Illumina vs. Ion Torrent in Cancer Diagnostics: A 2025 Comparative Guide for Precision Oncology
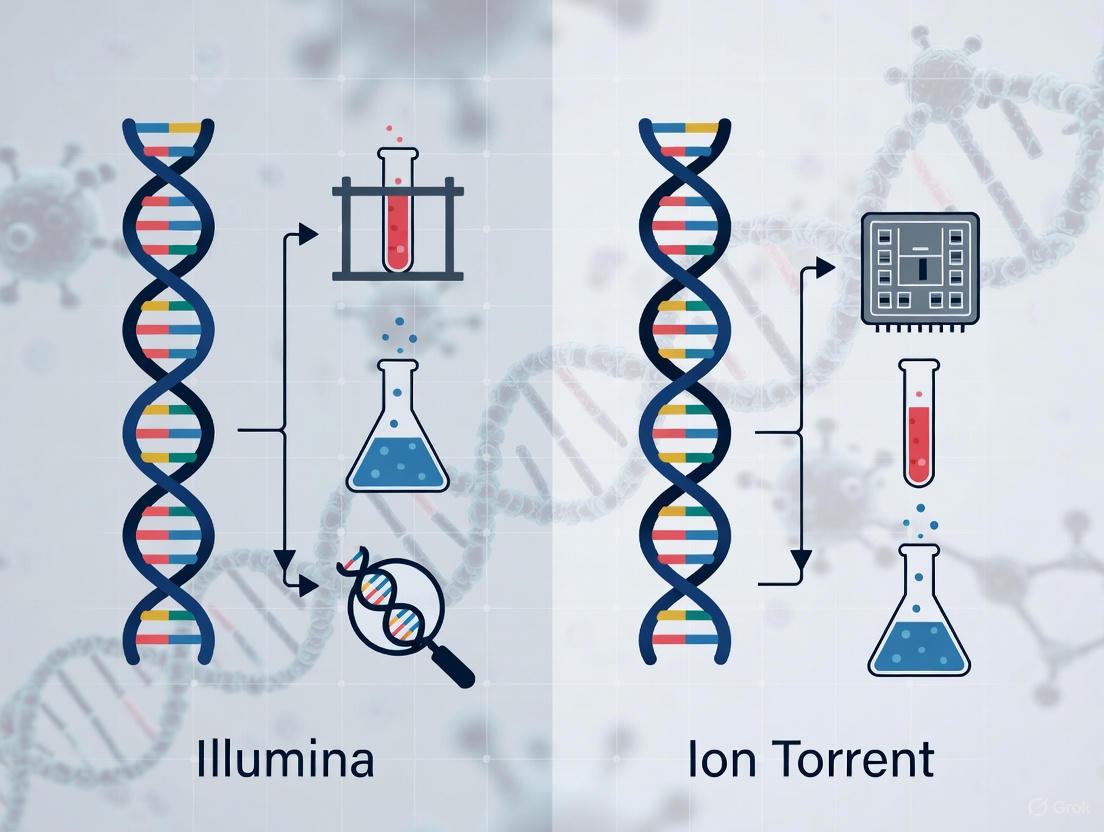
This article provides a comprehensive comparison of Illumina and Ion Torrent next-generation sequencing (NGS) platforms for cancer diagnostics, tailored for researchers, scientists, and drug development professionals.
Illumina vs. Ion Torrent in Cancer Diagnostics: A 2025 Comparative Guide for Precision Oncology
Abstract
This article provides a comprehensive comparison of Illumina and Ion Torrent next-generation sequencing (NGS) platforms for cancer diagnostics, tailored for researchers, scientists, and drug development professionals. It explores the foundational technologies, including Illumina's sequencing-by-synthesis and Ion Torrent's semiconductor-based detection. The scope covers methodological applications in comprehensive genomic profiling and liquid biopsy, addresses troubleshooting for GC-bias and variant calling, and presents validation data on sensitivity, specificity, and concordance. By synthesizing recent clinical studies and technological advances, this guide aims to inform strategic platform selection to optimize precision oncology workflows, biomarker discovery, and companion diagnostic development.
Core Technologies Unveiled: The Fundamental Principles of Illumina and Ion Torrent Sequencing
Next-generation sequencing (NGS) has become the cornerstone of modern genomic research, enabling the rapid, high-throughput analysis of DNA and RNA that is critical for applications like cancer diagnostics. While several platforms exist, two distinct technological approaches dominate the short-read sequencing market: Illumina's fluorescent dye-terminator method and Ion Torrent's semiconductor pH detection. These technologies represent fundamentally different solutions to the challenge of deciphering genetic code, each with unique strengths and limitations. For researchers designing studies in oncology, where accurately identifying somatic variants can dictate therapeutic decisions, understanding the core mechanics, performance characteristics, and practical implications of these two chemistries is paramount. This guide provides a detailed, objective comparison to inform platform selection for cancer diagnostics research.
Core Technology and Mechanism
The fundamental difference between these platforms lies in their method of base detection—one relies on optics and the other on electronics.
Illumina: Sequencing by Synthesis with Fluorescent Dyes
Illumina's technology is based on sequencing-by-synthesis (SBS) with reversible dye-terminators [1] [2].
- Workflow: DNA fragments are attached to a flow cell and amplified in situ via bridge PCR to create clusters of identical sequences [2].
- Base Detection: Each cycle introduces fluorescently labeled, reversibly terminated nucleotides. DNA polymerase incorporates a single complementary base per cluster. A laser excites the fluorophore, and a camera captures the specific fluorescent signal (color) to identify the base (A, C, G, or T) [2].
- Key Feature: After imaging, the fluorescent dye and terminator are chemically cleaved, allowing the next cycle to begin [2]. This process enables paired-end sequencing, where both ends of a DNA fragment are sequenced, providing superior alignment accuracy, especially in complex genomic regions [3] [2].
Ion Torrent: Semiconductor Sequencing via pH Sensing
Ion Torrent's technology bypasses optical detection entirely, translating chemical signals directly into digital data [2].
- Workflow: DNA libraries are amplified via emulsion PCR on microscopic beads. Each DNA-coated bead is deposited into a millions-strong array of microscopic wells on a proprietary semiconductor chip [2].
- Base Detection: The sequencer sequentially flows unmodified nucleotides (A, C, G, T) over the chip. When a nucleotide is incorporated into a growing DNA strand by polymerase, a hydrogen ion (H+) is released as a natural byproduct. This release causes a minute, localized pH change [2].
- Key Feature: An ion-sensitive field-effect transistor (ISFET) beneath each well detects this pH shift [2]. The magnitude of the pH change is theoretically proportional to the number of nucleotides incorporated in a single flow, which is crucial for interpreting homopolymer regions (e.g., a run of "AAAA") [2].
The following diagram illustrates the core biochemical detection mechanisms for each technology.
Performance Comparison in Cancer Research
For cancer diagnostics, key performance metrics include accuracy, throughput, and the ability to detect variants. The following table summarizes quantitative data critical for platform evaluation.
| Performance Metric | Illumina (Fluorescent Dye-Terminator) | Ion Torrent (Semiconductor pH Detection) |
|---|---|---|
| Primary Error Type | Low substitution error rate (~0.1-0.5%) [2] | Higher insertion-deletion (indel) rate, specifically in homopolymer regions [2] |
| Reported Raw Error Rate | Typically below 1% (often 0.1-0.5%) [2] | Approximately 1% (roughly double Illumina's rate) [2] |
| Read Structure | Uniform length; supports paired-end reads [3] [2] | Variable length; single-end reads only [3] [2] |
| Typical Read Lengths | Up to 2x300 bp (paired-end) [2] | Up to 400-600 bp (single-end) [2] |
| Throughput (per run) | Scalable from millions to billions of reads [2] | Millions to tens of millions of reads [2] |
| Speed | ~24-48 hours for a high-output run [2] | As fast as a few hours for smaller runs [2] |
| Alignment Concordance | Strong gene-level Spearman correlation (0.938-0.974) with Ion Torrent in controlled studies [3] | Strong gene-level correlation with Illumina, though alignment rates can vary by software [3] |
Experimental Data from Direct Comparisons
A 2017 study directly compared the platforms using a treatment/control experimental design, which mirrors the subtle transcriptional differences sought in cancer research [3].
- Gene Quantification: The study found a strong Spearman correlation (ranging from 0.9380 to 0.9737) between gene-level read counts from the Illumina HiSeq and Ion Torrent Proton platforms, indicating good agreement in measuring gene expression levels [3].
- Differential Expression: There was a moderate level of concordance when identifying differentially expressed genes (DEGs) between the two conditions. However, the biological conclusions at the pathway level were nearly identical, suggesting both platforms can effectively identify core biological mechanisms [3].
- Alignment Sensitivity: A critical finding was a strong interaction between the sequencing platform and the choice of alignment software. For instance, the STAR aligner showed a lower percentage of uniquely mapped reads for Ion Torrent data, potentially due to its variable read length [3]. This highlights the need to optimize bioinformatic tools for the specific platform.
The Scientist's Toolkit: Essential Reagents and Materials
Successful NGS requires a suite of specialized reagents. The table below details key solutions for library preparation and sequencing.
| Research Reagent / Material | Function in Workflow | Platform Specificity |
|---|---|---|
| Flow Cell (Illumina) | Glass surface with patterned lanes for bridge amplification and sequencing-by-synthesis [2]. | Illumina |
| Semiconductor Chip (Ion Torrent) | Proprietary chip containing millions of ion-sensitive wells that house DNA beads and detect pH changes [2]. | Ion Torrent |
| Reversible Terminator Nucleotides | Nucleotides labeled with a specific fluorophore and blocked to allow single-base incorporation per cycle [2]. | Illumina |
| Unmodified Nucleotides | Natural nucleotides flowed sequentially; incorporation is detected via H+ release without optical labels [2]. | Ion Torrent |
| Library Preparation Kit | Kits for fragmenting DNA, ligating platform-specific adapters, and (optionally) amplifying the library [1]. | Both (but kit chemistry is platform-specific) |
| Emulsion PCR Reagents | Reagents for performing clonal amplification of DNA libraries on microscopic beads in water-in-oil emulsions [2]. | Ion Torrent |
| Bridge PCR Reagents | Reagents for clonal amplification of DNA libraries on a flat, solid-phase flow cell surface [2]. | Illumina |
The following diagram and protocol outline a typical differential gene expression (DGE) experiment, a common application in cancer research for comparing tumor to normal tissue.
Detailed Methodology for a DGE Study [3]:
Sample Preparation & Library Construction:
- Extract total RNA from matched patient samples (e.g., tumor and normal adjacent tissue).
- For Illumina: Libraries are typically prepared using poly-A selection to enrich for mRNA, followed by fragmentation and adapter ligation.
- For Ion Torrent: Platform-specific adapters are ligated, and libraries are amplified via emulsion PCR on beads.
Sequencing:
- Load the prepared libraries onto their respective platforms—a flow cell for Illumina or a semiconductor chip for Ion Torrent.
- Perform sequencing according to the manufacturer's protocol. A common depth for DGE is 20-40 million reads per sample.
Data Analysis:
- Quality Control: Assess raw read quality using tools like FastQC.
- Read Alignment: Map sequencing reads to a reference genome.
- The choice of aligner is critical. The cited study used GSNAP and STAR for both platforms but found a sequential alignment strategy (STAR followed by Bowtie2) performed well for Ion Torrent data [3].
- Gene Quantification: Count the number of reads mapping to each gene using tools like featureCounts or HTSeq.
- Differential Expression: Identify genes significantly differentially expressed between tumor and normal groups using statistical packages like DESeq2 or edgeR. The cited study used a Mann-Whitney U test with Benjamini-Hochberg correction [3].
- Pathway Analysis: Input the list of DEGs into pathway analysis tools (e.g., DAVID, GSEA) to identify affected biological processes.
The choice between Illumina and Ion Torrent sequencing chemistries involves a trade-off between accuracy, throughput, speed, and cost. For large-scale projects like whole-genome sequencing or where detecting single-base substitutions is critical, Illumina's fluorescent-based system offers superior accuracy and the strategic advantage of paired-end reads. For rapid, lower-throughput targeted sequencing (e.g., monitoring a specific cancer gene panel), Ion Torrent's semiconductor technology provides a fast, cost-effective solution, though users must be wary of errors in homopolymer regions that could confound indel calling.
Ultimately, as the 2017 comparison study concluded, both platforms have a similar capacity to reveal core biology in a treatment/control experiment, with pathway-level analysis being nearly identical [3]. The decision should be guided by the specific requirements of the cancer diagnostic application, available budget, and in-house bioinformatics expertise. As both technologies continue to evolve, the trend towards higher accuracy, longer reads, and lower costs will further empower precision oncology.
Next-generation sequencing (NGS) has revolutionized oncology by enabling comprehensive genomic profiling of tumors, facilitating personalized treatment plans that target specific mutations and improve patient outcomes [4]. In clinical cancer diagnostics, two major platforms dominate the landscape: Illumina and Ion Torrent. Each employs distinct technological approaches for library preparation, template generation, and sequence detection, leading to different performance characteristics that influence their application in research and clinical settings. Understanding these workflow differences is crucial for researchers, scientists, and drug development professionals seeking to implement NGS in precision oncology. This guide provides an objective comparison of Illumina and Ion Torrent platforms, focusing on their architectural principles, experimental workflows, and performance data in cancer genomics.
Platform Architecture and Core Technologies
The fundamental difference between Illumina and Ion Torrent platforms lies in their sequencing chemistry and detection methods. Illumina utilizes Sequencing-by-Synthesis (SBS) technology with fluorescently labeled nucleotides, detecting incorporation events optically through fluorescence imaging [5] [4]. In contrast, Ion Torrent employs semiconductor sequencing, detecting hydrogen ions released during DNA polymerization through pH changes, without requiring optical scanning or fluorescent dyes [6] [4]. This core architectural difference significantly impacts their workflow requirements, run times, and data characteristics.
Illumina's SBS chemistry involves bridge amplification on flow cells to create clusters of identical DNA fragments [4]. Each cycle incorporates fluorescently tagged nucleotides that emit specific signals when excited by lasers, with the fluorescence captured by imaging systems. The reversible terminator chemistry ensures single-base resolution through cyclic nucleotide incorporation and imaging. Recent advancements like XLEAP-SBS chemistry have further enhanced speed and quality [5].
Ion Torrent's semiconductor approach performs sequencing on microwells placed over ion sensors. When a nucleotide is incorporated into the DNA strand, a hydrogen ion is released, causing a pH change detectable by the ion sensor [6] [4]. The key advantage is the direct detection of chemical changes without cameras or scanners, potentially simplifying instrument design. However, homopolymer regions (stretches of identical consecutive bases) can be challenging as they produce a larger signal proportional to length, sometimes leading to interpretation errors [4].
Library Preparation Workflows Compared
Fundamental Library Construction Steps
Despite platform differences, NGS library preparation shares common steps across technologies. The process begins with nucleic acid extraction from samples (tissue, cells, or biofluids), followed by quality control using UV spectrophotometry and fluorometric methods [5]. Library construction then proceeds through these core steps:
- Fragmentation: Sample DNA is fragmented physically, enzymatically, or chemically to optimal sizes (typically 150-300 bp for Illumina, 200-400 bp for Ion Torrent) [4] [7].
- End Repair and A-tailing: Fragmented DNA with sticky ends is converted to blunt ends, and an adenosine base is added to 3' ends to facilitate adapter ligation (note: A-tailing is not required for Ion Torrent library preparation) [7].
- Adapter Ligation: Platform-specific adapters containing sequencing priming sites and barcodes are ligated to fragments [8] [9] [7].
- Library Amplification: Adapter-ligated fragments are amplified via PCR to generate sufficient material for sequencing [7].
- Purification and Quality Control: Final libraries are purified and quantified before sequencing [8].
Platform-Specific Library Preparation Methods
Illumina offers multiple library prep technologies, including:
- Bead-Linked Transposome Tagmentation: Uses bead-bound transposomes for simultaneous fragmentation and adapter insertion, providing more uniform reactions than in-solution methods [8].
- Adapter Ligation: Traditional method involving fragmentation followed by adapter ligation [8].
- Amplicon Library Prep: PCR-based workflow suitable for users new to NGS, enabling simultaneous measurement of thousands of targets [8].
Ion Torrent library preparation typically involves end repair of 3' and 5' ends, adaptor ligation, size-selection of adaptor-ligated molecules, and PCR amplification [9]. Libraries can be prepared manually or automated using systems like the AB Library Builder, which reduces hands-on time by more than 50% compared to manual methods [6].
Table 1: Library Preparation Method Comparison
| Parameter | Illumina | Ion Torrent |
|---|---|---|
| Fragmentation Methods | Bead-linked transposome tagmentation, enzymatic, physical | Enzymatic, physical |
| A-tailing Required | Yes | No [7] |
| Adapter Design | Platform-specific with priming sites | Platform-specific with priming sites |
| Automation Options | Hamilton, Beckman, Eppendorf, Tecan systems [10] | AB Library Builder, Ion Chef Systems [6] |
| Typical Hands-on Time | ~45 min to 3 hours depending on kit [8] | Varies; >50% reduction with automation [6] |
Diagram 1: Comparative NGS Library Preparation Workflow
Experimental Methodology for Platform Comparison
Study Design and Sample Processing
A recent comparative study provides objective performance data between Ion Torrent Genexus and Illumina-based FoundationOne systems in cancer diagnostics [11]. The research employed a methodological approach with six cancer patients (breast and head/neck cancers) using matched tissue and blood samples. For tissue analysis, researchers compared Genexus Oncomine Comprehensive Assay v3 (OCA) against FoundationOne CDx (F1), while for blood-based circulating tumor DNA analysis, Genexus Oncomine Precision Assay (OPA) was compared to FoundationOne Liquid (F1L) [11].
Sample processing followed standardized protocols: DNA and RNA from formalin-fixed paraffin-embedded (FFPE) tissue specimens were extracted using Maxwell RSC Instrument with FFPE-specific kits. Blood plasma was obtained through double centrifugation of EDTA-treated whole blood, with cell-free total nucleic acid extracted using Maxwell RSC miRNA Plasma and Serum Kit [11]. Nucleic acid concentrations were measured using fluorometric systems, with minimum quality thresholds established (>1.1 ng/μl for tissue DNA, >0.95 ng/μl for tissue RNA, >1.33 ng/μl for blood) to proceed with sequencing [11].
Bioinformatics and Analysis Parameters
The study analyzed 130 genes common between F1 and OCA, and 41 genes between F1L and OPA [11]. Variant concordance was assessed by comparing genomic alterations detected by both systems, with sensitivity and specificity calculated. The analysis focused on different variant types including single-nucleotide variants (SNVs), copy number alterations (CNAs), and gene fusions relevant to cancer diagnostics [11].
Performance Data and Comparison Results
Concordance Metrics and Variant Detection
The comparative study revealed that when analyzing common genes across both platforms, the Genexus system demonstrated 55% sensitivity and 99% specificity compared to FoundationOne as the reference [11]. Both platforms successfully detected several important cancer-related variants, including nine SNVs, one CNA, and one fusion gene [11].
However, platform-specific differences emerged in detection capabilities. The Ion Torrent Genexus system uniquely detected one SNV (MAP2K1 F53V), two CNAs (AKT3 and MYC), and one fusion (ESR-CCDC170) that were not identified by FoundationOne [11]. Conversely, FoundationOne detected two SNVs (TP53 Q331* and KRAS G12V) that were not found by Genexus [11]. These findings indicate that while the two cancer genome panels are largely equivalent, they are not perfectly aligned in detection capabilities, suggesting that different assays and analytical methods influence results.
Table 2: Performance Comparison in Cancer Genomic Profiling
| Performance Metric | Genexus (Ion Torrent) | FoundationOne (Illumina) |
|---|---|---|
| Sensitivity | 55% (vs. FoundationOne) | Reference standard |
| Specificity | 99% (vs. FoundationOne) | Reference standard |
| Commonly Detected Variants | 9 SNVs, 1 CNA, 1 fusion | 9 SNVs, 1 CNA, 1 fusion |
| Uniquely Detected SNVs | MAP2K1 F53V | TP53 Q331*, KRAS G12V |
| Uniquely Detected CNAs | AKT3, MYC | None reported |
| Uniquely Detected Fusions | ESR-CCDC170 | None reported |
| Genes Analyzed (Tissue) | 130 common genes | 324 total genes |
| Genes Analyzed (Blood) | 41 common genes | Not specified |
Practical Workflow Considerations
From an operational perspective, the Ion Torrent Genexus system offers automation advantages with minimal manual steps - requiring only a few pipetting iterations before loading prefilled reagents and consumables [11]. This automation potential makes NGS services feasible in routine clinical diagnostic laboratories, including regional hospitals with limited NGS experience [11]. The integrated workflow reduces technical variability and may facilitate implementation in resource-limited settings.
Illumina systems, while potentially requiring more specialized expertise, provide comprehensive bioinformatics solutions through their Connected Software portfolio, including push-button analysis options that make data interpretation accessible to users without extensive bioinformatics backgrounds [5]. The platform's versatility supports a broad range of applications and throughput needs, from small labs to genome centers [5] [8].
Essential Research Reagent Solutions
Table 3: Key Research Reagents for NGS Cancer Diagnostics
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| Nucleic Acid Extraction Kits | Maxwell RSC FFPE Plus DNA Kit, Maxwell RSC RNA FFPE Kit, Maxwell RSC miRNA Plasma and Serum Kit [11] | Isolation of high-quality DNA/RNA from various sample types including FFPE tissue and blood plasma |
| Library Preparation Kits | Illumina DNA Prep, Illumina RNA Prep, Ion AmpliSeq Library Kit, Ion Xpress Plus Fragment Library Kit [6] [8] | Convert nucleic acids to sequence-ready libraries with platform-specific adapters |
| Target Enrichment Panels | Oncomine Comprehensive Assay v3, Oncomine Precision Assay, FoundationOne CDx, AmpliSeq Cancer Hotspot Panel [11] [6] | Selectively target cancer-related genes for sequencing |
| Quantification Reagents | QuantiFluor ONE dsDNA System, QuantiFluor RNA System [11] | Accurate measurement of nucleic acid concentration and quality before sequencing |
| Automation Systems | Hamilton Microlab NGS STAR, Beckman Biomek i7, AB Library Builder, Ion Chef Systems [6] [10] | Standardize and scale library preparation, reducing hands-on time and variability |
| Quality Control Tools | Agilent 4200 TapeStation, Fragment Analyzer systems [11] [10] | Assess fragment length distribution and library quality before sequencing |
Diagram 2: Platform Architecture and Performance Relationship
The comparative analysis demonstrates that both Illumina and Ion Torrent platforms offer viable solutions for cancer genomic profiling, each with distinct advantages. The Ion Torrent Genexus system provides an automated, integrated workflow particularly suitable for clinical settings with limited NGS expertise, offering rapid turnaround times and minimal manual intervention [11]. Meanwhile, Illumina's FoundationOne delivers comprehensive genomic coverage with established performance metrics as a FDA-approved companion diagnostic [11].
The choice between platforms should be guided by specific research or clinical needs. For laboratories prioritizing automation and ease-of-use, Ion Torrent systems present an attractive option. For applications requiring the most extensive genomic coverage and regulatory approval, Illumina platforms may be preferable. Critically, the detection differences observed between platforms highlight the importance of considering each test's characteristics and the specific genetic variants relevant to the disease being studied [11]. As precision oncology continues to evolve, understanding these platform workflows and architectures becomes essential for optimizing cancer diagnostic strategies and advancing personalized treatment approaches.
Inherent Strengths and Limitations of Short-Read Sequencing Technologies
Next-generation sequencing (NGS) technologies have revolutionized genomic research by enabling massively parallel DNA sequencing that is faster, cheaper, and more accurate than traditional methods [2]. Among these technologies, short-read sequencing platforms—primarily Illumina and Ion Torrent—currently dominate the landscape for applications requiring high accuracy and throughput, particularly in cancer diagnostics research [2] [1]. These technologies are characterized by their ability to sequence DNA in small fragments typically ranging from 50 to 600 base pairs, with the resulting reads then computationally assembled against a reference genome [12].
The fundamental principle underlying these platforms is "sequencing by synthesis," though they employ distinct detection mechanisms [2] [1]. Illumina utilizes a fluorescence-based method with reversible terminators, while Ion Torrent relies on semiconductor technology to detect pH changes [2]. These methodological differences create complementary profiles of inherent strengths and limitations that researchers must carefully consider when selecting a platform for specific applications, especially in clinical cancer diagnostics where accuracy, turnaround time, and cost directly impact patient care [2] [11].
Technology Comparison: Core Methodologies
Illumina Sequencing-by-Synthesis Technology
Illumina's sequencing technology employs a fluorescence-based detection system that has become the gold standard for short-read sequencing [2] [13]. The process begins with DNA fragmentation and adapter ligation to create sequencing libraries. These libraries are loaded onto a flow cell where they undergo bridge amplification through solid-phase PCR, generating millions of clusters of identical DNA fragments [2] [1]. During sequencing, the system cycles through all four fluorescently labeled, reversibly terminated nucleotides. As DNA polymerase incorporates a complementary base at each cluster, a camera captures the fluorescent signal emitted [2]. A key advantage of this method is the reversible terminator chemistry, which ensures only one base is added per cycle before the terminator is removed to allow subsequent incorporation [13]. This approach provides highly accurate, base-by-base sequencing that virtually eliminates errors associated with strings of repeated nucleotides (homopolymers) [13]. Illumina platforms excel at generating paired-end reads, sequencing each DNA fragment from both ends, which effectively doubles the information per fragment and significantly aids in read alignment and detection of structural variants [2].
Ion Torrent Semiconductor Sequencing Technology
Ion Torrent platforms utilize a fundamentally different detection approach based on semiconductor technology [2]. Similar to other NGS platforms, DNA libraries are prepared through fragmentation and adapter ligation, but amplification occurs via emulsion PCR on microscopic beads [2] [1]. Each DNA-coated bead is deposited into a well on a semiconductor chip containing millions of wells. The sequencer cycles through each DNA base (A, C, G, T) sequentially, and when a complementary base is incorporated into the growing DNA strand, a hydrogen ion is released, causing a minute pH change [2]. This chemical signal is detected by an ion-sensitive sensor under each well and directly converted to digital data [2]. This electronic detection method eliminates the need for lasers or cameras, allowing for more compact instruments and potentially simpler maintenance [2]. However, a significant limitation of this technology is its difficulty with homopolymer regions—stretches of identical bases—where the cumulative proton release struggles to precisely count long runs of the same nucleotide, leading to insertion/deletion errors [2]. Additionally, Ion Torrent generates only single-end reads, which can be a disadvantage for certain analyses where paired-end reads provide critical information [2].
Diagram: Ion Torrent semiconductor sequencing workflow combines emulsion PCR with pH change detection.
Performance Comparison in Cancer Diagnostics
Technical Specifications and Operational Characteristics
The table below summarizes the key technical specifications and performance metrics for Illumina and Ion Torrent platforms, highlighting their fundamental differences for cancer diagnostics research:
Table 1: Platform Technical Specifications Comparison
| Parameter | Illumina Platforms | Ion Torrent Platforms |
|---|---|---|
| Sequencing Chemistry | Fluorescent reversible terminators [2] [13] | Semiconductor pH detection [2] |
| Detection Method | Optical (cameras) [2] | Electronic (ion sensors) [2] |
| Amplification Method | Bridge amplification [2] [1] | Emulsion PCR [2] [1] |
| Read Configuration | Paired-end available [2] | Single-end only [2] |
| Typical Read Lengths | Up to 2×300 bp (paired-end) [2] | Up to 400-600 bp (single-end) [2] |
| Raw Base Accuracy | ~99.9% (error rate: 0.1-0.5%) [2] | ~99% (error rate: ~1%) [2] |
| Homopolymer Performance | High accuracy [13] | Prone to indels [2] |
| Run Time (typical) | 18-48 hours [2] | 2-24 hours [2] |
| Throughput Range | Millions to billions of reads [2] | Millions to tens of millions of reads [2] |
Experimental Comparison in Clinical Cancer Profiling
A 2025 study directly compared the performance of Ion Torrent Genexus and Illumina-based FoundationOne CDx for comprehensive genomic profiling (CGP) in cancer diagnostics [11]. The research analyzed tissue and blood samples from patients with breast, head, and neck cancers, focusing on variant detection concordance between the two clinical NGS systems.
Experimental Protocol: The study utilized matched samples from six cancer patients. For tissue analysis, researchers compared Genexus Oncomine Comprehensive Assay v3 (OCAv3) with FoundationOne CDx, while for blood-based analysis, they compared Genexus Oncomine Precision Assay (OPA) with FoundationOne Liquid CDx [11]. DNA and RNA from formalin-fixed paraffin-embedded (FFPE) tissue specimens were extracted using the Maxwell RSC Instrument with specialized kits, while cell-free total nucleic acid was extracted from blood plasma [11]. The concentrations and fragment lengths of nucleic acids were rigorously quantified before proceeding to library preparation and sequencing according to manufacturers' protocols for each platform [11].
Results and Concordance Data: The analysis revealed 130 genes common between FoundationOne and OCAv3, and 41 genes common between the liquid biopsy assays [11]. When comparing FoundationOne to Genexus for common genes, the sensitivity and specificity were 55% and 99%, respectively [11]. The study identified nine single-nucleotide variants (SNVs), one copy number alteration (CNA), and one fusion gene detected by both platforms. However, several variants were platform-specific: one SNV (MAP2K1 F53V), two CNAs (AKT3 and MYC), and one fusion (ESR1-CCDC170) were detected only by Genexus, while two SNVs (TP53 Q331* and KRAS G12V) were detected only by FoundationOne [11]. The authors concluded that while the two cancer genome panels were broadly equivalent, they were not perfectly concordant, indicating that different assays and analytical methods influenced the results [11].
Table 2: Variant Detection Concordance in Cancer Genomic Profiling
| Variant Category | Detected by Both Platforms | Detected Only by Genexus | Detected Only by FoundationOne |
|---|---|---|---|
| Single Nucleotide Variants (SNVs) | 9 | 1 (MAP2K1 F53V) | 2 (TP53 Q331*, KRAS G12V) |
| Copy Number Alterations (CNAs) | 1 | 2 (AKT3, MYC) | 0 |
| Gene Fusions | 1 | 1 (ESR1-CCDC170) | 0 |
| Overall Sensitivity | 55% | - | - |
| Overall Specificity | 99% | - | - |
Cross-Platform Compatibility in Genomic Analyses
The compatibility of data generated from different sequencing platforms represents a critical consideration for collaborative research and centralized analysis, particularly in outbreak investigation and genomic surveillance [14]. A 2025 study evaluated the compatibility of whole-genome sequencing data from Illumina and Ion Torrent devices for genomic analysis of Listeria monocytogenes, with implications for cancer genomics [14].
Experimental Protocol: Researchers performed WGS on 47 L. monocytogenes isolates using both Illumina and Ion Torrent platforms [14]. For Illumina sequencing, libraries were prepared using Nextera XT or DNA Prep Kits and sequenced on NextSeq 500 or MiSeq instruments in paired-end mode [14]. For Ion Torrent sequencing, libraries were prepared using the Ion Plus Fragment Library Kit and sequenced on Ion Torrent S5 instruments [14]. The raw sequences were trimmed, assembled, and quality-checked using the AQUAMIS pipeline with three different assemblers (MEGAHIT, SKESA, and SPAdes), followed by core genome multilocus sequence typing (cgMLST) analysis using a scheme containing 1,748 loci [14].
Results and Analytical Implications: The study found that only the SPAdes assembler delivered qualitatively comparable results between platforms [14]. In cgMLST analysis, the same-strain allele discrepancy between platforms averaged 14.5 alleles, well above the threshold of 7 alleles routinely used for cluster detection in L. monocytogenes [14]. The application of a strict frameshift filter reduced the mean discrepancy below this threshold but simultaneously reduced discriminatory power [14]. The platform's impact on read-based single nucleotide polymorphism (SNP) analysis was lower than on cgMLST, suggesting SNP analysis may be more robust for cross-platform studies [14]. The researchers concluded that while compatibility could be improved through bioinformatic filtering, perfect compatibility between platforms remained elusive [14].
Essential Research Reagent Solutions
The following table details key reagents and kits essential for implementing short-read sequencing workflows in cancer diagnostics research:
Table 3: Essential Research Reagents for Short-Read Sequencing Workflows
| Reagent/Kits | Primary Function | Application Notes |
|---|---|---|
| Maxwell RSC FFPE Plus DNA Kit [11] | DNA extraction from formalin-fixed paraffin-embedded (FFPE) tissue | Maintains DNA quality from archived clinical specimens; critical for cancer research |
| Maxwell RSC miRNA Plasma and Serum Kit [11] | Cell-free total nucleic acid extraction from blood plasma | Enables liquid biopsy approaches for cancer genomic profiling |
| Illumina Nextera XT DNA Library Prep Kit [14] | Library preparation for Illumina sequencing | Streamlines NGS library construction from limited input DNA |
| Ion Plus Fragment Library Kit [14] | Library preparation for Ion Torrent sequencing | Optimized for semiconductor sequencing workflow |
| xGen Amplicon Core Kit for SARS-CoV-2 [15] | Targeted amplicon sequencing (adaptable for cancer panels) | Enables focused sequencing of specific genomic regions |
| Qubit dsDNA BR Assay Kit [14] | Accurate DNA quantification | Essential for quality control before library preparation |
Short-read sequencing technologies from Illumina and Ion Torrent offer complementary profiles of strengths and limitations for cancer diagnostics research. Illumina platforms provide superior accuracy, particularly in homopolymer regions, higher throughput capabilities, and paired-end read configurations that benefit complex genomic analyses [2] [13]. Conversely, Ion Torrent systems offer faster turnaround times, simpler workflows with reduced hands-on time, and lower capital investment, making them attractive for laboratories with moderate throughput needs [2] [11].
The choice between these platforms ultimately depends on specific research requirements and clinical applications. For applications demanding the highest possible accuracy—such as variant calling for therapeutic decision-making—Illumina remains the gold standard [2] [11]. For rapid turnaround targeted sequencing or resource-constrained settings, Ion Torrent platforms provide a compelling alternative [2] [11]. As the 2025 experimental data demonstrates, understanding the nuanced performance characteristics of each platform is essential for selecting the appropriate technology and correctly interpreting results in cancer diagnostics research [11] [14].
The next-generation sequencing (NGS) landscape is dominated by Illumina and Thermo Fisher Scientific, whose Ion Torrent platform represents a key alternative. Their technologies operate on fundamentally different principles, leading to distinct performance characteristics that influence their application in cancer diagnostics.
Illumina's technology relies on sequencing-by-synthesis with fluorescently labeled, reversible-terminator nucleotides. Clonally amplified DNA templates are immobilized on a flow cell, and bases are incorporated over sequential cycles. Each incorporation is detected via fluorescence imaging, providing highly accurate base calling [16] [17]. This method produces reads of uniform length and supports paired-end sequencing, which is valuable for detecting complex genomic rearrangements in cancer [3].
In contrast, Ion Torrent's semiconductor sequencing detects the release of hydrogen ions as nucleotides are incorporated during DNA synthesis. Templated beads, prepared via emulsion PCR, are loaded into proton-sensing wells on a semiconductor chip. The key differentiator is that multiple identical nucleotides can be incorporated in a single cycle when traversing homopolymer regions, which is a major source of sequencing error for this platform [16] [17]. Ion Torrent typically generates variable-length reads and does not natively support paired-end sequencing [3].
The following diagram illustrates the core workflow differences between the two platforms.
Performance Comparison in Diagnostic Applications
Direct comparisons of these platforms reveal critical differences in performance metrics that directly impact their utility in cancer research, particularly in sensitivity for variant detection and coverage uniformity.
The table below summarizes key performance characteristics derived from comparative studies:
| Performance Metric | Illumina Platforms | Ion Torrent Platforms |
|---|---|---|
| Read Type | Uniform length, Paired-end capable [3] | Variable length, Single-end [3] |
| Typical Error Mode | Substitution errors [17] | Homopolymer indel errors [16] [17] |
| Library Prep Input | Higher input requirements [18] | Works well with limited DNA/RNA [18] |
| Sequencing Speed | Moderate to fast (hours to days) | Fast run times (hours) [18] |
| GC-Rich Region Performance | Good coverage uniformity [17] | Moderate coverage uniformity [17] |
| Extreme AT-Rich Region Performance | Good coverage uniformity [17] | Severe coverage loss (e.g., ~30% of P. falciparum genome) [17] |
Key Performance Differentiators in Cancer Genomics
Variant Calling Accuracy: For DNA sequencing, a study comparing the Illumina NextSeq 550Dx and Ion Torrent S5 XL using the same hybridization-capture panel found both platforms delivered high accuracy. The sensitivity for single-nucleotide variant (SNV) and indel calling was 98.53% for Illumina and 97.06% for Ion Torrent, with 100% specificity for both [18]. The slightly higher sensitivity for Illumina may be critical for detecting low-frequency tumor variants.
Coverage Bias: A fundamental difference emerges in sequencing genomes with extreme base compositions. While both platforms perform well on GC-neutral genomes, Ion Torrent shows a profound bias when sequencing extremely AT-rich genomes, with one study showing approximately 30% of the Plasmodium falciparum genome received no coverage [17]. This bias, attributed to the dual amplification steps in the Ion Torrent workflow, can be mitigated by using high-fidelity polymerases like Kapa HiFi during library preparation [17]. In cancer genomics, this could translate to missed mutations in specific genomic regions.
RNA-Seq Concordance: In differential gene expression (DGE) studies, a treatment/control experiment using mouse liver transcriptomes found a strong gene-level correlation (Spearman correlation 0.93-0.97) between Illumina HiSeq and Ion Torrent Proton data [3]. Despite this, the concordance in calling differentially expressed genes was only moderate, though pathway-level conclusions were nearly identical [3]. This suggests that while the overarching biological interpretation may be consistent, the specific gene lists generated can differ.
Experimental Protocols and Workflows
For cancer diagnostics, the choice between amplicon-based and hybridization-capture-based library preparation is a critical decision that interacts with the sequencing platform.
Hybridization-Capture-Based Sequencing for Hereditary Cancer Panels
A 2021 study provided a direct methodological comparison for detecting variants in hereditary cancer genes using both platforms with hybridization capture [18].
Methodology:
- Samples: 31 clinical samples (28 blood, 3 tumor tissues) and the NA12878 reference material with confirmed variants.
- Library Preparation: A hybridization-based hereditary cancer predisposition (HCP) panel was used. Platform-specific adapters were ligated for sequencing on the Illumina NextSeq 550Dx and the Ion Torrent S5 XL.
- Sequencing: Libraries were run on their respective platforms according to manufacturer protocols.
- Data Analysis: Reads were aligned, and variants were called. Sensitivity, specificity, and accuracy were calculated against known variants.
Key Workflow Diagram:
Results: The study demonstrated that a hybrid-capture panel could be successfully implemented on both platforms. The on-target rate was higher for the Ion S5 XL system (82.3% vs. 56.8%), but both systems achieved high analytical performance, making them viable for clinical cancer panel testing [18].
RNA-Seq for Differential Gene Expression Analysis
A 2017 study compared the platforms in the context of a biomedical research experiment: profiling the hepatic inflammatory response in mice [3].
Methodology:
- Experimental Design: Ten male mice were treated with either IL-1β (n=5) or saline (n=5). Liver RNA was extracted from all animals.
- Library Prep and Sequencing: Platform-specific libraries were prepared from all ten RNA samples and sequenced on both an Illumina HiSeq 2500 and an Ion Torrent Proton.
- Alignment: Data were aligned using multiple aligners (GSNAP, STAR, STAR+Bowtie2) to assess platform-aligner interaction.
- Analysis: Gene-level counts were obtained, and differential expression between treatment and control groups was assessed using a Mann-Whitney U test with multiple-testing correction.
Key Findings: The greatest technical difference was observed at the read alignment level, which was influenced by the choice of alignment software. The interaction between platform and aligner was particularly important for resolving complex regions like gene-pseudogene pairs [3].
Essential Research Reagent Solutions
The following table details key reagents and their functions, as utilized in the cited comparative studies.
| Reagent / Kit | Function in Workflow | Application Context |
|---|---|---|
| Hybridization Capture HCP Panel | Enriches for specific genomic targets (e.g., hereditary cancer genes) prior to sequencing [18]. | Targeted DNA sequencing for germline and somatic variant detection. |
| Ion Xpress Fragment Library Kit | Uses a "Fragmentase" formulation for enzymatic shearing of DNA, streamlining library prep [17]. | Rapid library preparation for Ion Torrent sequencing. |
| Kapa HiFi Polymerase | A high-fidelity enzyme used for amplification during library prep; reduces GC-bias [17]. | Mitigating sequence-specific bias, crucial for Ion Torrent sequencing of AT-rich regions. |
| PORT (Pipeline Of RNA-Seq Transformations) | A bioinformatics tool for normalization and quantification of aligned RNA-Seq reads [3]. | Downstream analysis of gene expression data from both platforms. |
| AmpliSeq Panel Kits | Amplification-based library prep for targeted sequencing on Ion Torrent systems [18]. | Fast, highly multiplexed targeted sequencing for limited sample input. |
Strategic Considerations for Cancer Diagnostics
The choice between Illumina and Ion Torrent is not a simple matter of superiority but depends on the specific requirements of the research or diagnostic application.
For Comprehensive Genomic Profiling: Illumina's platform, with its high accuracy and low homopolymer error rate, is generally the default choice for whole-genome and whole-exome sequencing in cancer, where detecting all variant types across the entire genome is critical.
For Rapid, Targeted Sequencing: Ion Torrent's shorter run times and scalability make it suitable for targeted panels where speed is essential, such as profiling a known set of cancer hotspots for rapid therapeutic decision-making [18]. Its performance with hybridization capture panels also reduces the risk of allele dropout [18].
For RNA-Seq and Pathway Analysis: Both platforms can yield biologically congruent results at the pathway level, which is often the primary goal of transcriptomic studies in cancer biology [3]. However, researchers should be aware that the specific lists of differentially expressed genes may vary, and the choice of alignment software interacts significantly with the platform [3].
For Low-Input and Challenging Samples: Ion Torrent has demonstrated capability with limited amounts of input DNA or RNA, a common scenario in clinical cancer samples [18].
From Bench to Bedside: Implementing NGS in Clinical Oncology and Biomarker Research
Comprehensive Genomic Profiling (CGP) represents a transformative approach in cancer diagnostics and research, enabling the simultaneous analysis of hundreds of cancer-related genes to identify targetable mutations, genomic signatures, and resistance mechanisms. As precision medicine continues to reshape oncology care, CGP has become indispensable for therapy selection research, clinical trial enrollment, and advancing our understanding of tumor biology. The current CGP landscape is predominantly shaped by two major sequencing platforms: Illumina's TruSight Oncology portfolio and Thermo Fisher Scientific's Oncomine portfolio. These solutions offer researchers distinct technological approaches, with Illumina utilizing sequencing-by-synthesis chemistry and Thermo Fisher employing semiconductor-based sequencing on Ion Torrent platforms. Understanding their comparative performance, technical capabilities, and practical implementation requirements is essential for research laboratories aiming to establish robust genomic profiling programs. This comparison guide examines these platforms through analytical performance data, workflow efficiency, and practical considerations for research implementation, providing scientists with evidence-based insights for platform selection.
Technical Specifications Comparison
The technical architecture and genomic content of CGP assays directly influence their research applications and biomarker detection capabilities. The TruSight Oncology 500 v2 and Oncomine Comprehensive Assay Plus represent the latest iterations of their respective portfolios, incorporating expanded gene content and enhanced biomarker assessment features.
Table 1: Comprehensive Technical Specifications Comparison
| Specification | Illumina TruSight Oncology 500 v2 | Thermo Fisher Oncomine Comprehensive Assay Plus |
|---|---|---|
| Total Genes | 523 genes (DNA), 55 genes (RNA) [19] | 517 genes [20] [21] |
| Panel Size | 1.94 Mb [19] | Information missing |
| Variant Types | SNVs, indels, CNVs, fusions, splice variants [19] | SNVs, indels, CNVs, fusions [20] [21] |
| Genomic Signatures | TMB, MSI, HRD (with GIS) [19] | TMB, MSI, HRD [20] [21] |
| Input Requirements | 30 ng DNA (10 ng min), 40 ng RNA (20 ng min) [19] | 20-30 ng DNA, 20 ng RNA [20] |
| Key Technological Differentiators | Integrated HRD with Myriad GIS algorithm; Unique molecular indices (UMIs) [22] [19] | Ion Ampliseq technology with minimal sample input [20] [21] |
Both panels demonstrate comprehensive coverage of major variant classes, though their technological approaches differ significantly. The TruSight Oncology 500 v2 incorporates unique molecular indices (UMIs) to reduce errors and improve variant calling accuracy, particularly for low-frequency variants [23]. The inclusion of homologous recombination deficiency (HRD) status using the Myriad Genomic Instability Score represents a significant advancement, providing researchers with a clinically validated biomarker for predicting response to PARP inhibitors [22] [19]. The Oncomine Comprehensive Assay Plus leverages Ion Ampliseq technology, which enables robust performance with potentially degraded samples commonly encountered in FFPE tissue workflows [20]. This technology demonstrates particular strength in copy number variation detection and fusion identification, with studies reporting a 94% success rate based on this amplification method [20].
Analytical Performance and Experimental Data
Independent comparative studies provide critical insights into the real-world performance characteristics of these CGP assays. A 2022 study published in the Journal of Personalized Medicine directly compared the Oncomine Comprehensive Assay Plus (OCAP) on the Ion Torrent platform with the TruSight Oncology 500 (TSO500) on the Illumina platform using 19 small cell lung cancer (SCLC) diagnostic samples and standardized control material [23].
Table 2: Analytical Performance Comparison from Independent Validation Study
| Performance Metric | TruSight Oncology 500 | Oncomine Comprehensive Assay Plus |
|---|---|---|
| Sequencing Quality | High mean read coverage and coverage uniformity [23] | Comparable mean read coverage and coverage uniformity [23] |
| Variant Detection Sensitivity | 100% of variants in clinical samples; 80% in AcroMetrix control [23] | 100% of variants in clinical samples; 80% in AcroMetrix control [23] |
| Variant Allele Frequency Correlation | Highly similar VAF reporting between platforms [23] | Highly similar VAF reporting between platforms [23] |
| TMB Concordance | 74% of samples classified in same TMB category [23] | 74% of samples classified in same TMB category [23] |
| Key Performance Differentiator | Sensitive variant calling in difficult genomic regions [22] | Robust performance with low DNA input (20 ng) [20] |
The study demonstrated that both panels achieved highly comparable next-generation sequencing quality metrics, including mean read coverage and coverage uniformity across target regions [23]. Both assays detected 100% of variants present in the clinical SCLC samples and approximately 80% of variants in the AcroMetrix assessment sample, which contains over 500 mutations with variant allele frequencies ranging from 5-35% [23]. The reported variant allele frequencies showed strong correlation between platforms, indicating consistent variant detection despite their different sequencing chemistries. For tumor mutation burden assessment, which has emerged as a critical biomarker for immunotherapy response prediction, the assays showed 74% concordance in TMB categorization across the sample set [23].
Experimental Protocol for Performance Validation
The comparative study employed rigorous methodology to ensure unbiased performance assessment [23]:
- Sample Selection: 19 diagnostic FFPE tumors from SCLC patients (17 limited disease stage, 2 extensive disease stage) were retrieved from a regional biobank, alongside AcroMetrix Oncology Hotspot Control with 521 somatic mutations.
- Nucleic Acid Extraction: DNA and RNA were extracted using Allprep DNA/RNA FFPE kit (Qiagen) or separately using QIAamp DNA FFPE Tissue Kit for DNA and RNeasy FFPE kit for RNA.
- Quality Assessment: Nucleic acid concentration was measured fluorometrically by Qubit Fluorometric Quantification. DNA quality was assessed by quantitative real-time PCR of a 300 bp fragment in FCGR3B.
- Library Preparation: For OCAP, 20-100 ng DNA was treated with Uracil DNA Glycosylase to remove deaminated cytosines. For TSO500, 65-100 ng DNA was fragmented to 90-250 bp using Covaris M220.
- Sequencing: OCAP libraries were sequenced on Ion GeneStudio S5 Prime System with Ion 550 chips. TSO500 libraries were sequenced on Illumina platforms following manufacturer specifications.
- Data Analysis: Variants were annotated using Ion Reporter Software v.5.18.2.1 for OCAP and DRAGEN pipeline for TSO500.
Workflow and Practical Implementation
The practical implementation of CGP assays in research settings requires careful consideration of workflow efficiency, automation capabilities, and integration with existing laboratory infrastructure. Both platforms offer distinct advantages depending on the research context and operational priorities.
Table 3: Workflow Efficiency and Practical Implementation
| Workflow Aspect | TruSight Oncology 500 v2 | Oncomine Comprehensive Assay Plus |
|---|---|---|
| Total Assay Time | 3-4 days from sample to results [19] | 1-3 days (varies by instrument) [20] |
| Hands-on Time | ~7 hours (manual workflow) [19] | 20-60 minutes (varies by instrument) [20] |
| Automation Options | Liquid handling robots (~50% hands-on time reduction) [19] | Fully automated on Genexus System [21] |
| Instrument Compatibility | NextSeq 550, NextSeq 1000/2000, NovaSeq 6000, NovaSeq X Series [19] | Ion Torrent Genexus System, Ion GeneStudio S5 Series [20] |
| Sample Throughput Range | 8-960 samples per run (depending on instrument) [19] | Scalable based on chip configuration [1] |
| Data Analysis Integration | DRAGEN secondary analysis, Illumina Connected Insights, Velsera CGW [19] | Ion Reporter Software with integrated analysis pipelines [20] |
The TruSight Oncology 500 v2 features a streamlined workflow that reduces turnaround time and hands-on time compared to previous versions, with new kit configurations featuring 50% less packaging and 70% fewer tubes to improve usability [22] [19]. The platform offers broad compatibility across Illumina's sequencing portfolio, from mid-throughput NextSeq systems to high-throughput NovaSeq platforms, providing scalability for research laboratories with varying sample volumes [19]. The integration of DRAGEN secondary analysis enables rapid, accurate variant calling, with options for on-premise or cloud-based analysis through Illumina Connected Analytics [19].
The Oncomine Comprehensive Assay Plus emphasizes rapid turnaround times, potentially delivering results in as little as one day when implemented on the Genexus System [21]. This system provides complete end-to-end automation, requiring only 20-60 minutes of hands-on time, which can significantly reduce operational complexity and technical variability [20]. The Ion Torrent platforms demonstrate particular strength in laboratories prioritizing simplicity of operation and rapid results, with studies reporting a 94% success rate based on the Ampliseq technology [20].
The Scientist's Toolkit: Essential Research Reagent Solutions
Implementing comprehensive genomic profiling requires specialized reagents, controls, and consumables to ensure reproducible results. The following table outlines essential components for establishing a robust CGP workflow in research settings.
Table 4: Essential Research Reagent Solutions for CGP Implementation
| Reagent Category | Specific Product Examples | Research Application & Function |
|---|---|---|
| Nucleic Acid Extraction | Allprep DNA/RNA FFPE kit (Qiagen) [23] | Simultaneous DNA/RNA extraction from precious FFPE samples |
| Quality Assessment | Qubit Fluorometric Quantification [23] | Accurate nucleic acid quantification prior to library prep |
| Library Preparation | TruSight Oncology 500 v2 Library Prep Kit [19] | Target enrichment and library construction for Illumina platforms |
| Library Preparation | Oncomine Comprehensive Assay Plus [20] | Automated library preparation for Ion Torrent systems |
| Quality Control | Bioanalyzer High Sensitivity DNA Analysis [23] | Fragment size distribution analysis and library QC |
| Reference Materials | AcroMetrix Oncology Hotspot Control [23] | Process control with 521 known mutations for assay validation |
| Indexing | Illumina DNA/RNA UD Indexes [19] | Sample multiplexing for efficient sequencing runs |
| Sequencing Consumables | Ion 550 chips (Thermo Fisher) [23] | Semiconductor sequencing chips for Ion Torrent platforms |
| Sequencing Consumables | NextSeq 1000/2000 XLEAP-SBS Reagent Kit [19] | Sequencing reagents for Illumina benchtop systems |
The comparative analysis of TruSight Oncology 500 v2 and Oncomine Comprehensive Assay Plus reveals two highly capable CGP platforms with distinct strengths for research applications. The TruSight Oncology 500 v2 demonstrates advantages in comprehensive biomarker content, with integrated HRD assessment using the validated Myriad GIS algorithm and sensitive variant calling across difficult genomic regions [22] [19]. Its broad instrument compatibility and scalable throughput make it suitable for research programs with diverse sample volumes. The Oncomine Comprehensive Assay Plus offers exceptional workflow efficiency, particularly when implemented on the Genexus System, with minimal hands-on time and rapid turnaround potentially as quick as one day [21]. Its performance with low-input samples (as little as 20 ng DNA) provides valuable flexibility for research involving limited or precious specimens [20].
For research laboratories selecting between these platforms, the decision framework should consider several key factors: sample volume and throughput requirements, available sequencing infrastructure, technical expertise, and specific research applications. Research programs requiring comprehensive immuno-oncology biomarker assessment including HRD status may prioritize the TruSight Oncology 500 v2, while programs emphasizing rapid turnaround and operational simplicity may favor the Oncomine Comprehensive Assay Plus on automated systems. Both platforms demonstrate strong analytical performance in comparative studies, with 100% concordance on clinical samples and 74% TMB categorization agreement, providing researchers with confidence in either technological approach [23]. As CGP continues to evolve, both platforms represent mature solutions for advancing precision oncology research through comprehensive molecular profiling.
Liquid biopsy, the analysis of tumor-derived components in bodily fluids, has emerged as a transformative approach in cancer diagnostics and management. Among its various analytes, circulating tumor DNA (ctDNA)—fragments of DNA released into the bloodstream by tumor cells through apoptosis, necrosis, or active release—holds particular promise for revolutionizing cancer care [24] [25]. The clinical utility of ctDNA analysis spans the entire cancer continuum, from early detection and diagnosis to monitoring treatment response and detecting minimal residual disease (MRD) [24] [26]. The short half-life of ctDNA in circulation (ranging from 16 minutes to 2.5 hours) makes it a dynamic biomarker for real-time monitoring of disease burden and treatment efficacy [25].
Analyzing ctDNA presents significant technical challenges due to its low concentration in the total cell-free DNA (cfDNA) pool, often constituting less than 1% of total cfDNA, especially in early-stage cancers or low-shedding tumors [27] [28]. This biological constraint necessitates highly sensitive detection methods capable of identifying rare mutant alleles against a high background of wild-type DNA. The field has responded with a diverse array of technological platforms and approaches, each with distinct strengths, limitations, and optimal use cases. Next-generation sequencing (NGS) technologies, including those from Illumina and Ion Torrent, have become cornerstone platforms for comprehensive ctDNA analysis, enabling the detection of a broad range of genomic alterations including single nucleotide variants (SNVs), insertions/deletions (indels), copy number alterations (CNAs), and structural variants (SVs) [24] [4]. This guide provides an objective comparison of current ctDNA detection platforms, focusing on their analytical performance, methodologies, and applications within cancer diagnostics research.
Key Analytical Technologies for ctDNA Detection
ctDNA detection methodologies can be broadly categorized into targeted approaches, which interrogate a predefined set of known mutations, and untargeted approaches, which broadly screen the genome for novel or unexpected alterations [25]. PCR-based methods, including digital PCR (dPCR) and BEAMing (beads, emulsion, amplification, magnetics), offer high sensitivity for detecting single or a few well-characterized mutations and are particularly suitable for monitoring known variants during treatment [24] [25]. In contrast, next-generation sequencing (NGS) methods provide a more comprehensive profiling capability, detecting a broad spectrum of genomic alterations across multiple genes simultaneously [24] [4].
The evolution of NGS technologies has addressed many limitations of earlier sequencing methods, offering massive parallel sequencing capabilities that significantly reduce time and cost compared to traditional Sanger sequencing [4]. Among NGS platforms, Illumina and Ion Torrent have emerged as leading technologies for ctDNA analysis, each with distinct sequencing chemistries and detection methods. Illumina sequencing utilizes fluorescence-based detection of nucleotide incorporation, while Ion Torrent employs semiconductor-based detection of hydrogen ions released during DNA polymerization [4]. These fundamental technological differences influence key performance parameters including sensitivity, specificity, throughput, and cost—critical considerations for researchers selecting appropriate platforms for specific applications.
Performance Comparison of Detection Platforms
Recent studies have directly compared the analytical performance of various ctDNA detection platforms, revealing important differences in sensitivity, specificity, and variant detection capabilities. A comprehensive 2024 evaluation of nine ctDNA assays using standardized reference samples found substantial variability in performance, particularly at lower ctDNA inputs and variant allele frequencies (VAFs) below 0.5% [27]. The sensitivity for SNV detection at VAF of 0.5% reached approximately 0.95 for most assays, with some platforms demonstrating superior performance for specific variant types including indels, CNAs, and fusions [27].
Table 1: Comparative Analytical Performance of ctDNA Detection Platforms
| Platform/Assay | Technology Type | Sensitivity (SNV at 0.5% VAF) | Variant Types Detected | Optimal Input | Key Limitations |
|---|---|---|---|---|---|
| UltraSEEK Lung Panel v2 [28] | MassARRAY-based panel | >82% concordance with tissue NGS | 78 SNVs/indels in 5 genes | 2 mL plasma | Does not cover fusions; limited gene content |
| FoundationOne Liquid CDx [11] [28] | NGS-based liquid biopsy | High concordance with tissue | SNVs, indels, CNAs, fusions | >20 ng ctDNA | Higher cost; longer turnaround |
| Guardant360 CDx [29] | NGS-based liquid biopsy | FDA-approved for multiple indications | SNVs, indels, CNAs, fusions | >20 ng ctDNA | Higher cost; requires specialized infrastructure |
| Oncomine Precision Assay (Ion Torrent) [11] | NGS-based targeted sequencing | 55% sensitivity vs. FoundationOne | SNVs, indels, CNAs, fusions | >20 ng ctDNA | Lower sensitivity for some variants |
| Bridge Capture [30] | Novel targeted NGS | Detects lowest VAF among compared methods | SNVs, indels, CNAs | Low input requirements | Limited clinical validation data |
A 2025 comparative study of comprehensive genomic profiling tests highlighted specific performance differences between the Ion Torrent Genexus system and FoundationOne (Illumina-based) platforms [11]. When comparing these platforms for common genes across tissue and liquid biopsy analyses, the Ion Torrent Oncomine Comprehensive Assay (OCA) and Oncomine Precision Assay (OPA) demonstrated 55% sensitivity and 99% specificity compared to FoundationOne [11]. The study identified specific variants that were differentially detected between platforms, with some SNVs, CNAs, and fusions detected only in Genexus, while other SNVs were detected only in FoundationOne, indicating that different assays and analytical methods can influence variant detection [11].
Table 2: Concordance Study Results: Ion Torrent Genexus vs. FoundationOne* [11]
| Variant Category | Detected by Both Platforms | Detected Only by Genexus | Detected Only by FoundationOne |
|---|---|---|---|
| Single Nucleotide Variants (SNVs) | 9 SNVs | 1 SNV (MAP2K1 F53V) | 2 SNVs (TP53 Q331*, KRAS G12V) |
| Copy Number Alterations (CNAs) | 1 CNA | 2 CNAs (AKT3, MYC) | None |
| Gene Fusions | 1 fusion | 1 fusion (ESR-CCDC170) | None |
Emerging technologies continue to push the boundaries of ctDNA detection sensitivity. The novel Bridge Capture technology, for example, has demonstrated superior sensitivity compared to established methods like Archer LIQUIDPlex and AmpliSeq CHP version 2, detecting lower variant allele frequencies while maintaining strong correlation (R² = 0.995 with Archer LIQUIDPlex) [30]. This method also showed high reproducibility across independent laboratories and minimal performance impact with increased panel size, suggesting promising multiplexing capabilities for broader genomic coverage [30].
Experimental Protocols for Platform Evaluation
Standardized Workflow for ctDNA Analysis
Robust evaluation of ctDNA detection platforms requires standardized methodologies from sample collection through data analysis. The typical workflow begins with blood sample collection in specialized tubes such as Cell-Free DNA BCTs (Streck), which preserve ctDNA integrity by preventing white blood cell lysis and subsequent release of genomic DNA that would dilute the tumor-derived fraction [28]. Processing within 48 hours of collection is generally recommended, with initial centrifugation at 1,600 × g for 10 minutes to separate plasma from cellular components, followed by a second centrifugation at 16,000 × g for 10 minutes to ensure complete removal of residual cells [28].
Cell-free DNA extraction typically employs silica membrane-based methods such as the QiaAMP Circulating Nucleic Acid Kit (Qiagen), with elution volumes standardized to ensure consistent concentration across samples [28]. Accurate quantification of extracted cfDNA is critical, with common approaches including fluorometric methods like the Qubit dsDNA HS Assay (Thermo Fisher Scientific) and fragment analysis using the LiquidIQ Panel (Agena Bioscience), which have demonstrated strong correlation (Pearson's r² = 0.75) in comparative studies [28]. The extracted cfDNA is then subjected to platform-specific library preparation protocols, which typically involve fragment end-repair, adapter ligation, and amplification steps optimized for the low-input, highly fragmented nature of ctDNA.
Method-Specific Experimental Procedures
Ion Torrent Oncomine Precision Assay: This targeted NGS approach utilizes multiplex PCR amplification for target enrichment. Library preparation begins with a small volume of extracted cfDNA (as low as 5-20 ng) followed by two successive PCR reactions—initial amplification of target regions followed by barcoding with Ion Code adapters. The templating process uses Ion Chef instruments with Ion 530 chips, and sequencing is performed on Ion GeneStudio S5 systems [11]. The automated workflow enables rapid turnaround times, with library preparation and sequencing completed within 24 hours.
Illumina-Based FoundationOne Liquid CDx: This hybrid capture-based NGS approach uses biotinylated oligonucleotide probes to enrich for targeted genomic regions spanning 324 genes. Library preparation involves end-repair, A-tailing, and adapter ligation followed by hybridization with capture probes. Sequencing is performed on Illumina sequencing platforms, typically achieving high sequencing depths (>5,000x) to enable sensitive detection of low-frequency variants [11] [28]. The comprehensive nature of this assay provides broad genomic coverage but requires longer turnaround times (10-21 days) compared to targeted panels.
UltraSEEK Lung Panel on MassARRAY System: This mass spectrometry-based detection system employs an initial multiplex PCR reaction followed by a single base extension reaction that incorporates mass-modified terminators. The resulting products are dispensed onto a silicon chip array (SpectroCHIP) and analyzed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry [28]. This technology offers a cost-effective approach for focused mutation profiling with rapid turnaround (within 48 hours) but is limited to predefined mutation hotspots.
Quality Control Metrics
Robust quality control measures are essential throughout the experimental workflow. For NGS-based methods, key metrics include sequencing depth (deduplicated mean depth >5,000x recommended for sensitive ctDNA detection), on-target rate (≥50% considered acceptable), and uniformity of coverage [27]. Sample-level QC should include cfDNA quantity and fragment size distribution, with typical ctDNA fragments ranging from 70-200 base pairs [25]. For quantitative comparisons, reference standards with known variant allele frequencies (e.g., Seraseq ctDNA Mutation Mix, Horizon Discovery) provide essential calibration for determining analytical sensitivity and specificity across platforms [27].
Research Reagent Solutions for ctDNA Analysis
Successful ctDNA analysis requires a comprehensive suite of specialized reagents and tools at each stage of the workflow. The following table details essential research reagent solutions and their specific functions in ctDNA detection protocols.
Table 3: Essential Research Reagents for ctDNA Analysis Workflows
| Reagent/Tool | Manufacturer/Provider | Primary Function | Application Notes |
|---|---|---|---|
| Cell-Free DNA BCTs | Streck | Preserves blood samples by preventing white blood cell lysis | Enables sample stability for up to 48 hours post-collection [28] |
| QiaAMP Circulating Nucleic Acid Kit | Qiagen | Silica membrane-based extraction of cfDNA from plasma | High recovery efficiency for low-abundance ctDNA; elution in AVE buffer [28] |
| Qubit dsDNA HS Assay | Thermo Fisher Scientific | Fluorometric quantification of double-stranded DNA | Essential for accurate input measurement; superior sensitivity for low-concentration samples [28] |
| LiquidIQ Panel | Agena Bioscience | Fragment size analysis and quantification of cfDNA | Assesses cfDNA quality and quantity; correlates strongly with Qubit (r²=0.75) [28] |
| Oncomine Precision Assay | Thermo Fisher Scientific | Targeted NGS panel for ctDNA mutation detection | Designed for Ion Torrent platforms; covers key cancer genes [11] |
| FoundationOne Liquid CDx | Foundation Medicine | Comprehensive NGS-based liquid biopsy test | Hybrid capture-based; covers 324 genes; FDA-approved companion diagnostic [11] [29] |
| UltraSEEK Lung Panel v2 | Agena Bioscience | Mass spectrometry-based mutation detection panel | Focused panel for 78 SNVs/indels in 5 lung cancer genes; cost-effective [28] |
Additional specialized reagents include unique molecular identifiers (UMIs) for error correction in NGS workflows, hybrid capture probes for target enrichment in whole-genome approaches, and various library preparation kits optimized for the low-input, degraded nature of ctDNA. The selection of appropriate reagents significantly impacts assay performance, particularly for detecting low-frequency variants at allele frequencies below 0.5% where technical artifacts become increasingly problematic [27].
Advanced Applications and Emerging Methodologies
Multimodal and Fragmentomic Approaches
Beyond mutation detection, advanced ctDNA analysis methods leverage additional molecular features to enhance sensitivity and clinical utility. Fragmentomics analysis examines the size distribution and end motifs of cfDNA fragments, with tumor-derived DNA typically exhibiting shorter fragment lengths and distinct cleavage patterns compared to DNA from healthy cells [24]. The DELFI (DNA evaluation of fragments for early interception) method uses machine learning models applied to genome-wide fragmentation profiles, achieving a sensitivity of cancer detection of 91% when combined with mutation-based analyses [24].
Methylation profiling represents another powerful approach for ctDNA analysis, exploiting cancer-specific epigenetic alterations. Bisulfite conversion followed by sequencing enables identification of hypermethylated or hypomethylated regions characteristic of malignant cells. Recent advancements include bisulfite-free methods such as chromatin immunoprecipitation sequencing (ChIP-Seq) and methylated DNA immunoprecipitation sequencing (MeDIP-Seq) that overcome the limitations of DNA degradation associated with traditional bisulfite treatment [24]. Integrating multiple analytical approaches—combining genomic, fragmentomic, and epigenetic analyses—has demonstrated significant improvements in detection sensitivity, with one study reporting a 25-36% increase in sensitivity for recurrence detection when epigenomic signatures were added to genomic alterations alone [24].
Minimal Residual Disease and Early Detection Applications
The extreme sensitivity required for MRD detection has driven development of increasingly sophisticated ctDNA technologies. Tumor-informed approaches, which first sequence the tumor tissue to identify patient-specific mutations then monitor for these specific variants in plasma, currently provide the highest sensitivity for MRD assessment. In the VICTORI study on colorectal cancer, this approach demonstrated that 87% of recurrences were preceded by ctDNA positivity, while no ctDNA-negative patients relapsed [26].
Novel methods continue to push detection limits further. The MUTE-Seq (Mutation tagging by CRISPR-based Ultra-precise Targeted Elimination in Sequencing) method utilizes engineered advanced-fidelity FnCas9 to selectively eliminate wild-type DNA, enabling highly sensitive detection of low-frequency cancer-associated mutations for MRD evaluation in NSCLC and pancreatic cancer [26]. Similarly, the neXT Personal MRD detection assay has shown utility in predicting outcomes in patients with stage II-IIIb, EGFR-mutated NSCLC receiving neoadjuvant therapy [29] [26].
Multi-cancer early detection (MCED) tests represent one of the most promising applications of ctDNA analysis, with several tests currently in clinical development. These tests typically combine multiple biomarker classes including mutations, methylation patterns, and fragmentomic profiles to detect dozens of cancer types simultaneously. Recent studies have demonstrated specificities exceeding 98.5% with sensitivities ranging from 59.7% for all cancers to 84.2% for late-stage tumors [26]. The ongoing Vanguard Study, part of the NCI Cancer Screening Research Network, is evaluating the feasibility of implementing MCED tests in real-world settings, having enrolled over 6,200 participants with high adherence across diverse populations [26].
The rapidly evolving landscape of ctDNA detection technologies offers researchers and clinicians an expanding arsenal of platforms for cancer detection and monitoring. Each platform presents distinct advantages—from the focused sensitivity of PCR-based methods for known variants to the comprehensive genomic profiling capability of NGS-based approaches. The choice among Illumina, Ion Torrent, and other platforms depends heavily on the specific research context, including required sensitivity, breadth of genomic coverage, throughput needs, and cost considerations.
Current evidence demonstrates that no single platform excels across all performance parameters. The Ion Torrent Genexus system offers advantages in workflow automation and rapid turnaround, while Illumina-based FoundationOne provides more comprehensive genomic coverage. Emerging technologies like Bridge Capture and MUTE-Seq show promise for pushing detection sensitivity to new lows, potentially enabling earlier cancer detection and more sensitive MRD monitoring. For researchers, the optimal approach often involves leveraging multiple complementary technologies—using broad NGS panels for initial discovery and focused, highly sensitive assays for longitudinal monitoring.
As the field advances, key areas of development include standardization of pre-analytical and analytical processes, integration of multi-omics data, and validation of clinical utility through large-scale prospective trials. The growing emphasis on multimodal approaches that combine genomic, fragmentomic, and epigenetic analyses represents a particularly promising direction, potentially overcoming the limitations of any single methodology. These continued innovations in ctDNA detection platforms will further establish liquid biopsy as an indispensable tool in cancer research and clinical management, ultimately advancing toward more personalized and effective cancer care.
Next-generation sequencing (NGS) has revolutionized cancer diagnostics by enabling comprehensive genomic profiling that identifies actionable biomarkers for targeted therapy and immunotherapy. Within oncology, two major sequencing platforms—Illumina and Ion Torrent—provide complementary approaches for detecting critical genomic alterations. This comparison guide evaluates their performance in identifying three cornerstone biomarkers in oncology: KRAS, EGFR, and Tumor Mutational Burden (TMB). These biomarkers play pivotal roles in directing treatment strategies for non-small cell lung cancer (NSCLC) and other malignancies, with KRAS and EGFR representing the most frequently altered oncogenes and TMB serving as a key predictor of immunotherapy response. Understanding the technical capabilities and limitations of each sequencing platform is therefore essential for researchers and clinicians aiming to optimize biomarker detection accuracy, assay sensitivity, and ultimately, patient stratification for targeted therapies.
Biomarker Prevalence and Co-alteration Landscape
The clinical utility of genomic biomarkers depends fundamentally on their prevalence across patient populations and their patterns of co-occurrence with other genomic alterations. Large-scale genomic studies have revealed significant ancestry-associated differences in the prevalence of major driver mutations. In non-squamous NSCLC, KRAS is the most frequently altered oncogene in patients of European (38.9%) and African (32.5%) ancestry, whereas EGFR alterations predominate in East Asian (53.4%), South Asian (36.4%), and Admixed American (30.3%) populations [31]. KRAS G12C is the most prevalent KRAS substitution across all ancestry groups, occurring in 15.2% of European and 11.9% of African patients, but only 4-6% of East Asian, South Asian, and Admixed American patients [31].
The co-alteration landscape further refines our understanding of tumor biology. KRAS-altered tumors frequently harbor co-occurring alterations in TP53 (48.8% in European, 60.7% in African), STK11, and KEAP1 [31]. These co-alterations have significant clinical implications; for instance, STK11/LKB1 co-mutations with KRAS are associated with reduced response to immunotherapy (7.4% objective response rate) compared to TP53/KRAS co-alterations (35.7% response rate) [32]. Different KRAS mutation subtypes also associate with distinct TMB profiles; KRAS p.Q61X mutations demonstrate lower TMB, while KRAS p.A59X and p.G13X show higher TMB [32].
Table 1: Prevalence of KRAS and EGFR Alterations Across Ancestry Groups in Non-Squamous NSCLC
| Ancestry Group | KRAS Alteration Prevalence | EGFR Alteration Prevalence | Most Common KRAS Subtype |
|---|---|---|---|
| European | 38.9% | 14.6% | G12C (15.2%) |
| African | 32.5% | 15.7% | G12C (11.9%) |
| East Asian | 15.0% | 53.4% | G12C (4-6%) |
| South Asian | 20.5% | 36.4% | G12C (4-6%) |
| Admixed American | 23.1% | 30.3% | G12C (4-6%) |
Platform Performance Comparison: Illumina vs. Ion Torrent
Illumina and Ion Torrent employ fundamentally different sequencing chemistries that impact their performance in biomarker detection. Illumina utilizes sequencing-by-synthesis with fluorescently labeled, reversible-terminator nucleotides, while Ion Torrent relies on semiconductor technology that detects pH changes during nucleotide incorporation [2]. These methodological differences translate into distinct performance characteristics with direct implications for oncology applications.
A direct comparison of comprehensive genomic profiling tests revealed that the Ion Torrent Oncomine Comprehensive Assay (OCA) and FoundationOne (Illumina-based) showed 55% sensitivity and 99% specificity when analyzing common genes across tissue and liquid biopsies [11]. Both platforms successfully detected key single-nucleotide variants, copy number alterations, and fusions, though some variants were platform-specific [11]. For instance, one SNV (MAP2K1 F53V), two CNAs (AKT3 and MYC), and one fusion (ESR-CCDC170) were detected only by Ion Torrent, whereas two SNVs (TP53 Q331* and KRAS G12V) were identified exclusively by FoundationOne [11].
Error profiles differ substantially between platforms. Ion Torrent exhibits higher error rates in homopolymer regions due to challenges in precisely counting identical consecutive bases, with a raw error rate approximately double that of Illumina (~1% vs. ~0.1-0.5%) [2]. Illumina's reversible terminator chemistry provides more accurate homopolymer resolution but typically requires longer run times [2].
Table 2: Platform Performance Characteristics for Biomarker Detection
| Parameter | Illumina | Ion Torrent |
|---|---|---|
| Sequencing Chemistry | Fluorescent reversible terminators | Semiconductor pH detection |
| Read Configuration | Paired-end | Single-end |
| Raw Error Rate | 0.1-0.5% | ~1% |
| Homopolymer Accuracy | High | Moderate |
| Sensitivity (vs. reference) | N/A | 55% |
| Specificity (vs. reference) | N/A | 99% |
| Typical Run Time | 24-48 hours | <24 hours |
Concordance in Clinical Applications
Studies directly comparing variant detection between Illumina and Ion Torrent platforms in clinical cancer samples demonstrate generally high concordance for actionable variants. In pancreatic cancer, a comparison of whole genome sequencing (Illumina) and targeted sequencing (Ion Torrent Oncomine Comprehensive Assay Plus) showed 81% concordance across all variants and 100% concordance for variants relevant to targeted therapy [33]. Both techniques reliably identified common driver mutations in KRAS, TP53, SMAD4, and CDKN2A, which are critical for pancreatic cancer pathogenesis [33].
In differential gene expression analysis, both platforms showed strong correlation (Spearman correlation coefficients of 0.938-0.974) in gene-level read counts and identified similar pathway perturbations in treatment/control experiments [34]. However, the optimal alignment tools differed between platforms, with STAR and GSNAP performing better for Illumina data, while a combination of STAR and Bowtie2 was more effective for Ion Torrent data [34].
For liquid biopsy applications, the Ion Torrent Oncomine Precision Assay demonstrated comparable performance to FoundationOne Liquid, though some variants were detected by only one platform, highlighting the importance of understanding platform-specific biases in ctDNA analysis [11].
Experimental Protocols for Biomarker Detection
Sample Preparation and Library Construction
Robust biomarker detection begins with standardized sample preparation protocols. For tissue-based comprehensive genomic profiling, DNA and RNA are typically co-extracted from formalin-fixed paraffin-embedded (FFPE) tissue sections with minimum tumor cellularity of 30% [11] [33]. For liquid biopsy applications, cell-free total nucleic acid (cfTNA) is extracted from blood plasma obtained through double centrifugation of EDTA-blood samples (4°C, 2,000 × g, 10 minutes) [11].
For Ion Torrent sequencing using the Oncomine Comprehensive Assay, DNA is treated with Uracil-DNA glycosylase to remove deaminated bases arising from formalin fixation, while RNA is converted to complementary DNA using the SuperScript VILO kit [33]. Library preparation utilizes a multiplex PCR approach with inputs of 5-40 ng DNA and 20 ng RNA according to manufacturer specifications (Manual MAN0018490) [33]. For Illumina-based FoundationOne CDx, similar extraction methods are followed, though library preparation employs different adapter ligation and amplification techniques optimized for their platform [11].
Sequencing and Data Analysis
For Ion Torrent sequencing, templating is performed via emulsion PCR on Ion Sphere Particles, which are then deposited on semiconductor chips [2] [33]. Sequencing occurs through sequential flows of nucleotides, with pH changes indicating incorporation events. The Ion Torrent Genexus system automates the workflow from sample to result in approximately 14-24 hours [2].
For Illumina sequencing, libraries are loaded onto flow cells where bridge amplification creates clusters of identical sequences [2]. Sequencing-by-synthesis occurs through cycles of nucleotide incorporation, fluorescence imaging, and terminator removal, typically requiring 24-48 hours for completion [2].
Bioinformatic analysis pipelines differ between platforms. Ion Torrent data analysis includes signal processing, base calling, and alignment to reference genomes, with specialized algorithms to address homopolymer errors [2]. Illumina data utilizes platforms like DRAGEN for secondary analysis, including alignment, variant calling, and annotation [11]. For both platforms, variant calling thresholds typically require a minimum of 5% variant allele frequency for single nucleotide variants and 10% for insertions/deletions [11].
Signaling Pathways and Biomarker Biology
The EGFR and KRAS signaling pathways represent critical oncogenic drivers in multiple cancer types, particularly NSCLC. EGFR is a transmembrane receptor tyrosine kinase that modulates cellular processes including proliferation, growth, and apoptosis inhibition through downstream pathways including MAPK, PI3K/AKT, and JAK/STAT [35]. Oncogenic mutations, such as exon 19 deletions or L858R mutations, result in constitutive activation independent of ligand binding [35].
KRAS functions as a GTPase downstream of EGFR and other receptor tyrosine kinases, regulating cell growth and survival through MAPK and PI3K/AKT signaling cascades [35]. Mutations at codons 12, 13, and 61, particularly G12C, impair GTP hydrolysis, maintaining KRAS in a constitutively active state [35]. These mutations are strongly associated with tobacco exposure and demonstrate distinct TMB profiles compared to EGFR mutations [35].
Figure 1: EGFR and KRAS Signaling Pathways. This diagram illustrates the central role of EGFR and KRAS in key oncogenic signaling pathways that drive cell proliferation, survival, and apoptosis inhibition in cancer cells.
The relationship between these biomarkers and the tumor immune microenvironment explains their differential responses to immunotherapy. KRAS-mutant tumors typically exhibit higher TMB, elevated PD-L1 expression, and more immunologically active microenvironments, making them more responsive to immune checkpoint inhibitors [35]. In contrast, EGFR-mutant tumors generally demonstrate non-inflamed microenvironments and poorer responses to immunotherapy [35]. This biological distinction underscores the importance of accurate biomarker detection for optimal treatment selection.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for NGS-Based Biomarker Detection
| Reagent/Kits | Function | Example Application |
|---|---|---|
| Maxwell RSC DNA/RNA FFPE Kits | Nucleic acid extraction from formalin-fixed paraffin-embedded tissue | Simultaneous DNA/RNA extraction from FFPE sections with >30% tumor cellularity [11] [33] |
| Maxwell RSC miRNA Plasma/Serum Kit | Cell-free nucleic acid extraction from blood plasma | Isolation of cell-free total nucleic acid from liquid biopsy samples [11] |
| Oncomine Comprehensive Assay | Targeted multiplex PCR panel for cancer-relevant genes | Simultaneous detection of SNVs, indels, CNAs, and fusions in 501 genes [33] |
| FoundationOne CDx | Comprehensive genomic profiling test | Detection of substitutions, indels, CNAs, and rearrangements in 324 genes [11] |
| SuperScript VILO cDNA Kit | Reverse transcription of RNA to cDNA | RNA conversion for gene fusion detection in sequencing assays [33] |
| Ion Torrent Genexus Sequencer | Automated NGS system from library prep to final report | Automated sequencing workflow with minimal hands-on time [11] |
| Illumina MiSeq/NovaSeq | Benchtop and production-scale sequencing systems | High-throughput sequencing with reversible terminator chemistry [2] |
Both Illumina and Ion Torrent platforms provide effective solutions for detecting critical biomarkers like KRAS, EGFR, and TMB, albeit with distinct technical strengths. Illumina offers superior accuracy, particularly in homopolymer-rich regions, and higher throughput capabilities, making it ideal for large-scale genomic studies. Ion Torrent provides faster turnaround times, lower instrument costs, and streamlined automated workflows suitable for clinical environments requiring rapid results. The choice between platforms should be guided by specific research needs, weighing factors such as required throughput, budget constraints, turnaround time requirements, and the specific genomic regions of interest. As precision oncology continues to evolve, both platforms will play crucial roles in advancing biomarker discovery and validation, ultimately improving patient stratification and treatment outcomes.
Next-generation sequencing (NGS) has become a cornerstone of modern cancer diagnostics research, with Illumina and Ion Torrent emerging as two leading platforms for short-read sequencing. For researchers, scientists, and drug development professionals, selecting the appropriate technology involves careful consideration of workflow efficiency, which directly impacts laboratory throughput, operational costs, and time-to-result for critical experiments. This guide provides an objective comparison of Illumina and Ion Torrent platforms, focusing specifically on hands-on time, automation capabilities, and total turnaround time, with supporting experimental data from published studies to inform platform selection for cancer diagnostics research.
Illumina and Ion Torrent employ fundamentally different sequencing technologies that inherently influence their workflow characteristics. Illumina utilizes a fluorescence-based sequencing-by-synthesis approach with reversible dye-terminators, where DNA fragments are amplified on a flow cell via bridge PCR and bases are detected optically through fluorescent signals [2] [36]. In contrast, Ion Torrent employs semiconductor technology that detects hydrogen ions (pH changes) released during nucleotide incorporation, with DNA fragments amplified via emulsion PCR on microscopic beads deposited into semiconductor chip wells [2] [17].
These core technology differences translate to distinct workflow implications. Illumina's optical detection system requires sophisticated imaging hardware, while Ion Torrent's electronic detection eliminates the need for lasers and cameras, contributing to a more compact instrument design [2]. Additionally, Illumina platforms typically generate uniform read lengths and offer paired-end sequencing capabilities, whereas Ion Torrent produces variable-length reads and is generally limited to single-end sequencing [3] [2].
Experimental Protocols and Benchmarking Methodologies
Cross-platform comparisons require carefully designed experiments to ensure equitable assessment. The following methodologies represent approaches used in published benchmarking studies:
Hybridization Capture-Based Cancer Panel Evaluation
A 2021 study directly compared the Ion Torrent S5 XL and Illumina NextSeq 550Dx systems using a hybridization capture-based hereditary cancer predisposition panel [18]. The experimental protocol involved:
- Sample Preparation: 31 clinical samples (28 peripheral blood buffy coats and 3 fresh tumor tissues) with previously confirmed variants and NA12878 reference material.
- Library Preparation: Hybridization-based capture libraries were prepared using platform-specific kits designed for each system, with identical input DNA.
- Sequencing Parameters: Each platform was operated according to manufacturer specifications using the same samples for direct comparison.
- Analysis Metrics: On-target reads, uniformity of coverage, variant calling sensitivity, specificity, and accuracy were assessed bioinformatically.
SARS-CoV-2 Sequencing Protocol Comparison
A 2023 multi-center benchmarking study compared five sequencing protocols, including Illumina and Ion Torrent platforms, using clinical samples [37]. The methodology included:
- Sample Selection: 26 SARS-CoV-2 PCR-positive clinical samples with a range of cycle threshold values (13.9-33.6) to assess performance across different viral loads.
- Standardized Extraction: Identical nucleic acid extraction methodology for all protocols to eliminate variability.
- Platform-Specific Protocols: Each platform was used according to manufacturer instructions with appropriate library prep kits.
- Performance Metrics: Genome coverage, depth of coverage, amplicon distribution, variant calling, hands-on time, and total turnaround time were systematically recorded.
Workflow Efficiency Metrics and Comparative Data
Sequencing Run Metrics and Performance
Table 1: Sequencing Performance Metrics from Hybridization Capture-Based Cancer Panel Study
| Parameter | Ion Torrent S5 XL | Illumina NextSeq 550Dx |
|---|---|---|
| Mean On-Target Reads | 82.3% | 56.8% |
| Uniformity of Coverage | 92.5% | 95.7% |
| Variant Calling Sensitivity | 97.06% | 98.53% |
| Variant Calling Specificity | 100% | 100% |
| Variant Calling Accuracy | 100% | 100% |
Data source: [18]
The study demonstrated that both platforms achieved high performance standards for clinical cancer panel testing, with the Ion Torrent S5 XL showing higher on-target efficiency while Illumina demonstrated slightly better variant calling sensitivity and coverage uniformity [18].
Workflow Efficiency and Turnaround Time
Table 2: Workflow Efficiency Comparison Across Sequencing Platforms
| Parameter | Ion Torrent | Illumina |
|---|---|---|
| Typical Library Prep Time | 4-8 hours [17] | 4-8 hours [17] |
| Additional Template Prep | 2-hour emulsion PCR [17] | Cluster generation on flow cell |
| Sequencing Run Time | Several hours to same-day results [2] | 24-48 hours for high-output runs [2] |
| Total Hands-on Time | Lower [18] | Higher [18] |
| Automation Compatibility | Integrated systems available (e.g., Genexus) [2] | Compatible with automated liquid handlers |
| Rapid Turnaround Systems | Genexus: ~14-24 hours sample-to-result [2] | Illumina DNA Prep: ~8.5 hours library prep [36] |
Ion Torrent systems generally offer faster sequencing run times compared to Illumina platforms, contributing to shorter overall turnaround times [2]. The Ion Torrent Genexus system automates the entire workflow from sample to result in approximately 14-24 hours, representing a significant advantage for time-sensitive applications [2]. Illumina workflows typically require longer sequencing times, particularly for high-output runs, which can extend to 24-48 hours [2].
Hands-on Time Comparison
The SARS-CoV-2 benchmarking study specifically evaluated hands-on time across platforms and found that protocols optimized for both Illumina and Ion Torrent could minimize manual intervention [37]. However, the study noted that the EasySeq (Illumina) and Oxford Nanopore Technologies protocols required the least hands-on time, suggesting that workflow efficiency depends significantly on the specific library preparation method employed rather than just the sequencing technology itself [37].
Platform Characteristics and Technical Specifications
Table 3: Platform Characteristics and Technical Specifications
| Characteristic | Ion Torrent | Illumina |
|---|---|---|
| Sequencing Chemistry | Semiconductor pH detection [2] | Fluorescent reversible terminators [2] |
| Amplification Method | Emulsion PCR [2] | Bridge PCR [2] |
| Read Structure | Single-end, variable length [3] | Paired-end available, uniform length [3] |
| Error Profile | Higher in homopolymer regions [2] [18] | Lower overall error rate [2] |
| Capital Cost | Generally lower [2] | Generally higher [2] |
| Throughput Range | Moderate (millions to tens of millions of reads) [2] | Broad (millions to billions of reads) [2] |
Visualization of Sequencing Workflows
The following workflow diagrams illustrate the procedural differences between Ion Torrent and Illumina sequencing platforms, highlighting key steps that impact hands-on time and turnaround.
Diagram 1: Illumina sequencing workflow characterized by bridge PCR cluster generation and fluorescent detection, typically requiring longer sequencing run times but offering paired-end reads.
Diagram 2: Ion Torrent workflow utilizing emulsion PCR and semiconductor pH detection, offering faster sequencing times but limited to single-end reads.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Research Reagent Solutions for NGS Workflows
| Reagent/Kit | Function | Platform Compatibility |
|---|---|---|
| Hybridization Capture-Based Panels | Target enrichment for specific gene regions; reduces allele dropout in GC-rich regions [18] | Both (platform-specific designs) |
| AmpliSeq Panels | Amplification-based target enrichment; rapid library prep for focused gene sets [37] | Both (platform-specific versions) |
| Emulsion PCR Reagents | Clonal amplification of DNA fragments on beads for Ion Torrent sequencing [2] | Ion Torrent |
| Bridge PCR Reagents | Cluster generation on flow cell surface for Illumina sequencing [2] | Illumina |
| Magnetic Bead Clean-up Kits | Library purification and size selection; critical for reducing background [37] | Both |
| Universal Blocking Oligos | Reduce non-specific binding in hybridization capture protocols [18] | Both |
| Index/Barcode Adapters | Sample multiplexing; enable pooling of multiple libraries [37] | Both |
Discussion and Platform Selection Considerations
The comparative data presented indicates that both Illumina and Ion Torrent platforms can deliver high-quality sequencing results for cancer diagnostics research, but with different efficiency profiles that may advantage particular use cases.
For Ion Torrent, the key efficiency advantages include faster sequencing run times, lower hands-on time with integrated automated systems like Genexus, and generally lower capital equipment costs [2]. These characteristics make it particularly suitable for laboratories requiring rapid turnaround for time-sensitive applications, such as targeted cancer panel testing in clinical diagnostics settings [18]. However, researchers should consider the technology's limitations in homopolymer-rich regions and lower overall throughput compared to high-end Illumina systems [2] [18].
For Illumina, the strengths lie in higher overall throughput capabilities, superior accuracy particularly in challenging genomic contexts, and the availability of paired-end sequencing which provides advantages for complex variant detection and structural variant analysis [3] [2]. These features make Illumina platforms well-suited for comprehensive genomic analyses in cancer research, including whole-exome and whole-genome sequencing applications where maximum data quality is prioritized over speed [2].
The choice between platforms for cancer diagnostics research should consider specific application requirements, including desired throughput, turnaround time expectations, budget constraints, and the genomic contexts of target regions. Both platforms continue to evolve, with ongoing improvements in workflow automation and efficiency benefiting researchers in both basic and translational cancer research.
Navigating Technical Challenges and Optimizing Performance in Cancer NGS
In next-generation sequencing (NGS) for cancer diagnostics, sequence context bias, particularly GC content bias, presents a significant technical challenge that can compromise data accuracy and clinical interpretation. This bias refers to the uneven sequencing coverage resulting from variations in the proportion of guanine (G) and cytosine (C) nucleotides across genomic regions. Both GC-rich regions (>60% GC) and AT-rich regions (<40% GC) frequently exhibit reduced sequencing efficiency, leading to uneven read depth and potential gaps in genomic coverage [38]. This bias is particularly problematic in oncology applications where comprehensive and accurate detection of somatic variants is critical for treatment decisions. The bias manifests as a unimodal relationship between fragment count and GC content, where both extremely GC-rich and AT-rich fragments are underrepresented in sequencing results [39]. This underrepresentation can lead to false negatives in variant calling or false positives arising from sequencing artifacts, ultimately affecting the reliability of cancer genomic profiling.
The persistence of GC bias across different NGS platforms, including both Illumina and Ion Torrent technologies, necessitates careful consideration in clinical cancer genomics. Understanding the sources, impacts, and mitigation strategies for this bias is essential for researchers and clinicians utilizing these technologies for precision oncology. This article examines how GC bias affects two major sequencing platforms and provides evidence-based recommendations for optimal implementation in cancer research and diagnostics.
Experimental Evidence of Platform-Specific GC Bias
Comparative Performance in Clinical Studies
Recent clinical studies directly comparing Illumina and Ion Torrent platforms reveal important differences in how GC bias manifests and impacts variant detection. A 2025 study by Fujiyoshi et al. comprehensively compared the Ion Torrent Genexus Sequencer with Illumina's FoundationOne CDx for comprehensive genomic profiling using both tissue and blood-based biopsies. The researchers analyzed six patients with breast, head, and neck cancers, examining concordance between the platforms for somatic variants in common genes [11].
The results demonstrated that while both platforms identified clinically relevant variants, notable discrepancies existed. When comparing FoundationOne to Genexus for common genes, the sensitivity and specificity were 55% and 99%, respectively. Specifically, nine single-nucleotide variants (SNVs), one copy number alteration (CNA), and one fusion were detected by both platforms. However, one SNV (MAP2K1 F53V), two CNAs (AKT3 and MYC), and one fusion (ESR-CCDC170) were detected only in Genexus, whereas two SNVs (TP53 Q331* and KRAS G12V) were detected only in FoundationOne [11]. These findings indicate that different assays and analytical methods influence variant detection, particularly in challenging genomic regions.
Table 1: Key Experimental Findings from Fujiyoshi et al. Comparison Study
| Metric | Ion Torrent Genexus | Illumina FoundationOne |
|---|---|---|
| Sensitivity | 55% (compared to FoundationOne as reference) | Reference standard |
| Specificity | 99% | Reference standard |
| Exclusive SNVs Detected | MAP2K1 F53V | TP53 Q331*, KRAS G12V |
| Exclusive CNAs Detected | AKT3, MYC | None reported |
| Exclusive Fusions Detected | ESR-CCDC170 | None reported |
Another 2025 study by Hvidtfeldt et al. compared whole genome sequencing (Illumina) to targeted sequencing (Ion Torrent Oncomine Comprehensive Assay Plus) in pancreatic cancer tissue. This research found 81% concordance across all variants between the technologies, with 100% concordance for variants relevant to targeted therapy [33]. This suggests that while overall technical performance differs, both platforms can reliably identify the most clinically actionable mutations when properly optimized.
Molecular Basis of GC Bias Differences
The fundamental technologies underlying Illumina and Ion Torrent platforms contribute to their different bias profiles. Illumina sequencing relies on sequencing-by-synthesis with fluorescently labeled nucleotides, while Ion Torrent uses semiconductor-based detection of hydrogen ions released during DNA polymerization [4]. Both technologies are susceptible to GC bias, though the mechanisms and severity differ.
Research indicates that the GC content of the entire DNA fragment – not just the sequenced read – most significantly influences fragment count in Illumina sequencing. The bias follows a unimodal pattern: both GC-rich and AT-rich fragments are underrepresented, with optimal representation occurring at intermediate GC levels. This pattern strengthens the hypothesis that PCR amplification during library preparation is a primary contributor to GC bias, as fragments with extreme GC content amplify less efficiently [39].
Ion Torrent technologies face additional challenges in homopolymer regions – stretches of identical bases – which can lead to misinterpretation of indel variants, though this is distinct from GC bias per se. The platform-specific chemistries and detection methods interact differently with sequence context, resulting in the variant detection discrepancies observed in clinical comparisons.
Methodologies for GC Bias Assessment and Mitigation
Experimental Protocols for Bias Evaluation
Standardized experimental protocols are essential for evaluating and comparing GC bias across platforms. The Fujiyoshi et al. study employed a rigorous methodology that can serve as a template for bias assessment [11]:
Sample Preparation and Nucleic Acid Extraction:
- Tissue samples: DNA and RNA were extracted from FFPE tissue specimens using the Maxwell RSC Instrument with FFPE-specific kits (Maxwell RSC FFPE Plus DNA kit and RNA FFPE kit).
- Blood samples: Cell-free total nucleic acid (cfTNA) was extracted from blood plasma using the Maxwell RSC instrument with a miRNA Plasma and Serum Kit.
- Quality control: Nucleic acid concentrations were quantified using the QuantiFluor ONE dsDNA System and QuantiFluor RNA System, with minimum quality thresholds established (>1.1 ng/μl for DNA, >0.95 ng/μl for RNA from tissues; >1.33 ng/μl from blood).
Library Preparation and Sequencing:
- Ion Torrent Genexus: Utilized Oncomine Comprehensive Assay v3 (OCAv3) for tissue and Oncomine Precision Assay (OPA) for blood with automated library preparation and sequencing on the Genexus platform.
- Illumina FoundationOne: Employed the standard FDA-approved FoundationOne CDx for tissue and FoundationOne Liquid CDx for blood according to manufacturer specifications.
Data Analysis and Variant Calling:
- Bioinformatics pipelines specific to each platform were used for variant calling.
- Common genes between platforms (130 for tissue, 41 for blood) were analyzed for concordance.
- Sensitivity and specificity calculations were performed using FoundationOne as the reference standard.
Mitigation Strategies for GC Bias
Several methodological approaches can reduce GC bias in both platforms:
PCR-Free Library Preparation: Utilizing PCR-free workflows significantly reduces amplification biases, though this requires higher input DNA (typically >100ng) [38]. This approach is particularly effective for Illumina platforms.
Unique Molecular Identifiers (UMIs): Incorporating UMIs before amplification helps distinguish true biological duplicates from PCR duplicates, providing crucial mitigation when PCR-free workflows are impractical [38].
Mechanical Fragmentation: Sonication-based fragmentation demonstrates improved coverage uniformity across varying GC content compared to enzymatic fragmentation methods, which can exhibit sequence-dependent biases [38].
Bioinformatic Correction: Computational normalization approaches can correct sequencing biases post-hoc. These algorithms adjust read depth based on local GC content, improving uniformity and accuracy in downstream analyses [39] [38].
Table 2: Comparison of GC Bias Mitigation Strategies
| Strategy | Mechanism | Applicability | Limitations |
|---|---|---|---|
| PCR-Free Library Prep | Eliminates amplification bias | Both platforms | Requires high DNA input |
| UMIs | Distinguishes PCR duplicates | Both platforms | Adds complexity, cost |
| Mechanical Fragmentation | Reduces sequence-specific bias | Both platforms | Requires specialized equipment |
| Bioinformatic Correction | Computationally normalizes coverage | Both platforms | May obscure true biological signals |
| Modified Polymerases | Engineered for GC-rich templates | Platform-dependent | Limited availability |
Technical Workflows and Visualization
The following diagram illustrates the comparative workflows of Illumina and Ion Torrent platforms, highlighting critical points where GC bias may be introduced and mitigated:
Essential Research Reagents and Solutions
The following toolkit outlines essential reagents and materials referenced in the studies discussed, particularly focusing on their application in managing sequence context bias:
Table 3: Research Reagent Solutions for GC Bias Management
| Reagent/Kit | Platform | Function in Bias Management |
|---|---|---|
| Maxwell RSC FFPE Plus DNA Kit | Both | Optimized DNA extraction from challenging FFPE samples with minimal degradation |
| Maxwell RSC RNA FFPE Kit | Both | High-quality RNA extraction from archived tissue for fusion detection |
| Maxwell RSC miRNA Plasma Kit | Both | Cell-free nucleic acid extraction for liquid biopsy applications |
| Oncomine Comprehensive Assay | Ion Torrent | Targeted panels with optimized coverage of clinically relevant genes |
| FoundationOne CDx | Illumina | Comprehensive genomic profiling with validated performance across GC ranges |
| Uracil-DNA Glycosylase | Both | Treatment for deaminated bases in damaged samples (e.g., FFPE) |
| QuantiFluor Quantification Systems | Both | Accurate nucleic acid quantification for optimal library input |
The management of GC-rich and AT-rich genome regions remains a critical consideration in cancer genomics, with significant implications for variant detection accuracy and clinical decision-making. Both Illumina and Ion Torrent platforms demonstrate strengths and limitations in handling sequence context bias, with comparative studies revealing platform-specific detection patterns [11] [33]. The observed discrepancies in variant calling between platforms highlight the importance of understanding each technology's bias profile when interpreting results, particularly for clinical applications.
For researchers and clinicians, several evidence-based recommendations emerge: First, implement platform-specific validation for genes known to have extreme GC content. Second, employ bias-aware bioinformatic pipelines that account for GC content in variant calling. Third, consider orthogonal validation for critical biomarkers in challenging genomic regions, particularly when results inform therapeutic decisions. As NGS continues to evolve as the foundation of precision oncology, acknowledging and addressing technical limitations like GC bias will be essential for maximizing its clinical utility and improving patient outcomes.
The accurate identification of genomic variants, including single nucleotide variants (SNVs) and insertions/deletions (indels), is a cornerstone of cancer diagnostics and research. Next-generation sequencing (NGS) platforms, primarily Illumina and Ion Torrent, enable this discovery, but each introduces distinct technical artifacts that influence downstream variant calling [17]. The bioinformatics pipeline, particularly the step of read alignment, serves as a critical bridge between raw sequencing data and high-confidence variant calls. The choice of alignment software can either mitigate or amplify the inherent biases of a sequencing platform, making it a key factor in optimizing diagnostic accuracy [34].
This guide objectively compares the performance of Illumina and Ion Torrent sequencing platforms within the context of cancer genomics, with a focused examination of how aligner selection impacts variant calling efficacy. We present supporting experimental data from controlled studies to inform researchers and clinicians in designing robust bioinformatics workflows.
Platform Comparison: Illumina vs. Ion Torrent
Illumina and Ion Torrent platforms employ fundamentally different sequencing chemistries. Illumina uses fluorescence-based reversible terminator chemistry, while Ion Torrent relies on semiconductor technology to detect hydrogen ions released during nucleotide incorporation [17]. This fundamental difference leads to distinct error profiles and performance characteristics.
Table 1: Key Technical Specifications and Performance Characteristics of Illumina and Ion Torrent Platforms
| Feature | Illumina | Ion Torrent | Impact on Variant Calling |
|---|---|---|---|
| Sequencing Chemistry | Fluorescent reversible terminators [17] | Semiconductor, detection of H+ ions [17] | Ion Torrent is more prone to homopolymer errors [16] [40] |
| Typical Read Length | Up to 2x300 bp (MiSeq) [41] | Up to 400 bp [16] | Longer reads can improve alignment in complex regions |
| Read Structure | Fixed length, supports paired-end [34] | Variable length, typically single-end [34] | Paired-end reads (Illumina) facilitate better alignment and SV detection |
| Inherent GC Bias | Low to moderate [17] | Can be severe for extreme AT-rich genomes [42] [17] | Ion Torrent may have significant coverage gaps in low-GC regions |
| Error Profile | Substitution errors [17] | Homopolymer indel errors [16] [40] | Directly influences false positive and false negative variant types |
| Library Amplification | Bridge PCR (on flowcell) | Emulsion PCR (offline) [17] | Additional amplification bias possible with Ion Torrent |
Table 2: Comparison of Somatic Variant Caller Performance on Ion Torrent Data
| Variant Caller | Number of SNVs Called | Variant Allele Frequency (VAF) Profile | Key Characteristics |
|---|---|---|---|
| Torrent Variant Caller (TVC) | 7,634 (lowest count) [43] | Wide, uniform distribution (mean VAF ~0.16) [43] | Lower number of calls but higher validation rate; less overcalling [43] |
| MuTect2 | 301,959 (highest count) [43] | Skewed to very low VAF (mean VAF ~0.019) [43] | High number of low-VAF calls; potential for many false positives [43] |
| VarScan2 | 12,119 (moderate count) [43] | Skewed to high VAF (mean VAF ~0.42) [43] | By default, does not call variants with VAF < 0.2; may miss low-frequency variants [43] |
| Ion Reporter (IR) | Not directly comparable | Not specified | Offered a reasonable trade-off, capturing 83% of validated variants with a 50% validation rate [40] |
The Critical Role of the Aligner
Following sequencing, reads must be accurately aligned to a reference genome. Different alignment algorithms use varied strategies to handle mismatches, indels, and spliced alignment (for RNA-Seq), which interacts with platform-specific error profiles.
The diagram above outlines the foundational steps of a sequencing data analysis pipeline. A key study investigating differential gene expression found a strong interaction between the sequencing platform and the choice of aligner [34]. For example, while the STAR aligner achieved a high percentage of uniquely mapped reads for both Illumina and Ion Torrent data, its performance was comparatively lower for Ion Torrent, likely due to that platform's variable read lengths [34]. In contrast, a sequential alignment strategy using STAR followed by Bowtie2 was found to be particularly well-suited for Ion Torrent RNA-Seq data, yielding some of the highest percentages of uniquely aligned reads [34].
Experimental Data: Performance Benchmarks
Somatic Variant Calling in Cancer
The performance of variant callers must be evaluated in the context of the sequencing platform. A comprehensive study on ovarian cancer samples sequenced with Ion Torrent revealed a startlingly low concordance among three popular somatic callers: Torrent Variant Caller (TVC), MuTect2, and VarScan2 [43]. As shown in Table 2, they shared only 0.5% of SNVs and 0.02% of INDELs across all samples. This highlights that the choice of caller, not just the platform and aligner, dramatically impacts the final set of identified variants. The same study concluded that the intersection of calls from multiple methods or the use of the platform-specific Torrent Variant Caller alone provided more reliable results [43].
Another study on non-small cell lung cancer emphasized the challenge of achieving high-confidence variant calls from Ion Torrent data. After visual validation, only 30% of putative variant calls from several algorithms were deemed valid. The proprietary Ion Reporter method was found to offer a reasonable balance, capturing 83% of all discovered variants, with half of its calls being visually validated [40].
Differential Gene Expression (RNA-Seq)
The platform-aligner interaction is also critical in transcriptomic studies. Research comparing Illumina HiSeq and Ion Torrent Proton for profiling the mouse liver transcriptome found that the greatest difference occurred at the level of read alignment [34] [44]. However, despite these technical discrepancies, the downstream biological interpretation was remarkably consistent. The study reported a high Spearman correlation (0.94 to 0.97) for gene-level read counts between platforms, and nearly identical results at the level of pathway analysis of differentially expressed genes [34]. This suggests that while alignment differences exist, both platforms can converge on the same core biological insights.
The Scientist's Toolkit: Key Research Reagents and Software
Table 3: Essential Tools for Sequencing and Analysis
| Item Name | Function/Description | Relevance to Pipeline Optimization |
|---|---|---|
| AmpliSeq for Illumina Library PLUS Kit | Targeted library preparation for Illumina platforms [41] | Ensures compatibility and optimized performance for targeted sequencing on Illumina. |
| Ion AmpliSeq Comprehensive Cancer Panel (CCP) | Targeted panel of 409 cancer-related genes for Ion Torrent [40] | Standardized panel for cancer mutation profiling, allowing cross-study comparisons. |
| Trimmomatic | Pre-alignment read trimming tool to remove adapters and low-quality bases [45] | Critical pre-processing step that improves subsequent alignment quality and variant calling accuracy. |
| HISAT2 | Aligner commonly used for DNA sequencing, particularly with the SARS-CoV-2 Illumina pipeline [45] | A specific aligner optimized for efficient and accurate mapping of DNA reads. |
| GATK HaplotypeCaller | Variant caller for germline SNPs and indels, part of a standard Illumina pipeline [45] | A widely adopted, robust tool for variant discovery in DNA sequencing data. |
| SnpEff | Variant annotation tool for predicting functional effects of sequence variants [45] | Downstream annotation is crucial for interpreting the biological and clinical impact of called variants. |
Detailed Experimental Protocols
Protocol 1: Somatic Variant Calling from Ion Torrent Data
This protocol is adapted from a study investigating somatic variants in non-small cell lung cancer using the Ion AmpliSeq Comprehensive Cancer Panel on the Ion Torrent Proton platform [40].
Library Preparation & Sequencing:
- Isolate genomic DNA from matched tumor-normal pairs (e.g., tumor tissue and blood).
- Prepare barcoded sequencing libraries from 40 ng of DNA per sample using the Ion AmpliSeq Library Kit 2.0.
- Perform emulsion PCR using the Ion Proton Template OT2 kit for clonal amplification.
- Sequence the templated beads on an Ion Proton chip.
Data Generation & Primary Analysis:
- The Torrent Suite software performs base calling and generates aligned BAM files for each sample. The mean coverage depth in the cited study was >1400X [40].
Variant Calling (Multi-Tool Approach):
- Apply multiple somatic variant callers to the tumor-normal BAM pairs. The cited study used:
- Ion Reporter (IR): The proprietary, platform-specific caller.
- VarScan2 (VS): Run with a modified p-value filter of p>10⁻⁶ to reduce false positives.
- "Poor Man's Caller" (PM): A simple subtraction method using variants called by the Torrent Variant Caller in the normal sample from those called in the tumor.
- Annotate the resulting variant calls from all methods using a tool like ANNOVAR.
- Apply multiple somatic variant callers to the tumor-normal BAM pairs. The cited study used:
Data Filtration & High-Confidence Call Selection:
- Filter out variants found in population databases (e.g., 1000 Genomes, ExAC) to remove common germline polymorphisms.
- Exclude variants in intronic and intergenic regions if the focus is on exonic mutations.
- Visually validate a subset of the calls using a tool like IGV. The cited study found that aggregating results from multiple callers or using the IR caller alone provided the most reliable results [40].
Protocol 2: Comparing Platform-Specific Aligners for RNA-Seq
This protocol is based on a study designed to compare Illumina and Ion Torrent platforms for differential gene expression analysis [34].
Experimental Design & Sequencing:
- Obtain RNA from biological samples (e.g., mouse liver from treatment/control groups).
- Prepare platform-specific libraries (Illumina HiSeq and Ion Torrent Proton).
- Sequence the libraries according to the manufacturers' protocols.
Alignment with Multiple Algorithms:
- Align the raw sequencing data from both platforms using several aligners. The cited study used:
- STAR
- GSNAP
- A sequential strategy: STAR first, followed by Bowtie2 for unmapped reads.
- Use the same genome indexes for both platforms to ensure a direct comparison.
- Align the raw sequencing data from both platforms using several aligners. The cited study used:
Quantification and Differential Expression Analysis:
- Use a tool like PORT (Pipeline Of RNA-Seq Transformations) to normalize and quantify the aligned reads from each platform-aligner combination.
- Perform differential expression analysis using a statistical test like the Mann-Whitney U test with multiple-testing correction.
Concordance Assessment:
- Calculate the Spearman correlation between gene-level read counts from the two platforms.
- Compare the lists of differentially expressed genes (DEGs) identified from each platform-aligner combination.
- Perform pathway enrichment analysis on the DEGs from each dataset to evaluate concordance at the biological level.
The integration of sequencing platform characteristics, aligner performance, and variant calling algorithms is a complex but essential consideration for robust cancer genomics. Illumina platforms generally offer higher base-level accuracy, while Ion Torrent can be more prone to homopolymer errors [16]. The choice of aligner is not one-size-fits-all; it must be tailored to the platform and the biological question, as certain aligner-platform combinations are better suited for specific tasks, such as disentangling gene expression in complex genomic regions [34].
For the critical task of somatic variant detection, reliance on a single variant caller, especially for Ion Torrent data, is inadvisable. The low concordance between callers necessitates a strategy that combines multiple callers or carefully uses the platform-optimized caller after rigorous validation [40] [43]. Ultimately, a thoughtfully optimized pipeline that accounts for these interactions is paramount for generating reliable, actionable data in cancer diagnostics and research.
Next-generation sequencing (NGS) has fundamentally transformed oncology research and diagnostic practices, enabling comprehensive genomic profiling that guides personalized cancer therapy. However, the sample quality challenges associated with formalin-fixed paraffin-embedded (FFPE) tissues and low-input nucleic acids present significant barriers to reliable data generation. These challenges are particularly acute when comparing major sequencing platforms like Illumina and Ion Torrent, as their performance can vary substantially with suboptimal sample inputs.
FFPE specimens represent a vast and invaluable resource for cancer research, with an estimated 50-80 million solid tumor samples archived globally that are potentially suitable for NGS analysis [46]. Despite their abundance, the formalin fixation process introduces extensive chemical modifications to DNA, including cross-links, fragmentation, and base damage such as cytosine deamination (leading to C>T/G>A artifacts) [46]. These modifications can result in false positive variants, reduced library complexity, and sequencing failures that compromise data integrity. Similarly, low-input samples from limited tissue biopsies or liquid biopsies present obstacles related to limited template material and potential amplification biases.
Understanding how different NGS platforms perform with these challenging samples is essential for researchers and drug development professionals seeking to implement robust genomic profiling workflows. This guide provides an objective comparison of Illumina and Ion Torrent systems specifically for low-input and FFPE-derived nucleic acids, supported by experimental data and detailed methodological approaches.
Platform Performance Comparison with Challenging Samples
Direct Performance Metrics with FFPE and Liquid Biopsy Samples
A 2025 study directly compared the Ion Torrent Genexus system (using Oncomine Comprehensive Assay v3 and Oncomine Precision Assay) with FoundationOne products (which utilize Illumina-based sequencing) for both tissue and blood-based analyses [11]. The research examined six patients with breast, head, and neck cancers using tissue and circulating tumor DNA biopsies.
Table 1: Variant Concordance Between Ion Torrent Genexus and FoundationOne (Illumina)
| Performance Metric | Ion Torrent Genexus | FoundationOne (Illumina) |
|---|---|---|
| Sensitivity for Common Genes | 55% | Reference standard |
| Specificity for Common Genes | 99% | Reference standard |
| Concordantly Detected Variants | 9 SNVs, 1 CNA, 1 fusion | 9 SNVs, 1 CNA, 1 fusion |
| Variants Detected Only by Platform | 1 SNV (MAP2K1 F53V), 2 CNAs (AKT3, MYC), 1 fusion (ESR-CCDC170) | 2 SNVs (TP53 Q331*, KRAS G12V) |
| Genes Common Between Panels | 130 genes (tissue), 41 genes (blood) | 130 genes (tissue), 41 genes (blood) |
The study concluded that while the two platforms showed equivalence, they were not perfectly concordant, indicating that different assays and analytical methods influence variant detection capabilities [11]. This has critical implications for researchers working with precious limited samples who must select the most appropriate platform for their specific variant detection needs.
Broader Platform Performance Characteristics
Beyond direct comparisons, understanding the general performance characteristics of each platform with challenging samples is essential for experimental planning.
Table 2: General Platform Characteristics Relevant to Challenging Samples
| Characteristic | Illumina Platforms | Ion Torrent Platforms |
|---|---|---|
| Primary Sequencing Chemistry | Sequencing-by-synthesis with fluorescently labeled nucleotides [47] | Semiconductor-based detection of hydrogen ions released during DNA polymerization [4] |
| Typical Read Lengths | Short reads (75-300 bp) [47] | Short to medium reads |
| Error Profile | Low error rates (0.1-0.6%), primarily substitution errors [47] | Higher error rates in homopolymer regions [4] |
| FFPE DNA Damage Mitigation | DRAGEN secondary analysis with error correction; specialized FFPE kits [48] | Automated library preparation with integrated damage mitigation [11] |
| Reported Sensitivity for Low-Frequency Variants | ~1% variant allele frequency [47] | Similar sensitivity in practice [11] |
| Hands-on Time | Varies by workflow | Minimal hands-on time with automated library prep [11] |
Illumina has demonstrated particular strength in challenging genomic regions, with the NovaSeq X Series maintaining high coverage and variant calling accuracy in repetitive regions, GC-rich sequences, and homopolymers longer than 10 base pairs [49]. This capability is crucial for comprehensive cancer genomics, as many clinically relevant regions contain these challenging sequences.
Experimental Protocols and Methodologies
Nucleic Acid Extraction from FFPE Samples
Proper extraction is the critical first step in ensuring sample quality. The following protocols are recommended based on current research:
DNA Extraction from FFPE Tissues:
- Deparaffinization: Remove paraffin wax using xylene (or substitutes) and ethanol washes, or through direct incubation with specialized additives that penetrate and lift away wax [50].
- Proteolytic Digestion: Perform enzymatic proteolysis under optimized conditions. Typical conditions include incubation with proteinase K at 50-60°C for 15-60 minutes, followed by higher temperature incubation (80°C) to reverse cross-links [50].
- Nucleic Acid Purification: Use solid-phase extraction on glass-fiber filters or magnetic beads. Magnetic bead-based systems enable higher-throughput processing and are more amenable to automation [50].
- Quality Assessment: Measure DNA concentration using fluorometric methods and assess fragment size distribution using capillary electrophoresis systems [11].
RNA Extraction from FFPE Tissues:
- Follow similar deparaffinization and digestion steps as for DNA.
- Use specialized kits designed for RNA preservation, as RNA is more susceptible to degradation during fixation and extraction.
- Include DNase digestion steps to remove genomic DNA contamination.
- Assess RNA quality using methods appropriate for degraded RNA, as traditional metrics like RNA Integrity Number (RIN) may not be meaningful for FFPE-derived RNA [48].
DNA Library Preparation Protocols
For Illumina Platforms with FFPE-DNA:
- DNA Repair: Treat with specialized repair mixes that target damaged bases. The NEBNext FFPE DNA repair mix selectively excises damaged portions from single-stranded DNA, while double-strand damage undergoes base excision repair mechanisms [51].
- Library Construction: Use specialized FFPE-compatible library prep kits such as the Illumina TruSight Oncology 500 or TruSight Tumor 15, which are optimized for degraded material [48].
- Target Enrichment: Perform hybrid capture-based or amplicon-based enrichment depending on the panel design. Illumina's TruSight Oncology 500 uses a hybrid capture approach to target multiple variant types [48].
- Quality Control: Assess library quality and quantity using capillary electrophoresis and qPCR methods specifically validated for FFPE libraries [48].
For Ion Torrent Platforms with FFPE-DNA:
- Automated Library Preparation: Utilize the integrated Genexus system which automates library construction, templating, and sequencing with minimal hands-on steps [11].
- Panel-Specific Optimization: Use the Oncomine Comprehensive Assay or Oncomine Precision Assay, which have been specifically optimized for FFPE and liquid biopsy samples [11].
- Template Preparation: Perform emulsion PCR or automated templating on the Genexus instrument.
- Sequencing: Run on the Ion Torrent sequencer with semiconductor-based detection [11].
Diagram 1: FFPE Nucleic Acid Workflow
Technical Solutions for Sample Quality Challenges
DNA Damage Repair Strategies
FFPE-induced DNA damage requires specialized repair approaches to ensure accurate sequencing results:
Enzymatic Repair Treatments:
- Uracil-DNA Glycosylase (UDG) Treatment: Removes uracil residues resulting from cytosine deamination, significantly reducing C>T/G>A artifacts [46].
- DNA Repair Mixes: Comprehensive enzyme cocktails that address multiple damage types including nicks, gaps, abasic sites, and oxidized bases [51].
- Fragmentation Control: Modern enzymatic fragmentation methods provide consistent sizing without overfragmentation concerns, even with highly degraded FFPE-DNA [51].
Bioinformatic Correction:
- Error Correction Algorithms: Sophisticated computational methods that distinguish true variants from FFPE-induced artifacts by analyzing strand bias, context-specific error patterns, and damage signatures [46].
- Duplicate Marking: Identification and removal of PCR duplicates that can amplify artifacts and skew variant allele frequency calculations [46].
- Damage-Aware Variant Calling: Specialized variant callers that incorporate models of FFPE damage patterns to improve specificity [46].
Low-Input and Degraded Sample Workflows
Whole Genome Amplification (WGA) Methods:
- Multiple Displacement Amplification (MDA): Provides high yields from minimal input but can introduce coverage biases and false positives [51].
- PCR-Based Amplification: More uniform coverage but limited by amplification errors and shorter fragment lengths [51].
Target Enrichment Optimization:
- Hybrid Capture Approaches: Better for highly fragmented DNA as they can capture smaller fragments, though with lower on-target efficiency compared to intact DNA [48].
- Amplicon-Based Approaches: Higher efficiency with good-quality samples but can struggle with extremely degraded DNA where primer binding sites may be compromised [48].
Table 3: Research Reagent Solutions for Challenging Samples
| Reagent Type | Specific Products | Function | Considerations for Challenging Samples |
|---|---|---|---|
| Nucleic Acid Extraction Kits | MagMAX FFPE DNA/RNA Ultra Kit [50], RecoverAll Total Nucleic Acid Isolation Kit [50] | Simultaneous extraction of DNA and RNA from FFPE samples | Magnetic bead-based systems enable higher throughput processing |
| DNA Repair Mixes | NEBNext FFPE DNA repair mix [51] | Targeted repair of FFPE-induced DNA damage | Specifically excises damaged bases while preserving true mutations |
| Library Prep Kits | NEBNext UltraShear FFPE DNA Library Prep Kit [51], TruSight Oncology 500 [48] | Library preparation optimized for damaged DNA | Enzymatic fragmentation provides consistent sizing without overfragmentation |
| Target Enrichment Panels | Oncomine Comprehensive Assay [11], TruSight Oncology 500 [52] | Capture of cancer-relevant genes | Designed for performance with FFPE and liquid biopsy samples |
| QC Assays | TapeStation [11], Qubit [11] | Quality and quantity assessment | Fluorometric methods more accurate than spectrophotometry for degraded samples |
Implementation in Cancer Research Workflows
Practical Considerations for Platform Selection
Choosing between Illumina and Ion Torrent platforms for challenging samples involves multiple considerations:
Turnaround Time and Automation: The Ion Torrent Genexus system offers significant advantages in workflow integration, automating the entire process from library preparation to sequencing with minimal hands-on time [11]. This can be particularly valuable in clinical research settings with limited technical staff. Illumina workflows typically require more discrete steps but offer greater flexibility in sample batch sizes and processing approaches.
Coverage Uniformity and GC Bias: Illumina platforms demonstrate superior performance in GC-rich regions, with minimal coverage drop-off compared to competing technologies [49]. This is particularly important for cancer panels that include GC-rich genes. Ion Torrent systems may show more variability in coverage uniformity across regions with extreme GC content.
Variant Type Performance: Both platforms effectively detect single nucleotide variants (SNVs) and small indels, but show differences in specific variant types. The FoundationOne (Illumina) system demonstrated better detection of some SNVs (TP53 Q331* and KRAS G12V) in comparative studies, while the Ion Torrent system uniquely detected certain copy number alterations and fusions [11].
Multi-Institutional Validation Data
A 2025 Italian multi-institutional study evaluating in-house NGS testing for non-small cell lung cancer (NSCLC) samples provides valuable real-world performance data [53]. The study, which utilized targeted sequencing of 50 genes from 283 NSCLC samples, demonstrated:
- 99.2% sequencing success rate for DNA and 98% for RNA across participating institutions [53]
- Detection of 285 relevant variants (81.1% SNVs/indels, 9.8% copy number variants, and 9.1% gene fusions) [53]
- Median turnaround time of 4 days from sample processing to molecular report [53]
- Identification of co-mutations with potential clinical relevance in 20.5% of samples positive for main oncogenic drivers [53]
This large-scale implementation demonstrates that robust, reproducible results are achievable with challenging clinical samples when appropriate quality control measures are implemented.
Diagram 2: FFPE DNA Damage and Mitigation
Ensuring sample quality with low-input and FFPE-derived nucleic acids remains a critical challenge in cancer genomics research, with significant implications for platform selection and experimental design. Both Illumina and Ion Torrent platforms offer viable solutions, but with distinct performance characteristics that must be aligned with research objectives.
The Ion Torrent Genexus system provides advantages in automation and workflow integration, potentially reducing turnaround time and technical variability. Meanwhile, Illumina platforms demonstrate strengths in coverage uniformity, particularly in challenging genomic regions, and have established extensive partnerships for companion diagnostic development [52].
Successful implementation requires a comprehensive approach addressing pre-analytical sample quality, specialized library preparation methods, DNA damage repair strategies, and bioinformatic correction for FFPE-specific artifacts. By understanding the capabilities and limitations of each platform with challenging samples, researchers can make informed decisions that maximize data quality and reliability, ultimately advancing cancer diagnostics and therapeutic development.
As NGS technologies continue to evolve, improvements in damage tolerance, library efficiency from minimal inputs, and computational artifact removal will further enhance our ability to extract meaningful biological insights from even the most challenging clinical samples.
Minimizing False Positives and Negatives in Somatic Variant Detection
Somatic variant detection is a cornerstone of cancer genomics, enabling the identification of tumor-specific mutations that drive cancer progression and inform targeted therapy selection. The accuracy of this process, defined by the minimization of false positives (erroneous variant calls) and false negatives (missed true variants), is critical for both research and clinical applications. A primary source of technical variation stems from the choice of sequencing platform, with Illumina and Ion Torrent representing two widely used technologies. These platforms differ fundamentally in their chemistry—Illumina using sequencing-by-synthesis with fluorescently labeled nucleotides and Ion Torrent relying on the detection of hydrogen ions released during DNA polymerization. These underlying differences influence library preparation, error profiles, and the subsequent bioinformatic analysis required for reliable variant calling [43] [40]. This guide objectively compares the performance of these platforms in the context of somatic variant detection, synthesizing experimental data to highlight strategies for optimizing accuracy.
Performance Comparison of Sequencing Platforms and Bioinformatic Tools
The interplay between sequencing technology and choice of bioinformatic algorithm significantly impacts the final set of reported somatic variants. Studies consistently reveal substantial differences in the output of different variant callers, and these discrepancies are often more pronounced on specific platforms.
Concordance of Variant Callers
A comprehensive study on Ion Torrent data from 208 paired ovarian cancer samples demonstrated alarmingly low concordance between three popular somatic variant callers: Torrent Variant Caller (TVC), MuTect2, and VarScan2. As detailed in Table 1, only a minute fraction of called single nucleotide variants (SNVs) and insertions/deletions (INDELs) were identified by all three methods. MuTect2, while exhibiting high sensitivity, generated the vast majority of unique calls, potentially contributing to a high false-positive rate if not properly filtered [43].
Table 1: Concordance of Somatic Variant Callers on Ion Torrent Data (208 paired tumour-blood samples)
| Variant Type & Metric | Torrent Variant Caller (TVC) | MuTect2 | VarScan2 |
|---|---|---|---|
| Total SNVs Called | 7,634 | 301,959 | 12,119 |
| Unique SNVs (% of total) | 2,419 (0.8%) | 295,746 (94.3%) | 8,929 (2.8%) |
| SNVs Common to All Three | 1,524 (0.5% of all SNVs) | 1,524 (0.5% of all SNVs) | 1,524 (0.5% of all SNVs) |
| Total INDELs Called | Information missing | 272,063 | Information missing |
| Unique INDELs (% of total) | Information missing | 268,642 (96.3%) | Information missing |
| INDELs Common to All Three | 51 (0.02% of all INDELs) | 51 (0.02% of all INDELs) | 51 (0.02% of all INDELs) |
This lack of consensus is not merely a software issue but is linked to platform-specific error profiles. Ion Torrent is notably more vulnerable to errors within homopolymer regions (stretches of identical consecutive bases), which can lead to false INDEL calls [40]. Furthermore, the baseline quality of base calling is generally lower compared to Illumina, necessitating robust quality control and tailored bioinformatic parameters [40].
Impact of Sequencing Depth and Variant Allele Frequency
The ability to detect a somatic variant is fundamentally constrained by its Variant Allele Frequency (VAF) and the sequencing depth at its genomic position. Lower VAFs, often resulting from tumor heterogeneity or low tumor purity, present a significant challenge.
Theoretical modeling and empirical data, primarily from Illumina-based studies, provide clear guidelines. Modeling suggests that a sequencing depth of 30x is sufficient to confidently detect variants with VAFs ≥ 0.2, but deeper sequencing (75x-100x) is necessary for variants at VAF ≤ 0.1 [54]. A systematic evaluation of sequencing depth and mutation frequency performance is shown in Table 2.
Table 2: Somatic Variant Calling Performance at Different Sequencing Depths and VAFs (Illumina WES, using Strelka2 and MuTect2)
| Sequencing Depth | Mutation Frequency (VAF) | Recall Rate | Precision Rate | Recommended Application |
|---|---|---|---|---|
| 100X | 1% | 2.7 - 34.5% | >95% | Insufficient for low-VAF variants |
| 200X | ≥20% | ≥95% | >95% | Cost-effective for moderate VAF |
| 500X | 5-10% | 50 - 96% | >95% | Improved recall for lower VAF |
| 800X | 1% | 23 - 37% (Strelka2) / 32 - 50% (MuTect2) | >93% | Research settings requiring low-VAF sensitivity |
This study also revealed that for very low mutation frequencies (≤10%), simply increasing sequencing depth yields diminishing returns. At a 1% VAF, even 800x depth achieved a recall of only 32-50%, indicating that improvements in wet-lab methods or error suppression are more effective than deeper sequencing alone [55].
Experimental Protocols for Optimal Variant Detection
To achieve high-confidence somatic variant calling, a rigorous and multi-faceted experimental protocol is essential. The following methodologies, derived from the cited literature, provide a framework for minimizing errors.
Wet-Lab Methods: Library Preparation and UMI Integration
The foundation of accurate variant calling is laid during sample and library preparation.
- Sample Type Considerations: Formalin-Fixed Paraffin-Embedded (FFPE) specimens, while common in clinical practice, introduce DNA fragmentation and artifacts that can mimic true variants. Using matched normal tissue (e.g., blood) is crucial for distinguishing somatic from germline variants, though tumor-only sequencing can be performed with stringent population frequency filtering (e.g., using gnomAD, 1000 Genomes) to remove common polymorphisms [43] [40].
- Unique Molecular Identifiers (UMIs): For liquid biopsy or any application requiring ultra-sensitive detection of low-frequency variants, the use of UMIs is critical. UMIs are short random oligonucleotide tags ligated to each DNA fragment prior to amplification. After sequencing, bioinformatic tools like the DRAGEN platform collapse all reads sharing the same UMI into a single consensus sequence. This process effectively removes PCR duplicates and random sequencing errors, dramatically reducing noise. Studies show that UMI collapsing with DRAGEN can increase the error-free position fraction from 36.3% to 92.9% and reduce the mean error rate from 0.062% to 0.003%, effectively raising base quality scores from Q30 to Q50-60 [56].
The following diagram illustrates the core experimental workflow for high-accuracy somatic variant detection, integrating both wet-lab and computational best practices.
Bioinformatic Analysis and Validation Strategies
The computational pipeline is where false positives and negatives are most effectively controlled.
- Multi-Caller Consensus Approach: Given the low concordance shown in Table 1, relying on a single variant caller is not advisable. A robust strategy involves using multiple callers (e.g., MuTect2, Strelka2, VarScan2, or platform-specific callers like TVC) and taking the intersection of their outputs. This intersection set demonstrably shows better performance, with higher correlation to known mutational signatures and greater overlap with the COSMIC database [43] [40].
- Platform-Specific Tuning: Default parameters for callers like MuTect2 and VarScan2 are often tuned for Illumina data. Applying them to Ion Torrent data without modification can lead to poor performance. Parameter adjustment, such as filtering VarScan2 calls by a stricter p-value threshold, is necessary to achieve optimal results [40].
- Systematic Quality Region Filtering: The genome contains regions that are systematically problematic for sequencing due to issues with mapping or base quality. These regions can be identified by aggregating metrics like base quality, mapping quality, and depth across many samples. Studies show that 86-89% of false-positive SNVs cluster in these "low-quality" regions, which constitute about 10% of the genome. Filtering out these regions can dramatically reduce false positives with a minimal impact on sensitivity [54].
The logical relationship between the primary sources of error and the corresponding mitigation strategies is summarized in the following diagram.
The Scientist's Toolkit: Essential Reagents and Software
This section details key reagents, tools, and software solutions referenced in the experimental data, which form the essential toolkit for robust somatic variant detection.
Table 3: Research Reagent Solutions and Key Tools for Somatic Variant Detection
| Item Name | Function/Description | Application Context |
|---|---|---|
| TruSight Oncology 500 ctDNA Kit | Library prep kit with duplex UMIs for error correction. | Ultra-sensitive liquid biopsy analysis; enables low-frequency variant detection in ctDNA [56]. |
| Ion AmpliSeq Comprehensive Cancer Panel | Targeted gene panel for Ion Torrent sequencing. | Targeted deep sequencing of known cancer genes; requires careful bioinformatic handling of homopolymer errors [40]. |
| DRAGEN Somatic Pipeline | Secondary analysis platform with optimized somatic callers and UMI processing. | Provides highly accurate variant calling with integrated error suppression; shown to outperform other UMI tools like fgbio [56] [57]. |
| MuTect2 | Bayesian variant caller for somatic SNVs and indels. | Widely used, high-specificity caller; part of the GATK toolkit. Performs well on Illumina data and with low-VAF variants [43] [55]. |
| Strelka2 | Fast, mixture-model based somatic variant caller. | Top-performing caller known for speed and high precision, especially at higher VAFs (≥20%) [55]. |
| Torrent Variant Caller | Platform-specific variant caller for Ion Torrent data. | Default caller for Ion Torrent; shows a higher percentage of validated calls despite lower total output, suggesting good precision [43]. |
Minimizing false positives and negatives in somatic variant detection is an integrated process that spans wet-lab protocols and computational analysis. The choice between Illumina and Ion Torrent involves trade-offs; while Illumina's lower error rate provides a more robust foundation, especially in homopolymer regions, Ion Torrent data can yield highly reliable results when processed with platform-optimized pipelines and multi-caller consensus. The universal strategies for maximizing accuracy include: the mandatory use of UMIs for detecting low-frequency variants, employing a multi-caller consensus approach to leverage the strengths of different algorithms, and understanding the relationship between sequencing depth and VAF to set realistic detection limits. As technologies evolve, with improvements in long-read sequencing and deep learning-based callers like DeepSomatic [58], the standards for accuracy will continue to rise. By adopting the rigorous, multi-layered strategies outlined here, researchers can ensure the generation of high-confidence somatic variant data, thereby enhancing the reliability of their cancer diagnostics and research outcomes.
Data-Driven Platform Assessment: Concordance, Sensitivity, and Clinical Utility
Next-generation sequencing (NGS) has fundamentally transformed cancer diagnostics and personalized treatment strategies, with platforms from Illumina and Thermo Fisher's Ion Torrent representing two dominant technologies in clinical oncology. The convergence of rapid technological advancement with escalating clinical demand for comprehensive genomic profiling (CGP) has intensified the need for rigorous, head-to-head performance comparisons across platforms. Clinical concordance studies provide essential empirical evidence regarding the reliability, strengths, and limitations of each system, forming a critical knowledge base for molecular pathologists, clinical researchers, and drug developers. These evaluations extend beyond mere technical specifications to assess real-world performance in detecting clinically actionable variants—including single-nucleotide variants (SNVs), insertions/deletions (indels), copy number alterations (CNAs), and gene fusions—each presenting distinct detection challenges. Within this framework, this review synthesizes recent evidence from comparative studies evaluating Illumina and Ion Torrent platforms, with particular emphasis on analytical sensitivity, specificity, and their combined impact on clinical decision-making in oncology.
Comparative Analytical Performance Across Sequencing Platforms
Tissue-Based Comprehensive Genomic Profiling: Genexus vs. FoundationOne CDx
A recent 2025 study conducted by Fujiyoshi et al. provided a direct comparative analysis of two automated CGP systems: the Ion Torrent Genexus with Oncomine Comprehensive Assay v3 (OCA) and the Illumina-based FoundationOne CDx (F1) for tissue biopsies [11]. The investigation analyzed tumor tissues from patients with breast, head, and neck cancers, focusing on 130 genes common to both panels.
The results demonstrated that the Genexus system (OCA) exhibited a sensitivity of 55% and a specificity of 99% when compared against the FoundationOne benchmark for common genes [11]. The study identified several variants that were differentially detected between platforms: nine SNVs, one CNA, and one fusion were detected by both systems; one SNV (MAP2K1 F53V), two CNAs (AKT3 and MYC), and one fusion (ESR-CCDC170) were detected exclusively by Genexus; while two SNVs (TP53 Q331* and KRAS G12V) were detected only by FoundationOne [11]. This variant distribution pattern underscores a critical finding—while overall concordance is high, particularly for common SNVs, neither platform detected all variants, suggesting that technological differences can influence mutation detection in clinically relevant genes.
Table 1: Key Performance Metrics from Tissue-Based CGP Comparison [11]
| Performance Parameter | Ion Torrent Genexus (OCA) | FoundationOne CDx (F1) |
|---|---|---|
| Sensitivity | 55% | Benchmark |
| Specificity | 99% | Benchmark |
| Commonly Detected Variants | 9 SNVs, 1 CNA, 1 Fusion | 9 SNVs, 1 CNA, 1 Fusion |
| Exclusively Detected Variants | 1 SNV, 2 CNAs, 1 Fusion | 2 SNVs |
| Genes in Common Panel | 130 | 130 |
Liquid Biopsy Performance: Plasma-Based Mutation Detection
The analytical performance of NGS platforms extends to liquid biopsy applications, where detection sensitivity becomes increasingly critical due to low circulating tumor DNA (ctDNA) fraction. Fujiyoshi et al. further evaluated the blood-based counterparts—Genexus Oncomine Precision Assay (OPA) versus FoundationOne Liquid (F1L)—focusing on 41 common genes [11]. The comparative analysis revealed parallel performance characteristics with the tissue-based evaluation, with OPA demonstrating 55% sensitivity and 99% specificity against the F1L benchmark [11].
A separate 2025 retrospective study by Leduc et al. investigating liquid biopsy performance in advanced non-small cell lung cancer (aNSCLC) provided additional insights into platform-specific performance differences across different assay methodologies [59]. This study compared four different ctDNA NGS assays, including both amplicon-based and hybrid capture-based approaches, highlighting substantial differences in fusion detection capability. Hybrid capture-based assays demonstrated superior performance in detecting structurally variant alleles, identifying 7-8 gene fusions compared to only 2 detected by amplicon-based assays [59]. Additionally, the hybrid capture methodology showed enhanced capability for detecting MET amplifications, identifying 12 such events, five of which were confirmed by fluorescence in situ hybridization (FISH) but missed by tissue-based NGS [59].
Table 2: Liquid Biopsy Assay Performance in aNSCLC Detection [59]
| Assay Characteristic | Amplicon-Based Assays | Hybrid Capture-Based Assays |
|---|---|---|
| Positive Percent Agreement with Tissue | 56% (Assay 2) | Up to 79% (Assay 4) |
| Fusion Detection Capability | 2 fusions detected | 7-8 fusions detected |
| MET Amplification Detection | Limited | 12 MET amplifications detected |
| Input DNA Requirements | Lower | Higher |
| Turnaround Time | Faster | More lengthy |
Methodological Frameworks for Platform Validation
Experimental Protocols for Cross-Platform Benchmarking
The validation methodologies employed in recent concordance studies provide robust frameworks for cross-platform NGS evaluation. The Fujiyoshi et al. study implemented a standardized sample processing protocol where each patient specimen was analyzed in parallel on both platforms [11]. For tissue samples, DNA and RNA were co-extracted from formalin-fixed paraffin-embedded (FFPE) tissue specimens using the Maxwell RSC Instrument with dedicated FFPE kits, followed by quantification using the QuantiFluor ONE dsDNA System [11]. For liquid biopsy analysis, cell-free total nucleic acid was extracted from blood plasma using the Maxwell RSC instrument with the miRNA Plasma and Serum Kit [11]. This methodological consistency ensured that pre-analytical variables were minimized, enabling direct platform performance comparison.
The Leduc et al. study implemented a similar matched-pair design for liquid biopsy validation, with 102 matched tissue and liquid biopsy samples from aNSCLC patients collected at diagnosis [59]. Their protocol involved collecting 18mL of blood into EDTA or Streck tubes, followed by double centrifugation to isolate plasma within 2 hours of collection [59]. Cell-free DNA was extracted from 8mL of plasma using the QIAamp Circulating Nucleic Acid Kit, with quantification performed via Qubit Fluorometer with the dsDNA HS Assay Kit [59]. This meticulous attention to pre-analytical conditions reflects the growing recognition that sample quality profoundly impacts downstream sequencing performance, particularly for liquid biopsy applications where analyte concentration is limited.
Bioinformatic Processing and Variant Calling
Divergences in bioinformatic processing pipelines contribute significantly to platform-specific variant detection differences. The 2025 review by Satam et al. emphasized that NGS data analysis involves multiple critical steps: quality control, adapter trimming, read alignment, normalization, and variant calling [4]. Each platform employs proprietary algorithms with different thresholds for variant calling, particularly for challenging variant types like indels in homopolymer regions, where Ion Torrent has historically demonstrated limitations [4] [60]. FoundationOne's bioinformatic pipeline incorporates sophisticated noise reduction algorithms and personalized reference standards to distinguish true somatic variants from background artifacts, while the Genexus system utilizes Torrent Suite and Ion Reporter software with unique filtering parameters [11].
Diagram 1: NGS Analysis Workflow with Platform Variation Points
Technological Bases of Platform Divergence
Fundamental Sequencing Chemistry Differences
The observed differences in variant detection between Illumina and Ion Torrent platforms originate in their fundamentally distinct sequencing chemistries. Illumina technology employs reversible dye-terminator sequencing by synthesis (SBS) chemistry, wherein fluorescently-labeled nucleotides are incorporated and imaged during each cycle [60]. This approach generates high-resolution sequencing data with low substitution error rates, though read lengths are typically shorter. Conversely, Ion Torrent utilizes semiconductor sequencing technology that detects hydrogen ions released during nucleotide incorporation rather than optical signals [60]. While this methodology enables faster run times and lower instrument costs, it presents challenges in homopolymer regions where accurate quantification of consecutive identical nucleotides can be problematic, potentially leading to indel errors [60].
Library Preparation Methodologies: Amplicon vs. Hybrid Capture
Library preparation methodology represents another significant source of technical variation between NGS platforms and specific assay implementations. Amplicon-based approaches (commonly used in targeted panels like some Ion Torrent assays) employ PCR with primers flanking regions of interest to enrich targeted sequences [59]. This method is efficient and requires less input DNA, but may struggle with uniform coverage and structural variant detection. In contrast, hybrid capture-based methods (utilized in FoundationOne and other comprehensive panels) use biotinylated probes to pull down target sequences from fragmented genomic DNA [59] [61]. This approach typically delivers more uniform coverage and better performance for detecting CNAs and fusions, though at the cost of greater input DNA requirements and more complex workflows [59].
Diagram 2: Library Preparation Methodologies Comparison
Essential Research Reagents and Solutions
The consistent implementation of NGS-based cancer profiling requires standardized research reagents and solutions across platforms. The following table summarizes critical components utilized in the evaluated studies, providing researchers with a reference for experimental replication.
Table 3: Essential Research Reagents for NGS Cancer Profiling
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Nucleic Acid Extraction Kits | Maxwell RSC FFPE Plus DNA Kit; QIAamp Circulating Nucleic Acid Kit | Standardized recovery of high-quality nucleic acids from different sample types (FFPE, plasma) [11] [59] |
| Quantification Assays | QuantiFluor ONE dsDNA System; Qubit dsDNA HS Assay | Accurate nucleic acid quantification critical for library preparation input requirements [11] [59] |
| Library Preparation Kits | Oncomine Comprehensive Assay; FoundationOne CDx | Target enrichment and library construction with specific chemistry dependencies [11] |
| Reference Standards | Multiplex cfDNA Reference Standard HD786 | Process control for assay validation and quality monitoring [59] |
| Sequence Capture Reagents | Biotinylated probes (hybrid capture); Target-specific primers (amplicon) | Target enrichment methodology determines variant detection capabilities [59] [61] |
Clinical Implications and Future Directions
The accumulating evidence on platform concordance has substantial implications for clinical practice and translational research in oncology. The observation that neither platform detects all variants suggests that orthogonal confirmation may be warranted for negative results in genes with high clinical relevance when the initial test fails to align with clinical presentation [11]. This is particularly important for liquid biopsy applications, where the combination of low ctDNA fraction and platform-specific limitations can yield false-negative results [59]. The incorporation of tumor fraction estimation methods, such as cfDNA methylation analysis or copy number variation assessment, provides valuable context for interpreting negative liquid biopsy results [59].
Future methodology development will likely focus on hybrid approaches that leverage the complementary strengths of both platforms. The ongoing development of pan-cancer panels with demonstrated clinical utility for therapy selection—such as those targeting KRAS mutations across multiple cancer types—highlights the industry movement toward standardized, globally distributable tests [62]. Additionally, the integration of artificial intelligence and machine learning approaches into bioinformatic pipelines promises to enhance variant calling accuracy and interpretation across platforms [63]. As NGS technology continues to evolve, with emerging third-generation sequencing platforms offering long-read capabilities, the framework for platform comparison established in these concordance studies will remain essential for validating new methodologies against current standards [64].
Recent head-to-head clinical concordance studies between Illumina and Ion Torrent sequencing platforms reveal a complex landscape of technological complementarity rather than simple superiority of one system over another. The evidence demonstrates that while overall concordance is high for common SNVs, platform-specific differences in sequencing chemistry, library preparation, and bioinformatic processing can lead to divergent detection of certain variant types, particularly CNAs, fusions, and indels in challenging genomic contexts. These findings underscore the importance of understanding platform limitations when interpreting clinical genomic results and highlight the potential value of orthogonal confirmation in specific clinical scenarios. As precision oncology continues to evolve, ongoing platform comparisons will remain essential for optimizing clinical test selection, interpretation, and ultimately, patient care.
Within molecular diagnostics and cancer research, next-generation sequencing (NGS) has become an indispensable tool for precision medicine. The establishment of robust, reliable, and reproducible analytical methods is critical for both research validity and clinical application. This guide provides an objective comparison of two major NGS platforms—Illumina and Ion Torrent—focusing on the core performance metrics of sensitivity, specificity, and reproducibility within the context of cancer diagnostics research. Understanding these metrics empowers researchers, scientists, and drug development professionals to select the most appropriate technology for their specific experimental and clinical needs, ensuring that detected genomic variations truly reflect biological reality rather than technological artifact.
Core Performance Metrics: Definitions and Importance
In the context of NGS platform evaluation, key analytical performance metrics have specific and critical meanings. Sensitivity refers to the probability that the test will correctly detect a true genomic variant (e.g., a single nucleotide variant or SNV) when it is present. A highly sensitive test minimizes false-negative results. Specificity is the probability that the test will correctly indicate the absence of a variant when it is truly not present, thereby minimizing false-positive results. Reproducibility measures the consistency of results between technical replicates, different sequencing runs, or across various sites. High reproducibility ensures that findings are reliable and not subject to unpredictable technical noise. For clinical oncology, where treatment decisions may hinge on identifying a low-frequency mutant allele, optimizing all three metrics is paramount.
Direct Platform Comparison: Sensitivity, Specificity, and Reproducibility
Data from controlled studies provide a direct, quantitative comparison of Illumina and Ion Torrent platforms. The following tables summarize key findings on these critical metrics from multiple independent investigations.
Table 1: Comparison of Sensitivity and Specificity
| Platform/Test Combination | Metric | Performance | Context & Notes |
|---|---|---|---|
| Illumina NextSeq 550Dx (Hybridization Capture HCP Panel) | Sensitivity | 98.53% [18] | For SNV and indel calling |
| Ion Torrent S5 XL (Hybridization Capture HCP Panel) | Sensitivity | 97.06% [18] | For SNV and indel calling |
| Illumina NextSeq 550Dx (Hybridization Capture HCP Panel) | Specificity | 100% [18] | For SNV and indel calling |
| Ion Torrent S5 XL (Hybridization Capture HCP Panel) | Specificity | 100% [18] | For SNV and indel calling |
| Ion Torrent PGM (AmpliSeq BRCA Panel) | Sensitivity | High (>99% confirmation) [65] | For germline BRCA1/2 mutations; required Sanger confirmation |
| Ion Torrent Genexus (Oncomine Comprehensive Assay v3) | Sensitivity | 55% [11] | Compared to FoundationOne (tissue); based on limited (n=6) patient samples |
| Ion Torrent Genexus (Oncomine Comprehensive Assay v3) | Specificity | 99% [11] | Compared to FoundationOne (tissue) |
Table 2: Comparison of Reproducibility and Concordance
| Platform/Method | Metric | Performance | Context & Notes |
|---|---|---|---|
| Illumina NextSeq vs. Ion Proton | SNV Concordance | 89% [66] | For SNVs with mutant allele frequency ≥5% in FFPE samples |
| Illumina NextSeq vs. Ion Proton | indel Concordance | 100% [66] | For small insertions and deletions |
| Small RNA-Seq (Illumina) | Reproducibility (CV) | 8.2% [67] [68] | Coefficient of variation for technical replicates |
| EdgeSeq | Reproducibility (CV) | 6.9% [67] [68] | Coefficient of variation for technical replicates |
| FirePlex | Reproducibility (CV) | 22.4% [67] [68] | Coefficient of variation for technical replicates |
| RNA-Seq Pipelines (with svaseq correction) | Reproducibility of DE Calls | >80% [69] | Reproducibility for genome-scale differential expression surveys |
Key Findings from Comparative Data
- High Concordance in DNA Sequencing: When comparing the Illumina NextSeq and Ion Torrent Ion Proton for DNA-based mutation detection in oncology, studies show high concordance. One study found that the Ion Proton platform identified 89% of the single nucleotide variants (SNVs) detected by NextSeq, with an almost perfect correlation (R² = 0.973) in mutant allele frequencies between the two platforms [66]. Concordance for small insertions and deletions (indels) was reported at 100% [66].
- Varying Sensitivity in Different Contexts: A more recent 2025 study comparing the Ion Torrent Genexus system to the established FoundationOne test (which uses Illumina sequencing) found a tissue-based sensitivity of 55% and specificity of 99% [11]. This indicates that while the platforms are largely equivalent, the specific assay and analytical methods can influence variant detection, and they are not perfectly interchangeable [11].
- Reproducibility Across RNA Platforms: A comparative study of miRNA quantification platforms demonstrated that the coefficient of variation (CV) for technical replicates was lowest for HTG Molecular EdgeSeq (6.9%) and small RNA-seq (typically performed on Illumina platforms) (8.2%), indicating high reproducibility. In contrast, the FirePlex platform showed higher technical variation (CV of 22.4%) [67] [68].
Detailed Experimental Protocols from Key Studies
To critically assess the data, understanding the underlying experimental methodologies is essential. Below are the detailed protocols from several pivotal studies cited in this comparison.
- Study Aim: To compare the analytic performance of Illumina's NextSeq 550Dx and Thermo Fisher's Ion S5 XL using a hybridization capture-based target enrichment method, which is less prone to allele dropout than amplicon-based methods.
- Sample Preparation: 31 clinical samples (28 peripheral blood buffy coats and 3 fresh tumor tissues) with known variants and the NA12878 reference material were used. Variants were pre-confirmed by Sanger sequencing.
- Library Preparation & Sequencing: A hybridization-based capture Hereditary Cancer Predisposition (HCP) panel was designed for each platform. Libraries were prepared according to the manufacturer's instructions for each system and sequenced on their respective platforms.
- Data Analysis: Sequencing data were processed using the default pipelines for each platform. The sensitivity, specificity, and accuracy of SNV and indel calling were calculated against the known Sanger-verified variants.
- Study Aim: To compare the reproducibility, accuracy, sensitivity, and specificity of four miRNA profiling platforms: small RNA-seq (Illumina), FirePlex, EdgeSeq, and nCounter.
- Sample Types: The study utilized both a synthetic pool of miRNAs and biological samples (plasma from pregnant and non-pregnant women).
- Experimental Process: Each sample was processed according to the specific protocols for the four platforms. For the synthetic miRNA pool, technical replicates were run to assess reproducibility.
- Data Analysis: Coefficients of variation (CV) were calculated for technical replicates. Receiver operating characteristic (ROC) analysis was performed to evaluate the accuracy of distinguishing present vs. absent miRNAs. The ability of each platform to detect expected biological differences (placenta-associated miRNAs in pregnancy) was also assessed.
- Study Aim: To evaluate the performance of the Ion Torrent PGM platform with the AmpliSeq BRCA1/2 panel for routine rapid detection of germline mutations compared to conventional methods (dHPLC and Sanger sequencing).
- Sample Cohorts: The study included a validation cohort of 33 patients with previous conventional genetic results and a prospective cohort of 29 newly diagnosed patients.
- Library Preparation & Sequencing: Libraries were prepared using the Ion AmpliSeq BRCA1 and BRCA2 panel and sequenced on the Ion PGM.
- Variant Confirmation: All variants identified by NGS in the prospective cohort were confirmed by Sanger sequencing. The study then calculated the concordance between NGS and traditional methods.
Platform Selection and Validation Workflow
The following diagram illustrates the critical decision points and validation steps a researcher should undertake when choosing and implementing an NGS platform for cancer diagnostics.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents and consumables critical for performing the NGS experiments and comparisons discussed in this guide.
Table 3: Essential Research Reagents and Materials for NGS Comparison Studies
| Item | Function in the Experimental Workflow | Example from Search Results |
|---|---|---|
| Formalin-Fixed Paraffin-Embedded (FFPE) Tissue Samples | Common source of clinical oncology DNA samples; presents challenges due to DNA fragmentation and cross-linking. | Used in performance comparison of NextSeq and Ion Proton [66]. |
| Reference DNA/RNA Materials | Provide a known ground truth for benchmarking platform accuracy, sensitivity, and specificity. | NA12878 reference material used in hybridization capture study [18]; Synthetic miRNA pools used in miRNA platform comparison [67] [68]. |
| Hybridization Capture-Based Panels | Use long biotinylated probes to capture genomic regions of interest; tolerate mismatches better than amplicon-based methods, reducing allele dropout. | Used in the hereditary cancer panel comparison between Illumina NextSeq and Ion S5 XL [18]. |
| Amplicon-Based Panels | Use PCR primers to amplify target regions; can be faster but are vulnerable to allele dropout from variants in primer binding sites. | Ion AmpliSeq Cancer Hotspot Panel v2 and BRCA1/2 Panel used with Ion Torrent PGM [70] [65]. |
| Library Preparation Kits | Reagent sets for fragmenting, indexing, and preparing nucleic acids for sequencing. Specific kits are often platform-specific. | Maxwell RSC kits for nucleic acid extraction from FFPE and plasma [11]. |
| Quality Control Assays | Essential for quantifying and qualifying input nucleic acids (e.g., DNA/RNA concentration, fragment size) to ensure they meet sequencing standards. | QuantiFluor ONE dsDNA System and Agilent 4200 TapeStation system [11]. |
The direct comparison between Illumina and Ion Torrent sequencing platforms reveals a landscape of high performance with nuanced differences. For DNA-based somatic variant detection in cancer, both platforms demonstrate high sensitivity, specificity, and concordance, making them suitable for clinical research [66] [18]. The choice between them may depend on factors such as required throughput, turnaround time, and existing laboratory infrastructure. For RNA expression analyses, sensitivity and reproducibility can vary significantly based on the specific profiling method used, with small RNA-seq on Illumina platforms showing superior accuracy [67] [68]. Ultimately, researchers must align their platform selection with their specific project goals, and regardless of the platform chosen, orthogonal validation of key findings remains a cornerstone of rigorous genomic research [65].
Next-generation sequencing (NGS) has revolutionized cancer diagnostics and research by enabling comprehensive profiling of genomic alterations that drive oncogenesis. Among short-read sequencing technologies, platforms from Illumina and Ion Torrent (Thermo Fisher Scientific) have emerged as the dominant clinical tools for detecting key variant types including single nucleotide variants (SNVs), copy number variations (CNVs), gene fusions, and insertions/deletions (indels). Understanding the performance characteristics of these platforms is essential for researchers and clinicians seeking to optimize their genomic testing workflows. While both platforms utilize sequencing-by-synthesis principles, their underlying detection chemistries—optical for Illumina and semiconductor for Ion Torrent—impart distinct advantages and limitations across different variant types and genomic contexts [71] [2]. This guide provides an objective, data-driven comparison of these platforms specifically for variant detection in cancer research, synthesizing evidence from recent analytical validation studies to inform platform selection and experimental design.
The fundamental difference between Illumina and Ion Torrent platforms lies in their detection mechanisms. Illumina employs a fluorescence-based method using reversible terminator chemistry, wherein each incorporated nucleotide is identified by its fluorescent tag before the terminator is cleaved to allow subsequent incorporation [2]. This process occurs on DNA clusters generated by bridge amplification on a flow cell. In contrast, Ion Torrent utilizes semiconductor sequencing, detecting pH changes from hydrogen ions released during nucleotide incorporation without optical imaging [1] [2]. DNA templates are amplified via emulsion PCR on beads that are deposited into semiconductor chip wells. This core methodological difference drives variations in read characteristics, error profiles, and optimal applications.
Table 1: Fundamental Platform Characteristics
| Feature | Illumina | Ion Torrent |
|---|---|---|
| Detection Method | Fluorescence (optical) | pH change (semiconductor) |
| Amplification | Bridge amplification on flow cell | Emulsion PCR on beads |
| Read Structure | Paired-end available | Single-end only |
| Typical Read Lengths | Up to 2×300 bp (MiSeq) [34] | 200-600 bp [2] |
| Run Times | ~24-48 hours for high-output runs [2] | As fast as same-day results [2] |
| Upfront Instrument Cost | Generally higher [2] | Generally lower for comparable capacity [2] |
Performance Comparison Across Variant Types
Single Nucleotide Variants (SNVs) and Insertions/Deletions (Indels)
Both platforms demonstrate strong capability for SNV detection, though with important distinctions in accuracy profiles. Illumina typically exhibits lower raw error rates (approximately 0.1-0.5% per base) compared to Ion Torrent (approximately ~1% error per base) [2]. The most significant difference emerges in homopolymer regions, where Ion Torrent's measurement of cumulative proton release struggles to precisely count identical consecutive nucleotides, leading to higher indel error rates in these contexts [2]. A 2025 comparative study of comprehensive genomic profiling tests found 55% sensitivity for Ion Torrent Genexus compared to FoundationOne (Illumina-based) for overlapping genes, with 99% specificity [11]. The same study identified specific SNVs (TP53 Q331* and KRAS G12V) detected only by the Illumina-based platform, while other SNVs (MAP2K1 F53V) were unique to Ion Torrent [11].
Copy Number Variations (CNVs) and Gene Fusions
Detection of structural variants including CNVs and fusions presents distinct challenges for both platforms. The 2025 comparative study reported that one CNA (AKT3 and MYC) and one fusion (ESR1-CCDC170) were detected only by the Ion Torrent Genexus system, suggesting potential platform-specific differences in structural variant detection [11]. Ion Torrent's limitations in homopolymer-rich regions can impact fusion detection when breakpoints occur in repetitive sequences. Illumina's paired-end reading capability provides an advantage for structural variant detection by generating mapping information from both ends of DNA fragments [2]. For CNV detection, both platforms require sufficient and uniform coverage depth for accurate quantification, with Illumina typically providing more consistent coverage across regions with varying GC content [17].
Table 2: Variant Detection Performance Comparison
| Variant Type | Illumina Performance | Ion Torrent Performance | Key Observations |
|---|---|---|---|
| SNVs | High accuracy (0.1-0.5% error rate) [2] | Good accuracy (~1% error rate), homopolymer-associated errors [2] | Some SNVs platform-specific in comparative studies [11] |
| Indels | Excellent detection in homopolymers [2] | Reduced accuracy in homopolymer regions [2] | Ion Torrent struggles with identical consecutive bases |
| CNVs | Good detection with uniform coverage [11] | Capable detection, some platform-specific calls [11] | AKT3 and MYC CNAs detected only by Ion Torrent in one study [11] |
| Fusions | Good detection with paired-end reads [2] | Capable detection, some platform-specific calls [11] | ESR1-CCDC170 fusion detected only by Ion Torrent in one study [11] |
| Overall Concordance | - | - | 55% sensitivity, 99% specificity for Ion Torrent vs Illumina-based test [11] |
Experimental Designs and Methodologies
Direct Platform Comparison Studies
Recent studies have employed rigorous experimental designs to enable head-to-head performance comparisons. A 2025 study compared the Ion Torrent Genexus Sequencer with FoundationOne (Illumina-based) using six patients with breast, head, and neck cancers [11]. The methodology included parallel analysis of both tissue and circulating tumor DNA biopsies, with DNA extraction from FFPE tissue specimens using the Maxwell RSC Instrument and cell-free total nucleic acid extraction from blood plasma [11]. This study evaluated 130 common genes between FoundationOne and Oncomine Comprehensive Assay v3 (OCAv3) for tissues, and 41 common genes between FoundationOne Liquid and Oncomine Precision Assay for blood [11]. Another study comparing whole-genome sequencing data from both platforms for Listeria monocytogenes surveillance emphasized the importance of bioinformatic tools, finding that only the SPAdes assembler delivered qualitatively comparable results between platforms [14].
Bioinformatics Considerations
The choice of bioinformatic pipelines significantly impacts variant calling accuracy and platform performance. The 2017 RNA-Seq comparison study found a strong interaction between sequencing platform and aligner choice, with different aligner and platform combinations better suited to probing different genomic features [34]. For Ion Torrent data, special consideration is needed for homopolymer-related errors, which may require specialized filtering approaches. In core genome multilocus sequence typing (cgMLST), the same-strain allele discrepancy between Illumina and Ion Torrent platforms averaged 14.5 alleles—above the threshold of 7 alleles routinely used for cluster detection in L. monocytogenes [14]. Application of strict frameshift filters could reduce this discrepancy but at the cost of reduced discriminatory power [14].
Experimental and Analysis Workflow for NGS-Based Variant Detection
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for NGS-Based Variant Detection
| Reagent/Kit | Function | Platform Compatibility |
|---|---|---|
| Maxwell RSC FFPE Plus DNA Kit (Promega) | DNA extraction from FFPE tissue specimens [11] | Platform-agnostic |
| Maxwell RSC miRNA Plasma and Serum Kit (Promega) | Cell-free total nucleic acid extraction from blood plasma [11] | Platform-agnostic |
| Oncomine Comprehensive Assay v3 (Thermo Fisher) | Targeted sequencing panel for comprehensive genomic profiling [11] | Ion Torrent |
| Oncomine Precision Assay (Thermo Fisher) | Targeted sequencing panel for liquid biopsy analysis [11] | Ion Torrent |
| FoundationOne CDx (Foundation Medicine) | FDA-approved tissue-based comprehensive genomic profiling test [11] | Illumina |
| AmpliSeq (Thermo Fisher) | Amplicon-based library preparation technology [71] | Ion Torrent |
| SureSelect (Agilent) | Hybrid capture-based library preparation [71] | Illumina |
| QuantiFluor ONE dsDNA System (Promega) | DNA quantification for quality control [11] | Platform-agnostic |
Illumina and Ion Torrent platforms both offer robust capabilities for detecting key variant types in cancer genomics, but with complementary strengths and limitations. Illumina generally provides higher base-level accuracy and paired-end reads advantageous for structural variant detection, while Ion Torrent offers faster turnaround times and lower instrument costs [2]. The choice between platforms should be guided by specific research priorities: applications requiring high sensitivity for indels in homopolymer regions may favor Illumina, whereas studies prioritizing rapid results for SNV detection may lean toward Ion Torrent. As both technologies continue to evolve, improvements in read lengths, error correction, and analysis pipelines will further enhance their variant detection capabilities. The emerging paradigm in cancer genomics will likely involve leveraging the strengths of each platform based on specific variant detection requirements rather than seeking a universal solution.
Platform Selection Guide Based on Research Requirements
Cost-Benefit Analysis and Scalability for High-Throughput Diagnostic Labs
Next-generation sequencing (NGS) has become the cornerstone of precision oncology, enabling clinicians and researchers to identify targetable genomic alterations that guide therapeutic decisions. Within this landscape, two major short-read sequencing platforms—Illumina and Ion Torrent (Thermo Fisher Scientific)—dominate the clinical diagnostic market [2] [1]. Both technologies offer distinct approaches to sequencing, leading to important differences in performance, cost, and operational workflow that directly impact their suitability for high-throughput diagnostic laboratories. A thorough cost-benefit analysis is therefore essential for labs seeking to implement or scale their genomic profiling capabilities, particularly in the context of cancer diagnostics where accuracy, turnaround time, and comprehensive genomic coverage are paramount.
This guide provides an objective comparison of Illumina and Ion Torrent platforms, focusing on their application in cancer diagnostics research. We present supporting experimental data from direct comparison studies, detailed methodologies, and analytical frameworks to inform decision-making for researchers, scientists, and drug development professionals.
Fundamental Technological Differences
Illumina and Ion Torrent platforms employ fundamentally different detection mechanisms. Illumina utilizes a fluorescence-based sequencing-by-synthesis approach with reversible terminators, enabling highly accurate base detection [2]. This optical method sequences DNA fragments from both ends (paired-end sequencing), which improves alignment accuracy and facilitates detection of complex genomic rearrangements [2] [41].
In contrast, Ion Torrent employs semiconductor technology that detects hydrogen ions released during nucleotide incorporation, translating chemical signals directly into digital data without requiring cameras or fluorescent dyes [2]. This electronic detection method contributes to a more compact instrument design and faster run times but presents specific challenges with homopolymer regions where multiple identical bases occur in sequence [2] [14] [16].
Direct Performance Comparison Data
Recent studies directly comparing these platforms reveal critical performance differentials. A 2025 clinical study comparing comprehensive genomic profiling tests for cancer diagnostics found that while there was substantial agreement between Ion Torrent Genexus and Illumina FoundationOne systems, significant differences emerged in specific alteration detection [11].
Table 1: Performance Comparison in Clinical Cancer Profiling
| Performance Metric | Illumina FoundationOne | Ion Torrent Genexus |
|---|---|---|
| Sensitivity | Reference Standard | 55% [11] |
| Specificity | Reference Standard | 99% [11] |
| Concordant Variants Detected | 9 SNVs, 1 CNA, 1 fusion | 9 SNVs, 1 CNA, 1 fusion [11] |
| Platform-Specific Variants | 2 SNVs (TP53 Q331*, KRAS G12V) | 1 SNV (MAP2K1 F53V), 2 CNAs (AKT3, MYC), 1 fusion (ESR-CCDC170) [11] |
| Total Common Genes (Tissue) | 130 genes (vs. OCA) | 130 genes (vs. F1) [11] |
Further comparative studies in other applications highlight foundational performance characteristics. Research on bacterial whole-genome sequencing for molecular surveillance found that data compatibility between platforms was assembler-dependent, with SPAdes being the only assembler delivering qualitatively comparable results [14]. In 16S rRNA amplicon sequencing, the Ion Torrent platform demonstrated higher error rates and a pattern of premature sequence truncation that resulted in organism-specific biases [16].
Table 2: Fundamental Platform Characteristics
| Characteristic | Illumina | Ion Torrent |
|---|---|---|
| Sequencing Chemistry | Fluorescence-based (reversible terminators) [2] | Semiconductor (pH detection) [2] |
| Read Structure | Paired-end [2] | Single-end only [2] |
| Error Rate | ~0.1-0.5% per base (Q30-Q20) [2] [41] | ~1% per base (Q20) [2] |
| Error Profile | Mainly substitution errors [2] | Homopolymer indels predominant [2] [14] |
| Maximum Output | Billions to trillions of reads (production-scale) [2] | Tens of millions to ~130 million reads [2] |
| Typical Run Time | 24-48 hours (larger outputs) [2] | Few hours to one day [2] |
Experimental Protocols and Methodologies
Direct Comparative Study Design for Cancer Genomic Profiling
A 2025 study by Fujiyoshi et al. provides a robust methodological framework for directly comparing NGS platforms in a cancer diagnostic context [11]. The experimental design offers a template for laboratories seeking to validate platform performance.
Sample Preparation:
- Cohort: Six patients with breast, head, and neck cancers [11]
- Sample Types: Three primary tumor tissues (FFPE) and three peripheral blood samples for circulating tumor DNA analysis [11]
- DNA Extraction:
- Quality Control: DNA/RNA concentration measured using QuantiFluor ONE dsDNA System and QuantiFluor RNA System; fragment length evaluation via Agilent 4200 TapeStation [11]
Sequencing Protocols:
- Illumina System: FoundationOne CDx (F1) for tissue and FoundationOne Liquid (F1L) for blood [11]
- Ion Torrent System: Genexus Oncomine Comprehensive Assay v3 (OCA) for tissue and Genexus Oncomine Precision Assay (OPA) for blood [11]
- Analysis: Comparison of variant concordance for somatic variants across 130 common genes for tissue and 41 common genes for blood [11]
Bioinformatic Analysis:
- Sensitivity and specificity calculations for variant detection [11]
- Direct comparison of detected single-nucleotide variants, copy number alterations, and gene fusions [11]
- Assessment of platform-specific variants that were uniquely detected by each system [11]
Cross-Platform Compatibility Assessment Protocol
A 2025 study on whole-genome sequencing compatibility provides additional methodological insights for comparing data analysis pipelines across platforms [14].
Experimental Design:
- Sample Set: 47 Listeria monocytogenes isolates from the German National Reference Laboratory collection [14]
- Sequencing: Each isolate sequenced on both Illumina and Ion Torrent platforms [14]
- Platform Parameters:
Bioinformatic Processing:
- Assembly: Three different assemblers tested (MEGAHIT, SKESA, SPAdes) [14]
- cgMLST Analysis: Core genome multilocus sequence typing using a scheme containing 1,748 loci [14]
- Frameshift Filtering: Application of relative (0.1 fraction) and absolute (9 bp) frameshift filters to address homopolymer errors [14]
- SNP Analysis: Single nucleotide polymorphism calling compared across platforms [14]
Cost-Benefit Analysis for High-Throughput Labs
Operational Cost Considerations
High-throughput diagnostic laboratories must evaluate both direct and indirect costs when selecting NGS platforms. While specific pricing varies by configuration and regional factors, general cost differentials emerge from published comparisons and platform characteristics.
Capital Investment:
- Illumina benchtop instruments historically carried higher upfront costs than comparable Ion Torrent systems [2]
- Ion Torrent machines offered lower capital investment, with early benchtop models approximately 40% less expensive than Illumina equivalents [2]
Consumables and Reagent Costs:
- Both platforms utilize proprietary consumables with costs dependent on application and throughput requirements
- Ion Torrent's semiconductor sequencing eliminates fluorescent dyes and optical detection systems, potentially reducing reagent costs [2]
Operational Efficiency:
- Ion Torrent platforms generally offer faster turnaround times, with the Genexus system delivering results in approximately one day with minimal hands-on time [11] [1]
- Illumina workflows may require longer run times (24-48 hours for larger outputs) but generate substantially more data per run [2]
Labor Costs:
- Ion Torrent systems typically require fewer manual steps and less specialized technical expertise for operation [2]
- Illumina's more complex workflow may necessitate higher levels of technical training and involvement [2]
Throughput and Scalability Analysis
Scalability requirements significantly influence platform selection for growing diagnostic laboratories.
Table 3: Scalability and Infrastructure Considerations
| Factor | Illumina | Ion Torrent |
|---|---|---|
| Data Output Scalability | Highly scalable from millions to billions of reads [2] | Moderate scalability, maximum ~130 million reads [2] |
| Multiplexing Capability | High (up to 96 samples with barcoding) [41] | Moderate multiplexing capabilities |
| IT Infrastructure Needs | Significant data storage and computing resources required [2] | More modest data management needs [2] |
| Workflow Automation | Option for full automation with integrated systems | Genexus offers automated specimen-to-report workflow [11] [1] |
| Laboratory Space | Larger instrument footprint for production-scale systems | More compact benchtop designs [2] |
Laboratory Implementation and Workflow Integration
Laboratory Information Management System (LIMS) Integration
Effective integration with Laboratory Information Management Systems (LIMS) is crucial for high-throughput diagnostic operations. Modern molecular diagnostics LIMS platforms must handle complex sample genealogies where single specimens generate multiple derivatives across different analytical platforms [72].
Key LIMS Considerations:
- Sample Tracking: Systems must maintain clear relationships between primary specimens and derivatives through extraction, purification, amplification, and analysis [72]
- Instrument Integration: Bidirectional communication with analytical platforms is essential; specialized LIMS like Scispot offer pre-built connectors for both Illumina and Ion Torrent sequencers [72]
- Workflow Automation: Rules-based reflex testing that automatically triggers additional tests based on initial results optimizes laboratory efficiency [72]
- Compliance Tools: Comprehensive audit trails, electronic signatures, and configurable permission controls are essential for regulated environments [72] [73]
According to industry research, laboratories using dedicated molecular diagnostics LIMS report 40% fewer tracking errors compared to those using general LIMS for genetic testing workflows [72].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of NGS platforms requires specific reagent systems and supporting technologies.
Table 4: Essential Research Reagent Solutions for NGS Implementation
| Reagent/Technology | Function | Platform Compatibility |
|---|---|---|
| Maxwell RSC Extraction Kits | Nucleic acid extraction from FFPE tissue and plasma | Both platforms [11] |
| AmpliSeq for Illumina | Targeted amplicon sequencing library preparation | Illumina [41] |
| Oncomine Comprehensive Assay | Comprehensive genomic profiling panel | Ion Torrent [11] |
| Qubit dsDNA Assay Kits | Accurate DNA quantification for library QC | Both platforms [11] [41] |
| Agencourt AMPure Beads | PCR purification and size selection | Both platforms [16] |
| Ion Xpress Barcodes | Sample multiplexing identification | Ion Torrent [16] |
| Nextera XT DNA Library Kit | Library preparation for whole-genome applications | Illumina [14] |
Discussion and Concluding Recommendations
The choice between Illumina and Ion Torrent platforms for high-throughput diagnostic laboratories involves balancing multiple factors including accuracy requirements, throughput needs, budget constraints, and application focus.
Platform Selection Guidelines
Choose Illumina when:
- Maximum sequencing accuracy is paramount, particularly for variant detection in cancer diagnostics [11] [2]
- Highest throughput and scalability are required for population-scale sequencing [2]
- Paired-end sequencing is necessary for complex structural variant detection [2] [41]
- Applications require detection of methylation states (with 5-base chemistry) [1]
Choose Ion Torrent when:
- Rapid turnaround time is critical for clinical decision-making [11] [1]
- Capital budget constraints are significant [2]
- Laboratory space is limited, requiring compact benchtop instruments [2]
- Technical staff expertise favors simpler workflow automation [2]
Emerging Trends and Future Considerations
The NGS landscape continues to evolve with new technologies promising to reshape the competitive dynamics. Roche's anticipated Sequencing by Expansion (SBX) technology, slated for commercial launch in 2026, may offer new alternatives for high-throughput sequencing [74] [1]. Similarly, emerging platforms from Element Biosciences and MGI Tech are increasing competition, potentially driving down costs and increasing innovation [74] [1].
For high-throughput diagnostic laboratories, the decision between Illumina and Ion Torrent remains nuanced. While Illumina generally offers superior accuracy and throughput, Ion Torrent provides compelling advantages in speed, cost, and operational simplicity. The optimal choice depends on specific laboratory priorities, with the understanding that platform selection should align with primary diagnostic applications, scalability requirements, and operational constraints. As the technology continues to advance, regular re-evaluation of this cost-benefit analysis will be essential for maintaining competitive, effective genomic diagnostic services.
Conclusion
The choice between Illumina and Ion Torrent in cancer diagnostics is not a matter of absolute superiority but of strategic alignment with clinical and research objectives. Illumina platforms often deliver exceptional accuracy and high throughput for broad genomic discovery, while Ion Torrent systems offer streamlined, automated workflows with rapid turnaround times, as demonstrated by the Genexus system. Key differentiators include performance in challenging genomic regions, variant detection capabilities, and integration into clinical workflows. Future directions will be shaped by the convergence of NGS with artificial intelligence for data interpretation, the rise of multi-omics approaches, and the increasing demand for decentralized, point-of-care testing. For the field to advance, ongoing, independent validation studies and standardized reporting will be crucial to fully harness the power of both platforms in delivering personalized cancer care.
References
- 1. DNA Sequencing: How to Choose the Right Technology [https://frontlinegenomics.com/dna-sequencing-how-to-choose-the-right-technology/]
- 2. Comparing NGS Platforms: Ion Torrent vs. Illumina [https://www.biotechveritas.com/blog/ngs]
- 3. A comparison of Illumina and Ion Torrent sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5553782/]
- 4. Next-generation sequencing in cancer diagnosis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12009796/]
- 5. NGS Workflow Steps - Next-Generation Sequencing [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/ngs-workflow.html]
- 6. Ion Torrent Next-Generation Sequencing Construct Library [https://www.thermofisher.com/us/en/home/life-science/sequencing/next-generation-sequencing/ion-torrent-next-generation-sequencing-workflow/ion-torrent-next-generation-sequencing-construct-library.html]
- 7. The Workflow of NGS Library Preparation Kit [https://www.celemics.com/resources/blogs/the-workflow-of-ngs-library-preparation-kit/]
- 8. NGS Library Preparation - 3 Key Technologies [https://www.illumina.com/techniques/sequencing/ngs-library-prep.html]
- 9. Library Preparation for Ion Torrent [https://www.neb.com/en-us/products/next-generation-sequencing-library-preparation/library-preparation-for-ion-torrent?srsltid=AfmBOooeGaIRGY4iYevWAZxwYpGG4mbk7-zpR5g92QoErR-EWPHm74Iw]
- 10. Library prep automation | Automated NGS protocols [https://www.illumina.com/techniques/sequencing/ngs-library-prep/automation.html]
- 11. Comparison of Comprehensive Genomic Profiling Testing “ ... [https://ar.iiarjournals.org/content/45/8/3479]
- 12. Short-Read Sequencing vs. Long- ... [https://www.zymoresearch.com/blogs/blog/short-read-vs-long-read-sequencing?srsltid=AfmBOor8ROJtonnnF6m-5prdlYSwBCUsPStGxaLRMIt0KGM3YPl3EQ-D]
- 13. Sequencing Technology | Sequencing by synthesis [https://www.illumina.com/science/technology/next-generation-sequencing/sequencing-technology.html]
- 14. Compatibility of whole-genome sequencing data from Illumina ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12046094/]
- 15. Short-Read and Long-Read Whole Genome Sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12031342/]
- 16. Performance Comparison of Illumina and Ion Torrent Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4249215/]
- 17. A tale of three next generation sequencing platforms ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-13-341]
- 18. Evaluation of a hybridization capture-based hereditary ... [https://www.sciencedirect.com/science/article/abs/pii/S0009898121002588]
- 19. TruSight Oncology 500 v2 | Enable CGP utilizing DNA and ... [https://www.illumina.com/products/by-type/clinical-research-products/trusight-oncology-500.html]
- 20. Oncomine Comprehensive Genomic Profiling NGS ... [https://www.thermofisher.com/us/en/home/clinical/preclinical-companion-diagnostic-development/oncomine-oncology/oncomine-cancer-research-panel-workflow.html]
- 21. Thermo Fisher Scientific Introduces the Oncomine ... [https://www.biospace.com/press-releases/thermo-fisher-scientific-introduces-the-oncomine-comprehensive-assay-plus-on-the-ion-torrent-genexus-system-to-help-advance-the-future-of-precision-medicine]
- 22. Illumina enhances flagship assay to accelerate access to ... [https://www.illumina.com/company/news-center/press-releases/2025/249e5ce3-32b2-4e97-98fb-4a40323b92fa.html]
- 23. Assessment of Two Commercial Comprehensive Gene Panels ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9863125/]
- 24. A Review of Circulating Tumor DNA (ctDNA) and the Liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12032849/]
- 25. Clinical Circulating Tumor DNA Testing for Precision Oncology [https://pmc.ncbi.nlm.nih.gov/articles/PMC10101787/]
- 26. Cancer in a Drop: Liquid biopsy insights from AACR 2025 [https://pmc.ncbi.nlm.nih.gov/articles/PMC12223554/]
- 27. Analytical evaluation of circulating tumor DNA sequencing ... [https://www.nature.com/articles/s41598-024-54361-w]
- 28. Detection of actionable mutations in circulating tumor DNA ... [https://www.nature.com/articles/s43856-025-00921-8]
- 29. Liquid Biopsy | September Round-Up 2025 [https://www.decibio.com/insights/liquid-biopsy-september-round-up-2025]
- 30. Bridge Capture permits cost-efficient, rapid and sensitive ... [https://www.sciencedirect.com/science/article/pii/S1525157825002491]
- 31. Ancestry-associated co-alteration landscape of KRAS and ... [https://www.nature.com/articles/s41698-024-00644-4]
- 32. The relationship between different subtypes of KRAS and... [https://tlcr.amegroups.org/article/view/61437/html]
- 33. Paired Comparison of Whole Genome Sequencing and ... [https://ar.iiarjournals.org/content/45/2/605]
- 34. A comparison of Illumina and Ion Torrent sequencing ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-4011-0]
- 35. Shaping the battlefield: EGFR and KRAS tumor mutations' role ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12170712/]
- 36. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 37. A comparison of five Illumina, Ion Torrent, and nanopore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10075175/]
- 38. Reducing GC & PCR Bias in Whole-Genome Sequencing [https://www.revvity.com/blog/understanding-impact-gc-and-pcr-biases-whole-genome-sequencing]
- 39. Summarizing and correcting the GC content bias in high- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3378858/]
- 40. Strategies for identification of somatic variants using the Ion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5753459/]
- 41. Transitioning from Ion Torrent to Illumina NGS Platforms [https://support.illumina.com/support-content/Ion-Torrent-to-IlluminaNGS.html]
- 42. A tale of three next generation sequencing platforms: comparison of... [https://pubmed.ncbi.nlm.nih.gov/22827831/]
- 43. Comparison of somatic variant detection algorithms using Ion ... [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-019-0636-y]
- 44. A comparison of Illumina and Ion Torrent sequencing platforms in the context of differential gene expression - PubMed [https://pubmed.ncbi.nlm.nih.gov/28797240/]
- 45. Illumina Variant Calling Pipeline - NCBI - NIH [https://www.ncbi.nlm.nih.gov/sra/docs/sars-cov-2-illumina-variant-calling-pipeline/]
- 46. A critical spotlight on the paradigms of FFPE-DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10415133/]
- 47. Next-Generation Sequencing: A Review of Its Transformative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523276/]
- 48. FFPE Sample Analysis with Sequencing and Arrays [https://www.illumina.com/science/education/ffpe-sample-analysis.html]
- 49. Ultima Genomics UG 100 platform vs. Illumina NovaSeq X ... [https://www.illumina.com/science/genomics-research/articles/ultima-ug-100-vs-illumina-novaseq-x-wgs-data.html]
- 50. Extraction of Nucleic Acids from FFPE Samples [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rna-isolation/general-articles/extraction-of-nucleic-acids-from-ffpe-samples.html]
- 51. Overcoming challenges in FFPE DNA library prep [https://www.neb.com/en-us/nebinspired-blog/overcoming-challenges-in-ffpe-dna-library-prep?srsltid=AfmBOopmjuhDYigy2O-3BidQzS9c7qprRU2hv2hOWdMfcpd3S1pGbCjE]
- 52. Illumina advances personalized cancer care with new ... [https://www.illumina.com/company/news-center/press-releases/press-release-details.html?newsid=cafb84d4-ffe1-431b-ab7c-712a24857022]
- 53. Impact and Reproducibility of In-House Targeted Next ... [https://www.sciencedirect.com/science/article/pii/S152515782500042X]
- 54. Population-based metrics to stratify somatic variant calling ... [https://www.illumina.com/science/genomics-research/articles/population-metrics-stratify-somatic-variant.html]
- 55. Systematic comparison of somatic variant calling ... [https://www.nature.com/articles/s41598-020-60559-5]
- 56. The challenge of somatic variant detection accuracy in ... [https://www.illumina.com/science/genomics-research/articles/dragen-liquid-biopsy-variant-detection.html]
- 57. New DRAGEN v4.4 - Comprehensive genomic analysis ... [https://developer.illumina.com/news-updates/dragen-v4-4]
- 58. DeepSomatic: Accurate somatic small variant discovery for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11370364/]
- 59. Detecting actionable mutations from matched plasma-based ... [https://jeccr.biomedcentral.com/articles/10.1186/s13046-025-03480-x]
- 60. Recent advances in the methods and clinical applications ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11830917/]
- 61. Clinical Validation of a Next-Generation Sequencing ... [https://www.sciencedirect.com/science/article/pii/S1525157816301945]
- 62. Illumina advances personalized cancer care with new ... [https://investor.illumina.com/news/press-release-details/2025/Illumina-advances-personalized-cancer-care-with-new-pharma-development-partnerships/default.aspx]
- 63. an integrated pipeline from NGS to AI-based target discovery [https://link.springer.com/article/10.1007/s13353-025-01024-9]
- 64. Why Are Long-Read Sequencing Methods Revolutionizing ... [https://www.mdpi.com/2076-2607/13/8/1861]
- 65. Evaluation of the Ion Torrent PGM sequencing workflow for ... [https://www.sciencedirect.com/science/article/abs/pii/S0014480016302970]
- 66. Performance comparison of NextSeq and Ion Proton ... [https://pubmed.ncbi.nlm.nih.gov/28127742/]
- 67. Comparison of Reproducibility, Accuracy, Sensitivity ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/31851944/]
- 68. Comparison of Reproducibility, Accuracy, Sensitivity, and ... [https://www.sciencedirect.com/science/article/pii/S2211124719315694]
- 69. Sensitivity, specificity, and reproducibility of RNA-Seq ... [https://biologydirect.biomedcentral.com/articles/10.1186/s13062-016-0169-7]
- 70. Performance characteristics of the AmpliSeq Cancer ... [https://pubmed.ncbi.nlm.nih.gov/26387931/]
- 71. Bioinformatics and Computational Tools for Next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7019349/]
- 72. Best Molecular Diagnostics LIMS in 2026: A Real-World ... [https://www.scispot.com/blog/best-molecular-diagnostics-lims-a-real-world-guide]
- 73. Guide to LIMS: Core Functions & 2025 System Comparison [https://intuitionlabs.ai/articles/lims-system-guide-2025]
- 74. Is Illumina's Moat Drying up? Inside the Next Generation ... [https://www.signifyresearch.net/insights/is-illuminas-moat-drying-up-inside-the-next-generation-sequencing-ngs-market/]