TMB calculation: Number of eligible variants within 1.44 Mb panel size [51]
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents and Their Applications in NGS for Cancer Genomics
| Reagent/Kit | Manufacturer | Primary Function | Quality Control Parameters |
|---|---|---|---|
| QIAamp DNA FFPE Tissue Kit | Qiagen | DNA extraction from challenging FFPE tissue specimens | DNA integrity, A260/A280 ratio (1.7-2.2), minimum 20 ng input [51] |
| Qubit dsDNA HS Assay | Invitrogen | Accurate DNA quantification | Concentration measurements, compatibility with fluorometer systems [51] |
| SureSelectXT Target Enrichment | Agilent | Hybrid capture-based library preparation | Library size (250-400 bp), concentration (≥2 nM/μL) [51] |
| Agilent High Sensitivity DNA Kit | Agilent | Library quality assessment | Fragment size distribution, adapter dimer detection [51] |
| Q20+ Kit14 | Oxford Nanopore | Duplex sequencing chemistry | Read accuracy (>99.9%), homopolymer resolution [6] |
| HiFi Chemistry | Pacific Biosciences | Circular consensus sequencing | Read length (10-25 kb), accuracy (Q30-Q40) [6] |
As NGS technologies continue to evolve and their applications in cancer research expand, quality management systems must adapt to new challenges and opportunities. Emerging areas requiring quality framework development include validation of machine learning algorithms for variant calling, standardization for agnostic pathogen detection in immuno-oncology, and quality assurance for liquid biopsy approaches that analyze circulating tumor DNA [9]. The rapid pace of technological advancement, with new platforms from companies such as Element Biosciences offering increasing accuracies at lower costs, necessitates continual reassessment of validation strategies and quality metrics [9].
The future of QMS for NGS in cancer research will likely involve more dynamic, data-driven approaches that leverage the sequencing data itself for real-time quality assessment. Meanwhile, harmonization efforts by international organizations such as the Global Alliance for Genomics and Health (GA4GH) aim to establish consistent standards that facilitate data sharing and collaboration while maintaining rigorous quality standards [100]. For cancer researchers, implementing and maintaining a comprehensive QMS is not merely a regulatory obligation but a fundamental scientific requirement to ensure that NGS-generated insights driving therapeutic development are reliable, reproducible, and ultimately beneficial to patients.
The widespread adoption of Next-Generation Sequencing (NGS) has revolutionized cancer molecular profiling, enabling comprehensive genomic characterization of tumors. However, a significant challenge persists in the clinical interpretation of genomic variants, particularly Variants of Unknown Significance (VUS). These variants represent alterations with undetermined biological or clinical impact, creating uncertainty in therapeutic decision-making. In clinical practice, VUS are distinct from both pathogenic (P)/likely pathogenic (LP) variants, which are associated with human disease and may be clinically actionable, and benign (B)/likely benign (LB) variants, which are not disease-contributing [101]. The accurate classification of VUS is therefore critical for realizing the full potential of precision oncology, as it ensures patients receive appropriate molecularly-guided treatments while avoiding unnecessary interventions based on incorrectly interpreted variants.
VUS Classification Frameworks and Standards
Established Classification Systems
Multiple professional organizations have developed standardized frameworks for variant classification to address inconsistencies in clinical interpretation. The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) established a five-tier classification system: Pathogenic (P), Likely Pathogenic (LP), Variant of Unknown Significance (VUS), Likely Benign (LB), and Benign (B), where "likely" corresponds to >90% confidence in the classification [101]. Subsequently, a joint consensus from AMP, American Society of Clinical Oncology (ASCO), and College of American Pathologists (CAP) proposed a four-tiered system focusing on clinical significance: Tier I (strong clinical significance), Tier II (potential clinical significance), Tier III (unknown significance), and Tier IV (benign/likely benign) [101]. More recently, the Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC) released guidelines specifically addressing the classification of somatic variant oncogenicity in cancer [101].
Clinical Actionability Threshold
In clinical practice, a practical actionability threshold exists between the LP and VUS classifications, where only P/LP variant classes should typically inform patient management decisions [101]. This creates a binary decision point for therapeutic intervention, underscoring the critical need for accurate VUS classification to determine whether a variant crosses this actionability threshold.
Table 1: Variant Classification Systems in Oncology
| ACMG/AMP System | AMP/ASCO/CAP System | Clinical Implications | Actionability Threshold |
|---|---|---|---|
| Pathogenic (P) | Tier I (Strong clinical significance) | Well-understood, clinically actionable | Therapeutic intervention appropriate |
| Likely Pathogenic (LP) | Tier I/II | Potential clinical significance | Therapeutic intervention appropriate |
| Variant of Unknown Significance (VUS) | Tier III (Unknown significance) | Uncertain association with disease | Avoid clinical decision-making |
| Likely Benign (LB) | Tier IV (Benign/Likely benign) | Not disease-contributing | No clinical action |
| Benign (B) | Tier IV (Benign/Likely benign) | Not disease-contributing | No clinical action |
Methodologies for VUS Actionability Assessment
Multidimensional Evidence Evaluation
Determining variant pathogenicity requires a holistic approach that evaluates multiple evidence domains. A comprehensive assessment should integrate data from literature reviews, genomic data repositories, computational predictive algorithms, and functional studies [101]. The weighting of evidence across these domains remains challenging, with different data types carrying varying levels of predictive value for clinical actionability. Key considerations include variant location within functional protein domains, proximity to known oncogenic variants, and biological context distinguishing tumor suppressors from oncogenes [101].
Rule-Based Actionability Classification
The MD Anderson Precision Oncology Decision Support (PODS) team developed a systematic approach for VUS actionability classification that categorizes VUS as either "Unknown" or "Potentially" actionable based on specific molecular characteristics [102]. This framework first determines whether the variant occurs in a therapeutically actionable gene, then assigns Functional Significance and Variant Actionability based on domain knowledge and proximity to known oncogenic variants.
Functional Validation Using Genomic Platforms
Functional genomics platforms provide empirical data on variant oncogenicity through standardized cell-based assays. The MD Anderson platform utilizes MCF10A (human mammary epithelial cells) and Ba/F3 (murine pro-B cells) cell lines to measure an alteration's impact on cell viability under growth factor-independent conditions [102]. These assays determine whether variants demonstrate gain-of-function activity indicative of oncogenic potential, providing functional evidence to reclassify VUS.
Table 2: Functional Genomics Platform Components
| Component | Description | Utility in VUS Assessment |
|---|---|---|
| MCF10A Cell Line | Human mammary epithelial cells | Measures oncogenic transformation in human cell context |
| Ba/F3 Cell Line | Murine pro-B cells (IL-3 dependent) | Detects growth factor-independent proliferation |
| Cell Viability Assays | Quantitative measures of proliferation | Determines functional impact of variants |
| Wild-type Controls | Reference comparison for each gene | Establishes baseline for functional assessment |
| Oncogenic Classification Threshold | Statistical significance in viability increase | Objective criteria for oncogenic designation |
Quantitative Evidence for Actionability Classification
Validation of Rule-Based Classification
The PODS actionability classification system demonstrates significant correlation with functional outcomes. In a study of 438 VUS, variants categorized as "Potentially actionable" were significantly more likely to be functionally oncogenic (37%) compared to those categorized as "Unknown" (13%) with an odds ratio of 3.94 (p = 4.08e-09) [102]. This represents nearly a three-fold enrichment in identifying functionally significant variants through the rule-based system. Application of the same classification scheme to an independent set of 777 variants showed even more pronounced enrichment, with 44% of "Potentially actionable" variants demonstrating oncogenic activity compared to only 8% of "Unknown" variants (odds ratio: 9.50, p = 4.719e-16) [102].
Table 3: Functional Validation of VUS Actionability Classification
| PODS Classification | Total Variants Tested | Oncogenic in Functional Assays | Oncogenic Percentage | Odds Ratio |
|---|---|---|---|---|
| Potentially Actionable | 204 | 76 | 37% | 3.94 |
| Unknown | 230 | 30 | 13% | Reference |
| Independent Set - Potentially Actionable | 659 | 290 | 44% | 9.50 |
| Independent Set - Unknown | 118 | 9 | 8% | Reference |
Real-World Clinical Impact of NGS Testing
Implementation of NGS testing in clinical practice demonstrates the substantial presence of VUS in patient populations. A study of 990 patients with advanced solid tumors found that 86.8% of patients carried tier II variants (potential clinical significance), while 26.0% harbored tier I variants (strong clinical significance) [51]. Among patients with tier I variants, 13.7% received NGS-based therapy, with varying response rates across cancer types. Patients with measurable lesions who received NGS-matched therapy showed promising outcomes, with 37.5% achieving partial response and 34.4% achieving stable disease [51]. The median treatment duration was 6.4 months (95% CI, 4.4-8.4), demonstrating the clinical utility of proper variant interpretation.
Research Reagent Solutions for VUS Investigation
Table 4: Essential Research Reagents for VUS Functional Characterization
| Reagent/Resource | Function | Application in VUS Research |
|---|---|---|
| SNUBH Pan-Cancer Panel | Targeted NGS platform (544 genes) | Comprehensive genomic profiling of tumor specimens [51] |
| QIAamp DNA FFPE Tissue Kit | DNA extraction from archival samples | Nucleic acid isolation from clinical specimens [51] |
| Agilent SureSelectXT Target Enrichment | Library preparation and target enrichment | Sequence capture for targeted NGS [51] |
| Illumina NextSeq 550Dx | NGS sequencing platform | High-throughput sequencing [51] |
| MCF10A Cell Line | Human mammary epithelial cells | Functional assessment of variant oncogenicity [102] |
| Ba/F3 Cell Line | Murine pro-B cells | Detection of growth factor-independent proliferation [102] |
| ngs.plot Software | Visualization of NGS enrichment patterns | Mining and visualization of NGS data [103] |
| dbSNP Database | Catalog of genetic variants | Population frequency data for pathogenicity assessment [101] |
| Genome Aggregation Database (gnomAD) | Population genome variant data | Filtering of common polymorphisms [101] |
Integrated Workflow for VUS Interpretation and Functional Validation
The interpretation of Variants of Unknown Significance represents a critical challenge in realizing the full potential of NGS-based cancer molecular profiling. A multidimensional approach integrating rule-based classification informed by functional domain knowledge, systematic functional validation using high-throughput platforms, and clinical correlation with patient outcomes provides a robust framework for VUS actionability assessment. The demonstrated enrichment of functionally oncogenic variants through systematic classification (37% in Potentially actionable vs. 13% in Unknown categories) enables more effective prioritization of variants for therapeutic targeting [102]. As NGS continues to transform cancer care, overcoming VUS interpretation challenges through integrated computational and functional approaches will be essential for advancing precision oncology and delivering molecularly-guided therapies to appropriate patient populations.
Next-generation sequencing (NGS) has revolutionized cancer molecular profiling, enabling the detection of low-frequency genetic variants critical for diagnosis, treatment selection, and disease monitoring. However, the accuracy of these analyses is fundamentally constrained by errors introduced throughout the NGS workflow. In cancer genomics, where identifying rare subclonal populations or minimal residual disease can dictate clinical decisions, distinguishing true biological variants from technical artifacts becomes paramount [104]. A systematic error-based approach is therefore essential for advancing the precision of cancer genomic applications, particularly as the field moves toward detecting variants at increasingly lower allele frequencies [105] [106].
This technical guide provides a comprehensive framework for identifying, quantifying, and mitigating analytical errors within NGS workflows focused on cancer genomics. We present detailed experimental protocols for error quantification, structured data on error rates and sources, visualization of complex workflows, and a curated toolkit of research reagents. By implementing this systematic approach, researchers and drug development professionals can enhance the reliability of their NGS data, ultimately supporting more accurate cancer molecular profiling.
Errors in NGS data originate from multiple steps in the analytical pipeline, each with distinct characteristics and implications for cancer variant detection. A typical NGS workflow involves sample collection, nucleic acid extraction, library preparation, target enrichment, sequencing, and bioinformatic analysis, with each stage contributing uniquely to the final error profile [104] [107].
Different steps of the NGS workflow contribute differently to the overall error burden. Sample handling and DNA damage can introduce specific substitution patterns, while library preparation and PCR amplification can both generate and amplify errors [104]. The sequencing process itself introduces platform-specific errors, with the overall error rate being a composite of these individual contributions.
Table 1: Quantitative Error Rates Across NGS Workflow Steps
| Workflow Step | Error Type | Reported Error Rate | Primary Impact on Cancer Variants |
|---|---|---|---|
| Sample Handling | C>A/G>T Transversions | ~10⁻⁵ [104] | False positive SNVs in oxidative damage-prone regions |
| Library Preparation | Polymerase Errors | Varies by enzyme (Taq vs. PWO) [107] | Increased background mutation rate |
| Target Enrichment PCR | All Substitutions | ~6-fold overall increase [104] | Reduced sensitivity for low-frequency variants |
| Sequencing-by-Synthesis | Substitutions (All Types) | 0.24% ± 0.06% per base [107] | Uniform background across all variants |
| Phasing Effects | Insertions/Deletions | Position-dependent (increases along read) [107] | False indels in homopolymer regions |
Context-Dependent Error Patterns
Beyond technical steps, error profiles exhibit significant sequence context dependencies. C>T/G>A substitutions show strong sequence context dependency, particularly in CpG islands, which is critical for cancer methylome studies [104]. Furthermore, persistent pre-phasing effects throughout sequencing runs can cause artificial insertions and deletions, especially problematic in homopolymer regions common in cancer genomes [107]. Different nucleotide substitutions occur at characteristic frequencies, with A>G/T>C changes occurring at approximately 10⁻⁴, while A>C/T>G, C>A/G>T, and C>G/G>C changes occur at lower frequencies of ~10⁻⁵ [104].
Computational Error Mitigation Strategies
Computational approaches can significantly suppress NGS errors, enhancing the detection of low-frequency variants crucial for cancer research.
Error Suppression and Correction Techniques
Through evaluation of read-specific error distributions, the substitution error rate can be computationally suppressed to 10⁻⁵ to 10⁻⁴, representing a 10- to 100-fold improvement over generally accepted rates of 10⁻³ [104]. This level of suppression enables detection of more than 70% of hotspot variants at 0.1-0.01% allele frequency, which is critical for identifying rare subclonal populations in tumors [104] [108].
Error-corrected sequencing (ECS) strategies employing unique molecular identifiers (UMIs) have demonstrated particularly robust error suppression. By tagging individual DNA molecules with UMIs before amplification, PCR and sequencing errors can be bioinformatically identified and removed, enabling detection limits of ≥0.001 for both single nucleotide variants and structural variants [105]. This approach is especially valuable for minimal residual disease monitoring in leukemia, where it can identify FLT3 internal tandem duplications and novel gene fusions at clinically relevant sensitivities [105].
Table 2: Error-Corrected Sequencing Performance in Cancer Applications
| Application | Method | Limit of Detection | Clinical Utility |
|---|---|---|---|
| SNV Detection | UMI-based ECS | ≥0.001 [105] | Subclonal mutation tracking |
| Structural Variants | AMP technology + ECS | ≥0.001 [105] | FLT3-ITD monitoring |
| Gene Fusions | RNA-ECS | Single mRNA molecule [105] | Novel fusion discovery |
| Liquid Biopsy | UMI + Targeted Sequencing | Part-per-million level [109] | Early resistance detection |
Artificial Intelligence-Enhanced Error Reduction
The integration of artificial intelligence (AI) and machine learning (ML) into NGS analysis represents a transformative approach for error mitigation. AI-driven tools such as DeepVariant apply deep neural networks to improve variant calling accuracy, surpassing traditional heuristic-based methods [110]. These models can learn complex patterns of technical artifacts and distinguish them from true biological signals, significantly reducing false positive rates in cancer mutation profiling [110].
AI methods are particularly valuable for addressing platform-specific error profiles. For third-generation sequencing technologies, AI models have been developed for more accurate basecalling and epigenetic modification detection, overcoming some of the inherent higher error rates of these platforms [110]. Furthermore, AI-powered laboratory automation systems can provide real-time quality control during library preparation, detecting procedural errors such as pipetting inaccuracies that might otherwise manifest as systematic biases in sequencing data [110].
Experimental Protocols for Error Quantification
Robust error profiling requires standardized experimental approaches. Below we detail two essential protocols for comprehensive error assessment in cancer NGS workflows.
Protocol 1: Substitution Error Rate Measurement
Purpose: To quantify position-specific substitution error rates in targeted NGS panels used for cancer mutation profiling.
Materials:
- High-quality genomic DNA from well-characterized cancer cell lines (e.g., COLO829/COLO829BL matched pair)
- Targeted amplicon or hybrid capture panel
- NGS library preparation kit
- NGS platform (Illumina HiSeq/NovaSeq recommended)
- Bioinformatics pipeline for variant calling
Method:
- Sample Preparation: Use matched cancer/normal cell lines or synthetic DNA standards with known mutation profiles. For dilution studies, spike-in cancer DNA into normal DNA at defined ratios (e.g., 1:1000, 1:5000) to create low-frequency variant standards [104].
- Sequencing: Sequence samples to high depth (>100,000X coverage) to ensure statistical power for low-frequency error detection [104].
- Bioinformatic Analysis:
- Align sequences to reference genome (hg19/GRCh38) using BWA-MEM or similar aligner [111]
- Identify positions in flanking sequences known to be devoid of genetic variations
- Calculate error rate for each genomic site i using the formula: [ \text{error rate}_i(g>m) = \frac{#\ \text{reads with nucleotide}\ m\ \text{at position}\ i}{\text{Total}\ #\ \text{reads at position}\ i} ] where g indicates the reference allele and m represents each of the three possible substitutions [104]
- Data Interpretation: Calculate error rates by substitution type and sequence context. Compare error profiles between different polymerases (e.g., Q5 vs. Kapa), library preparation methods, and sequencing platforms [104].
Protocol 2: False-Negative Error Assessment in Cancer Cell Lines
Purpose: To evaluate false-negative rates in highly multiplexed NGS panels by comparing targeted sequencing results with database mutations.
Materials:
- 35 cancer cell lines common to GDSC and CCLE databases
- Targeted NGS panel covering 151+ cancer genes
- Validated PCR primers for Sanger sequencing confirmation
- Access to GDSC and CCLE mutation databases
Method:
- Database Analysis: Retrieve mutation calls for the 35 cell lines from GDSC and CCLE databases [111].
- Targeted Sequencing: Perform deep targeted sequencing (>1000X median coverage) of the same cell lines using a validated cancer gene panel [111].
- Variant Calling: Use established bioinformatics pipelines (GATK Best Practices) including:
- FastQC for sequence quality assessment
- BWA for alignment to reference genome
- Picard for duplicate removal
- GATK for base quality recalibration and variant calling [111]
- Mutation Validation: Select discordant mutations for validation by Sanger sequencing across the 35 cell lines [111].
- Error Rate Calculation:
- Define possible false-negative (P-FN) rates under the assumption that mutation calls with mutant allele frequency ≥10% in targeted sequencing represent true positives
- Calculate P-FN rate as: [ \text{P-FN rate} = \frac{\text{Number of hmAF calls} - \text{Database-specific calls}}{\text{Number of hmAF calls}} ] where hmAF represents high mutant allele frequency (≥10%) [111]
The Scientist's Toolkit: Essential Reagents and Methods
Table 3: Research Reagent Solutions for NGS Error Mitigation
| Reagent/Method | Function | Application in Error Reduction |
|---|---|---|
| Unique Molecular Identifiers (UMIs) | Tags individual DNA molecules before amplification | Enables bioinformatic error correction by consensus building [105] |
| Q5 High-Fidelity DNA Polymerase | PCR amplification during library prep | Reduces polymerase-induced errors compared to standard Taq [104] |
| Matched Cancer/Normal Cell Lines (COLO829/COLO829BL) | Positive controls for error rate determination | Provides ground truth for somatic mutation detection benchmarks [104] |
| ArcherDX VariantPlex/FusionPlex | Targeted enrichment using AMP technology | Maintains molecular integrity while capturing targeted regions [105] |
| High-Fidelity Library Prep Kits (e.g., Illumina DNA Prep) | Fragment end-repair, A-tailing, adapter ligation | Minimizes artifacts during library construction [109] |
| CleanNGS MagBeads | Solid-phase reversible immobilization purification | Reduces adapter dimer formation and improves library quality [107] |
Visualizing NGS Error Analysis Workflows
Comprehensive NGS Error Assessment Workflow
Error-Corrected Sequencing for Low-Frequency Variant Detection
The field of NGS error mitigation is rapidly evolving, with several promising trends emerging. The integration of artificial intelligence and machine learning into NGS analysis is revolutionizing error detection and correction, with AI models now capable of identifying subtle error patterns that escape traditional statistical methods [109] [110]. The ongoing development of third-generation sequencing technologies with increasingly accurate long-read capabilities promises to address errors associated with short-read sequencing, particularly in complex genomic regions relevant to cancer [109]. Multiomic approaches that integrate genomic, epigenomic, and transcriptomic data from the same sample are creating new opportunities for cross-validation and error reduction through concordance analysis [109] [110].
For cancer molecular profiling research, these advancements translate to increasingly precise detection of low-frequency variants, enabling earlier cancer detection, more accurate monitoring of treatment response, and improved identification of resistant subclones. By implementing the systematic error-based approach outlined in this guide—incorporating rigorous experimental design, comprehensive error profiling, computational error suppression, and appropriate reagent selection—researchers can significantly enhance the reliability of their NGS data, ultimately supporting more confident biological conclusions and clinical applications in cancer genomics.
Ensuring Clinical Grade Results: Validation Frameworks and Comparative Effectiveness of NGS
The integration of Next-Generation Sequencing (NGS) into cancer molecular profiling represents a cornerstone of precision oncology, enabling the identification of targetable genomic alterations that inform therapeutic decisions [36] [73]. Analytical validation provides the critical foundation that ensures NGS test results are reliable, accurate, and reproducible for clinical research and diagnostic purposes. This process formally establishes the performance characteristics of an assay—specifically its sensitivity, specificity, and reproducibility—under defined operating conditions [36] [112]. In the context of a broader thesis on NGS in cancer research, rigorous analytical validation is the indispensable link that transforms innovative sequencing technologies into trusted tools for drug development and clinical application. Without it, the potential of precision oncology to match patients with effective treatments based on their tumor's genomic profile cannot be fully realized. These guidelines outline the core principles and methodologies for establishing the analytical validity of NGS assays within oncological research settings.
Core Performance Metrics for NGS Assays
Analytical validation of an NGS assay requires the precise quantification of key performance metrics through controlled experiments. These metrics define the operational boundaries and reliability of the test [36] [113].
- Sensitivity and Specificity: Also referred to as Positive Percent Agreement (PPA) and Negative Percent Agreement (NPA) in validation studies, these metrics measure the assay's ability to correctly identify true-positive and true-negative variants, respectively, when compared to a validated orthogonal method or reference standard [113] [114]. For example, a validation study for an RNA-based fusion detection assay demonstrated a PPA of 98.28% and an NPA of 99.89% [114].
- Reproducibility and Repeatability: Reproducibility (inter-run precision) assesses the consistency of results across different operators, instruments, and days. Repeatability (intra-run precision) measures consistency when the same sample is tested multiple times in the same run. A well-validated assay will show high concordance (e.g., 100% for pre-defined fusions) across all replicates [113] [114].
- Limit of Detection (LoD): The LoD is the lowest variant allele frequency or input quantity at which the assay can consistently detect a genomic variant with high confidence. This is crucial for detecting somatic variants present in a low proportion of cells due to tumor heterogeneity or low tumor purity [36]. LoD studies often involve titrating input nucleic acid or diluting positive control samples to determine the minimal requirements for reliable detection [113] [114].
Table 1: Key Analytical Performance Metrics and Their Definitions
| Metric | Technical Term | Definition | Example from Literature |
|---|---|---|---|
| Sensitivity | Positive Percent Agreement (PPA) | The proportion of true positives correctly identified by the assay. | 98.28% for fusion detection in an RNA-seq assay [114]. |
| Specificity | Negative Percent Agreement (NPA) | The proportion of true negatives correctly identified by the assay. | 99.89% for fusion detection in an RNA-seq assay [114]. |
| Precision | Reproducibility & Repeatability | The consistency of results under varied (reproducibility) or identical (repeatability) conditions. | 100% reproducibility for 10 target fusions across 9 replicates [114]. |
| Limit of Detection (LoD) | Analytical Sensitivity | The lowest variant allele frequency or input amount reliably detected. | LoD determined with 1.5-30 ng RNA input and 21-85 supporting reads for fusions [114]. |
Experimental Design and Methodologies for Validation
A robust validation requires careful planning and execution of experiments to characterize the assay's performance across its intended use.
Sample Selection and Characterization
The foundation of a strong validation is a well-characterized sample set.
- Sample Types: Use samples that reflect the real-world specimens the assay will encounter, such as Formalin-Fixed Paraffin-Embedded (FFPE) tissue blocks, which are common in oncology [113] [114].
- Tumor Content: For solid tumors, pathologist review of hematoxylin and eosin (H&E)-stained slides is mandatory to estimate tumor cell fraction and mark areas for macrodissection to enrich tumor content. This is critical for accurate interpretation of mutant allele frequencies and copy number alterations [36].
- Reference Materials: Well-characterized reference cell lines, commercially available reference standards, and residual clinical samples with prior orthogonal testing data are essential for accuracy studies. These materials should cover the variant types the assay is designed to detect (e.g., SNVs, indels, CNAs, fusions) [36] [113] [114].
Establishing Accuracy and Concordance
Accuracy is demonstrated by comparing the NGS assay results to a validated reference method.
- Orthogonal Testing: Compare results from the NGS assay against those from established, non-NGS methods like fluorescence in situ hybridization (FISH), Sanger sequencing, or digital PCR for specific variants [113] [114].
- Data Analysis: Calculate PPA and NPA for each variant type (SNV, indel, fusion, etc.) separately, as assay performance can vary significantly across alteration types [36].
Table 2: Example Experimental Plan for Analytical Validation
| Validation Component | Recommended Sample Number & Type | Methodology | Output Metrics |
|---|---|---|---|
| Accuracy/Concordance | 100+ clinical samples with prior orthogonal data [113] [114]. | Compare NGS results to outcomes from FISH, Sanger, or other NGS assays. | PPA, NPA for each variant type. |
| Precision (Reproducibility) | 10+ samples, tested in multiple replicates (e.g., 3x3 design) [114]. | Run samples across different days, by different operators, and on different instruments. | Percent concordance across all replicates. |
| Limit of Detection (LoD) | Dilution series from 5+ fusion- or mutation-positive cell lines [114]. | Titrate input nucleic acid (e.g., 1-50 ng) or dilute positive samples to low tumor purity. | Minimum input and supporting reads; lowest detectable VAF. |
| Analytical Specificity | Samples with high homology regions or known cross-reactive sequences. | Assess performance in challenging genomic contexts. | False positive and false negative rates. |
Workflow for Targeted NGS Assay Validation
The following diagram illustrates the key stages in the development and analytical validation of a targeted NGS assay for oncology applications.
Bioinformatics and Computational Validation
The bioinformatics pipeline is a critical component of the NGS assay and requires its own rigorous validation to ensure variant calls are accurate and reliable [36] [115].
Variant Calling and Filtering Strategies
Somatic variant calling from tumor samples is complex and benefits from a multi-faceted approach.
- Variant Caller Diversity: Different algorithms (e.g., MuTect2, Strelka2, VarScan2) use distinct methodologies (haplotype-based, allele frequency-based, etc.), and none are perfect for all variant types [115]. Combining multiple callers can improve sensitivity and specificity [116].
- Machine Learning for Validation: Machine learning approaches, including Convolutional Neural Networks (CNNs), can be trained to mimic expert manual review of variant calls, helping to distinguish true somatic variants from sequencing artefacts. This increases throughput and standardization [115].
- Automated Pipelines: Integrated software solutions like AMLVaran provide flexible, reproducible pipelines that combine multiple variant callers, annotate variants, and generate clinical reports, which is vital for a standardized clinical research setting [116].
Data Analysis Workflow
The process from raw sequencing data to a finalized variant list involves multiple, validated steps, as shown in the computational workflow below.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following reagents and materials are fundamental for developing and validating a targeted NGS assay in oncology.
Table 3: Essential Research Reagents for NGS Assay Validation
| Category | Item | Specific Function in Validation |
|---|---|---|
| Reference Standards | Characterized Cell Lines (e.g., fusion-positive) [114] | Provide a source of known, reproducible variants for determining LoD, accuracy, and precision. |
| Clinical Samples | FFPE Tissue Blocks with Orthogonal Data [113] [114] | Serve as real-world benchmarks for establishing concordance and clinical relevance. |
| NGS Library Prep | Hybrid-Capture or Amplicon-Based Kits [36] [117] | Enable targeted enrichment of genomic regions of interest prior to sequencing. |
| Automation Tools | Integrated Analysis Software (e.g., AMLVaran) [116] | Standardize the variant calling and filtering process, ensuring reproducibility and traceability. |
| Computational Tools | Machine Learning Models (e.g., deepCNNvalid) [115] | Automate the refinement of variant calls, reducing the need for manual review and increasing throughput. |
The establishment of rigorous analytical validation guidelines is paramount for the reliable application of NGS in cancer molecular profiling research. By systematically defining and verifying performance metrics such as sensitivity, specificity, and reproducibility, researchers and drug developers can ensure that the genomic data generated is of the highest quality and fit-for-purpose. This, in turn, builds a foundation of trust in the data that accelerates drug discovery, supports regulatory submissions for companion diagnostics [112] [118], and ultimately advances the field of precision oncology. As NGS technologies and our understanding of cancer genomics continue to evolve, so too must these validation frameworks, adapting to new challenges and opportunities to better serve patient care.
Next-generation sequencing (NGS) has fundamentally transformed oncology research and drug development by enabling comprehensive genomic characterization of tumors. The successful translation of NGS-based discoveries into clinically meaningful insights, however, depends entirely on the accuracy, reproducibility, and reliability of the generated data. For researchers and drug development professionals, reference materials and proficiency testing provide the critical benchmarks that validate assay performance, ensure data integrity, and support regulatory submissions. These quality control tools have become indispensable in a research landscape increasingly focused on precision oncology, where identifying low-frequency variants, complex biomarkers, and novel genomic signatures directly impacts therapeutic development and patient stratification strategies.
Within cancer molecular profiling research, reference materials allow laboratories to establish analytical validation parameters including accuracy, precision, sensitivity, limit of detection, and specificity across various genomic alterations. Meanwhile, proficiency testing provides external verification that assays perform consistently within and across research institutions. This technical guide examines the current frameworks, materials, and methodologies that underpin robust NGS assay validation in oncology research, with particular emphasis on their application throughout the drug development pipeline.
Reference Materials: Foundations for Assay Validation
Definitions and Classifications
Reference materials are standardized substances with one or more sufficiently homogeneous and well-established properties for use in assay calibration, validation, or quality control. In NGS-based cancer research, they serve as ground-truth benchmarks containing known genomic variants at defined allele frequencies, enabling researchers to quantify assay performance metrics objectively. These materials are categorized based their composition, source, and intended application, each offering distinct advantages for different stages of research and development.
Table 1: Classification of Reference Materials for NGS Oncology Assays
| Classification Basis | Material Type | Key Characteristics | Primary Research Applications |
|---|---|---|---|
| Source/Composition | Cell Line-derived | Clonal origin, renewable supply, well-characterized | Assay development, analytical validation, reproducibility studies |
| Synthetic Oligos | Precisely engineered sequences, high multiplexing capability | Verification of specific variant types, panel optimization | |
| Patient-derived | Authentic genetic background, natural fragmentation | Clinical trial assay validation, biomarker discovery | |
| Format | Purified Nucleic Acids | Ready-to-sequence, minimal processing | Bioinformatics pipeline validation, cross-platform comparisons |
| Formalin-Fixed Paraffin-Embedded (FFPE) | Mimics common specimen type, includes artifacts | Pre-analytical variable assessment, extraction protocol optimization | |
| Circulating Tumor DNA (ctDNA) in plasma | Natural fragmentation, low allele frequencies | Liquid biopsy assay development, minimal residual disease detection | |
| Traceability | Certified Reference Materials (CRMs) | Metrological traceability, value-assigned | Regulatory submissions, method harmonization across sites |
| Research Use Only (RUO) | Flexible specifications, rapidly available | Early-stage assay development, exploratory studies |
Key Technical Specifications and Applications
Effective utilization of reference materials requires careful alignment of their technical specifications with research objectives. Variant representation should encompass the full spectrum of genomic alterations relevant to cancer research, including single nucleotide variants (SNVs), insertions and deletions (indels), copy number variations (CNVs), gene fusions, and complex biomarkers such as microsatellite instability (MSI) and tumor mutational burden (TMB) [119]. The allele frequency becomes particularly critical for liquid biopsy applications, where detecting variants at frequencies below 0.5% is often necessary for monitoring treatment response and emerging resistance mechanisms [120].
Matrix composition must closely mimic real patient samples to properly evaluate pre-analytical and analytical variables. For solid tumor profiling, FFPE-formatted references containing authentic DNA damage patterns are essential, while liquid biopsy applications require reference materials with ctDNA fragmented to ~167 base pairs in a background of wild-type cell-free DNA [119]. The availability of multiplexed reference materials containing dozens to hundreds of variants across multiple genes enables comprehensive validation of large NGS panels while maximizing resource efficiency [119].
For drug development pipelines, reference materials with regulatory compliance support facilitate smoother transitions from research to clinical trials and ultimately to companion diagnostic development. Materials manufactured under quality standards such as ISO 13485 and with regulatory acceptance (e.g., by the New York State Department of Health or FDA) provide greater confidence for submissions [119].
Table 2: Technical Specifications for NGS Reference Materials in Cancer Research
| Parameter | Specification Range | Impact on Research Applications |
|---|---|---|
| Variant Types | SNVs, Indels, CNVs, Fusions, MSI, TMB | Determines breadth of assay validation possible with a single material |
| Allele Frequency | 0.1% - 50% for ctDNA; 5% - 100% for tissue | Enables validation of limit of detection and quantitative accuracy |
| Material Format | Purified DNA/RNA, FFPE, ctDNA in plasma | Affects applicability for different sample types and pre-analytical steps |
| Variant Verification Method | dPCR, orthogonal NGS, Sanger sequencing | Impacts confidence in ground-truth values and measurement uncertainty |
| Gene Coverage | Panels (dozens of genes) to exome/genome | Should align with or exceed the scope of the research assay |
| Manufacturing Standards | ISO 13485, cGMP, regulatory acceptance | Critical for regulated research and diagnostic development |
Proficiency Testing: Ensuring Ongoing Assay Quality
Frameworks and Program Structures
Proficiency testing (PT) provides external quality assessment through which laboratories analyze distributed samples and report results to an organizing body for evaluation against pre-established criteria. In NGS-based cancer research, PT programs have evolved to address the complexity of genomic testing, assessing not only variant detection but also interpretation and reporting capabilities. The College of American Pathologists (CAP) with representation from the Association for Molecular Pathologists (AMP) has developed structured worksheets that guide the entire life cycle of an NGS test, with a focus on establishing quality management systems for ongoing assay monitoring [121].
These frameworks address the pre-analytical, analytical, and post-analytical phases of NGS testing, recognizing that errors can occur at multiple points in the workflow. For the pre-analytical phase, monitoring includes specimen acceptability, nucleic acid extraction efficiency, and library preparation quality. Analytical phase monitors encompass sequencing metrics (e.g., coverage uniformity, on-target rates, quality scores), while post-analytical monitors include variant interpretation concordance and report accuracy [121].
Implementation in Research Settings
For research laboratories, participation in formal PT programs provides critical data on assay robustness and inter-laboratory concordance, especially important for multi-center clinical trials and collaborative studies. The CAP/CLSI guidelines recommend that PT challenges should mirror the complexity of actual research samples, including varieties of variant types, allele frequencies, and sample matrices that reflect real-world scenarios [121].
Successful PT implementation requires establishing performance criteria prior to testing, with acceptance thresholds based on the intended research application. For example, drug development programs focusing on liquid biopsy may require higher sensitivity thresholds (e.g., ≥95% for variants at 0.5% allele frequency) compared to discovery-phase research [120]. Documentation of PT results, including any deviations from expected performance and subsequent corrective actions, provides evidence of assay reliability for publications and regulatory submissions.
Experimental Protocols for NGS Assay Validation
Analytical Validation Study Design
Robust validation of NGS assays for cancer research requires a structured approach that evaluates performance across all relevant variant types and sample conditions. The following protocol outlines a comprehensive validation framework suitable for targeted NGS panels in oncology applications:
1. Reference Material Selection and Preparation
- Select reference materials that encompass SNVs, indels, CNVs, and gene fusions relevant to the cancer type(s) under investigation [119]
- Include materials at multiple allele frequencies (e.g., 1%, 5%, 10%, 25%, 50%) to establish limit of detection and quantitative accuracy
- Incorporate both high-quality DNA and FFPE-formatted materials to assess pre-analytical variables
- For liquid biopsy assays, include ctDNA reference materials with variant frequencies as low as 0.1%-0.5% [120]
2. Experimental Replication Design
- Perform minimum of three replicates per reference material across multiple separate runs
- Include different operators, reagent lots, and sequencing instruments when applicable
- Incorporate negative controls (e.g., human genomic DNA without known variants) to assess specificity
- Consider using commercially available multiplexed reference materials such as Seraseq NGS Reference Materials to maximize information per sequencing run [119]
3. Sequencing and Data Generation
- Process reference materials alongside routine research samples to mimic real-world conditions
- Follow established library preparation protocols with careful quality control at each step
- Sequence to a minimum depth that supports the intended sensitivity (e.g., ≥500x for tissue; ≥10,000x for liquid biopsy)
- Include positive control materials with known performance characteristics in each run
Performance Metric Calculation and Acceptance Criteria
Following data generation, analytical performance should be quantified using standardized calculations for each critical performance parameter:
1. Sensitivity and Specificity
- Sensitivity = TP/(TP+FN) × 100, where TP=true positives, FN=false negatives
- Specificity = TN/(TN+FP) × 100, where TN=true negatives, FP=false positives
- Calculate separately for each variant type and across different allele frequency ranges
- Establish acceptance criteria based on research application (e.g., ≥95% sensitivity for variants at 5% allele frequency in tissue; ≥98% specificity across all variant types) [120]
2. Precision and Reproducibility
- Intra-run precision: Consistent variant detection across replicates within the same sequencing run
- Inter-run precision: Consistent detection across different sequencing runs
- Inter-operator/instrument precision: Consistent performance across different operators or instruments
- Calculate coefficient of variation for quantitative measurements (e.g., variant allele frequency)
3. Limit of Detection (LoD) Determination
- Test reference materials with progressively lower allele frequencies
- LoD = lowest allele frequency at which ≥95% of expected variants are detected
- Requires sufficient replication at each allele frequency level (minimum n=5 recommended)
4. Accuracy and Concordance
- Compare measured allele frequencies to reference values using linear regression (R²)
- For orthogonal validation, compare NGS results to digital PCR or other established methods
- In recent studies, well-validated NGS assays have demonstrated high concordance (R² = 0.9786) with orthogonal methods [122]
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of NGS cancer profiling assays requires access to specialized reagents and materials that ensure reliable, reproducible results. The following table details essential components of the quality control toolkit for researchers in this field.
Table 3: Essential Research Reagent Solutions for NGS Cancer Profiling
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Reference Materials | Seraseq NGS Reference Materials, multiplexed ctDNA controls | Analytical validation, quality control, assay monitoring |
| Extraction Controls | Exogenous spike-in DNA (e.g., phage DNA), internal reference standards | Monitoring extraction efficiency, normalization |
| Library Prep Controls | Unique molecular identifiers (UMIs), adapter-specific spike-ins | Quantifying library complexity, detecting cross-contamination |
| Sequencing Controls | PhiX control library, platform-specific control reagents | Monitoring sequencing quality, base calling accuracy |
| Bioinformatics Standards | Genome in a Bottle (GIAB) reference data, synthetic FASTQ files | Pipeline validation, algorithm performance assessment |
| Proficiency Test Materials | CAP PT programs, commercial PT schemes (e.g., Seraseq PT) | External quality assessment, inter-laboratory comparison |
Visualization of NGS Quality Management Workflow
The following diagram illustrates the integrated relationship between reference materials and proficiency testing within the complete NGS quality management workflow for cancer profiling:
NGS Quality Management Workflow
Implementation Challenges and Future Directions
Despite the clear importance of reference materials and proficiency testing, several challenges persist in their implementation within cancer research settings. Cost and accessibility of commercially available reference materials can be prohibitive for academic research laboratories, potentially leading to inadequate validation. Additionally, the rapidly expanding landscape of cancer biomarkers—including complex signatures like TMB, MSI, and genomic rearrangements—outpaces the development of corresponding reference materials. There is also a significant need for reference materials representing rare cancer types and underrepresented ancestral backgrounds to ensure equitable advances in precision oncology.
Future developments in this field will likely focus on multiplexed, multi-omics reference materials that simultaneously enable validation of genomic, transcriptomic, and epigenomic assays. Third-generation sequencing technologies are also driving demand for long-read reference materials with characterized structural variants and epigenetic modifications. For drug development professionals, the integration of artificial intelligence tools with standardized reference datasets will enable more sophisticated assay optimization and quality control approaches. Finally, international harmonization of reference materials and proficiency testing standards will facilitate global collaboration and accelerate the translation of cancer genomics research into innovative therapeutics.
The continued evolution of reference materials and proficiency testing frameworks remains essential for realizing the full potential of NGS in cancer research. By establishing rigorous benchmarks for assay performance, these quality assurance tools empower researchers to generate reliable, reproducible genomic data that advances our understanding of cancer biology and accelerates the development of targeted therapies.
The paradigm of cancer treatment has fundamentally shifted from a histology-based approach to one driven by a deep understanding of the molecular alterations within a tumor. This transition to precision oncology necessitates comprehensive genomic profiling to identify targetable mutations and guide therapeutic decisions [90]. For years, standard-of-care (SoC) testing methods, including single-gene tests, fluorescence in situ hybridization (FISH), and immunohistochemistry (IHC), have formed the diagnostic backbone. However, the rapid discovery of clinically actionable biomarkers has exposed the limitations of these sequential testing approaches [123] [124].
Next-generation sequencing (NGS) has emerged as a transformative technology capable of interrogating hundreds of cancer-related genes simultaneously from a single tissue sample [90]. This in-depth technical guide examines the comparative effectiveness of NGS versus SoC testing paradigms within the broader thesis on the role of NGS in cancer molecular profiling research. We synthesize current evidence from economic models, clinical validation studies, and emerging applications to provide researchers, scientists, and drug development professionals with a definitive resource on optimized genomic testing strategies in oncology.
Methodological Approaches: NGS Versus SOC Testing
Fundamental Principles and Technical Comparison
Next-generation sequencing represents a fundamental departure from traditional Sanger sequencing and SoC molecular techniques. Its core principle lies in massively parallel sequencing, which enables the concurrent analysis of millions of DNA fragments, in contrast to the serial processing of single fragments in Sanger sequencing [90]. This architectural difference confers significant advantages in throughput, sensitivity, and discovery power.
Table 1: Technical Comparison of Genomic Testing Methodologies
| Aspect | Sanger Sequencing | Single-Gene Tests (IHC, FISH, PCR) | Next-Generation Sequencing |
|---|---|---|---|
| Throughput | Single DNA fragment | 1-3 biomarkers per test | Millions of fragments simultaneously; hundreds of genes |
| Sensitivity (Detection Limit) | Low (~15-20%) | Variable; ~89-94% for FISH/PCR [124] | High (down to ~1-3% VAF) [35] |
| Primary Applications | Validation of NGS results, single-gene analysis | Targeted detection of specific, known alterations | Comprehensive genomic profiling, novel variant discovery |
| Turnaround Time | Weeks for multiple genes | Weeks for a full biomarker panel (sequential) | ~4-7 days for a comprehensive panel [35] |
| Variant Detection Capability | Single-nucleotide variants (SNVs) | Limited to designed target (e.g., fusion, protein expression) | SNVs, indels, CNVs, SVs, fusions, TMB, MSI |
| Tissue Consumption | Low per test, but high for full panel | High due to multiple sequential tests | Low (single test for all biomarkers) |
| Cost-Effectiveness | Costly for large numbers of targets | High aggregate cost for full biomarker panel | Superior for comprehensive profiling [123] [124] |
Key Experimental Protocols in Comparative Studies
Research comparing NGS to SoC paradigms relies on rigorous experimental designs. The following protocols are representative of key studies in this field.
Protocol for Integrated Mutation and Rearrangement Detection in NSCLC
A 2019 study directly compared an integrated NGS platform with IHC for detecting EGFR, ALK, and ROS1 alterations in 107 NSCLC samples [125].
- Sample Preparation: DNA and RNA were co-extracted from Formalin-Fixed, Paraffin-Embedded (FFPE) tumor tissues using commercial kits (QIAamp DNA FFPE Tissue Kit, RNeasy FFPE Kit). DNA quality was assessed via fluorometry (Qubit), with a requirement of >20 ng total mass and fragment sizes >500 bp.
- SoC Testing (Comparator): IHC was performed on 4-µm tissue sections using mutation-specific antibodies for EGFR (L858R, E746-A750del) and fusion-specific antibodies for ALK (D5F3) and ROS1 (D4D6). Positive interpretation was defined as moderate-to-strong staining in >10% of tumor cells.
- NGS Testing: DNA libraries were prepared using a targeted kit (SGI OncoAim) designed to capture all exons of ten genes (e.g., EGFR, ALK, KRAS, TP53) and fusion intronic regions for ALK, ROS1, and RET. Sequencing was performed on an Illumina NextSeq 500 with 150 bp paired-end reads.
- Bioinformatic Analysis: Reads were aligned to GRCh37 (hg19). Variant calling and genotyping were performed with a minimum confidence threshold of 5%. Fusion calls required a minimum of 5 supporting reads.
- Statistical Analysis: Concordance between IHC and NGS was calculated using Cohen's κ coefficient.
Protocol for Evaluating Diagnostic Yield in Pediatric ALL
A 2025 study benchmarked emerging genomic approaches, including targeted NGS (t-NGS), against SoC for diagnosing pediatric Acute Lymphoblastic Leukemia (pALL) in 60 patients [126].
- SoC Baseline: Immunophenotyping by flow cytometry, chromosome banding analysis (G-banding), and FISH with commercial probes for recurrent fusions (e.g., BCR::ABL1, KMT2A).
- Emerging Methods:
- t-NGS: The ALLseq panel was used with 10 ng of gDNA and RNA on an Ion Chef system. Sequencing was performed on an Ion S5 sequencer, with variants called at >3% allelic frequency.
- RNA Sequencing (RNA-seq): Total RNA was extracted, and libraries were prepared for sequencing on Illumina platforms to detect fusion genes and expression outliers.
- Optical Genome Mapping (OGM): Ultra-high molecular weight DNA was labeled and run on a Saphyr system (Bionano Genomics) to detect structural variants at high resolution.
- Analysis: The diagnostic yield of each method, both individually and in combination, was assessed for the detection of structurally variant drivers and copy number alterations.
Critical Analysis of Comparative Data
Economic and Operational Effectiveness
Economic models and real-world studies consistently demonstrate the superiority of NGS from a cost-effectiveness and workflow efficiency perspective.
Table 2: Economic and Operational Outcomes of NGS vs. SoC Testing
| Metric | Single-Gene Testing (SoC) | Next-Generation Sequencing (NGS) | Context / Study |
|---|---|---|---|
| Cost per Correctly Identified Patient (CCIP) | €1,983 (non-squamous NSCLC) | €658 (non-squamous NSCLC) | Sequential SGT vs. NGS [124] |
| Health Plan Savings | - | $1.4M - $2.1M (Medicare); $127k - $250k (Commercial) | Per 1 million members [123] |
| Turnaround Time | Several weeks for full biomarker profile | ~4 days for a 61-gene panel [35] | In-house NGS vs. outsourced testing |
| Tissue Utilization | High (multiple slides for sequential tests) | Low (single test conserves tissue) | Clinical practice observation [123] [125] |
| Actionable Mutation Detection Rate | Lower (limited scope) | Higher (comprehensive scope) | Identifies more patients for clinical trials [123] |
A pivotal economic model presented at ASCO 2018 revealed that using NGS for metastatic NSCLC testing saved between $1.4 million and $2.1 million for Medicare health plans and between $127,402 and $250,842 for commercial health plans per million members compared to multiple other testing strategies [123]. The model also highlighted a faster turnaround time, enabling patients to start appropriate therapy 2.8 weeks earlier than with some SoC approaches.
Further validating this, a 2023 study introduced the metric "cost per correctly identified patient" (CCIP). For non-squamous NSCLC, the CCIP was €1,983 for sequential SGT versus €658 for NGS, underscoring a dramatic threefold cost reduction with NGS. This trend held across other cancer types, including colorectal, breast, and gastric cancers [124].
Diagnostic and Clinical Performance
Beyond cost, the comprehensive nature of NGS directly impacts diagnostic accuracy and therapeutic decision-making.
- Superior Detection of Actionable Alterations: In NSCLC, NGS proved more reliable than IHC for certain alterations. It provided more precise information on EGFR mutations, particularly in exon 19, and increased the positive rate of ALK rearrangements while decreasing false positives for ROS1 rearrangements observed with IHC [125]. The "one-test-fits-all" approach of NGS identifies a higher percentage of patients with targetable genomic alterations [123].
- Resolution of Complex and Rare Cancers: In pediatric ALL, a combination of dMLPA and RNA-seq detected clinically relevant alterations in 95% of cases, compared to only 46.7% with SoC techniques (CBA and FISH). OGM as a standalone test resolved 15% of non-informative cases [126]. In sarcomas, a highly heterogeneous disease, NGS led to a reclassification of diagnosis in 4 out of 81 patients, demonstrating its power as a diagnostic tool [14].
- Identification of Novel Biomarkers: NGS panels can simultaneously assess complex genomic signatures like Tumor Mutational Burden (TMB) and Microsatellite Instability (MSI), which are critical biomarkers for immunotherapy [90] [14]. However, studies note that different NGS methods and bioinformatic algorithms can impact TMB calculation, highlighting the need for standardization, especially near clinical cut-offs [127].
Diagram: Comparative Testing Workflows. The parallel, comprehensive NGS pathway contrasts with the sequential, limited SoC pathway.
The Scientist's Toolkit: Essential Research Reagents & Platforms
Table 3: Key Research Reagents and Platforms for NGS-Based Comparative Studies
| Category | Item / Platform | Specific Example / Vendor | Critical Function in Research |
|---|---|---|---|
| Nucleic Acid Extraction | DNA/RNA FFPE Kits | QIAamp DNA FFPE Kit (Qiagen), RNeasy FFPE Kit (Qiagen) | Isolate high-quality nucleic acids from challenging clinical samples. |
| Targeted NGS Panels | Hybridization-Capture Panels | OncoAim Lung Panel (Singlera), TTSH-oncopanel (61 genes) [125] [35] | Focus sequencing on clinically relevant genomic regions; enable high coverage at lower cost. |
| Library Preparation | Automated Library Prep Systems | MGI SP-100RS, Ion Chef System (Thermo Fisher) | Standardize and automate library construction, reducing human error and variability. |
| Sequencing Platforms | Benchtop Sequencers | Illumina NextSeq 500, Ion S5, MGI DNBSEQ-G50RS | Perform massively parallel sequencing; platform choice affects read length, error profile, and cost. |
| Variant Calling & Analysis | Bioinformatic Pipelines | Sophia DDM, Ion Reporter, BWA-GATK | Align sequences to a reference genome and identify somatic variants with high accuracy. |
| Validation Technologies | Orthogonal Assays | IHC (Ventana, Cell Signaling), FISH, ARMS-PCR | Provide independent confirmation of key NGS findings, essential for assay validation. |
Emerging Frontiers and Future Directions
The evolution of NGS continues to address initial limitations and expand its clinical, and research applications. Key emerging areas include:
- Liquid Biopsy: The use of cell-free DNA (cfDNA) for non-invasive genomic profiling enables real-time monitoring of treatment response and resistance mechanisms, which is central to adaptive therapy strategies in drug development [90].
- Enhanced Sequencing Architectures: Techniques like error-corrected sequencing (e.g., NanoSeq) achieve ultra-low error rates (<5 errors per billion base pairs), allowing for the detection of very low-frequency clones in normal tissues, thus providing a window into early carcinogenesis [128].
- Expanded Target Capture: Strategies that extend WES beyond coding regions to include introns, untranslated regions (UTRs), and mitochondrial DNA in a cost-effective manner can improve diagnostic yield without resorting to more expensive whole-genome sequencing (WGS) [129].
- Multi-Omics Integration: Combining DNA sequencing with RNA-seq (for transcriptomics) and other modalities provides a more holistic view of tumor biology, enabling the identification of novel fusion genes and therapeutic vulnerabilities [90] [126].
Diagram: Key Oncogenic Signaling Pathway. NGS can identify mutations in multiple pathway genes (e.g., EGFR, KRAS, PIK3CA) simultaneously, guiding combination targeted therapy.
The body of comparative effectiveness research unequivocally establishes that next-generation sequencing represents a technically superior and economically advantageous paradigm over traditional standard-of-care testing methods. For researchers and drug development professionals, the implementation of NGS is not merely an incremental improvement but a fundamental enabler of precision oncology. It accelerates biomarker discovery, rationalizes resource utilization, and provides the comprehensive genomic landscape necessary to develop and guide next-generation targeted therapies. Future advancements in sequencing sensitivity, bioinformatic analysis, and multi-omic integration will further solidify its role as the cornerstone of cancer molecular profiling research and clinical practice.
Within the paradigm of precision oncology, Next-Generation Sequencing (NGS) has transitioned from a research tool to a cornerstone of clinical cancer management. The core value proposition of NGS lies in its ability to comprehensively profile the molecular landscape of a tumor, thereby enabling therapeutic strategies matched to its specific genomic alterations. However, the integration of NGS into standard clinical practice and drug development pipelines is contingent upon robust evidence generation for its clinical utility—demonstrating that its use leads to improved patient outcomes and survival. This whitepaper provides an in-depth technical guide for researchers and drug development professionals on the frameworks, methodologies, and metrics essential for validating the impact of NGS-driven cancer profiling.
Clinical Impact of NGS-Informed Therapy: Survival Outcomes
The most direct evidence of clinical utility comes from studies comparing survival metrics between patients who received NGS-informed therapy and those who did not. A comprehensive literature review of 31 publications evaluated progression-free survival (PFS) and overall survival (OS) in patients with advanced cancer who received NGS testing [130].
Key Quantitative Findings on Survival Endpoints
Table 1: Summary of Clinical Survival Outcomes from NGS-Informed Therapy Across Multiple Cancers
| Outcome Measure | Number of Publications with Significant Findings | Reported Hazard Ratio (HR) Range | Mean HR | Clinical Interpretation |
|---|---|---|---|---|
| Progression-Free Survival (PFS) | 11 publications | 0.24 - 0.67 | 0.47 | Patients receiving NGS-matched therapy had significantly longer time before their cancer progressed. |
| Overall Survival (OS) | 16 publications | Not Specified | Not Specified | Patients receiving NGS-matched therapy had significantly longer overall survival. |
| Therapy Matching Rate | 24 publications (calculated) | 2% - 66% of tested patients | 29% (mean) | A substantial proportion of patients have actionable targets identified by NGS. |
This analysis concluded that NGS-informed treatment is associated with significantly longer PFS and OS across a spectrum of tumor types, providing strong aggregate evidence for its clinical utility [130]. Real-world evidence from a South Korean study of 990 patients with advanced solid tumors further supports this, showing that 13.7% of patients with Tier I alterations (strong clinical significance) received NGS-based therapy, resulting in a 37.5% partial response rate and a median treatment duration of 6.4 months [131].
Actionable Biomarkers and Tumor-Agnostic Approaches
A critical step in demonstrating clinical utility is establishing the actionability of NGS findings—the frequency with which testing identifies biomarkers linked to approved or investigational therapies.
A pan-cancer study of 1,166 tissue samples from an Asian cohort found that 62.3% of samples harbored at least one actionable biomarker [132]. When classified using the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT), which ranks biomarkers from Tier I (approved standard-of-care) to Tier V (preclinical evidence), 12.7% of samples contained Tier I alterations [132].
Tumor-agnostic biomarkers—molecular alterations that are actionable regardless of tumor histology—are a key area of NGS utility. The same study identified at least one such biomarker in 8.4% of samples across 26 different cancer types [132].
Table 2: Prevalence of Key Tumor-Agnostic and Actionable Biomarkers
| Biomarker Category | Specific Biomarker | Prevalence in Pan-Cancer Cohort | Example Cancer Types with High Prevalence |
|---|---|---|---|
| Established Tumor-Agnostic | TMB-High | 6.6% | Lung (15.4%), Endometrial (11.8%) |
| MSI-High | 1.4% | Endometrial (5.9%), Gastric (4.7%) | |
| NTRK Fusions | 0.3% | Pancreatic, Gastric, Colorectal | |
| BRAF V600E | ~1.0% | Colorectal, Melanoma, Thyroid | |
| Emerging/Other Actionable | HRD (Homologous Recombination Deficiency) | 34.9% | Breast (50%), Colorectal (49%), Ovary (42.2%) |
| ERBB2 Amplification | 3.6% | Breast (15%), Endometrial (11.8%), Ovarian (8.9%) |
This high prevalence of actionable targets, including in rare and unexpected cancer types, underscores the role of comprehensive genomic profiling in expanding treatment options beyond histology-based paradigms.
Technical Protocols for Evidence Generation
Generating robust evidence requires stringent technical protocols. The following outlines key methodological components for NGS-based clinical and prognostic studies.
NGS Wet-Lab and Bioinformatics Protocol
The clinical validation of a novel DNA/RNA assay for blood cancers, such as the Duoseq assay, provides a template for a robust NGS workflow [133].
Sample Preparation & Sequencing:
- Input Material: Formalin-Fixed Paraffin-Embedded (FFPE) tumor specimens or fresh tissue [133] [131].
- DNA Extraction: Use of kits such as QIAamp DNA FFPE Tissue kit (Qiagen). Quality control requires a minimum of 20 ng DNA with an A260/A280 ratio between 1.7 and 2.2 [131].
- Library Preparation: Hybrid capture-based method (e.g., Agilent SureSelectXT) for target enrichment. Library quantity and size distribution are assessed using systems like the Agilent 2100 Bioanalyzer [131].
- Sequencing: Performance on platforms such as Illumina NovaSeq or NextSeq 550Dx, with a mean depth of coverage >500x often required for reliable variant detection [131].
Bioinformatic Analysis:
- Alignment: Reads are aligned to a reference genome (e.g., GRCh37/hg19).
- Variant Calling: Tools like Mutect2 are used for single nucleotide variants (SNVs) and small insertions/deletions (indels). Copy number variations (CNVs) are identified with tools like CNVkit, and gene fusions are detected with tools like LUMPY [131].
- Variant Annotation and Filtering: Variants are annotated and filtered based on population frequency databases (e.g., gnomAD), clinical databases (e.g., ClinVar), and internal quality thresholds (e.g., variant allele frequency ≥ 2% for tissue; lower for liquid biopsy) [131].
- Tumor-Agnostic Biomarkers: Microsatellite instability (MSI) status can be determined using tools like mSINGs, and Tumor Mutational Burden (TMB) is calculated as mutations per megabase [131].
Protocol for Prognostic Marker Discovery
For discovering novel prognostic markers, as in a study on recurrent IDH wild-type gliomas, a multi-omics computational approach is essential [134] [135].
Data Acquisition and Pre-processing:
- Data Source: Utilize large-scale consortium data (e.g., GLASS for gliomas) or public repositories like The Cancer Genome Atlas (TCGA). Data modalities include gene expression (GE), miRNA expression (ME), DNA methylation (DM), and copy number variation (CNV) [134] [135].
- Pre-processing: Filter genes with excessive zero expression values. Normalize data using a
log2(x + 1)transformation followed by z-score normalization. For survival analysis, ensure clinical data includes vital status and time to event (death/last follow-up) [135].
Feature Selection and Survival Modeling:
- High-Dimensionality Reduction: Apply a univariate log-rank test to select top survival-associated genes (e.g., top 1,000) [135].
- Network-Based Discovery (e.g., netSurvival): Construct a gene-gene interaction network where edges represent shared patient clusters. Use random walk algorithms to identify paths of connected nodes with significant survival differences [135].
- Regularized Cox Models: Employ models like Lasso Cox (L1 regularization), Ridge Cox (L2 regularization), or Elastic Net Cox (combined L1/L2) to handle high-dimensional data and prevent overfitting. The objective function for Lasso Cox is:
L(β) = ℓ(β) + λ∑|βj|, whereℓ(β)is the Cox log-likelihood andλis the regularization parameter [135]. - Validation: Use cross-validation (e.g., 5-fold) to assess model performance and ensure generalizability.
Diagram 1: Workflow for multi-omics prognostic marker discovery, integrating computational biology and survival analysis.
NGS in Minimal Residual Disease (MRD) and Early Detection
Evidence of clinical utility extends to cancer management in the adjuvant and screening settings.
NGS for Measurable Residual Disease (MRD)
In Acute Myeloid Leukemia (AML), NGS-based MRD assessment provides powerful prognostic stratification. A 2025 study of 69 AML patients used a targeted 47-gene panel to track mutations during consolidation therapy and 2-year monitoring [136].
- Key Finding: Patients with a mean variant allele frequency (VAF) of somatic mutations (excluding CHIP) ≤0.004 at the consolidation phase had a better prognosis [136].
- Combined Modality: Integrating NGS-MRD with multiparameter flow cytometry (MFC) further refined risk assessment. Patients negative by both methods had significantly longer survival than those positive by either method alone [136].
This approach is also vital in solid tumors. Exact Sciences' Oncodetect, a tumor-informed NGS-based MRD test for colorectal cancer, uses whole-genome sequencing to track patient-specific variants. Data from the Beta-CORRECT study showed that patients with ctDNA-positive results after therapy had a 24-fold increased risk of recurrence [137]. Next-generation versions of this test aim to achieve a limit of detection below 1 part per million, enabling ultra-early recurrence detection [137].
NGS in Early Cancer Screening
The application of NGS in early cancer screening via liquid biopsy represents a frontier in preventive oncology. The global market for NGS-based early cancer screening is projected to grow from USD 591.6 million in 2025 to USD 2,393.5 million by 2035, reflecting intense development and anticipated clinical adoption [138].
The primary technological driver is cfDNA methylation sequencing, which is projected to account for 55% of the NGS early cancer screening market due to its high sensitivity and specificity for multi-cancer early detection and ability to predict the tissue of origin [138].
Diagram 2: Liquid biopsy workflow for NGS-based multi-cancer early detection, highlighting the path from blood draw to clinical decision.
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Research Reagents and Solutions for NGS-Based Clinical Utility Studies
| Tool / Reagent | Specific Example(s) | Primary Function in Workflow |
|---|---|---|
| Nucleic Acid Extraction Kits | QIAamp DNA FFPE Tissue Kit (Qiagen) | Isolation of high-quality genomic DNA from challenging FFPE tissue samples. |
| Targeted Gene Panels | SNUBH Pan-Cancer v2.0 (544 genes); 47-gene AML panel; UNITED DNA/RNA panel | Focused sequencing of clinically relevant genes to maximize depth and cost-efficiency. |
| Library Prep & Enrichment | Agilent SureSelectXT (Hybrid Capture); Illumina Nextera | Preparation of sequencing libraries and enrichment for target regions of the genome. |
| NGS Platforms | Illumina NovaSeq, NextSeq 550Dx | High-throughput sequencing to generate raw read data with required coverage. |
| Bioinformatic Tools | Mutect2 (SNVs/Indels), CNVkit (CNVs), LUMPY (Fusions), mSINGs (MSI) | Detection and annotation of genomic variants from raw sequencing data. |
| Unique Molecular Identifiers (UMIs) | Commercial UMI kits | Tagging original DNA molecules to correct for PCR errors and enable ultra-sensitive detection. |
The generation of high-grade evidence for the clinical utility of NGS is a multi-faceted endeavor, requiring rigorous technical protocols, robust statistical and bioinformatic analyses, and validation through clinically relevant endpoints like overall survival and minimal residual disease detection. The consistent demonstration that NGS-informed therapy improves patient outcomes across cancer types solidifies its role in modern oncology. For researchers and drug developers, mastering these evidence-generation frameworks is paramount for advancing precision medicine, validating new biomarkers, and ultimately delivering more effective, personalized cancer therapies to patients.
The evolution of precision oncology has established a new paradigm in cancer therapy, one increasingly dependent on the synergistic development of targeted therapeutics and their corresponding companion diagnostics (CDx). Next-generation sequencing (NGS) has emerged as the technological cornerstone of this paradigm, enabling comprehensive genomic profiling that informs treatment selection [10]. The U.S. Food and Drug Administration (FDA) regulates both therapeutic products and the CDx devices essential for identifying patients likely to benefit from these targeted treatments [139]. The drug-diagnostics co-development model, first exemplified by the simultaneous approval of trastuzumab and the HercepTest in 1998, has become a standard regulatory pathway [140]. This framework ensures that the diagnostic tool essential for identifying the appropriate patient population is available concurrently with the drug, thereby maximizing therapeutic efficacy and patient safety. Within this context, NGS-based CDxs have grown increasingly sophisticated, progressing from single-gene tests to complex multi-analyte panels that can guide therapy with remarkable precision [140] [51].
The Regulatory Framework for NGS-Based Companion Diagnostics
Definition and Significance of Companion Diagnostics
The FDA defines a companion diagnostic device as an in vitro diagnostic (IVD) device or an imaging tool that provides information essential for the safe and effective use of corresponding therapeutic products [139]. These devices are not merely adjunctive but are integral to the therapeutic product's labeling, as their use is stipulated in the instructions for both the diagnostic device and the corresponding drug [139]. The fundamental purpose of a CDx is to identify a biomarker-defined patient subgroup that is most likely to respond favorably to a specific targeted therapy, while also identifying patients who may experience serious adverse effects, thereby enabling improved risk-benefit assessment [140].
Pathways to Market for NGS-Based IVDs
The regulatory pathway for an NGS-based CDx depends on its intended use, risk profile, and the existence of legally marketed predicate devices. The FDA has established three primary pathways for these devices [141]:
- 510(k) Pre-market Notification: Appropriate when substantial equivalence to a predicate device can be demonstrated. This pathway is typically used for lower to moderate-risk devices.
- De Novo Classification: For novel devices of low to moderate risk with no predicate. This pathway establishes a new device classification and special controls for future devices.
- Pre-market Approval (PMA): Required for higher-risk Class III devices, such as those that direct critical treatment decisions for life-threatening illnesses. Most CDxs fall under this category.
For NGS-based CDxs specifically, the PMA pathway is most common due to their critical role in patient selection for oncology therapeutics. The choice of pathway is ultimately determined by the device's risk-based classification and the novelty of its technology and intended use [141].
Analytical Performance Validation
Robust analytical validation is a fundamental requirement for FDA compliance of NGS-based IVDs. The validation process must demonstrate that the test performs reliably and accurately according to its claimed specifications. Key validation parameters include [141]:
- Accuracy: Measured through Positive Percent Agreement (PPA) and Negative Percent Agreement (NPA), which quantify the test's ability to correctly identify true positive and true negative variants, respectively.
- Limit of Detection (LOD): The minimum amount of DNA or the lowest variant allele frequency that the test can reliably detect, which must be validated for all variant types covered by the test.
- Precision: Encompasses both repeatability (same conditions) and reproducibility (different conditions, such as multiple operators, instruments, and reagent lots) to ensure consistent results.
- Bioinformatics Pipeline Validation: The computational workflow, including variant calling, annotation, and filtering algorithms, must undergo thorough validation to ensure the conversion of raw sequencing data into clinically reliable results.
Table 1: Essential Analytical Performance Metrics for NGS-Based CDx
| Performance Parameter | Definition | Validation Requirement |
|---|---|---|
| Accuracy | Measure of correct identification of genetic variants | PPA, NPA, and Technical PPV against a reference method |
| Limit of Detection (LOD) | Lowest variant allele frequency reliably detected | Established for all variant types across relevant DNA inputs |
| Precision | Consistency of results under varying conditions | Testing across multiple runs, days, operators, and instrument lots |
| Specificity | Ability to correctly identify wild-type sequences | Demonstration of low false-positive rate in negative samples |
| Bioinformatics Validation | Reliability of data analysis pipeline | Verification of variant calling, annotation, and filtering algorithms |
The Evolving Landscape of FDA Approvals in Oncology
Current Trends in Drug and CDx Approvals
The landscape of FDA oncology approvals has shifted dramatically toward targeted therapies and their corresponding CDxs. An analysis of FDA approvals from 1998 to 2024 reveals that of 217 new molecular entities (NMEs) approved for oncological and hematological malignancies, 78 (36%) were linked to one or more CDx assays [140]. The trend has accelerated significantly over time; from 1998-2010, only 7 NMEs (15% of approvals) had associated CDxs, compared to 71 NMEs (42% of approvals) from 2011-2024 [140]. Kinase inhibitors represent the therapeutic class most frequently paired with a CDx, with 48 (60%) of the 80 approved kinase inhibitors having a corresponding diagnostic [140].
Recent approvals in the third quarter of 2025 exemplify this trend, with the FDA simultaneously approving new targeted therapies and their companion diagnostics. These include zongertinib for HER2-mutated non-small cell lung cancer (NSCLC) and sunvozertinib for EGFR exon 20 insertion-mutated NSCLC, both accompanied by the Oncomine Dx Express Test to identify eligible patients [142]. Similarly, the approval of imlunestrant for ESR1-mutated breast cancer was paired with approval of the Guardant360 CDx assay to detect ESR1 mutations [142].
The Rise of Tissue-Agnostic Indications
A significant evolution in precision oncology has been the emergence of tissue-agnostic (or "histology-agnostic") drug approvals, which are based solely on specific molecular biomarkers regardless of tumor origin. Since 1998, the FDA has granted tissue-agnostic indications to nine NMEs, all of which are associated with a CDx [140]. These approvals represent a fundamental shift from organ-based to genomics-based cancer classification.
However, a notable challenge in this area has been the frequent delay in CDx approval relative to the corresponding therapeutic. For tissue-agnostic indications, the mean delay between drug approval and CDx approval has been 707 days, ranging from 0 to 1,732 days [140]. This disconnect can create clinical confusion and barriers to patient access, as clinicians must identify alternative testing methods to determine patient eligibility during the interim period.
Table 2: FDA-Approved Tissue-Agnostic Therapies and Companion Diagnostics
| Drug | Therapeutic Class | Biomarker | Drug Approval Date | CDx Approval Date | Approval Delay (Days) |
|---|---|---|---|---|---|
| Pembrolizumab | Antibody | MSI-H/dMMR, TMB-H | May 23, 2017 | June 16, 2022 | 1,732 |
| Larotrectinib | Kinase Inhibitor | NTRK gene fusion | November 26, 2018 | October 23, 2020 | 697 |
| Entrectinib | Kinase Inhibitor | NTRK gene fusion | August 15, 2019 | June 7, 2022 | 1,027 |
| Dostarlimab | Antibody | dMMR | August 17, 2021 | February 23, 2023 | 555 |
| Trastuzumab deruxtecan | Antibody-drug conjugate | HER2 (IHC 3+) | April 5, 2024 | December 31, 2024 | 270 |
| Dabrafenib | Kinase Inhibitor | BRAF V600E | June 22, 2022 | December 31, 2024 | 923 |
| Trametinib | Kinase Inhibitor | BRAF V600E | June 22, 2022 | December 31, 2024 | 923 |
Implementation of NGS in Clinical Practice: Methodologies and Workflows
NGS Testing Workflow
The clinical implementation of NGS-based genomic profiling follows a standardized workflow that transforms patient tumor samples into clinically actionable reports. The following diagram illustrates this multi-step process:
Figure 1. NGS Clinical Testing Workflow. The process begins with sample preparation from FFPE tissue or blood, followed by library construction, target enrichment, sequencing, bioinformatic analysis, clinical interpretation, and final clinical reporting.
Key Experimental Protocols
Sample Preparation and Library Construction
The initial phase of NGS testing requires meticulous sample preparation. For tissue samples, formalin-fixed paraffin-embedded (FFPE) tumor specimens are sectioned and subjected to manual microdissection to ensure sufficient tumor cellularity (typically >20%) [51]. DNA extraction is performed using specialized kits (e.g., QIAamp DNA FFPE Tissue kit), with quality assessment measuring DNA concentration and purity (A260/A280 ratio between 1.7-2.2) [51]. A minimum of 20ng of DNA is typically required for library generation.
Library construction involves fragmenting genomic DNA to approximately 300bp, followed by adapter ligation. These synthetic oligonucleotides with specific sequences are essential for attaching DNA fragments to the sequencing platform and for subsequent amplification [10]. The three primary methods for nucleic acid fragmentation are physical, enzymatic, and chemical, with the choice of method affecting library quality and reproducibility [10].
Target Enrichment and Sequencing
For targeted NGS panels, either hybrid capture or amplicon-based approaches are used for target enrichment. The hybrid capture method, using products such as the Agilent SureSelectXT Target Enrichment Kit, involves biotinylated probes that hybridize to regions of interest, which are then pulled down using streptavidin-coated magnetic beads [51]. Following enrichment, libraries are quantified and sequenced on platforms such as Illumina's NextSeq 550Dx, with a minimum mean depth of 500-1000x recommended for reliable variant detection [51].
Bioinformatic Analysis and Interpretation
The bioinformatic workflow begins with base calling and quality control, followed by alignment to a reference genome (e.g., hg19) [51]. Variant calling utilizes specialized algorithms: Mutect2 for single nucleotide variants (SNVs) and small insertions/deletions (INDELs); CNVkit for copy number variations; and LUMPY for gene fusions [51]. Only variants with a variant allele frequency (VAF) above a established threshold (typically ≥2-5%) are considered for clinical reporting [51].
Critical to clinical implementation is variant classification according to established guidelines such as the Association for Molecular Pathology (AMP) system, which categorizes variants into four tiers [51]:
- Tier I: Variants of strong clinical significance (FDA-approved or professional guideline-recognized)
- Tier II: Variants of potential clinical significance (may predict response to investigational therapies)
- Tier III: Variants of unknown clinical significance
- Tier IV: Benign or likely benign variants
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for NGS-Based Companion Diagnostic Development
| Reagent/Material | Function | Example Products |
|---|---|---|
| Nucleic Acid Extraction Kits | Isolation of high-quality DNA from FFPE tissue or blood | QIAamp DNA FFPE Tissue Kit |
| DNA Quantitation Assays | Precise measurement of DNA concentration and quality | Qubit dsDNA HS Assay, NanoDrop Spectrophotometer |
| Library Preparation Kits | Fragmentation, end-repair, and adapter ligation | Agilent SureSelectXT, Illumina Nextera |
| Target Enrichment Probes | Capture of genomic regions of interest | IDT xGen Pan-Cancer Panel, Agilent SureSelect |
| Sequencing Reagents | Cluster generation and nucleotide incorporation | Illumina SBS Kits, Ion Torrent Chef Reagents |
| Bioinformatic Tools | Variant calling, annotation, and interpretation | Mutect2, CNVkit, SnpEff, OncoKB |
Barriers to Clinical Implementation and Future Directions
Challenges in Widespread NGS Adoption
Despite the demonstrated clinical utility of NGS-based molecular profiling, significant barriers impede its optimal implementation in clinical practice. A multi-stakeholder survey revealed that inconsistent payer coverage, high out-of-pocket costs for patients, and challenges in managing reimbursement and prior authorization processes lead to suboptimal utilization of NGS [26]. This subsequently results in suboptimal treatment decisions where approved targeted therapies exist but cannot be appropriately directed to eligible patients.
Interestingly, 33% of payers surveyed were not aware of the current somatic biomarker testing recommendations from National Comprehensive Cancer Network (NCCN) guidelines [26]. Payers identified the lack of clear clinical guidelines (74% ranked as top 3 barrier), lack of internal consensus on which NGS tests to cover (45%), and absence of internal expertise on NGS (39%) as primary hurdles for broader NGS access [26]. These findings suggest that widespread education of healthcare professionals and payers on clinical guidelines is crucial for enhanced adoption of NGS-based molecular profiling.
Regulatory Developments and Future Directions
The FDA has recently undergone significant regulatory evolution regarding laboratory-developed tests (LDTs), which include many NGS-based assays. In May 2024, the FDA issued a final rule amending the definition of "in vitro diagnostic products" to explicitly include LDTs; however, this rule was vacated by a federal district court in March 2025, and the FDA issued a final rule in September 2025 reverting to the previous regulatory text [143]. This regulatory uncertainty presents ongoing challenges for test developers.
Looking forward, the FDA's New Alternative Methods Program aims to spur the adoption of alternative methods for regulatory use that can improve predictivity of nonclinical testing [144]. This includes advancing computational modeling and simulation, microphysiological systems (organs-on-chips), and other innovative approaches that may eventually complement or replace aspects of traditional validation methods for CDxs [144].
The future of NGS in cancer diagnosis will likely see increased integration of liquid biopsies for non-invasive monitoring, single-cell sequencing for resolving tumor heterogeneity, and artificial intelligence for enhanced variant interpretation [10]. Furthermore, the development of consensus standards for analytical validation and clinical databases may simplify the regulatory process, enabling test developers to certify compliance without extensive premarket review [141]. As the field progresses, continued collaboration among regulators, industry leaders, and academic researchers will be essential to advance genomic diagnostics while ensuring patient safety.
The integration of next-generation sequencing (NGS) into oncology represents a paradigm shift in cancer care, moving from a one-size-fits-all approach to personalized treatment strategies. This transformation necessitates rigorous economic evaluations to determine the cost-effectiveness and value proposition of these advanced technologies within healthcare systems. Economic evaluations in genomic medicine systematically compare the costs and consequences of different NGS-based testing strategies to inform resource allocation decisions [145]. As precision oncology advances, with an increasing number of targeted therapies requiring companion diagnostics, the importance of these economic assessments has grown substantially.
The fundamental challenge in evaluating NGS technologies lies in their unique value proposition: unlike single-gene tests, NGS can simultaneously interrogate hundreds of genes from a single tissue sample or liquid biopsy, potentially eliminating the need for sequential testing and reducing time to appropriate treatment [146] [147]. However, this comprehensive approach comes with higher upfront costs and complexities in evidence generation that challenge conventional health technology assessment methodologies [148]. Furthermore, the rapid evolution of both sequencing technologies and their clinical applications creates a moving target for economic evaluations, requiring flexible frameworks that can adapt to this dynamic landscape.
Methodological Frameworks for Economic Evaluation of NGS
Core Health Economic Methodologies
Economic evaluations of NGS technologies in oncology primarily employ three methodological approaches, each with distinct advantages and limitations for assessing value in genomic medicine. Cost-effectiveness analysis (CEA) measures costs in monetary units and effectiveness in natural units (e.g., life-years gained), calculating an incremental cost-effectiveness ratio (ICER) to compare testing strategies [146]. Cost-utility analysis (CUA) extends this approach by measuring health outcomes in quality-adjusted life years (QALYs), which incorporate both quantity and quality of life, allowing comparison across different healthcare interventions [145]. Cost-benefit analysis (CBA) attempts to value both inputs and outcomes in monetary terms, though this approach is less common in healthcare due to ethical concerns about valuing human life in financial terms [148].
The selection of analytical perspective is crucial in NGS evaluation, as findings can vary significantly depending on whether the analysis adopts a healthcare system, payer, patient, or societal viewpoint. The time horizon must be sufficient to capture all relevant long-term clinical outcomes and costs, often requiring lifetime modeling for chronic conditions like cancer [146]. Discounting future costs and benefits is standard practice, typically at rates between 3-5% annually, to reflect time preference—the principle that people value present benefits more highly than future ones [145].
Special Considerations for NGS Evaluation
Economic evaluations of NGS technologies present unique methodological challenges that distinguish them from assessments of conventional medical technologies. The comprehensive nature of NGS testing generates information about multiple genetic alterations simultaneously, creating challenges for defining the appropriate comparator, which may consist of multiple sequential single-gene tests [146]. The dynamic clinical utility of NGS evolves as new biomarkers are discovered and targeted therapies developed, creating difficulties for economic modeling based on current clinical practice [148]. Complex value elements beyond direct health outcomes include the value of knowing ambiguous or negative results, option value (preserving future treatment possibilities), and public health value through improved understanding of cancer genetics [147].
Real-world evidence (RWE) is increasingly important for NGS evaluations, as traditional randomized controlled trials may not be feasible or sufficient to capture all relevant outcomes. RWE can inform parameters related to test performance, clinical utility, and long-term outcomes in diverse patient populations [148]. However, methodological standards for incorporating RWE into economic evaluations are still evolving, particularly regarding approaches to address potential biases and confounding in observational data.
Current Evidence on Cost-Effectiveness of NGS in Oncology
Tissue-Based Comprehensive Genomic Profiling
The cost-effectiveness evidence for tissue-based comprehensive genomic profiling (CGP) varies by cancer type, clinical context, and healthcare system. A 2018 systematic review identified only six cost-effectiveness studies of NGS in cancer care, highlighting the limited evidence base despite rapid clinical adoption [145]. The review found that NGS was an effective tool for identifying mutations in cancer patients, with 83% of successfully sequenced patients harboring at least one mutation, but concluded that more rigorous cost-effectiveness studies were needed to determine whether NGS improves patient outcomes cost-effectively [145].
A 2020 cost-effectiveness analysis compared NGS with sequential single-gene testing for advanced lung adenocarcinoma patients from the perspective of the Brazilian private health system [146]. The study found that NGS correctly identified 24% more true positive cases compared to sequential testing (96.3% vs 72.6%), with an ICER of $3,479 per additional correctly diagnosed case [146]. However, when evaluated in terms of QALYs gained, NGS was not cost-effective in this specific healthcare context, demonstrating how value propositions differ across settings [146].
Table 1: Key Cost-Effectiveness Studies of NGS in Oncology
| Cancer Type | Comparison | Key Findings | ICER | Reference |
|---|---|---|---|---|
| Advanced lung adenocarcinoma | NGS vs. sequential single-gene testing | NGS identified 24% more true positive cases; not cost-effective in QALYs in Brazilian private system | $3,479 per additional correct case | [146] |
| Multiple solid tumors | NGS panels vs. standard of care | 83% of sequenced patients had actionable mutations; insufficient cost-effectiveness evidence | Limited evidence | [145] |
| Central nervous system infections | mNGS vs. conventional culture | mNGS reduced turnaround time (1 vs. 5 days) and antibiotic costs | ¥36,700 per additional timely diagnosis | [149] |
Liquid Biopsy Applications
Liquid biopsy using NGS represents an emerging application with distinct economic considerations. The non-invasive nature of liquid biopsies reduces procedural risks and can be repeated longitudinally to monitor treatment response and resistance mechanisms [147]. A 2024 review highlighted that liquid biopsies are particularly valuable when tissue is insufficient or inaccessible, and for monitoring minimal residual disease [147]. The cost-effectiveness of liquid biopsy approaches depends on factors including the cancer type, stage, and the specific clinical question being addressed.
The detection rates of circulating tumor DNA (ctDNA) vary significantly across cancer types, influencing the economic value of liquid biopsy applications. ctDNA is detectable in more than 75% of patients with advanced pancreatic, colorectal, gastroesophageal, hepatocellular, and ovarian cancers, but in fewer than 50% of patients with primary brain, prostate, thyroid, and renal cancers [147]. This variability in detection rates directly impacts the diagnostic yield and consequent value proposition of liquid biopsy in different clinical contexts.
Experimental Design and Protocols for NGS Economic Evaluations
Decision-Analytic Modeling Approaches
Economic evaluations of NGS technologies typically employ decision-analytic modeling to compare testing strategies and their associated costs and outcomes. These models synthesize evidence from multiple sources to estimate long-term costs and health outcomes under conditions of uncertainty. The basic workflow for constructing such models follows a structured process, illustrated below:
The decision tree component typically models the initial testing strategy, capturing all possible test results and their probabilities, while the state-transition component models the subsequent disease course and treatment pathways [146]. Model parameters include test characteristics (sensitivity, specificity), clinical parameters (disease prevalence, treatment efficacy), cost parameters (testing, treatment, monitoring), and utility parameters (quality of life weights) [146]. Validation is a critical step, ensuring the model structure and outputs reflect clinical reality and align with observed data when available.
Clinical Study Designs for NGS Evaluation
Several experimental designs can generate evidence for economic evaluations of NGS technologies, each with distinct advantages and limitations. Pragmatic clinical trials embed NGS testing within routine care and collect prospective data on clinical outcomes and resource utilization, providing high-quality evidence for economic evaluation but requiring substantial time and resources [149]. Observational studies of patients receiving NGS testing in real-world settings can provide evidence on test performance, treatment patterns, and outcomes in diverse populations, though they are susceptible to confounding [148]. Model-based studies synthesize existing evidence from multiple sources to estimate cost-effectiveness, providing timely evidence to inform decision-making but dependent on the quality and applicability of available data [146].
A 2025 prospective randomized study of metagenomic NGS for central nervous system infections demonstrated how clinical trials can inform economic evaluations [149]. The study randomized 60 patients to mNGS plus conventional culture or conventional culture alone, finding that mNGS reduced turnaround time (1 vs. 5 days) and anti-infective costs, with an ICER of ¥36,700 per additional timely diagnosis [149]. While this study focused on infectious disease, similar designs can be applied to oncology settings.
Market Landscape and Value Proposition of NGS Technologies
Market Growth and Adoption Trends
The tumor profiling market is experiencing substantial growth, driven by technological advancements, rising cancer prevalence, and increasing adoption of personalized medicine approaches. The United States tumor profiling market is projected to grow from $3.41 billion in 2024 to $7.44 billion by 2033, achieving a compound annual growth rate (CAGR) of 9.05% [150]. The broader cancer tumor profiling market is expected to expand from $13.2 billion in 2025 to $36.0 billion by 2035, at a CAGR of 10.6% [151]. The NGS segment specifically demonstrates even more rapid growth, with the United States NGS market expected to increase from $3.88 billion in 2024 to $16.57 billion by 2033, representing a remarkable CAGR of 17.5% [17].
Genomics dominates the tumor profiling techniques segment with a 38.5% market share, while sequencing techniques lead the technology segment with 31.4% share [151]. Personalized cancer medicine applications drive primary demand at 41.2% market share, supported by increasing requirements for targeted therapy selection [151]. North America maintains the largest market share at 36.5%, benefiting from established precision oncology infrastructure and higher adoption rates of advanced diagnostic technologies [151].
Table 2: Tumor Profiling and NGS Market Projections
| Market Segment | 2024/2025 Value | 2033/2035 Projection | CAGR | Key Drivers |
|---|---|---|---|---|
| United States Tumor Profiling | $3.41 billion (2024) | $7.44 billion (2033) | 9.05% | Precision medicine, rising cancer prevalence [150] |
| Global Cancer Tumor Profiling | $13.2 billion (2025) | $36.0 billion (2035) | 10.6% | Comprehensive genomic panels, liquid biopsy [151] |
| United States NGS | $3.88 billion (2024) | $16.57 billion (2033) | 17.5% | Personalized medicine, automation advances [17] |
Technology Evolution and Future Directions
The NGS landscape is evolving toward more comprehensive genomic profiling, integration with artificial intelligence, and expanding applications beyond traditional tissue-based testing. Key trends shaping the future value proposition of NGS technologies include liquid biopsy maturation from specialized monitoring applications to routine screening tools, enabling non-invasive cancer detection in asymptomatic populations and minimal residual disease surveillance [151]. Multi-omics integration combining genomics, proteomics, and metabolomics data provides unprecedented insights into tumor biology and therapeutic vulnerabilities [147]. AI-enhanced interpretation of complex NGS data accelerates diagnostic turnaround times and improves clinical decision support while decreasing bioinformatics burdens [151].
The regulatory landscape is also evolving to accommodate these technological advances, with developing frameworks for tissue-agnostic drug approvals and basket trial designs that fundamentally change how oncologists approach treatment selection based on molecular features rather than tumor origin [151]. Reimbursement models are gradually shifting toward value-based structures that reward diagnostic tests demonstrating clear clinical utility and improved patient outcomes, though significant challenges remain in adequately capturing the comprehensive value of NGS technologies [148].
Challenges in Economic Evaluation of NGS Technologies
Methodological and Evidence Challenges
Economic evaluations of NGS face several methodological challenges that complicate value assessment and comparison across studies. Evidence generation complexity stems from the rapid pace of genomic discovery and therapeutic development, creating difficulties in establishing definitive clinical utility for many genomic markers [148]. Defining the appropriate comparator is complicated by variations in standard of care across institutions and the multi-gene nature of NGS, which may replace several sequential single-gene tests [146]. Capturing comprehensive benefits beyond survival, such as reduced diagnostic odyssey, avoidance of ineffective treatments, and value of information for family members, presents measurement challenges [147].
A 2024 review identified key challenges in economic evaluations of NGS in oncology, including defining the evaluative scope, managing evidentiary limitations including lack of causal evidence, incorporating preference-based utility, and assessing distributional and equity-based impacts [148]. These challenges reflect the difficulty of generating high-quality clinical effectiveness and real-world evidence for NGS-guided interventions, particularly as testing moves into earlier disease stages and screening populations [148].
Cost and Access Considerations
The high costs associated with NGS technologies present significant challenges for healthcare systems, particularly in resource-constrained settings. The substantial investments required for sequencing infrastructure, bioinformatics capabilities, and specialized personnel create barriers to widespread adoption [150]. Ongoing costs for reagents, system maintenance, and data storage further contribute to the economic burden [17]. Additionally, the complexity of NGS data interpretation requires significant expertise, potentially limiting access at community oncology practices without specialized support [150].
Economic constraints are particularly pronounced in low- and middle-income countries, where basic cancer care resources may be limited [147]. Even in high-income countries, reimbursement policies often lag behind technological advances, creating uncertainty about coverage for NGS testing [148]. These challenges are compounded by the absence of standardized frameworks for evaluating the cost-effectiveness of comprehensive genomic tests compared to traditional diagnostic pathways [148].
Research Reagents and Essential Materials for NGS Studies
The experimental workflow for NGS-based studies requires specialized reagents and materials at each processing stage, with selection directly impacting data quality and interpretation. The following table details key research reagent solutions essential for conducting NGS studies in cancer research:
Table 3: Essential Research Reagents for NGS Studies in Cancer
| Reagent Category | Specific Examples | Function in NGS Workflow | Technical Considerations |
|---|---|---|---|
| Nucleic Acid Extraction Kits | QIAamp DNA FFPE Kit (QIAGEN), Maxwell RSC ccfDNA Plasma Kit (Promega) | Isolation of high-quality DNA from various sample types (tissue, blood, FFPE) | Yield, fragment size distribution, inhibitor removal [147] |
| Library Preparation Kits | TruSight Oncology (Illumina), QIAseq Targeted Panels (QIAGEN) | Fragmentation, adapter ligation, and amplification of DNA for sequencing | Input DNA requirements, coverage uniformity, hands-on time [151] |
| Target Enrichment Reagents | xGen Lockdown Probes (IDT), SureSelect (Agilent) | Hybridization-based capture of specific genomic regions of interest | Panel design, off-target rate, coverage of difficult regions [147] |
| Sequencing Consumables | NovaSeq X Series Flow Cells (Illumina), SMRT Cells (Pacific Biosciences) | Template immobilization and nucleotide incorporation during sequencing | Read length, output capacity, error profiles [17] |
| Bioinformatics Tools | DRAGEN Platform (Illumina), CLC Genomics Workbench (QIAGEN) | Base calling, alignment, variant calling, and annotation | Computational requirements, automation capabilities [17] |
The selection of appropriate research reagents depends on multiple factors including the specific research question, sample type and quality, required sensitivity and specificity, and available budget. DNA extraction methods must be optimized for different source materials, with formalin-fixed paraffin-embedded (FFPE) tissue requiring specialized approaches to address cross-linking and fragmentation, while circulating tumor DNA from blood samples needs highly sensitive methods to capture low-abundance variants [147]. Library preparation approaches vary based on application, with hybrid capture-based methods generally providing more comprehensive coverage and amplicon-based methods offering lower input requirements and simpler workflows [151].
Economic evaluations of NGS technologies in cancer care face the dual challenge of keeping pace with rapid technological advances while adequately capturing their comprehensive value proposition. Traditional cost-effectiveness frameworks often struggle to accommodate the multi-faceted benefits of genomic testing, including the value of information for patients and families, the reduction of diagnostic odysseys, and the generation of knowledge that advances cancer biology understanding [148]. Future methodological developments should focus on standardized approaches for evaluating comprehensive genomic profiling, with attention to evolving clinical applications including minimal residual disease monitoring and early cancer detection [151].
The integration of real-world evidence and development of life-cycle health technology assessment approaches will be crucial for the sustainable implementation of NGS in oncology [148]. As healthcare systems worldwide face increasing cost pressures, demonstrating the value of NGS through rigorous economic evaluations will be essential for appropriate resource allocation. This will require collaboration among clinicians, researchers, policymakers, and patients to ensure that economic evaluations capture outcomes that matter most to stakeholders while maintaining methodological rigor. The ongoing evolution of NGS technologies promises to further transform cancer care, with parallel advances in economic evaluation methodologies needed to illuminate their optimal role in healthcare systems.
Conclusion
Next-generation sequencing has unequivocally established itself as a cornerstone technology in cancer research and precision oncology, enabling unprecedented insights into tumor biology and accelerating therapeutic development. The integration of NGS into research pipelines and clinical practice requires careful consideration of methodological approaches, rigorous validation, and proactive management of implementation barriers. As the field advances, emerging technologies including single-cell sequencing, spatial transcriptomics, liquid biopsies, and AI-driven bioinformatics promise to further refine our understanding of cancer genomics and resistance mechanisms. For researchers and drug development professionals, mastering NGS technologies and their applications is no longer optional but essential for driving the next wave of innovation in cancer care. Future success will depend on continued collaboration across disciplines to standardize practices, generate robust evidence of clinical utility, and ensure that the profound benefits of genomic medicine reach all cancer patients through both optimized drug development and enhanced clinical decision-making.
References
- 1. Next Generation Sequence Testing: Essential 2025 Guide [https://lifebit.ai/blog/next-generation-sequence-testing/]
- 2. A Feasibility Study of NGS-Based Variant Analysis of FFPE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384127/]
- 3. Sample Preparation for NGS – A Comprehensive Guide [https://frontlinegenomics.com/sample-preparation-for-ngs-a-comprehensive-guide/]
- 4. How-to: NGS Quality Control [https://frontlinegenomics.com/how-to-ngs-quality-control/]
- 5. The Complete Guide to NGS Data Types and Formats [https://ngs101.com/the-complete-guide-to-ngs-data-types-and-formats-from-raw-reads-to-analysis-ready-files/]
- 6. A Primer on NGS Technologies and Their Specs (2025) [https://blog.latch.bio/p/a-primer-on-ngs-technologies-and]
- 7. Introduction to Next Generation Sequencing (NGS) Data and ... [https://bioinformatics.ccr.cancer.gov/docs/bioinformatics-for-beginners-2025/Module2_RNA_Sequencing/Lesson7/]
- 8. Recommendations for bioinformatics in clinical practice [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-025-01543-4]
- 9. The Next-Generation Sequencing Quality Initiative and ... [https://wwwnc.cdc.gov/eid/article/31/13/24-1175_article]
- 10. Next-generation sequencing in cancer diagnosis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12009796/]
- 11. Next-generation sequencing: 2025 Revolution [https://lifebit.ai/blog/next-generation-sequencing/]
- 12. 2024 Whole Genome Sequencing Cost l ... [https://3billion.io/blog/price-of-whole-genome-sequencing-2024whole-genome-sequencing-cost-2023]
- 13. A nationwide comprehensive genomic profiling and ... [https://www.nature.com/articles/s41698-025-00858-0]
- 14. Next-generation sequencing-based genomic profiling of ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1627452/full]
- 15. Next-generation Sequencing Library Preparation Market ... [https://www.precedenceresearch.com/next-generation-sequencing-library-preparation-market]
- 16. Next Generation Sequencing Market Size & Forecast, 2025 ... [https://www.coherentmarketinsights.com/market-insight/next-generation-sequencing-market-158]
- 17. United States Next Generation Sequencing (NGS) Market ... [https://crisprmedicinenews.com/press-release-service/card/united-states-next-generation-sequencing-ngs-market-research-2025-2033-personalized-medicine-res/]
- 18. Characterization of cancer genomic heterogeneity by next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6333046/]
- 19. Tumor heterogeneity: next-generation sequencing enhances ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0463-6]
- 20. Integrative spatial and genomic analysis of tumor ... [https://www.nature.com/articles/s41467-024-53374-3]
- 21. a comparison of two molecular tumor boards - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9590222/]
- 22. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 23. Celebrating milestones and the future of oncology in China [https://www.illumina.com/company/news-center/feature-articles/CSCO-2025.html]
- 24. Structural variant detection in cancer genomes [https://www.nature.com/articles/s41698-021-00155-6]
- 25. Analysis of next-generation genomic data in cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC2953747/]
- 26. Perceived Barriers to NGS-Based Molecular Profiling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12564138/]
- 27. Article Complex structural variation is prevalent and highly ... [https://www.sciencedirect.com/science/article/pii/S2666979X24002945]
- 28. Advances in Precision Oncology: From Molecular Profiling to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607717/]
- 29. Precision oncology: current and future platforms for ... [https://www.sciencedirect.com/science/article/abs/pii/S2405803324001353]
- 30. Precision Oncology: What's Holding Us Back? [https://frontlinegenomics.com/precision-oncology-whats-holding-us-back/]
- 31. Integrating comprehensive genomic profiling in the ... [https://www.sciencedirect.com/science/article/pii/S2319417025000253]
- 32. Illumina Comprehensive Genomic Profiling Test for Cancer ... [https://www.illumina.com/company/news-center/press-releases/2025/586da3e1-460e-4a24-a018-0d82b7e35c01.html]
- 33. Clinical impact of single-gene vs. panel sequencing in ... [https://www.nature.com/articles/s41523-025-00805-z]
- 34. Tumor Profiling Market Growth Trends and Forecast Report ... [https://finance.yahoo.com/news/tumor-profiling-market-growth-trends-124100926.html]
- 35. A targeted next-generation sequencing panel for ... [https://www.nature.com/articles/s41598-025-08039-6]
- 36. Guidelines for Validation of Next-Generation Sequencing– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6941185/]
- 37. How our custom NGS panels are being used for clinical utility [https://nonacus.com/blog-how-our-custom-ngs-panels-are-being-used-for-clinical-utility/]
- 38. Clinical utility of custom-designed NGS panel testing in ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-019-0644-8]
- 39. Clinical Utility of a Next-Generation Sequencing Panel for ... [https://www.sciencedirect.com/science/article/pii/S1525157817306141]
- 40. Clinical Utility of Next-Generation Sequencing-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7961835/]
- 41. Advances in personalized medicine: translating genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12106117/]
- 42. Designing custom NGS panels | Panel Design Tool [https://nonacus.com/blog-get-great-coverage-for-the-genes-you-care-about/]
- 43. OGT expands MRD detection capabilities with new panel [https://www.ogt.com/about-us/news/ogt-expands-mrd-detection-capabilities-with-new-sureseq-myeloid-mrd-plus-ngs-panel/]
- 44. Next-Generation Sequencing: A Review of Its ... [https://www.mdpi.com/2075-4418/15/19/2425]
- 45. Capture-Seq VS. Amplicon-Seq Library Preparation Methods [https://www.cd-genomics.com/microbioseq/resource-capture-seq-vs-amplicon-seq-library-preparation-methods.html]
- 46. Hybridization-Based Target Enrichment for NGS [https://sequencing.roche.com/global/en/article-listing/hybridization-based-target-enrichment.html]
- 47. Hybridization capture vs amplicon sequencing | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/targeted-sequencing-hybridization-capture-or-amplicon-sequencing/]
- 48. Amplicon Sequencing vs. Other NGS Techniques [https://www.paragongenomics.com/amplicon-sequencing-vs-other-ngs-techniques-choosing-the-right-approach-for-your-research/]
- 49. NGS target enrichment method comparison [https://www.paragongenomics.com/cleanplex-technology-ultra-fast-simple-and-scalable-targeted-sequencing-solution-for-illumina-ion-torrent-and-mgiseq/]
- 50. Multicenter In-House Evaluation of an Amplicon-Based Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11860996/]
- 51. Clinical implementation of next-generation sequencing ... [https://www.nature.com/articles/s41598-024-84909-9]
- 52. Comprehensive Genomic Profiling (CGP) | Cancer genomic profiling benefits [https://sapac.illumina.com/areas-of-interest/cancer/ngs-in-oncology/cgp.html]
- 53. Clinical Value of Biomarker Testing in NSCLC [https://www.lungevity.org/learn-about-lungevity/precision-medicine/clinical-value-of-biomarker-testing-in-nsclc]
- 54. Editorial for Special Issue “Linking Genomic Changes with Cancer in the NGS Era, 2nd Edition” [https://www.mdpi.com/1467-3045/47/11/937]
- 55. Next-Generation Sequencing (NGS) in Drug Discovery & ... [https://www.corning.com/worldwide/en/products/life-sciences/resources/stories/the-cutting-edge/integrating-next-generation-sequencing-into-drug-discovery.html]
- 56. Next-Generation Sequencing in Drug Development [https://dscnextconference.com/next-generation-sequencing-in-drug-development-accelerating-precision-and-innovation/]
- 57. An NGS Workflow Blueprint for DNA Sequencing Data and Its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4827795/]
- 58. NGS is evolving: collaboration and tech lead the way [https://www.drugtargetreview.com/article/168383/ngs-is-evolving-collaboration-and-tech-lead-the-way/]
- 59. Next-Generation Sequencing in Drug Discovery Market ... [https://www.cervicornconsulting.com/next-generation-sequencing-in-drug-discovery-market]
- 60. Four Primary Steps and Tools for NGS Data Analyses [https://www.goldbio.com/blogs/articles/four-steps-for-ngs-data-analyses?srsltid=AfmBOop2YrKzf1z3A7q5VDz8W7umR1Rx9FnWu2jEK9BGcqtpE9dscgbC]
- 61. Randomized Clinical Trials With Biomarkers: Design Issues [https://pmc.ncbi.nlm.nih.gov/articles/PMC2911042/]
- 62. Stratification and Partial Ascertainment of Biomarker Value in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4130784/]
- 63. Biomarker-Driven Clinical Trials in Oncology [https://www.precisionformedicine.com/blog/biomarker-driven-clinical-trials-in-oncology-enrichment-stratification-all-comers-basket-when-to-use-what-and-why]
- 64. NGS Data Analysis for Illumina Platform—Overview and ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/next-generation-sequencing/ngs-data-analysis-illumina.html]
- 65. The diagnostic accuracy of next-generation sequencing in ... [https://pubmed.ncbi.nlm.nih.gov/40919126/]
- 66. The Diagnostic Accuracy of Next-Generation Sequencing ... [https://www.sciencedirect.com/science/article/pii/S2950195425000414]
- 67. Enhancing biomarker based oncology trial matching using ... [https://www.nature.com/articles/s41746-025-01673-4]
- 68. Circulating tumor DNA to monitor treatment response in ... [https://www.nature.com/articles/s41698-025-00876-y]
- 69. Liquid biopsy – a narrative review with an update on current ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12333414/]
- 70. Drug Discovery News — ctDNA monitoring is providing a ... [https://friendsofcancerresearch.org/news/drug-discovery-news-ctdna-monitoring-is-providing-a-smarter-way-to-track-and-treat-cancer/]
- 71. Next-generation sequencing in cancer diagnosis and ... [https://pubmed.ncbi.nlm.nih.gov/40253661/]
- 72. Next Generation Sequencing (NGS) and Cancer: New ... [https://www.frontiersin.org/research-topics/58203/next-generation-sequencing-ngs-and-cancer-new-steps-towards-personalized-medicine/magazine]
- 73. NGS for Precision Oncology: What Every Clinician Should ... [https://www.thermofisher.com/us/en/home/clinical/preclinical-companion-diagnostic-development/oncomine-oncology/education/ngs-oncologist.html]
- 74. Demonstrating the Clinical Utility of Foundation Medicine's ... [https://www.foundationmedicine.com/blog/q3-research-recap-blog?utm_source=linkedin&utm_medium=organic-social&utm_campaign=q3-research-recap&utm_content=corporate]
- 75. Cancer in a drop: Liquid biopsy highlights from the ... [https://www.sciencedirect.com/science/article/pii/S2950195425000360]
- 76. Circulating tumor DNA: a biomarker for oncology drug ... [https://pubmed.ncbi.nlm.nih.gov/40622305/]
- 77. Real-world application of tumor mutational burden-high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8717431/]
- 78. Can you use NGS panels to test for Tumor Mutational ... [https://nonacus.com/blog-can-you-use-ngs-panels-to-test-for-tumor-mutational-burden/]
- 79. Microsatellite instability and high tumor mutational burden ... [https://www.sciencedirect.com/science/article/abs/pii/S2210776224001558]
- 80. Tumor Microenvironment Analysis using NGS Workflows [https://www.qiagen.com/us/applications/cancer-research-and-oncology-solutions/tumor-microenvironment-analysis]
- 81. Crosstalk Between the MSI Status and Tumor ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2020.02039/full]
- 82. High tumor mutational burden assessed through next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11619254/]
- 83. Potential barriers to implementation of next-generation ... [https://pubmed.ncbi.nlm.nih.gov/39606845/]
- 84. Potential barriers to implementation of next-generation ... [https://www.openhealthgroup.com/publication-library/potential-barriers-to-implementation-of-next-generation-sequencing-in-cancer-management-a-u-s-physician-based-survey/]
- 85. Assessing the Cost-Effectiveness of Next-Generation ... [https://www.sciencedirect.com/science/article/pii/S109830152402357X]
- 86. Prevalence of druggable mutations and clinical trial ... [https://www.sciencedirect.com/science/article/pii/S0169500225004428]
- 87. Treatment choices based on multiplatform profiling platform ... [https://www.carislifesciences.com/research/publications/treatment-choices-based-multiplatform-profiling-platform-unlike-sequencing-alone-not-cause-cost-explosion-refractory-cancer-patients/]
- 88. 解读《肿瘤个体化治疗检测技术指南》 - ByDrug - 医药魔方 [https://bydrug.pharmcube.com/news/detail/f749ad4ec2330d9411b114784732fdc5]
- 89. Advances in Research on Circulating Tumor Cells in Lung ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5999834/]
- 90. Next-Generation Sequencing: A Review of Its Transformative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523276/]
- 91. 黏液性卵巢癌中HER2突变谱分析,德曲妥珠单抗或可作为 ... [https://www.medsci.cn/article/show_article.do?id=84fe9088039a]
- 92. Optimizing Bioinformatics Workflows: Why It Matters & ... [https://ardigen.com/optimizing-bioinformatics-workflows-why-it-matters-and-when-to-start-thinking-about-it/]
- 93. Clinical Oncology Next Generation Sequencing Market ... [https://www.novaoneadvisor.com/report/clinical-oncology-next-generation-sequencing-market]
- 94. Benchmarking of computational error-correction methods for ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-01988-3]
- 95. Key Strategies for Optimizing NGS Workflows [https://www.technologynetworks.com/genomics/articles/key-strategies-for-optimizing-ngs-workflows-395232]
- 96. Optimizing Next Generation Sequencing with Automation ... [https://seqwell.com/optimizing-next-generation-sequencing-with-automation-and-one-step-workflows/]
- 97. NGS: Six top tips to improve your sequencing library [https://www.ogt.com/ca/resources/ngs-resources-and-support/ngs-resources/ngs-six-top-tips-to-improve-your-sequencing-library/]
- 98. Standardization and quality management in next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5025460/]
- 99. QMS Tools and Resources | Laboratory Quality [https://www.cdc.gov/lab-quality/php/ngs-quality-initiative/qms-tools-resources.html]
- 100. Evolving Standards in Clinical NGS (updated) [https://www.euformatics.com/blog-post/evolving-standards-in-clinical-ngs]
- 101. Understanding variants of unknown significance and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11299939/]
- 102. Actionability classification of variants of unknown ... [https://www.nature.com/articles/s41698-023-00420-w]
- 103. ngs.plot: Quick mining and visualization of next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4028082/]
- 104. Analysis of error profiles in deep next-generation sequencing ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1659-6]
- 105. Error-corrected sequencing strategies enable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7057603/]
- 106. Error-corrected next generation sequencing – Promises ... [https://www.sciencedirect.com/science/article/pii/S1383574223000145]
- 107. Systematic evaluation of error rates and causes in short ... [https://www.nature.com/articles/s41598-018-29325-6]
- 108. Analysis of error profiles in deep next-generation sequencing ... [https://pubmed.ncbi.nlm.nih.gov/30867008/]
- 109. 2025 Trends: NGS [https://www.genengnews.com/topics/genome-editing/2025-trends-ngs/]
- 110. Integrating Artificial Intelligence in Next-Generation Sequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC12191491/]
- 111. False-negative errors in next-generation sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6742382/]
- 112. Companion Diagnostics Explained [https://www.foundationmedicine.com/blog/companion-diagnostics-explained-their-critical-role-cancer-care-and-our-latest-approvals]
- 113. Analytical validation (accuracy, reproducibility, limit of ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0329697]
- 114. Analytical validation (accuracy, reproducibility, limit of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12431237/]
- 115. Validation of genetic variants from NGS data using deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10116675/]
- 116. AMLVaran: a software approach to implement variant analysis ... [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-020-0668-3]
- 117. Computational Functional Genomics-Based AmpliSeq ... [https://www.mdpi.com/1422-0067/22/2/878]
- 118. Companion Diagnostics Development [https://www.illumina.com/areas-of-interest/genomics-in-drug-development/companion-dx-development.html]
- 119. Next Generation Sequencing, NGS | SeraCare Testing [https://www.seracare.com/Controls---Reference-Materials-NGS/]
- 120. Analytical Validation of a Pan-Cancer NGS Assay for In- ... [https://www.sciencedirect.com/science/article/pii/S152515782500220X]
- 121. Next Generation Sequencing ( NGS )… | College of American Pathologists [https://www.cap.org/member-resources/precision-medicine/next-generation-sequencing-ngs-worksheets]
- 122. Comparative performance of a targeted next-generation ... [https://www.sciencedirect.com/science/article/abs/pii/S1525157825002375]
- 123. Next-Generation Sequencing More Cost-Effective and ... [https://www.valuebasedcancer.com/categories/economics-value/next-generation-sequencing-more-cost-effective-and-faster-than-single-gene-testing]
- 124. NGS Testing More Cost-Effective Than SGT in Oncology [https://www.jons-online.com/web-exclusives/ngs-testing-more-cost-effective-than-sgt-in-oncology]
- 125. Comparison of next-generation sequencing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6988007/]
- 126. Benchmarking standard-of-care and emerging genomic ... [https://www.nature.com/articles/s41416-025-03204-0]
- 127. Different NGS identification methods of somatic mutation ... [https://pubmed.ncbi.nlm.nih.gov/40065248/]
- 128. Somatic mutation and selection at population scale [https://www.nature.com/articles/s41586-025-09584-w]
- 129. Augmenting cost-effectiveness in clinical diagnosis using ... [https://www.nature.com/articles/s10038-025-01403-4]
- 130. Comprehensive Review on the Clinical Impact of Next- Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10309568/]
- 131. implementation of next- Clinical sequencing testing and... generation [https://www.nature.com/articles/s41598-024-84909-9?error=cookies_not_supported&code=3edb5421-90b5-4531-a7f8-79547c51cf69]
- 132. High clinical actionability of a pan-cancer tissue-based ... [https://www.nature.com/articles/s41698-025-01126-x]
- 133. Clinical Validation of Duoseq, a novel assay for ... [https://pubmed.ncbi.nlm.nih.gov/41167303/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=10MSL6XygFtCE0XpaQpog-pEt8342fXkY9lzrR7q6H0wVRyEa2&fc=None&ff=20251031202225&v=2.18.0.post22+67771e2]
- 134. Multi-omics prognostic marker discovery and survival ... [https://www.nature.com/articles/s41598-025-20572-y]
- 135. A network-based discovery of prognostic markers in ... [https://www.frontiersin.org/article/1672015]
- 136. The clinical utility and prognostic value of next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12202931/]
- 137. Exact Sciences Announces Expanded Clinical Validation ... [https://www.exactsciences.com/newsroom/press-releases/exact-sciences-announces-expanded-clinical-validation-of-the-oncodetect-test]
- 138. Solution for Early NGS Screening Market | Global Market... Cancer [https://www.futuremarketinsights.com/reports/ngs-solution-for-early-cancer-screening-market]
- 139. List of Cleared or Approved Companion Diagnostic Devices [https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools]
- 140. An analysis of FDA drug approvals for oncological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507643/]
- 141. Next Generation Sequencing: 510k Regulatory ... [https://www.i3cglobal.com/next-generation-sequencing/]
- 142. FDA Approvals in Oncology: July-September 2025 | Blog [https://www.aacr.org/blog/2025/10/02/fda-approvals-in-oncology-july-september-2025/]
- 143. Laboratory Developed Tests [https://www.fda.gov/medical-devices/in-vitro-diagnostics/laboratory-developed-tests]
- 144. Implementing Alternative Methods [https://www.fda.gov/science-research/advancing-alternative-methods-fda/implementing-alternative-methods]
- 145. Application of next-generation sequencing to improve cancer ... [https://pubmed.ncbi.nlm.nih.gov/29265354/]
- 146. - Cost analysis comparing companion diagnostic tests for... effectiveness [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-020-07240-2]
- 147. Frontiers | Next-generation sequencing impact on cancer care... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2024.1420190/full]
- 148. Next-generation sequencing in oncology: challenges ... [https://pubmed.ncbi.nlm.nih.gov/39096135/]
- 149. Cost-effectiveness analysis of metagenomic next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12604022/]
- 150. United States Tumor Profiling Research Report 2025-2033 [https://finance.yahoo.com/news/united-states-tumor-profiling-research-130100611.html]
- 151. Tumor Cancer Market | Global Market Analysis Report - 2035 Profiling [https://www.factmr.com/report/cancer-tumor-profiling-market]