BEAMing Technology for ctDNA Analysis: A Comprehensive Guide for Precision Oncology Research
This article provides a comprehensive examination of BEAMing (Beads, Emulsion, Amplification, and Magnetics) technology and its application in circulating tumor DNA (ctDNA) analysis for precision oncology.
BEAMing Technology for ctDNA Analysis: A Comprehensive Guide for Precision Oncology Research
Abstract
This article provides a comprehensive examination of BEAMing (Beads, Emulsion, Amplification, and Magnetics) technology and its application in circulating tumor DNA (ctDNA) analysis for precision oncology. Tailored for researchers, scientists, and drug development professionals, it explores the foundational principles of BEAMing, detailed methodological workflows, and diverse clinical applications across cancer types including breast, colorectal, and lung cancers. The content addresses technical challenges, optimization strategies, and comparative performance against other digital PCR and sequencing platforms. With emphasis on clinical validation studies and emerging applications in treatment monitoring and minimal residual disease detection, this resource serves as both an introductory guide and technical reference for implementing BEAMing in cancer research and therapeutic development.
Understanding BEAMing Technology: Principles and Evolution in Liquid Biopsy
BEAMing (Beads, Emulsion, Amplification, and Magnetics) represents a powerful digital PCR technology that enables the highly sensitive detection and absolute quantification of rare circulating tumor DNA (ctDNA) mutations in a background of wild-type DNA. This methodology has emerged as a cornerstone technique in liquid biopsy analysis, addressing the critical challenge of detecting low-frequency somatic mutations with variant allele frequencies often below 0.1%. The fundamental principle of BEAMing involves compartmentalizing individual DNA molecules into water-in-oil emulsion microreactors, where clonal amplification occurs on magnetic bead surfaces, followed by flow cytometry analysis to distinguish mutant from wild-type alleles. This technology provides researchers with an indispensable tool for monitoring treatment response, identifying emerging resistance mutations, and detecting minimal residual disease (MRD) in cancer patients, making it particularly valuable for longitudinal studies in precision oncology [1] [2].
Within the broader thesis context of BEAMing technology for ctDNA analysis research, this methodology bridges the gap between conventional PCR and next-generation sequencing approaches, offering the sensitivity of digital PCR with the scalability for analyzing multiple mutations. The integration of BEAMing into clinical research protocols has accelerated the validation of ctDNA as a robust biomarker for therapy selection and disease monitoring in various malignancies, including non-small cell lung cancer (NSCLC), colorectal cancer, and breast cancer [3] [4]. As the field advances toward standardized liquid biopsy applications, BEAMing continues to provide the technical foundation for establishing analytical validation parameters essential for clinical translation.
Core BEAMing Methodology and Workflow
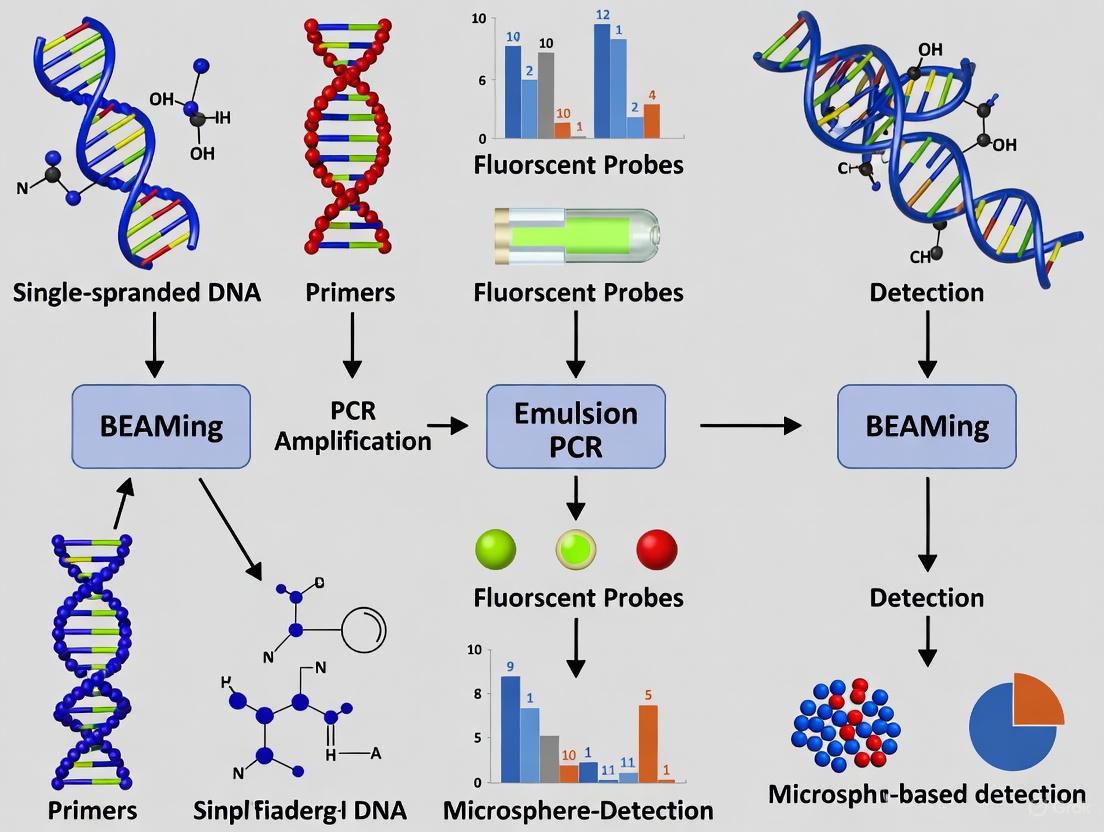
The BEAMing protocol transforms solution-based PCR into a surface-amplification system that physically links amplified products to microscopic beads, enabling precise enumeration of mutant molecules. The complete workflow integrates molecular biology, emulsion chemistry, and fluorescence detection technologies to achieve exceptional sensitivity and specificity for mutation detection.
Figure 1: Comprehensive BEAMing workflow for ctDNA mutation detection, illustrating the six major steps from sample input to data analysis.
Detailed Experimental Protocol
The following protocol outlines the standard BEAMing procedure for EGFR mutation detection in NSCLC patient plasma samples as described in the comparative methodological studies [3]:
Initial Primer Extension and Emulsion Setup
- Template DNA Preparation: Extract ctDNA from 1 mL of patient plasma using a DNA Micro Kit (Qiagen). Use 8 separate 25 μL PCR reactions, each containing template DNA from 250 μL of plasma, 5× Phusion High Fidelity PCR buffer (NEB), 1.5 U of HotStart Phusion polymerase (NEB), 0.2 μM of each primer, 0.25 mM of each dNTP, and 0.5 mM MgCl₂.
- Thermal Cycling Conditions: 98°C for 30 s (initial denaturation), followed by 35 cycles of: 98°C for 10 s (denaturation), 57°C for 10 s (annealing), and 72°C for 10 s (extension).
- Emulsion PCR Mixture: Prepare a 150 μL PCR mixture containing 18 pg of pooled template DNA, 40 U of Platinum Taq DNA polymerase (Invitrogen), 1× PCR buffer, 0.2 mM dNTPs, 5 mM MgCl₂, 0.05 μM Tag1 (5'-tcccgcgaaattaatacgac-3'), 8 μM Tag2 (5'-gctggagctctgcagcta-3'), and approximately 6×10⁷ magnetic streptavidin beads (MyOne, Invitrogen) coated with Tag1 oligonucleotide (5'-dual biotin-T-Spacer18-tcccgcgaaattaatacgac-3').
Emulsion Formation and Amplification
- Microemulsion Preparation: Combine 150 μL of PCR mixture with 600 μL of oil/emulsifier mixture (7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC) and one 5 mm steel bead in a 96-deep-well plate. Shake the plate in a TissueLyser for 10 s at 15 Hz followed by 7 s at 17 Hz to form uniform microemulsions.
- Verify aqueous compartment size under inverted microscope at 40× magnification to ensure proper bead distribution.
- Emulsion PCR Cycling: Dispense emulsions into eight PCR plates and run the following program: 94°C for 2 min; 3 cycles of 94°C for 10 s, 68°C for 45 s, 70°C for 75 s; 3 cycles of 94°C for 10 s, 65°C for 45 s, 70°C for 75 s; 3 cycles of 94°C for 10 s, 62°C for 45 s, and 70°C for 75 s; followed by 50 cycles of 94°C for 10 s, 57°C for 45 s, and 70°C for 75 s.
Post-Amplification Processing and Detection
- Emulsion Breaking: Add 150 μL of breaking buffer (10 mM Tris-HCl, pH 7.5; 1% Triton-X 100; 1% SDS; 100 mM NaCl; and 1 mM EDTA) to each well and mix with a TissueLyser at 20 Hz for 20 s. Recover beads by centrifugation at 3,200 × g for 2 min and remove the oil phase. Repeat breaking step twice.
- Bead Processing: Wash beads with 150 μL of wash buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl). Denature DNA on beads for 5 min with 0.1 M NaOH. Wash with 150 μL of wash buffer and resuspend in 150 μL of the same buffer.
- Allele-Specific Hybridization: Use fluorescently labeled probes complementary to mutant and wild-type DNA sequences (15-18 nt in length) for different EGFR mutations. Analyze hybridized beads using flow cytometry (FACSAria III) to detect mutant and wild-type populations.
Performance Characteristics and Validation Data
BEAMing technology demonstrates exceptional analytical performance for ctDNA mutation detection, as validated through extensive comparison with established methodologies.
Table 1: Analytical Performance of BEAMing PCR for EGFR Mutation Detection in NSCLC Patient Samples [3]
| Parameter | Exon 19 | Exon 20 | Exon 21 (L858R) | Exon 21 (L861Q) |
|---|---|---|---|---|
| Concordance with EMR-qPCR (%) | 98.8 | 98.9 | 95.5 | - |
| Concordance with Diatech qPCR (%) | 90.0 | 100 | 96.0 | 98.0 |
| Cohen's Kappa Agreement | Significant (p<0.001) | Significant (p<0.001) | Significant (p<0.001) | Significant (p<0.001) |
| Sensitivity Assessment | Detected 0.1% mutant DNA in wild-type background | Detected 0.1% mutant DNA in wild-type background | Detected 0.1% mutant DNA in wild-type background | Detected 0.1% mutant DNA in wild-type background |
Table 2: Clinical Applications of BEAMing in ctDNA Analysis Across Cancer Types [1] [4] [2]
| Application Domain | Utility | Cancer Types Validated | Technical Advantages |
|---|---|---|---|
| Treatment Monitoring | Real-time assessment of therapy response through mutation quantification | NSCLC, Colorectal Cancer, Breast Cancer | Rapid turnaround (24-48h), high sensitivity for early response detection |
| Minimal Residual Disease (MRD) Detection | Identification of molecular recurrence before radiographic progression | Colorectal Cancer, Breast Cancer, NSCLC | Capable of detecting mutant allele frequencies <0.1% |
| Resistance Mutation Identification | Detection of emerging resistance mechanisms during targeted therapy | NSCLC (EGFR T790M), Breast Cancer (ESR1 mutations) | Quantitative tracking of resistance mutation dynamics |
| Tumor Heterogeneity Assessment | Capture spatial and temporal genomic heterogeneity | Pan-cancer applications | Comprehensive mutation profiling from single blood draw |
The sensitivity and specificity validation experiments conducted using cell line models demonstrated BEAMing's capability to robustly identify specific mutations (from H1975 and PC9 cell lines) diluted in wild-type DNA background (A549 cell line) at concentrations as low as 0.1%, confirming the technology's utility for detecting rare mutant alleles in clinical samples [3].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for BEAMing Protocol Implementation
| Reagent/Category | Specific Product Examples | Function in BEAMing Workflow |
|---|---|---|
| Magnetic Beads | MyOne Streptavidin Beads (Invitrogen) | Solid support for oligonucleotide attachment and clonal amplification |
| Polymerase Systems | HotStart Phusion (NEB), Platinum Taq (Invitrogen) | High-fidelity amplification in initial extension and emulsion PCR phases |
| Emulsion Components | ABIL WE09, Mineral Oil, TegoSoft DEC | Formation of stable water-in-oil microemulsions for compartmentalization |
| Nucleic Acid Modifiers | Biotinylated dNTPs, Tagged Oligonucleotides | Incorporation of detection tags and binding moieties for downstream analysis |
| Buffer Systems | Phusion HF Buffer, Custom Breaking & Wash Buffers | Maintenance of optimal enzymatic activity and efficient post-amplification processing |
| Detection Reagents | Fluorescently Labeled Allele-Specific Probes | Differentiation of mutant and wild-type alleles during flow cytometry |
| Nucleic Acid Extraction | QIAamp DNA Micro Kit (Qiagen) | Isolation of high-quality ctDNA from plasma samples |
Technical Considerations and Optimization Guidelines
Successful implementation of BEAMing technology requires careful attention to several critical technical parameters that significantly impact assay performance and reliability.
Figure 2: Critical technical factors influencing BEAMing assay performance and their impacts on key outcome metrics.
Pre-analytical Considerations: Process blood samples within one hour of collection using double centrifugation (820 × g for 10 min followed by 16,000 × g for 10 min) to eliminate cellular contamination that contributes wild-type DNA and reduces mutant allele detection sensitivity. Store plasma at -80°C in 1 mL aliquots to prevent freeze-thaw degradation of cfDNA [3] [2].
Emulsion Optimization: Systematically optimize oil-to-aqueous phase ratios and emulsification parameters to achieve uniform microreactors of approximately 5-10 μm diameter. Verify emulsion quality microscopically before PCR amplification. Inadequate emulsion formation represents the most common technical failure point, leading to non-compartmentalized reactions and reduced sensitivity.
Analytical Validation: Establish limit of detection (LOD) and limit of quantification (LOQ) using serial dilutions of mutant DNA in wild-type background for each mutation target. Implement strict quality control measures including negative controls (no-template and wild-type only) and positive controls with known mutation frequencies in each run [3].
Comparative Methodological Advantages in ctDNA Analysis
BEAMing technology occupies a unique position in the liquid biopsy methodological landscape, offering distinct advantages for specific clinical research applications compared to other ctDNA analysis platforms.
Sensitivity Advantage Over Conventional Methods: BEAMing demonstrates superior sensitivity (0.1% variant allele frequency) compared to conventional qPCR methods (typically 1-5% sensitivity), enabling detection of rare resistance mutations and early MRD assessment. The compartmentalization approach reduces amplification bias and improves quantification accuracy of mutant fractions [3].
Targeted Application Scope: While NGS-based methods provide broader genomic coverage, BEAMing offers superior sensitivity for monitoring known mutations and is particularly suited for longitudinal tracking of specific variants during treatment. The rapid turnaround time (typically 24-48 hours) facilitates clinical decision-making in scenarios requiring timely intervention [4] [2].
Multiplexing Capabilities: Although primarily employed for single mutation detection, BEAMing can be adapted for parallel assessment of multiple mutations through incorporation of differentially labeled fluorescent probes, creating a balanced approach between targeted depth and genomic breadth for therapy selection and monitoring applications.
The robust performance characteristics and methodological precision of BEAMing technology establish it as an indispensable tool in the ctDNA research arsenal, particularly for studies requiring high-sensitivity detection of predefined mutations across the continuum of cancer management from early detection to therapy resistance monitoring.
Historical Development and Technological Evolution
The evolution of circulating tumor DNA (ctDNA) analysis represents a paradigm shift in oncology, moving from invasive tissue biopsies to minimally invasive liquid biopsies. BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) has emerged as a cornerstone in this evolution, providing the sensitivity required to detect rare tumor-derived DNA fragments in circulation [5]. This revolutionary approach addresses the critical challenge of identifying minute quantities of tumor-specific genetic material in blood, where ctDNA can represent less than 0.01% of total cell-free DNA [6] [7]. The development of BEAMing and related digital PCR technologies has enabled researchers and clinicians to monitor tumor dynamics in real-time, capturing spatial and temporal heterogeneity that traditional tissue biopsies often miss [1] [8]. This technological advancement has created new possibilities for cancer diagnosis, treatment selection, and monitoring throughout the patient journey.
Historical Timeline of Liquid Biopsy and BEAMing Technology
The conceptual foundation for liquid biopsy was established decades ago, with critical discoveries paving the way for current technologies. The timeline below summarizes key milestones in the development of BEAMing and ctDNA analysis:
Table 1: Historical Development of Liquid Biopsy and BEAMing Technology
| Year | Development Milestone | Significance |
|---|---|---|
| 1948 | Discovery of cell-free nucleic acids in plasma [9] [10] | First evidence of extracellular nucleic acids in bodily fluids |
| 1977 | Identification of elevated cfDNA in cancer patients [4] [9] | Established potential link between cfDNA levels and malignancy |
| 1994 | Detection of KRAS mutations in blood cfDNA of pancreatic cancer patients [9] | First demonstration of tumor-specific mutations in circulation |
| 2008 | BEAMing technology used to monitor ctDNA in colorectal cancer patients [9] | Demonstrated clinical utility for treatment monitoring |
| 2014 | EMA approval of ctDNA for EGFR mutation testing in NSCLC [4] [9] | First regulatory approval for clinical use of liquid biopsy |
| 2015-present | Rapid expansion and clinical validation of BEAMing applications [10] | Integration into clinical trials and oncology practice |
The period from 2015 to the present has witnessed exponential growth in liquid biopsy research and clinical application, with BEAMing technology playing a pivotal role in this expansion [10]. The OncoBEAM platform received CE marking as an in vitro diagnostic for RAS testing in colorectal cancer, representing a significant milestone in the standardization of BEAMing for clinical use [5]. Recent advances have focused on increasing multiplexing capabilities and lowering detection limits, with studies demonstrating BEAMing's ability to detect mutations at frequencies as low as 0.01% [5] [3]. These technological refinements have positioned BEAMing as a reference method for ultra-sensitive mutation detection in liquid biopsy applications.
Technical Evolution from PCR to BEAMing
The evolution of ctDNA analysis technologies has progressed from conventional PCR methods to increasingly sophisticated digital detection platforms. Conventional PCR methods, while useful for detecting abundant mutations, lacked the sensitivity required for most ctDNA applications due to the overwhelming background of wild-type DNA [6]. The development of digital PCR (dPCR) represented a significant advancement by partitioning samples into thousands of individual reactions, enabling absolute quantification and improved detection sensitivity [4] [6]. BEAMing technology built upon this digital concept by combining emulsion PCR with flow cytometry to create a highly sensitive and quantitative detection system [5].
Table 2: Evolution of ctDNA Detection Technologies
| Technology | Detection Limit | Advantages | Limitations |
|---|---|---|---|
| Conventional PCR | 1-10% | Simple, inexpensive, widely available | Low sensitivity, qualitative or semi-quantitative |
| Digital PCR (dPCR) | 0.001-0.01% | Absolute quantification, high sensitivity | Limited multiplexing, requires prior knowledge of mutations |
| BEAMing | 0.01% [5] | High sensitivity, precise quantification, visual validation via flow cytometry | Complex workflow, limited to known mutations |
| Next-Generation Sequencing (NGS) | 0.1-2.0% [5] | Broad genomic coverage, discovery of novel mutations | Higher cost, complex data analysis, longer turnaround |
BEAMing technology specifically addresses the challenge of detecting rare mutant alleles by combining emulsion PCR with magnetic beads and flow cytometry. This approach allows for the physical separation and individual amplification of DNA molecules, enabling precise quantification of mutation frequencies [5] [3]. The fundamental principle involves converting individual DNA molecules into magnetic beads covered with thousands of copies of the original DNA sequence, which can then be labeled with mutation-specific fluorescent probes and quantified using flow cytometry [5]. This process provides both digital quantification and visual validation of results, offering advantages over purely electronic detection systems.
Diagram 1: BEAMing technology combines emulsion PCR with flow cytometry to detect rare mutant alleles in ctDNA.
BEAMing Protocol for ctDNA Mutation Detection
Sample Collection and Plasma Preparation
Proper sample collection and processing are critical for successful ctDNA analysis. Blood samples should be collected in specialized tubes containing EDTA or cell-stabilizing additives to prevent leukocyte lysis and preserve ctDNA quality [7]. Within recommended timeframes (within 6 hours for EDTA tubes, up to several days for cell-stabilizing tubes), plasma must be separated through a two-step centrifugation process [7] [3]:
- Initial centrifugation: 1,200-1,600 × g for 10 minutes at room temperature to separate cellular components from plasma.
- Secondary centrifugation: 3,000-16,000 × g for 10 minutes to remove any remaining cellular debris.
The resulting plasma should be aliquoted and stored at -80°C to prevent degradation. ctDNA extraction can be performed using commercial kits specifically designed for cell-free DNA, such as the QIAamp Circulating Nucleic Acid Kit [11] [3]. DNA concentration should be quantified using fluorometric methods to ensure accurate input for subsequent analysis.
BEAMing Reaction Setup and Emulsion PCR
The core BEAMing protocol involves several meticulously optimized steps:
Initial Amplification: Set up initial PCR reactions using high-fidelity DNA polymerase to amplify target regions from ctDNA. Reaction conditions typically include:
- Template DNA from 250 μL of plasma
- Hot-start DNA polymerase with appropriate buffer
- 0.2 μM of each primer
- 0.25 mM dNTPs
- 0.5 mM MgCl₂
- Cycling conditions: 98°C for 30s, then 35 cycles of (98°C for 10s, 57°C for 10s, 72°C for 10s) [3]
Emulsion Preparation: Combine amplified products with:
- Approximately 6 × 10^7 magnetic streptavidin beads coated with specific oligonucleotides
- DNA polymerase with appropriate buffer and dNTPs
- Oil/emulsifier mixture (typically 7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC)
- Shake vigorously using a tissue lyser to create microemulsions (10s at 15 Hz, then 7s at 17 Hz) [3]
Emulsion PCR: Perform PCR amplification within the emulsion compartments with specialized cycling conditions:
- 94°C for 2 minutes
- 3 cycles of: 94°C for 10s, 68°C for 45s, 70°C for 75s
- 3 cycles of: 94°C for 10s, 65°C for 45s, 70°C for 75s
- 3 cycles of: 94°C for 10s, 62°C for 45s, 70°C for 75s
- 50 cycles of: 94°C for 10s, 57°C for 45s, 70°C for 75s [3]
Bead Recovery and Mutation Detection
Following emulsion PCR, the microemulsions are broken using a specialized breaking buffer (containing Triton-X-100, SDS, NaCl, and EDTA). The beads are recovered by centrifugation and washed to remove oil and debris. DNA on the beads is denatured using alkaline treatment (0.1 M NaOH) to prepare for hybridization [3].
Mutation detection is performed through allele-specific hybridization using fluorescently labeled probes complementary to mutant and wild-type sequences. The beads are analyzed by flow cytometry, which enables:
- Discrimination between mutant and wild-type beads based on fluorescence
- Quantification of mutation frequency by counting beads in each population
- Visual validation of the detection through direct observation of bead populations
The sensitivity of the assay should be validated using control samples with known mutation frequencies, typically by mixing DNA from mutant and wild-type cell lines in defined ratios [3].
Research Reagent Solutions for BEAMing Experiments
Table 3: Essential Research Reagents for BEAMing Protocols
| Reagent/Category | Specific Examples | Function in BEAMing Protocol |
|---|---|---|
| Blood Collection Tubes | EDTA tubes, PAXgene Blood ccfDNA tubes, Cell-free DNA BCT tubes [7] | Preserve blood sample integrity, prevent white blood cell lysis that dilutes ctDNA |
| DNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit [11] [3] | Isolate high-quality ctDNA from plasma samples with minimal contamination |
| Specialized Beads | Magnetic streptavidin beads (e.g., MyOne Streptavidin beads) [3] | Serve as solid support for DNA amplification and subsequent detection |
| PCR Enzymes | Hot-start Phusion polymerase, Platinum Taq DNA polymerase [3] | Provide DNA amplification with high fidelity and efficiency in emulsion |
| Emulsion Components | ABIL WE09, Mineral oil, TegoSoft DEC [3] | Create stable water-in-oil emulsions for compartmentalized PCR |
| Hybridization Probes | Fluorescently labeled allele-specific oligonucleotides [5] [3] | Detect mutant and wild-type sequences through specific hybridization |
| Control Materials | DNA from characterized cell lines (e.g., PC9, H1975 for EGFR) [3] | Validate assay sensitivity and specificity for mutation detection |
Applications and Validation of BEAMing Technology
BEAMing technology has been extensively validated across multiple cancer types, demonstrating high concordance with traditional tissue-based genotyping. In colorectal cancer, the OncoBEAM RAS assay showed 93.3% overall concordance with standard tissue testing in a multicenter evaluation across Europe [5]. This study demonstrated positive percent agreement of 92.6% and negative percent agreement of 94%, establishing BEAMing as a reliable method for determining RAS mutation status when tissue is unavailable or insufficient.
In non-small cell lung cancer (NSCLC), BEAMing has proven valuable for detecting EGFR mutations, with studies showing concordance rates of 98.8%, 98.9%, and 95.5% for exons 19, 20, and 21, respectively, when compared with standard qPCR methods [3]. This high degree of concordance, coupled with the method's sensitivity to detect mutations at allele frequencies as low as 0.01%, enables clinicians to identify targetable mutations and monitor treatment response through non-invasive blood collection.
The clinical utility of BEAMing extends beyond initial diagnosis to monitoring treatment response and detecting resistance mechanisms. Studies have demonstrated that changes in ctDNA mutation levels detected by BEAMing correlate with treatment response and can predict recurrence earlier than radiographic imaging [5] [8]. This capability for real-time monitoring provides a dynamic view of tumor evolution under therapeutic pressure, enabling more personalized treatment approaches.
Diagram 2: BEAMing technology applications span treatment selection, monitoring, and detection of resistance or minimal residual disease (MRD).
Current Challenges and Future Perspectives
Despite its demonstrated utility, BEAMing technology faces several challenges in clinical implementation. Pre-analytical factors including sample collection, processing, and DNA extraction methods require standardization to ensure reproducible results across laboratories [4] [7]. The detection of very low allele frequency mutations in early-stage cancers remains technically challenging, though approaches such as multimodal analysis combining mutation detection with epigenetic markers like methylation show promise for enhancing sensitivity [4].
The future evolution of BEAMing and related technologies will likely focus on increased multiplexing capabilities, reduced costs, and integration with complementary approaches such as fragmentomics analysis [4]. The combination of BEAMing's sensitivity for known mutations with next-generation sequencing's broad coverage represents a powerful approach for comprehensive tumor genotyping [8]. As clinical evidence accumulates, BEAMing technology is poised to expand beyond its current applications in treatment monitoring and resistance detection to include early cancer detection and minimal residual disease monitoring, potentially transforming cancer management across the diagnostic and therapeutic continuum.
The Role of ctDNA in Precision Oncology and Liquid Biopsy
Circulating tumor DNA (ctDNA) refers to the fraction of cell-free DNA (cfDNA) in the bloodstream that originates from tumor cells. This biomarker has emerged as a cornerstone of liquid biopsy, providing a non-invasive alternative to traditional tissue biopsies for cancer genotyping. Analysis of ctDNA enables real-time assessment of tumor burden, genomic heterogeneity, and therapeutic response, making it indispensable for precision oncology. The short half-life of ctDNA (ranging from 16 minutes to 2.5 hours) further enhances its value for dynamic monitoring of disease status and treatment efficacy. When integrated with highly sensitive detection technologies like BEAMing technology, ctDNA analysis facilitates unparalleled insights into cancer biology and patient management across the cancer care continuum.
ctDNA Detection Technologies and Methodologies
The reliable detection of ctDNA is technically challenging due to its low abundance in plasma, often constituting less than 0.1% of total cfDNA, particularly in early-stage cancers and minimal residual disease (MRD). This has driven the development of ultra-sensitive detection platforms.
Table 1: Key ctDNA Detection Technologies and Their Performance Characteristics
| Technology | Key Principle | Sensitivity | Key Advantages | Common Applications |
|---|---|---|---|---|
| Next-Generation Sequencing (NGS) | Massive parallel sequencing of DNA fragments [12] | Varies with input and panel; high sensitivity for VAF >0.5% [13] | Comprehensive genotyping; detects novel variants; high-throughput [14] [12] | Genomic profiling, MRD, therapy selection [15] [1] |
| Digital PCR (dPCR) | Partitioning of sample into thousands of individual reactions | Can detect VAFs as low as 0.001% in some applications [14] | Absolute quantification; high sensitivity and precision | Targeted mutation monitoring, therapy resistance [12] |
| BEAMing (Beads, Emulsion, Amplification, and Magnetics) | Combination of emulsion PCR, flow cytometry, and magnetic beads | High sensitivity for low-frequency variants | Excellent sensitivity for rare variant detection; digital quantification | Mutation detection, MRD monitoring [1] |
| Electrochemical Biosensors | Nanomaterial-based transduction of DNA-binding events [14] | Attomolar limits of detection [14] | Rapid results (e.g., 20 min); potential for point-of-care use [14] | Rapid diagnostics, potential for early detection [14] |
Advancing Sensitivity with Structural Variants and Fragmentomics
To overcome sensitivity limitations, novel approaches move beyond single nucleotide variants (SNVs):
- Structural Variant (SV)-Based Assays: These assays target tumor-specific chromosomal rearrangements (translocations, insertions, deletions), which are virtually absent in normal cells. This eliminates background noise from sequencing artifacts, enabling detection at parts-per-million sensitivity. In early-stage breast cancer, SV-based assays detected ctDNA in 96% of patients at baseline, with some VAFs below 0.01% [14].
- Fragmentomics and Size Selection: ctDNA fragments are typically shorter (90-150 base pairs) than non-tumor cfDNA. Library preparation methods that enrich for these shorter fragments can increase the fractional abundance of ctDNA in sequencing libraries, boosting sensitivity for low-frequency variants and making MRD detection more cost-effective [14].
The BEAMing Technology Workflow
BEAMing is a powerful technology for the digital detection and quantification of rare ctDNA variants. The following diagram illustrates its core workflow:
Detailed Experimental Protocol for BEAMing-based ctDNA Analysis
Sample Collection and Pre-processing:
- Collect peripheral blood (e.g., 10 mL) into EDTA or Streck Cell-Free DNA BCT tubes to prevent nucleated cell lysis.
- Process plasma within 2-6 hours of collection. Centrifuge blood at 800-1600 × g for 10-20 minutes to separate plasma. Transfer supernatant to a new tube and perform a second high-speed centrifugation (16,000 × g for 10 minutes) to remove residual cells.
- Store plasma at -80°C if not used immediately.
cfDNA Extraction:
- Extract cfDNA from 1-5 mL of plasma using commercially available kits (e.g., QIAamp Circulating Nucleic Acid Kit, MagMAX Cell-Free DNA Isolation Kit) according to manufacturer's instructions.
- Elute DNA in a low-EDTA TE buffer or nuclease-free water.
- Quantify cfDNA yield using fluorometric methods (e.g., Qubit dsDNA HS Assay). Assess DNA quality via capillary electrophoresis (e.g., Bioanalyzer, TapeStation).
BEAMing Reaction:
- Step 1: Primer Hybridization and Amplification: Design specific forward and reverse primers for the mutation of interest. The reverse primer is 5'-biotinylated. Perform a limited number of PCR cycles (e.g., 35-45 cycles) to amplify the target region from the extracted cfDNA.
- Step 2: Emulsion PCR: Mix the PCR products with streptavidin-coated magnetic beads and PCR reagents. Create a water-in-oil emulsion where each aqueous microdroplet contains, on average, less than one DNA molecule and one bead. Perform PCR amplification within the droplets, resulting in each bead being coated with thousands of copies of a single original DNA molecule.
- Step 3: Bead Recovery and Hybridization: Break the emulsion and recover the magnetic beads. Hybridize the bead-bound DNA with mutation-specific fluorescent probes (e.g., wild-type probe labeled with one fluorophore, mutant probe labeled with a different fluorophore) under stringent conditions.
- Step 4: Flow Cytometry Analysis: Analyze the beads using a flow cytometer. Beads are categorized as "mutant" (binding only the mutant probe), "wild-type" (binding only the wild-type probe), or "unlabeled" (no probe binding). The ratio of mutant beads to total beads (mutant + wild-type) provides the variant allele frequency.
Data Analysis:
- Calculate the mutant allele frequency:
(Number of mutant beads / (Number of mutant beads + Number of wild-type beads)) * 100. - Account for background error rates determined from control (non-template or wild-type) samples.
- Calculate the mutant allele frequency:
Clinical Applications and Utility
ctDNA analysis has demonstrated profound utility across multiple domains of cancer management, from early detection to monitoring advanced disease.
Minimal Residual Disease (MRD) and Recurrence Monitoring
The ability of ctDNA to detect MRD and predict recurrence is one of its most promising applications.
- In breast cancer, structural variant-based ctDNA assays can identify molecular relapse more than a year before clinical recurrence becomes evident, creating a window for early therapeutic intervention [14].
- In colorectal cancer, longitudinal ctDNA monitoring during and after adjuvant chemotherapy has proven to be a faster and more reliable indicator of recurrence than carcinoembryonic antigen (CEA) testing and imaging [14].
Therapy Selection and Resistance Monitoring
In advanced disease, ctDNA enables non-invasive genotyping to guide targeted therapy.
- For EGFR-mutant NSCLC, ctDNA analysis can detect the emergence of the T790M resistance mutation, allowing clinicians to switch patients to third-generation EGFR inhibitors without the need for a repeat tissue biopsy [14].
- Dynamic changes in ctDNA levels can predict radiographic response to therapy more accurately and rapidly than follow-up imaging [14] [16]. The MinerVa-Delta method, a novel approach to quantify ctDNA dynamics, has been validated to identify molecular responders to immunochemotherapy in lung squamous cell carcinoma, even among patients with radiologically stable disease [16].
Pan-Cancer Diagnostic and Prognostic Utility
Table 2: Clinical Utility of ctDNA Across Different Cancers
| Cancer Type | Key Clinical Applications | Representative Findings |
|---|---|---|
| Lung Cancer | EGFR mutation detection for TKI therapy, resistance monitoring, MRD [17] [15] | Tissue-liquid concordance in 36/96 cases in one cohort; EGFR mutations most frequent (44%) [15] |
| Colorectal Cancer | MRD detection, recurrence monitoring [14] | ctDNA monitoring more reliable than CEA/imaging for recurrence [14] |
| Breast Cancer | MRD detection, molecular relapse prediction [14] | SV-based assays detect ctDNA >1 year prior to clinical relapse [14] |
| Lymphoid Malignancies | MRD assessment post-immunochemotherapy [14] | ctDNA-based MRD more sensitive and informative than PET/CT imaging [14] |
| Gastroesophageal Cancers | Early detection via methylation profiling [14] | Tumor-agnostic methylation panels detect and quantify tumor development [14] |
The following diagram summarizes the key clinical decision points where ctDNA analysis informs patient management:
The Scientist's Toolkit: Essential Reagents and Materials
Successful ctDNA analysis requires careful selection of reagents and materials throughout the workflow.
Table 3: Research Reagent Solutions for ctDNA Analysis
| Item | Function/Purpose | Examples/Considerations |
|---|---|---|
| Blood Collection Tubes | Stabilize nucleated cells to prevent genomic DNA contamination during sample transport. | Streck Cell-Free DNA BCT, PAXgene Blood ccfDNA Tubes, CellSave Preservative Tubes |
| cfDNA Extraction Kits | Isolate high-quality, short-fragment cfDNA from plasma. | QIAamp Circulating Nucleic Acid Kit (Qiagen), MagMAX Cell-Free DNA Isolation Kit (Thermo Fisher), NEXTprep-Mag cfDNA Isolation Kit (Bioo Scientific) |
| NGS Library Prep Kits | Prepare sequencing libraries from low-input, fragmented cfDNA. | Kits with incorporation of unique molecular identifiers (UMIs) for error suppression; size selection capabilities are advantageous [14] [13] |
| Targeted Panels | Enrich for cancer-associated genes for deep sequencing. | Commercial (e.g., Roche AVENIO, Oncomine Precision Assay) or custom panels (e.g., SOPHiA Genetics) [15] [13] |
| PCR Reagents | Amplify target sequences for BEAMing or dPCR. | High-fidelity polymerases to minimize amplification errors; dPCR/ddPCR supermixes |
| Mutation-Specific Probes/Primers | Detect and quantify specific mutant alleles. | For BEAMing: 5'-biotinylated reverse primers, fluorescently labeled hybridization probes. For dPCR: TaqMan-style hydrolysis probes. |
Technical Challenges and Future Directions
Despite significant advances, several challenges remain for the widespread clinical implementation of ctDNA analysis.
- Pre-analytical Variability: Factors such as blood collection tube type, time-to-processing, and cfDNA extraction efficiency can significantly impact results. One study showed cfDNA extraction efficiency varied widely, with one assay having a mean efficiency of only 16% [13]. Standardization of pre-analytical protocols is critical.
- Clonal Hematopoiesis (CH): Age-acquired mutations in blood cells can be detected in cfDNA, leading to false-positive results. This is a significant confounder in tumor-naïve (agnostic) approaches. Strategies to mitigate this include sequencing matched white blood cells for subtraction or using in silico algorithms to infer the cellular origin of fragments [12].
- Assay Sensitivity and Specificity: While technologies have improved, sensitivity for very low VAFs (e.g., <0.1%) can be inconsistent across platforms, especially with low cfDNA input [13]. Reproducibility also remains a concern at these lower limits of detection.
- Tumor-Heterogeneity and Concordance: The genetic profile detected in ctDNA may not always perfectly mirror that of the primary tumor tissue due to spatial heterogeneity or differential shedding. Studies have reported poorly consistent mutations between ctDNA and tumor DNA, underscoring the need for further investigation into the biological and technical factors governing this relationship [17].
The future of ctDNA analysis is directed toward overcoming these hurdles and expanding applications. Emerging frontiers include the use of multiplexed CRISPR-based ctDNA assays, AI-based error suppression bioinformatic methods, and the integration of methylation and fragmentomic patterns for enhanced cancer detection and tissue-of-origin determination [14] [1]. As these technologies mature and standardization improves, ctDNA-based liquid biopsy is poised to become an even more integral component of precision oncology, enabling earlier detection and more dynamic, personalized cancer management.
BEAMing (Beads, Emulsion, Amplification, and Magnetics) represents a transformative digital PCR technology for circulating tumor DNA (ctDNA) analysis, addressing critical limitations of conventional tissue genotyping in oncology. This technology enables the highly sensitive and specific detection of somatic mutations in blood samples, providing a minimally invasive approach to monitor tumor dynamics and guide targeted therapies [5] [18]. The fundamental principle underpinning BEAMing's utility is its ability to partition individual DNA molecules across millions of microscopic emulsion droplets, thereby allowing for the precise quantification of rare mutant alleles against a background of wild-type DNA [5]. This technical capability positions BEAMing as an essential tool for researchers and drug development professionals pursuing precision oncology approaches.
The clinical relevance of BEAMing technology stems from the biological characteristics of ctDNA. Circulating tumor DNA consists of short DNA fragments released into the bloodstream primarily through apoptosis and necrosis of tumor cells [18]. These fragments typically constitute only a small fraction (often <0.01% to 1%) of the total cell-free DNA in circulation, creating a significant technical challenge for reliable detection [19]. BEAMing technology overcomes this limitation through its exceptional sensitivity, capable of detecting mutant alleles at frequencies as low as 0.01% [5], thereby enabling researchers to monitor tumor-specific genetic alterations throughout disease progression and treatment.
Performance Characteristics: Sensitivity, Specificity, and Quantification
Analytical Performance Metrics
BEAMing technology demonstrates exceptional performance characteristics that make it particularly suitable for ctDNA analysis in cancer research and drug development. The platform's analytical validity has been extensively evaluated through multiple studies comparing it with both tissue-based genotyping and alternative ctDNA detection methods.
Table 1: Key Analytical Performance Metrics of BEAMing Technology
| Performance Parameter | Specification | Experimental Support |
|---|---|---|
| Sensitivity | Detection limit of 0.01% mutant allele frequency [5] | Capable of identifying 1 mutant molecule in 10,000 wild-type molecules [5] |
| Specificity | >90% for RAS mutations in colorectal cancer [5] | High positive predictive value for mutation detection in clinical samples |
| Quantitative Range | Linear quantification from 0.01% to 100% mutant allele frequency [19] | Accurate monitoring of tumor dynamics during therapy [19] |
| Concordance with Tissue | 93.3% overall concordance for RAS mutations [5] | 92.6% positive agreement, 94% negative agreement with tissue reference [5] |
| Technical Reproducibility | High agreement with ddPCR (κ = 0.87-0.91) [20] | Minimal discordancy (3.9-5.0%) primarily at allele frequencies <1% [20] |
The sensitivity of BEAMing enables researchers to monitor minimal residual disease and emerging resistance mutations during targeted therapy. In a foundational study of colorectal cancer patients, BEAMing detected ctDNA in 100% of patients with metastatic disease, with median mutant DNA percentages of 0.18% in positive samples (10th-90th percentile range: 0.005-11.7%) [19]. This sensitivity proves particularly valuable for tracking resistance mechanisms, such as the emergence of EGFR T790M mutations in non-small cell lung cancer patients undergoing EGFR inhibitor therapy [18].
Comparative Method Performance
BEAMing demonstrates strong agreement with other sensitive detection methods, underscoring its reliability for clinical research applications. A comprehensive comparison between BEAMing and droplet digital PCR (ddPCR) using 363 baseline plasma samples from the PALOMA-3 trial in advanced breast cancer showed excellent concordance for both ESR1 (κ = 0.91) and PIK3CA (κ = 0.87) mutations [20]. The observed discordancy between methods (3.9% for ESR1, 5.0% for PIK3CA) primarily occurred at allele frequencies below 1%, likely resulting from stochastic sampling effects rather than technical limitations [20].
Table 2: Comparison of BEAMing with Other ctDNA Detection Platforms
| Methodology | Detection Limit | Advantages | Limitations |
|---|---|---|---|
| BEAMing | 0.01% [5] | High sensitivity, absolute quantification, clinical validation | Limited multiplexing capability, targeted approach |
| Droplet Digital PCR | ~0.01% [20] | High sensitivity, ease of use with available kits | Detection limited to predefined mutations |
| Next-Generation Sequencing | 0.1%-0.5% [21] | Broad mutational coverage, discovery capability | Higher cost, longer turnaround time, complex bioinformatics |
| Quantitative PCR | 1-10% | Rapid, low cost | Limited sensitivity, not suitable for low-frequency mutations |
When compared to next-generation sequencing approaches, BEAMing offers superior sensitivity for detecting predefined mutations but lacks the broad genomic coverage of NGS panels. While targeted NGS methods can achieve detection limits of 0.1% with sufficient sequencing depth, this requires ultra-deep sequencing (~20,000 unique reads per base) that remains prohibitively expensive for routine clinical research [21]. BEAMing thus occupies a crucial niche where high-sensitivity detection of specific mutations is required for studies of tumor dynamics or resistance monitoring.
Research Applications and Experimental Evidence
Monitoring Tumor Dynamics
BEAMing technology enables precise quantification of tumor burden changes in response to therapeutic interventions. In a landmark study of colorectal cancer patients undergoing surgical resection, BEAMing demonstrated a median 99% decrease in ctDNA levels following complete tumor resection, with this reduction detectable as early as 24 hours post-surgery [19]. Researchers calculated the half-life of ctDNA after surgery to be approximately 114 minutes, highlighting the dynamic nature of ctDNA turnover and the utility of BEAMing for real-time monitoring of tumor burden [19].
The technology's quantitative capabilities further enable correlation between ctDNA levels and traditional tumor markers. In the same study, measurements of mutant DNA fragments from two different genes in the same patient showed remarkable correlation (R² = 0.95), confirming the reliability of BEAMing for quantifying tumor-derived DNA [19]. This precise quantification provides drug development professionals with a powerful pharmacodynamic biomarker for assessing treatment response in early-phase clinical trials.
Guiding Targeted Therapy
BEAMing technology plays a crucial role in identifying patients who may benefit from targeted therapies and in monitoring emerging resistance. In colorectal cancer, BEAMing-based RAS mutation testing demonstrates 92.6% positive agreement and 94% negative agreement with standard tissue testing, enabling rapid identification of patients unlikely to benefit from anti-EGFR therapies [5]. The high concordance (93.3%) between plasma-based OncoBEAM RAS testing and tissue reference methods supports its utility for clinical research requiring expanded RAS profiling [5].
In breast cancer research, BEAMing has proven valuable for detecting ESR1 mutations associated with resistance to endocrine therapy. The technology's sensitivity enables identification of these mutations emerging under selective pressure of aromatase inhibition, allowing researchers to study resistance mechanisms and assess novel therapeutic approaches targeting ESR1-mutant clones [22] [20].
Research Reagent Solutions
Table 3: Essential Research Reagents for BEAMing Experiments
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Magnetic Streptavidin Beads | Solid support for DNA amplification | MyOne beads (Invitrogen) coated with Tag1 oligonucleotide [3] |
| High-Fidelity DNA Polymerase | Initial target amplification | HotStart Phusion polymerase (NEB) with proofreading activity [3] |
| Emulsion Formulation | Compartmentalization of PCR reactions | 7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC [3] |
| Allele-Specific Fluorescent Probes | Mutation detection and quantification | 15-18nt probes complementary to mutant/wild-type sequences [3] |
| Plasma DNA Extraction Kit | ctDNA isolation from blood samples | Qiagen DNA Micro Kit for 1mL plasma input [3] |
| Breaking Buffer | Emulsion disruption post-amplification | Contains Triton-X-100, SDS, NaCl, EDTA for efficient recovery [3] |
Detailed Experimental Protocol
Sample Preparation and DNA Extraction
Blood Collection and Processing: Collect peripheral blood into EDTA-containing tubes and process within one hour of collection. Centrifuge at 820 × g for 10 minutes to separate plasma, then transfer 1mL aliquots to clean tubes. Perform a second centrifugation at 16,000 × g for 10 minutes to pellet remaining cellular debris [3].
ctDNA Extraction: Isolate total genomic DNA from 1mL of plasma using a DNA Micro Kit (Qiagen) according to manufacturer's instructions. Elute DNA in a final volume of 20-50μL of provided elution buffer [3].
DNA Quantification: Measure isolated ctDNA concentration using a Nanodrop ND1000 spectrophotometer. Typical yields range from 1-50ng/mL plasma, depending on tumor type and stage [3].
BEAMing Reaction Setup
Initial Amplification: Set up eight separate 25μL PCR reactions, each containing:
- Template DNA from 250μL plasma equivalent
- 5× Phusion High Fidelity PCR buffer (NEB)
- 1.5U HotStart Phusion polymerase (NEB)
- 0.2μM each primer
- 0.25mM each dNTP
- 0.5mM MgCl₂ Cycling conditions: 98°C for 30s; 35 cycles of (98°C for 10s, 57°C for 10s, 72°C for 10s) [3]
Emulsion PCR Preparation: Prepare a 150μL PCR mixture containing:
- 18pg pooled template DNA
- 40U Platinum Taq DNA polymerase (Invitrogen)
- 1× PCR buffer
- 0.2mM dNTPs
- 5mM MgCl₂
- 0.05μM Tag1 primer
- 8μM Tag2 primer
- ~6×10⁷ magnetic streptavidin beads coated with Tag1 oligonucleotide [3]
Emulsion Formation: Combine 150μL PCR mixture with 600μL oil/emulsifier mixture (7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC) and one 5mm steel bead in a 96-deep-well plate. Create microemulsions by shaking the plate in a TissueLyser for 10s at 15Hz followed by 7s at 17Hz. Verify emulsion quality under microscope (40× magnification) [3].
Emulsion PCR Amplification: Dispense emulsions into PCR plates and run the following program:
- 94°C for 2min
- 3 cycles: 94°C for 10s, 68°C for 45s, 70°C for 75s
- 3 cycles: 94°C for 10s, 65°C for 45s, 70°C for 75s
- 3 cycles: 94°C for 10s, 62°C for 45s, 70°C for 75s
- 50 cycles: 94°C for 10s, 57°C for 45s, 70°C for 75s [3]
Detection and Analysis
Emulsion Disruption: Add 150μL breaking buffer (10mM Tris-HCl pH 7.5, 1% Triton-X-100, 1% SDS, 100mM NaCl, 1mM EDTA) to each well and mix with TissueLyser at 20Hz for 20s. Recover beads by centrifugation at 3,200 × g for 2min and remove oil phase. Repeat breaking step twice [3].
Hybridization: Wash beads with 150μL wash buffer (20mM Tris-HCl pH 8.4, 50mM KCl). Denature DNA on beads with 0.1M NaOH for 5min. Wash again with wash buffer and resuspend in 150μL of the same buffer [3].
Mutation Detection: Incubate beads with fluorescently labeled probes complementary to mutant and wild-type sequences (15-18nt). Analyze using flow cytometry to distinguish mutant and wild-type beads [3].
Data Analysis: Calculate mutant allele frequency as (number of mutant beads)/(number of mutant + wild-type beads) × 100. Determine absolute mutant DNA concentration by multiplying total DNA concentration by mutant allele frequency [3].
Workflow and Signaling Pathways
Diagram 1: BEAMing Workflow for ctDNA Analysis. This diagram illustrates the key steps in BEAMing technology, from sample processing through final quantification.
Technical Considerations and Limitations
While BEAMing offers exceptional sensitivity and specificity for ctDNA analysis, researchers must consider several technical aspects for optimal experimental design. The technology requires prior knowledge of specific mutations to be detected, making it less suitable for discovery applications compared to NGS approaches [5]. Additionally, the emulsion formation process demands technical expertise to achieve consistent compartmentalization, and the multi-step protocol requires careful quality control throughout [3].
The quantitative accuracy of BEAMing depends on adequate input DNA and proper normalization. Researchers should note that ctDNA yields vary significantly by cancer type, with lung cancers typically yielding lower amounts (∼5ng/mL plasma) compared to liver cancers (∼46ng/mL plasma) [21]. This variability impacts detection sensitivity, particularly for low-frequency mutations, and should inform sample size calculations and power analyses in research studies.
Recent advancements in BEAMing technology have expanded its applications to include monitoring of minimal residual disease and emerging resistance mutations. The incorporation of additional biomarkers, such as BRAF mutations, into BEAMing panels further enhances its utility for comprehensive genomic profiling in clinical research [5]. As the field advances, integration of BEAMing with complementary approaches like fragmentomics analysis may provide even deeper insights into tumor biology and treatment response [4].
Comparison with Traditional Tissue Biopsy Limitations
Tissue biopsy has long been the gold standard for tumor diagnosis, providing definitive pathological confirmation, cancer subtyping, and molecular characterization for targeted therapy selection [9] [4]. Its widespread clinical adoption is supported by high laboratory standardization, good result consistency, and established accuracy [9]. However, the invasive nature of tissue sampling and inherent tumor biological complexities present significant limitations in clinical practice, particularly for dynamic monitoring of cancer progression and treatment response [9] [2].
Liquid biopsy, particularly through analysis of circulating tumor DNA (ctDNA), has emerged as a minimally invasive alternative that addresses many constraints of traditional tissue sampling [1] [4]. BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) represents one of the most sensitive approaches for ctDNA mutation detection, enabling identification of rare tumor-derived DNA fragments in blood with variant allele frequencies as low as 0.01% [1] [4]. This application note provides a comprehensive comparison between traditional tissue biopsy and liquid biopsy approaches, with specific focus on technical and clinical limitations addressed by BEAMing technology in ctDNA analysis.
Comparative Analysis: Tissue vs. Liquid Biopsy
Table 1: Comprehensive comparison of key characteristics between traditional tissue biopsy and liquid biopsy
| Characteristic | Traditional Tissue Biopsy | Liquid Biopsy (ctDNA) |
|---|---|---|
| Invasiveness | Highly invasive surgical procedure | Minimally invasive (blood draw) [9] [1] |
| Tumor Representation | Limited to sampled site; may miss heterogeneity [2] | Captures contributions from all tumor sites [1] [2] |
| Sampling Frequency | Limited by patient risk and practicality | Enables frequent monitoring [9] [2] |
| Turnaround Time | Days to weeks (processing, sectioning, analysis) | Hours to days [23] |
| Sensitivity for MRD | Limited; cannot detect molecular residual disease | High sensitivity for MRD detection [1] [2] |
| Clinical Applications | Diagnosis, histopathological classification, initial molecular profiling | Early detection, treatment monitoring, resistance mechanism identification [1] [4] [2] |
| Tumor Evolution Tracking | Single snapshot in time | Dynamic, real-time monitoring capability [1] [2] |
| Spatial Heterogeneity | Limited to sampled region | Captures global tumor heterogeneity [1] |
| Complications Risk | Procedure-specific risks (bleeding, infection, pain) | Minimal risk (equivalent to blood draw) [9] |
Table 2: Quantitative performance comparison of detection technologies
| Technology | Sensitivity | Multiplexing Capacity | Throughput | Key Applications |
|---|---|---|---|---|
| BEAMing | 0.01% VAF [4] | Moderate (dozens of mutations) | Moderate | Mutation detection, therapy monitoring [4] |
| ddPCR | 0.001%-0.01% VAF [24] | Low (1-5 mutations) | High | MRD monitoring, resistance mutation detection [4] [24] |
| NGS Panels | 0.1%-0.5% VAF (standard); 0.02%-0.1% (ultrasensitive) [21] | High (hundreds of genes) | Variable | Comprehensive genomic profiling, novel alteration discovery [21] [23] |
| Tissue Biopsy | N/A (direct observation) | Limited by sample size | Low | Initial diagnosis, histopathological evaluation [9] |
Key Limitations of Traditional Tissue Biopsy
Invasiveness and Procedural Constraints
Tissue biopsy requires invasive surgical procedures that carry inherent risks including bleeding, infection, and patient discomfort [9]. Certain tumor locations (e.g., brain, lung, bone) present significant accessibility challenges, making tissue sampling difficult or contraindicated for some patients [9] [4]. These procedural constraints fundamentally limit the frequency of sampling, preventing dynamic monitoring of tumor evolution during treatment [2].
Tumor Heterogeneity and Sampling Bias
Intratumoral heterogeneity represents a fundamental limitation of single-site tissue biopsy. Malignant tumors contain geographically distinct subclones with divergent molecular profiles [2]. A single tissue sample captures only a limited snapshot of this heterogeneity, potentially missing resistant subclones or metastatic variants [2]. This sampling bias can lead to incomplete molecular characterization and suboptimal treatment selection.
Temporal Limitations and Clinical Utility
The static nature of tissue biopsy prevents real-time monitoring of treatment response and emerging resistance mechanisms [2]. Tumor evolution under therapeutic pressure occurs continuously, but tissue biopsy provides only a historical record of the tumor genome at the time of sampling [1] [2]. This temporal limitation is particularly significant for tracking acquired resistance mutations during targeted therapy, where timely intervention is critical [2].
BEAMing Technology: Technical Principles and Protocols
BEAMing technology combines emulsion PCR with flow cytometry to achieve ultra-sensitive detection of rare mutant DNA molecules in biological fluids [4]. The methodology transforms individual DNA molecules into magnetic beads containing thousands of copies of the original sequence, enabling precise enumeration of mutant alleles.
BEAMing Workflow Protocol
Table 3: Detailed BEAMing experimental protocol for ctDNA analysis
| Step | Process | Key Parameters | Quality Controls |
|---|---|---|---|
| Sample Preparation | Blood collection in EDTA or specialized BCTs; plasma separation via dual centrifugation | 2 × 10 mL blood; 1600g × 10 min → 16000g × 10 min [25] | Assess hemolysis; cfDNA concentration >0.5ng/μL |
| ctDNA Extraction | Silica membrane column or magnetic bead-based isolation | Input: 1-5 mL plasma; elution volume: 20-50 μL [25] | DNA fragment analysis (160-200 bp peak) |
| Primer Design | Mutation-specific primers for targets of interest | Amplicon size: 80-150 bp; TM: 60-65°C | Specificity validation against wildtype |
| Emulsion PCR | Water-in-oil emulsion formation; amplification on magnetic beads | 40-50 cycles; limiting dilution | Emulsion stability assessment |
| Bead Recovery & Hybridization | Emulsion breaking; fluorescent probe hybridization | Mutation-specific probes with fluorophores | Hybridization efficiency measurement |
| Flow Cytometry | Bead analysis and enumeration | 50,000-100,000 beads/sample | Gating controls for background |
| Data Analysis | VAF calculation | VAF = (mutant beads/total beads) × 100% | Background subtraction |
BEAMing Technology Schematic
BEAMing Workflow Schematic: Visual representation of the key procedural steps in BEAMing technology for ctDNA analysis.
Advantages of BEAMing in Addressing Tissue Biopsy Limitations
Overcoming Tumor Heterogeneity
BEAMing technology addresses spatial heterogeneity limitations by capturing tumor-derived DNA from all disease sites, providing a comprehensive molecular profile that transcends single-site sampling constraints [1] [2]. This approach enables detection of multiple metastatic subclones simultaneously, offering a more complete representation of the tumor genomic landscape than single-site tissue biopsy.
Enabling Dynamic Monitoring
The minimally invasive nature of blood collection facilitates frequent temporal monitoring of treatment response and resistance development [1] [2]. BEAMing's sensitivity for detecting mutant allele frequency changes enables real-time assessment of therapeutic efficacy, often weeks to months before radiographic evidence [4] [2]. This capability is particularly valuable for identifying emerging resistance mutations (e.g., EGFR T790M in NSCLC, ESR1 mutations in breast cancer) during targeted therapy [23] [2].
Technical Performance Characteristics
BEAMing technology achieves exceptional sensitivity down to 0.01% variant allele frequency, surpassing standard NGS approaches (0.1-0.5% LoD) and approaching the sensitivity of ddPCR [21] [4]. This performance enables reliable detection of minimal residual disease and early-stage cancers when tumor DNA fraction in total cfDNA is extremely low [1] [2]. The technology maintains high specificity through its combination of emulsion partitioning and fluorescence validation, minimizing false positives from PCR errors or background noise [4].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential research reagents and materials for BEAMing ctDNA analysis
| Reagent/Material | Function | Technical Specifications | Commercial Examples |
|---|---|---|---|
| cfDNA BCT Tubes | Preserves blood sample integrity during transport/storage | Prevents leukocyte lysis; stable up to 7 days at room temperature | Streck cfDNA BCT, PAXgene Blood ccfDNA, Roche cfDNA [25] [26] |
| Magnetic Beads | Solid support for emulsion PCR amplification | 1-5μm diameter; functionalized with oligonucleotides | Dynabeads, Sera-Mag Magnetic Beads |
| Emulsion Oil Phase | Creates microreactors for single-molecule PCR | Surfactant-stabilized; thermostable | Sigma Mineral Oil, Bio-Rad Droplet Generation Oil |
| Mutation-Specific Primers/Probes | Selective amplification/detection of target mutations | HPLC-purified; specific for hotspot mutations | Custom TaqMan assays, IDT PrimeTime qPCR assays |
| Digital PCR Master Mix | Optimized for emulsion PCR efficiency | Hot-start polymerase; optimized buffer | Bio-Rad ddPCR Supermix, Thermo Fisher Digital PCR Master Mix |
| Hybridization Buffers | Facilitates specific probe binding to amplified products | Stringency control; minimal background | SSC-based buffers, proprietary formulations |
| Flow Cytometry Reference Beads | Instrument calibration and quantification | Fluorescent reference standards; size-matched | Spherotech Alignment Beads, BD Calibrite Beads |
Integration of BEAMing in Cancer Research and Drug Development
BEAMing technology provides powerful applications in preclinical and clinical drug development, enabling pharmacodynamic monitoring and early response assessment in clinical trials [2]. The technology's quantitative capabilities support dose optimization studies by correlating mutant allele frequency changes with drug exposure levels [2]. In translational research, BEAMing facilitates biomarker discovery and validation through high-sensitivity detection of emerging resistance mechanisms across multiple cancer types [4] [2].
The integration of BEAMing with complementary approaches like NGS-based mutation panels creates a comprehensive liquid biopsy strategy - using NGS for broad mutation discovery and BEAMing for ultrasensitive monitoring of prioritized mutations in longitudinal studies [23] [2]. This combined approach maximizes both the breadth of genomic profiling and the sensitivity needed for minimal residual disease detection [2].
BEAMing technology addresses fundamental limitations of traditional tissue biopsy by providing a minimally invasive approach that captures global tumor heterogeneity and enables dynamic monitoring of cancer evolution. Its exceptional sensitivity and quantitative capabilities make it particularly valuable for detecting minimal residual disease, assessing treatment response, and identifying emerging resistance mechanisms during targeted therapy.
While tissue biopsy remains essential for initial diagnosis and histopathological characterization, BEAMing-enhanced liquid biopsy represents a complementary approach that extends our molecular profiling capabilities throughout the cancer treatment continuum. For researchers and drug development professionals, BEAMing technology offers a robust platform for pharmacodynamic studies, biomarker development, and clinical trial enrichment, ultimately supporting the advancement of precision oncology.
Circulating tumor DNA (ctDNA), a subset of cell-free DNA (cfDNA) originating from primary tumors and metastatic lesions, has emerged as a powerful biomarker for non-invasive cancer monitoring [27] [2]. BEAMing (Beads, Emulsion, Amplification, and Magnetics) represents a cornerstone technology for targeted ctDNA analysis, enabling the detection of rare mutations with high sensitivity and specificity [4] [28]. This technology is particularly valuable for monitoring treatment response, identifying emerging resistance mutations, and detecting minimal residual disease (MRD) in cancer patients [2]. As a digital PCR-based method, BEAMing allows for the absolute quantification of mutant allele fractions even at low frequencies (as low as 0.01%), making it suitable for analyzing ctDNA where tumor-derived DNA is heavily diluted by wild-type cfDNA from normal cells [2] [28]. This application note details the standardized protocols and methodologies for implementing BEAMing technology in ctDNA analysis from blood collection to mutation detection.
Pre-Analytical Phase: Blood Collection and Processing
The pre-analytical phase is critical for maintaining ctDNA integrity, as improper handling can significantly compromise downstream analysis [27] [25]. Standardized protocols must be established to ensure consistent and reliable results.
Blood Collection and Sample Preparation
Table 1: Blood Collection Tube Options for ctDNA Analysis
| Tube Type | Chemical Additive | Storage Stability | Advantages | Limitations |
|---|---|---|---|---|
| Standard EDTA | Ethylenediaminetetraacetic acid | ≤4 hours at 4°C [27] | Inhibits plasma DNase activity; widely available [27] | Limited stability; requires rapid processing [27] |
| Cell-Stabilizing Tubes (e.g., Streck, Roche, PAXgene) | Proprietary preservatives | Up to 5 days at 10-30°C [27] [25] | Prevents leukocyte lysis and gDNA contamination; enables transport [27] [25] | Higher cost; subtle performance differences between brands [27] |
Protocol: Blood Collection and Plasma Separation
- Collection: Draw blood into preferred collection tubes (cell-stabilizing tubes recommended for delayed processing) [27].
- Initial Centrifugation: Within specified stability timeframes, centrifuge at 800-1,900 × g for 10 minutes at room temperature to separate cellular components from plasma [27] [25].
- Secondary Centrifugation: Transfer supernatant to a new tube and perform high-speed centrifugation at 14,000-16,000 × g for 10 minutes to remove remaining cellular debris [27] [25].
- Aliquoting and Storage: Aliquot plasma into cryovials and store at -80°C until DNA extraction. Avoid more than three freeze-thaw cycles to prevent nucleic acid degradation [27].
ctDNA Extraction Techniques
Table 2: ctDNA Extraction Method Comparison
| Method | Principle | DNA Recovery | Processing Time | Suitability for BEAMing |
|---|---|---|---|---|
| Silica Spin Columns | DNA binding to silica membrane under high salt conditions | High for variable fragment sizes [27] | Moderate | Excellent [27] |
| Magnetic Beads | Silica-coated magnetic beads bind DNA | Efficient for small fragments; enables automation [27] | Fast | Excellent, especially for automated workflows [27] |
| Phase Isolation | Organic separation of nucleic acids | High purity | Lengthy | Good, but less practical for routine use [27] |
| Magnetic Ionic Liquid | Dispersive liquid-liquid microextraction | Superior enrichment factors [27] | Moderate | Promising emerging technology [27] |
Protocol: ctDNA Extraction Using Magnetic Beads
- Binding: Mix plasma with lysis buffer and magnetic beads under high salt conditions to promote DNA binding [27].
- Washing: Immobilize beads magnetically and wash twice with wash buffer to remove contaminants [27].
- Elution: Elute ctDNA in low-salt elution buffer or nuclease-free water [27].
- Quantification: Measure DNA concentration using fluorometric methods (e.g., Qubit) and assess fragment size distribution via bioanalyzer if needed [27].
BEAMing Workflow for Mutation Detection
BEAMing technology combines emulsion PCR with flow cytometry to detect and quantify specific mutations in ctDNA [4] [28]. The workflow transforms individual DNA molecules into bead-bound amplicons that can be analyzed statistically.
Core BEAMing Protocol
Step 1: Primer-Bead Preparation
- Use magnetic beads coated with streptavidin [28].
- Incubate with biotinylated primers specific to the genomic region of interest [28].
- Wash to remove unbound primers and resuspend in PCR reaction mix containing ctDNA template [28].
Step 2: Water-in-Oil Emulsion Formation
- Mix the aqueous PCR component with oil-surfactant solution [28].
- Vigorously vortex or use microfluidic devices to create monodisperse water-in-oil droplets [28].
- Each microreactor contains an average of <1 bead and <1 DNA molecule, ensuring clonal amplification [28].
Step 3: Emulsion PCR
- Perform thermal cycling with the following conditions:
- Initial denaturation: 95°C for 5-10 minutes
- 40-50 cycles of: 95°C for 30s, 55-65°C for 30s, 72°C for 30-60s
- Final extension: 72°C for 5-10 minutes [28]
Step 4: Emulsion Breakage and Hybridization
- Break emulsion using organic solvents or detergents [28].
- Wash beads to remove oil and PCR components [28].
- Hybridize with mutation-specific fluorescent probes:
Step 5: Flow Cytometry Analysis
- Analyze beads using flow cytometry to distinguish:
- Beads with mutant sequences (fluorophore A)
- Beads with wild-type sequences (fluorophore B)
- Beads with both sequences (both fluorophores) [28]
- Calculate mutant allele frequency: (mutant beads / total beads) × 100% [28]
Research Reagent Solutions
Table 3: Essential Reagents for BEAMing ctDNA Analysis
| Reagent Category | Specific Examples | Function | Technical Considerations |
|---|---|---|---|
| Blood Collection Systems | Streck Cell-Free DNA BCT, PAXgene Blood cDNA Tube | Preserves ctDNA integrity during storage/transport | Choose based on processing delays; stability varies 48h-5days [27] |
| DNA Extraction Kits | Silica membrane spin columns, Magnetic bead-based kits | Isolate ctDNA from plasma | Magnetic beads optimize small fragment recovery; spin columns provide consistent yield [27] |
| PCR Components | Thermostable polymerase, dNTPs, biotinylated primers | Amplify target sequences | Use high-fidelity enzymes; optimize primer concentrations [28] |
| Emulsion Reagents | Mineral oil, surfactants (Span 80, Tween 80) | Create stable water-in-oil compartments | Emulsion stability critical for compartmentalization [28] |
| Detection Probes | Fluorescently-labeled allele-specific probes | Distinguish mutant and wild-type sequences | Design with matched Tm; optimize hybridization stringency [28] |
| Magnetic Beads | Streptavidin-coated magnetic beads | Solid support for amplification | Uniform size distribution improves results [28] |
Quality Control and Data Interpretation
Implement rigorous quality control measures throughout the BEAMing workflow. Include control samples with known mutation status in each run to verify assay performance [28]. Determine the limit of detection (LOD) for each assay, typically achieving sensitivity down to 0.01%-0.1% mutant allele frequency [28]. For clinical applications, establish a threshold for positive calls that considers both analytical sensitivity and specific clinical context [2].
Data interpretation should account for biological and technical factors. The mutant allele frequency in ctDNA correlates with tumor burden but varies by cancer type and individual tumor biology [2]. Serial monitoring provides more clinically actionable information than single timepoint measurements, with decreasing levels indicating treatment response and rising levels suggesting progression or emerging resistance [2].
BEAMing technology provides a robust platform for precise ctDNA mutation detection, enabling researchers and clinicians to non-invasively monitor cancer dynamics. Following these standardized protocols ensures reliable, reproducible results that can inform both research hypotheses and clinical decision-making in precision oncology.
BEAMing in Practice: Technical Workflows and Clinical Implementation
Beads, Emulsion, Amplification, and Magnetics (BEAMing) is a highly sensitive digital PCR-based technology that enables the detection and quantification of rare somatic mutations, such as those found in circulating tumor DNA (ctDNA) from cancer patients. This protocol details the application of BEAMing for detecting epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer (NSCLC), a critical predictive biomarker for tyrosine kinase inhibitor therapy [3]. By combining emulsion PCR with flow cytometry, BEAMing allows for the absolute quantification of mutant alleles present at frequencies as low as 0.01% in a background of wild-type DNA, making it particularly suitable for liquid biopsy applications where ctDNA concentration is minimal [3] [1].
Materials and Equipment
Research Reagent Solutions
Table 1: Essential reagents and materials for BEAMing protocol
| Item | Function/Specification |
|---|---|
| EDTA Blood Collection Tubes | Plasma separation and cell-free DNA preservation |
| DNA Micro Kit (Qiagen) | Circulating tumor DNA extraction from plasma |
| HotStart Phusion Polymerase (NEB) | High-fidelity amplification in initial PCR |
| Platinum Taq DNA Polymerase (Invitrogen) | Emulsion PCR amplification |
| Magnetic Streptavidin Beads (MyOne, Invitrogen) | Solid support for amplification and separation |
| Oil/Emulsifier Mixture | Emulsion formation (7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC) |
| Allele-Specific Fluorescent Probes | Mutation detection and quantification (15-18 nt) |
| PC9, H1975, A549 Cell Lines | Positive and negative controls for validation |
Equipment
- Nanodrop ND1000 Spectrophotometer
- TissueLyser (Qiagen)
- Deep-well plates (1.2 ml; Abgene)
- Flow Cytometer (FACSAria III)
- Thermal cyclers
- Centrifuge capable of 16,000 × g
Experimental Workflow
Workflow Diagram
Pre-Analytical Phase: Sample Collection and Processing
Blood Collection and Plasma Separation
- Collect peripheral blood into 10 mL EDTA tubes from consented patients [3]
- Process within one hour of collection to prevent genomic DNA contamination
- Centrifuge at 820 × g for 10 minutes at room temperature to separate plasma from cellular components
- Transfer 1 mL aliquots of supernatant to fresh 1.5 mL tubes
- Centrifuge at 16,000 × g for 10 minutes to pellet remaining cellular debris
- Store plasma supernatant at -80°C until DNA extraction
ctDNA Isolation and Quantification
- Extract total cell-free DNA from 1 mL plasma using DNA Micro Kit (Qiagen) according to manufacturer's instructions
- Elute DNA in appropriate buffer (e.g., AE buffer or nuclease-free water)
- Quantify DNA concentration using Nanodrop ND1000 spectrophotometer
- Record concentration and purity (A260/A280 ratio) - expected yield typically ranges from 1-50 ng/μL depending on disease burden
BEAMing PCR Phase
Primary PCR Amplification
Table 2: Primary PCR reaction setup and conditions
| Component | Volume/Final Concentration |
|---|---|
| Template DNA (from 250 μL plasma) | Variable |
| 5× Phusion High Fidelity PCR Buffer | 5 μL |
| HotStart Phusion Polymerase | 1.5 U |
| Forward and Reverse Primers (0.2 μM each) | 1 μL each |
| dNTPs (0.25 mM each) | 1 μL |
| MgCl₂ (0.5 mM) | 0.5 μL |
| Nuclease-free water | To 25 μL |
Thermal Cycling Conditions:
- Initial denaturation: 98°C for 30 seconds
- 35 cycles of:
- Denaturation: 98°C for 10 seconds
- Annealing: 57°C for 10 seconds
- Extension: 72°C for 10 seconds
- Final extension: 72°C for 5 minutes
- Hold: 4°C
Post-Amplification: Pool eight separate 25 μL PCR reactions and quantify using Nanodrop spectrophotometer.
Emulsion PCR Mechanism
Emulsion PCR Setup
Prepare PCR mixture (150 μL total volume):
- Template DNA (18 pg from primary PCR)
- Platinum Taq DNA Polymerase (40 U)
- 1× PCR Buffer
- dNTPs (0.2 mM)
- MgCl₂ (5 mM)
- Tag1 oligonucleotide (0.05 μM)
- Tag2 oligonucleotide (8 μM)
- Magnetic streptavidin beads (~6×10⁷) coated with Tag1 oligonucleotide
Create microemulsions by combining:
- 150 μL PCR mixture
- 600 μL oil/emulsifier mixture
- One 5 mm steel bead
Shake in TissueLyser for 10 seconds at 15 Hz followed by 7 seconds at 17 Hz
Verify emulsion formation by checking aqueous compartments under inverted microscope (40× magnification)
Emulsion PCR Amplification
Thermal Cycling Conditions for Emulsion PCR:
- Initial denaturation: 94°C for 2 minutes
- 3 cycles of: 94°C for 10s, 68°C for 45s, 70°C for 75s
- 3 cycles of: 94°C for 10s, 65°C for 45s, 70°C for 75s
- 3 cycles of: 94°C for 10s, 62°C for 45s, 70°C for 75s
- 50 cycles of: 94°C for 10s, 57°C for 45s, 70°C for 75s
- Final hold: 4°C
Post-PCR Processing and Detection
Bead Recovery and Denaturation
- Disrupt emulsions by adding 150 μL breaking buffer (10 mM Tris-HCl pH 7.5, 1% Triton-X 100, 1% SDS, 100 mM NaCl, 1 mM EDTA) to each well
- Mix with TissueLyser at 20 Hz for 20 seconds
- Recover beads by centrifugation at 3,200 × g for 2 minutes
- Remove oil phase and repeat breaking step twice
- Wash beads with 150 μL wash buffer (20 mM Tris-HCl pH 8.4, 50 mM KCl)
- Denature DNA with 0.1 M NaOH for 5 minutes
- Wash and resuspend beads in 150 μL wash buffer
Mutation Detection by Hybridization
- Design fluorescently labeled probes complementary to mutant and wild-type sequences (15-18 nt)
- Hybridize probes to DNA bound to beads
- Analyze using flow cytometry (FACSAria III) to detect mutant and wild-type populations
Data Analysis and Interpretation
Quantitative Analysis
Table 3: Key analytical performance metrics of BEAMing for EGFR mutation detection
| Parameter | Performance Value | Notes |
|---|---|---|
| Sensitivity | Can detect 0.01% mutant alleles | Requires optimization of emulsion conditions |
| Concordance with Tissue qPCR | 90-100% for various exons | Exon 19: 90%, Exon 20: 100%, Exon 21: 96-98% [3] |
| Linear Range | 0.01% to 100% mutant allele frequency | Validated using cell line DNA mixtures |
| Sample Input | DNA from 250 μL plasma per PCR | Eight parallel reactions recommended |
Calculation of Mutant Allele Frequency
Mutant allele frequency (%) = (Number of mutant beads / Total number of beads) × 100
For clinical interpretation in NSCLC:
- Positive result: Detection of known EGFR mutations (e.g., exon 19 deletions, L858R)
- Negative result: No mutation detected above assay sensitivity threshold
- Reportable range: 0.01% - 100% mutant allele frequency
Troubleshooting Guide
Table 4: Common issues and solutions in BEAMing protocol
| Problem | Possible Cause | Solution |
|---|---|---|
| Low bead count | Inefficient emulsion formation | Verify shaking parameters; check emulsifier composition |
| High background | Non-specific hybridization | Optimize probe design; increase hybridization stringency |
| Poor sensitivity | Suboptimal DNA quality | Ensure rapid plasma processing; verify extraction method |
| Inconsistent results | Emulsion instability | Standardize shaking protocol; use fresh reagents |
The reliability of any circulating tumor DNA (ctDNA) analysis, including the highly sensitive BEAMing (Beads, Emulsion, Amplification, and Magnetics) technology, is fundamentally dependent on the quality of the pre-analytical phase. Pre-analytical variables encompass all steps from blood collection to the isolation of ctDNA and can significantly impact the integrity, purity, and yield of the extracted nucleic acids. For BEAMing, which is capable of detecting single-molecule mutations with high specificity, the presence of contaminating genomic DNA from lysed blood cells or degraded ctDNA can dramatically reduce the mutant allele fraction, thereby compromising assay sensitivity and potentially leading to false-negative results. This document outlines evidence-based protocols and considerations for standardizing the pre-analytical workflow to ensure optimal performance of downstream BEAMing applications in ctDNA analysis.
Blood Collection: The First Critical Step
The choice of blood collection tube and handling protocol immediately after venipuncture is crucial for preserving the quality of ctDNA and preventing the release of wild-type genomic DNA, which dilutes the already scarce ctDNA.
Blood Collection Tubes
Table 1: Comparison of Blood Collection Tubes for ctDNA Analysis
| Tube Type | Anticoagulant/Additive | Maximum Storage Time Before Processing (Room Temperature) | Key Considerations |
|---|---|---|---|
| K₂EDTA | Ethylenediaminetetraacetic acid | ≤ 6 hours [29] [30] | Requires rapid processing; inhibits DNases but does not prevent leukocyte lysis [27]. |
| Cell-Stabilizing Tubes (e.g., Streck cfDNA BCT) | Proprietary stabilizers | Up to 3-14 days [27] [29] | Prevents leukocyte lysis and preserves ctDNA integrity for extended periods, ideal for shipping [29]. |
| Sodium Citrate | Citrate | Up to 72 hours at 4°C [31] | Shows better cfDNA quality preservation compared to EDTA in some studies [31]. |
Recommendation: For most clinical research settings, cell-stabilizing tubes are preferred, especially when immediate sample processing is not feasible. They allow for room temperature storage for up to 3 days without significant gDNA contamination or loss of ctDNA integrity, enabling reliable mutation detection via BEAMing even after delayed processing [29]. If using K₂EDTA tubes, plasma separation must be completed within 4-6 hours of blood draw [27] [30].
Experimental Protocol: Blood Collection and Initial Handling
Objective: To collect whole blood suitable for ctDNA analysis while minimizing pre-analytical variability. Materials:
- Streck Cell-Free DNA BCT tubes (10 mL) or equivalent cell-stabilizing tubes.
- Tourniquet, needle, and blood collection set.
- Timer.
Procedure:
- Perform venipuncture using a standard technique. Minimize tourniquet time to reduce the risk of hemolysis.
- Fill each tube to the recommended 10 mL volume to ensure the correct blood-to-additive ratio [29].
- Immediately after collection, invert the tube 8-10 times gently to ensure complete mixing of the blood with the anticoagulant and stabilizers [30].
- Label the tubes and store them at room temperature (18-30°C) if using cell-stabilizing tubes until processing.
- Note: If K₂EDTA tubes are used, place the collected blood on wet ice and proceed to plasma separation within 4-6 hours [30].
Blood Processing and Plasma Separation
Efficient separation of plasma from cellular components is essential to obtain high-purity cfDNA.
Centrifugation Protocols
A two-step centrifugation process is widely recommended to ensure the removal of cells and cellular debris.
Table 2: Standardized Two-Step Centrifugation Protocol
| Centrifugation Step | Force (Gravity) | Time | Temperature | Purpose |
|---|---|---|---|---|
| First Spin | 1,600 - 2,000 × g | 10 minutes | Room Temperature | To separate plasma from blood cells and platelets [29]. |
| Second Spin | 6,000 - 16,000 × g | 10 minutes | Room Temperature | To remove any remaining cellular debris and platelets, yielding cell-free plasma [27] [29]. |
Recommendation: Following centrifugation, transfer the plasma carefully using a pipette into a fresh tube without disturbing the buffy coat (after the first spin) or the cell pellet (after the second spin). Aliquot the plasma into cryovials to avoid repeated freeze-thaw cycles and store at -80°C if DNA extraction is not performed immediately [27] [29].
Experimental Protocol: Plasma Preparation
Objective: To obtain cell-free plasma from whole blood. Materials:
- Centrifuge with swing-out rotor.
- Sterile pipettes and conical tubes.
- Cryovials for plasma aliquoting.
Procedure:
- First Centrifugation: Centrifuge the blood collection tube at 1,600 × g for 10 minutes at room temperature. Use a smooth braking profile to avoid disturbing the cell pellet [29].
- Plasma Transfer: Carefully transfer the supernatant (plasma) to a new 15 mL conical tube, leaving approximately 0.5 mL of plasma above the buffy coat to avoid cellular contamination.
- Second Centrifugation: Centrifuge the collected plasma at 6,000 × g for 10 minutes at room temperature, again with a smooth brake [29].
- Final Plasma Transfer: Transfer the clarified plasma into a fresh tube, leaving the bottom 0.3 mL to avoid any pellet. Gently mix the plasma by pipetting.
- Aliquoting and Storage: Aliquot the plasma into 2 mL cryotubes and store at -80°C for long-term preservation [29].
ctDNA Extraction Techniques
The extraction method directly influences the yield, fragment size representation, and purity of ctDNA, all of which are critical for the digital PCR principles underlying BEAMing.
Extraction Method Comparison
Table 3: ctDNA Extraction Methods
| Method | Principle | Advantages | Disadvantages |
|---|---|---|---|
| Silica Membrane Spin Columns | DNA binding to a silica membrane in the presence of a chaotropic salt. | High purity; effective for a range of fragment sizes; widely used and reliable [27]. | Potential loss of very short DNA fragments; manual or semi-automated. |
| Magnetic Bead-Based | DNA binding to silica-coated magnetic beads. | Higher recovery of small fragments; amenable to full automation; faster processing times [27] [31]. | Requires specialized equipment. |
| Magnetic Ionic Liquids (MIL) | Dispersive liquid-liquid microextraction using magnetic ionic liquids. | Superior enrichment factors and high recovery efficiency for multiple DNA fragments [27]. | Novel method, not yet widely adopted in clinical labs. |
Recommendation: For BEAMing applications, magnetic bead-based extraction systems are often superior due to their higher efficiency in recovering the short-fragmented ctDNA (typically ~166 bp), which can lead to improved detection sensitivity, especially for low-abundance mutations [31]. Studies have shown that switching from a silica membrane column to a magnetic bead system can overcome deterioration in mutation detection efficiency, even in long-term stored samples [31].
Experimental Protocol: ctDNA Extraction using Magnetic Beads
Objective: To isolate high-quality ctDNA from plasma with high yield and purity. Materials:
- Commercial cfDNA extraction kit based on magnetic beads (e.g., QIAamp Circulating Nucleic Acid Kit with bead-based automation).
- Microcentrifuge, vortex, and magnetic stand.
- Elution buffer (e.g., AVE buffer or 10 mM Tris-HCl).
Procedure:
- Thaw frozen plasma aliquots on ice or at 4°C.
- Follow the manufacturer's instructions for the selected kit. Typically, this involves: a. Digestion: Incubate plasma with a proteinase K and lysis buffer. For plasma from stabilizing tubes, extend the incubation time to 60 minutes at 60°C to ensure complete digestion [29]. b. Binding: Add magnetic beads and binding buffer to the lysate to allow cfDNA to bind to the beads. c. Washing: Place the tube on a magnetic stand to capture the beads. Remove the supernatant and wash the beads with wash buffers once or twice. d. Elution: Elute the pure cfDNA in a small volume (e.g., 40-100 µL) of elution buffer or nuclease-free water.
- Quantify the extracted cfDNA using a fluorescence-based method specific for double-stranded DNA (e.g., Qubit dsDNA HS Assay). Spectrophotometric methods (e.g., Nanodrop) are not recommended due to low sensitivity and inability to detect contamination reliably.
- Store the extracted cfDNA at 4°C for immediate use or at -20°C to -80°C for long-term storage.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Reagents and Materials for Pre-analytical ctDNA Workflow
| Item | Function | Example Product(s) |
|---|---|---|
| Cell-Free DNA Blood Collection Tubes | Prevents leukocyte lysis and preserves ctDNA for up to 3-14 days at RT. | Streck cfDNA BCT tubes [29] |
| Magnetic Bead-based cfDNA Extraction Kit | Efficient isolation of short-fragment ctDNA with high purity and yield. | QIAamp Circulating Nucleic Acid Kit (on automated systems like QIAsymphony) [29] [31] |
| Proteinase K | Enzymatic digestion of proteins during cell lysis in the extraction process. | Included in commercial kits [29] |
| Fluorometric DNA Quantification Assay | Accurate quantification of low-concentration, short-fragment cfDNA. | Qubit dsDNA HS Assay |
| Digital PCR System | Absolute quantification of mutant allele frequency for assay validation. | Droplet Digital PCR (ddPCR) |
Workflow Visualization
The following diagram summarizes the complete optimized pre-analytical workflow for ctDNA analysis:
Emulsion PCR and Microcompartmentalization Techniques
Emulsion PCR (ePCR) is a foundational technique that enables the amplification of individual DNA molecules within microscopic, water-in-oil emulsion droplets [32]. These droplets, typically ranging from 3 to 10 microns in diameter, function as independent micro-reactors, each ideally containing a single template molecule and all necessary reagents for PCR amplification [32] [33]. The core principle of this compartmentalization is to achieve a massive parallelization of biochemical reactions, facilitating the analysis of complex nucleic acid samples with high sensitivity and specificity [34].
Within the context of circulating tumor DNA (ctDNA) analysis, ePCR serves as the technological backbone for BEAMing (Beads, Emulsions, Amplification, and Magnetics), a highly sensitive digital PCR method [5] [33]. BEAMing technology addresses the critical challenge of detecting rare DNA sequences, such as somatic mutations present in ctDNA, against a high background of wild-type DNA [5]. The combination of ePCR with flow cytometry in the BEAMing process allows for the identification and quantification of specific mutations at a sensitivity threshold as low as 0.01% (or 0.001%, depending on the specific assay), making it particularly suited for liquid biopsy applications in oncology [5] [33]. This ultra-sensitive detection is quintessential for precision oncology, enabling real-time monitoring of tumor dynamics, treatment response, and the emergence of resistance mechanisms [2].
Principles of Microcompartmentalization
The Basis of In Vitro Compartmentalization (IVC)
In Vitro Compartmentalization (IVC) leverages water-in-oil emulsions to create cell-like volumes for conducting myriad biochemical reactions in parallel [34]. The power of IVC lies in its ability to miniaturize and parallelize assays. A single milliliter of emulsion can contain over 10^10 discrete compartments, each with a volume as low as one femtolitre [34]. This represents a reduction in reaction volume of between 10^4- and 10^10-fold compared to conventional assays conducted in microtitre plates, drastically reducing reagent consumption and cost while increasing throughput exponentially [34].
A key application of this compartmentalization is establishing a genotype-phenotype linkage, which is fundamental for directed evolution experiments [34]. By confining a single gene and the protein it encodes within a droplet, IVC allows for the selection of proteins based on their function (phenotype), as the gene responsible for that function is physically linked and can be subsequently recovered and amplified [34].
The Workflow of Emulsion PCR
The process of ePCR begins with the creation of a stable water-in-oil emulsion. The aqueous phase contains the template DNA, PCR primers, nucleotides, and a DNA polymerase, while the oil phase contains surfactants that stabilize the emulsion [35]. The two phases are mixed, typically with stirring or vortexing, to generate a vast number of microscopic aqueous droplets suspended in the continuous oil phase [35] [34]. The template DNA is diluted to a degree that, statistically, most droplets contain either zero or one template molecule [32].
Following emulsion preparation, the mixture is subjected to standard thermal cycling. Each droplet functions as an individual PCR microreactor. If a droplet contains a template molecule, that molecule is amplified clonally, resulting in thousands of copies confined within the same droplet [33]. After amplification, the emulsion is broken, and the amplified DNA products are recovered for downstream analysis [33] [35].
BEAMing Technology: A Detailed Protocol for ctDNA Analysis
BEAMing integrates ePCR with magnetic beads and flow cytometry to transform single DNA molecules into analytically quantifiable fluorescent particles [33]. The following protocol is adapted for the detection of somatic mutations, such as RAS mutations in metastatic colorectal cancer (mCRC), from plasma-derived ctDNA [5].
Step-by-Step Experimental Protocol
Step 1: Sample Collection and cfDNA Extraction
- Sample Collection: Collect peripheral blood (e.g., 10-20 mL) in cell-free DNA blood collection tubes (e.g., Streck cfDNA BCT) to prevent cellular lysis and preserve nucleic acid integrity [36]. Process samples within 24 hours of collection.
- Plasma Separation: Perform a two-step centrifugation protocol. First, centrifuge at 1,600 × g for 10 minutes at 4°C to separate plasma. Transfer the supernatant and perform a second centrifugation at 16,000 × g for 10 minutes at 4°C to remove any residual cells or debris [36].
- cfDNA Extraction: Extract cfDNA from 2-4 mL of plasma using a commercial cfDNA extraction kit (e.g., COBAS cfDNA Sample Preparation Kit) according to the manufacturer's instructions [36].
- Quality Control: Quantify the extracted cfDNA using a fluorescence-based assay (e.g., Qubit dsDNA HS Assay). Assess the fragment size distribution using a bioanalyzer system (e.g., Agilent TapeStation) to ensure a peak at ~160-170 bp, characteristic of mononucleosomal cfDNA [36].
Step 2: Pre-Amplification of Target Regions
- Primer Design: Design primers to amplify the genomic regions of interest (e.g., for the OncoBEAM RAS CRC test, this includes hotspots in KRAS and NRAS codons 12, 13, 59, 61, 117, and 146) [5].
- PCR Reaction: Perform a conventional PCR to pre-amplify the target regions from the purified cfDNA. This step increases the number of template molecules for the subsequent BEAMing process.
- Reaction Mix: 1X PCR buffer, 0.2 mM dNTPs, 0.2-0.4 µM of each forward and reverse primer, DNA polymerase, and template cfDNA.
- Cycling Conditions: Initial denaturation at 95°C for 5 min; 35-45 cycles of [94°C for 30 sec, 57-60°C for 30-40 sec, 68-72°C for 30-35 sec]; final extension at 68-72°C for 7 min [35].
Step 3: Emulsion PCR with Primer-Bound Magnetic Beads
- Bead Preparation: Use magnetic beads (e.g., streptavidin-coated beads) that are covalently bound to the 5'-biotinylated PCR primers via streptavidin-biotin interactions [33].
- Emulsion Formation:
- Prepare the aqueous phase containing the pre-amplified DNA templates, primer-bound magnetic beads, PCR reagents (buffer, dNTPs, high-fidelity DNA polymerase), and water.
- Prepare the oil-surfactant phase (e.g., 4.5% Span 80, 0.4% Tween 80, 0.05% Triton X-100, and 95.05% mineral oil) [35].
- Combine the two phases and emulsify by vigorous vortexing or stirring with a magnetic stir bar (e.g., at 1000 rpm for 5 minutes) to create a water-in-oil emulsion with droplets of 3-10 µm in diameter [33] [35].
- Emulsion PCR Amplification:
- Dispense the emulsion into PCR tubes and run the PCR under the following conditions: Initial denaturation at 94-95°C for 5 min; 35-50 cycles of [94°C for 30 sec, 57-60°C for 30 sec, 68-72°C for 30 sec]; final extension at 68-72°C for 7 min [35].
- During amplification, each DNA molecule is amplified clonally onto the surface of a single magnetic bead within its droplet, resulting in beads covered with thousands of copies of the original DNA fragment.
Step 4: Post-PCR Processing and Flow Cytometry
- Recovery and Purification: Break the emulsion by centrifugation or using chemical agents. Recover the magnetic beads bearing the amplified DNA and purify them using a magnet to separate them from the oil and aqueous phases [33].
- Hybridization with Fluorescent Probes: Incubate the beads with allele-specific fluorescent probes to distinguish mutant from wild-type sequences. For example, use one fluorophore-labeled probe that binds specifically to the wild-type DNA and another with a different fluorophore that binds to a specific mutant DNA [33].
- Flow Cytometry Analysis: Analyze the fluorescently labeled beads using a flow cytometer. Beads are categorized as:
- Wild-type: Beads that bind only the wild-type probe.
- Mutant: Beads that bind only the mutant probe.
- Mixed/Undetermined: Beads that bind both or neither probe.
- The ratio of mutant to wild-type beads is used to calculate the mutant allele fraction in the original sample [33].
Research Reagent Solutions
Table 1: Essential Reagents for BEAMing and ePCR Protocols
| Reagent/Material | Function/Description | Example Products/Components |
|---|---|---|
| cfDNA Stabilization Tubes | Prevents white blood cell lysis and preserves cfDNA integrity during blood transport. | Streck cfDNA BCT Tubes [36] |
| Magnetic Beads | Solid support for clonal amplification; provides a surface for primer attachment and subsequent analysis. | Streptavidin-coated magnetic beads [33] |
| Biotinylated Primers | Primers specific to target regions; biotin allows covalent binding to streptavidin-coated beads. | Custom synthesized primers [33] |
| Surfactant/Oil Mixture | Forms the stable water-in-oil emulsion; creates microscopic compartments for single-molecule PCR. | Mineral oil, Span 80, Tween 80, Triton X-100 [35] |
| High-Fidelity DNA Polymerase | Catalyzes DNA amplification with low error rates to minimize false-positive mutations during PCR. | Various commercial polymerases [33] |
| Allele-Specific Fluorescent Probes | Detect and distinguish wild-type from mutant sequences via flow cytometry. | TaqMan probes, Molecular Beacons [33] |
Performance Data and Clinical Validation
BEAMing technology has been rigorously validated in clinical studies, particularly for expanded RAS testing in metastatic colorectal cancer (mCRC). The table below summarizes key performance metrics from validation studies.
Table 2: Analytical and Clinical Performance of BEAMing for ctDNA Analysis
| Parameter | Performance Metric | Context / Notes |
|---|---|---|
| Sensitivity | 0.01% - 0.001% [5] [33] | Can detect 1 mutant molecule in 10,000 to 100,000 wild-type molecules. |
| Concordance with Tissue | 91.8% - 93.3% [5] | Concordance between plasma OncoBEAM RAS testing and standard tissue testing in mCRC. |
| Positive Percent Agreement (PPA) | 92.6% [5] | Plasma detected RAS mutations in 112 of 121 tissue-mutant cases. |
| Negative Percent Agreement (NPA) | 94.0% [5] | No RAS mutations in plasma for 110 of 117 tissue wild-type cases. |
| Mutation Coverage | 34 mutations in KRAS/NRAS [5] | OncoBEAM RAS CRC Kit covers codons 12, 13, 59, 61, 117, 146. |
| Throughput | Analyzes hundreds of millions of DNA molecules [33] | Enabled by compartmentalization in microemulsions. |
Applications in ctDNA Analysis and Precision Oncology
The primary application of BEAMing and related ePCR techniques in oncology is the analysis of ctDNA for liquid biopsy. This minimally invasive approach provides a dynamic snapshot of the tumor's molecular landscape, offering several key clinical applications:
- Treatment Selection: BEAMing is used to identify actionable mutations to guide targeted therapy. For example, detecting RAS mutations in mCRC determines eligibility for anti-EGFR therapy [5]. The high concordance with tissue testing makes it a reliable surrogate when tissue is unavailable or of poor quality.
- Monitoring Treatment Response and Resistance: The high sensitivity of BEAMing allows for the quantification of mutant allele fractions, which can be tracked serially to monitor tumor dynamics [2]. A decrease in ctDNA levels indicates a positive response to therapy, while the emergence or rise of specific mutations can signal acquired resistance [2].
- Detection of Minimal Residual Disease (MRD): Following curative-intent surgery, the presence of ctDNA (MRD) is a strong predictor of relapse [22] [2]. While highly sensitive, BEAMing's role in MRD is still under investigation in clinical trials, as current evidence is not yet sufficient for routine clinical use for this indication outside of trials [36].
Recent studies presented at ASCO 2025 highlight the evolving utility of ctDNA. The SERENA-6 trial demonstrated that switching therapies based on the emergence of ESR1 mutations detected in ctDNA improved progression-free survival and quality of life in patients with advanced breast cancer [22]. However, the DYNAMIC-III trial in stage III colon cancer showed that escalating adjuvant therapy based on a positive ctDNA result did not improve recurrence-free survival, suggesting that the effectiveness of ctDNA-guided strategies may depend on the context and availability of potent escalation therapies [22].
Troubleshooting and Technical Considerations
- Emulsion Stability: Unstable emulsions can lead to droplet coalescence, resulting in template mixing and loss of monoclonality. This can be mitigated by optimizing surfactant composition and concentration and ensuring thorough emulsification [35] [34].
- Template Concentration: Accurate dilution of the template DNA is critical. Overloading the emulsion will result in multiple templates per droplet, compromising the digital quantification. The template concentration must be carefully titrated to ensure a high proportion of droplets contain either zero or one molecule [32] [33].
- False Positives: PCR errors introduced during amplification can be misidentified as low-frequency mutations. Using a high-fidelity DNA polymerase is essential to limit this risk [33].
- Pre-analytical Variables: The quality of ctDNA analysis is highly dependent on sample collection and processing. Using dedicated cfDNA blood collection tubes, processing plasma within a strict timeframe, and rigorous quality control of extracted cfDNA are paramount for reliable results [36].
Flow Cytometry Analysis and Mutation Quantification
BEAMing (Beads, Emulsion, Amplification, and Magnetics) is a highly sensitive digital PCR-based technology designed for the precise detection and quantification of rare mutant alleles in circulating tumor DNA (ctDNA) [1]. ctDNA consists of short, double-stranded DNA fragments shed into the bloodstream by tumor cells through processes such as apoptosis and necrosis [37]. These fragments carry tumor-specific genetic alterations and typically constitute a very small fraction (often less than 0.1%) of the total cell-free DNA (cfDNA) in circulation, especially in early-stage cancer or minimal residual disease (MRD) [37] [38]. BEAMing technology enables the detection of these rare mutations with high specificity and sensitivity, making it a powerful tool for liquid biopsy in cancer research and drug development [1].
Experimental Protocol: BEAMing for ctDNA Mutation Quantification
Sample Collection and Plasma Processing
Proper sample collection and handling are critical for preserving the integrity of ctDNA, which is present at low concentrations and highly fragmented [38].
- Blood Collection: Draw blood using a 21-gauge butterfly needle to minimize hemolysis. Collect a minimum of 10 mL of blood per tube into Streck cfDNA BCT or similar cell-stabilizing blood collection tubes, which allow sample storage at room temperature for up to 7 days [38]. Avoid prolonged tourniquet use.
- Plasma Separation: Centrifuge tubes within 2-6 hours of collection if using EDTA tubes, or within the stabilizer's validated period for BCTs. Perform two-step centrifugation:
- First step: 380–3,000 × g for 10 minutes at room temperature to separate plasma from blood cells.
- Second step: Transfer the supernatant to a new tube and centrifuge at 12,000–20,000 × g for 10 minutes at 4°C to remove any remaining cellular debris [38].
- Plasma Storage: Aliquot the clarified plasma into small fractions and store at –80°C. Avoid freeze-thaw cycles to prevent DNA degradation [38].
cfDNA Extraction
Extract cfDNA from 1-5 mL of plasma using silica membrane-based kits, such as the QIAamp Circulating Nucleic Acid Kit, which generally provides higher yields than magnetic bead-based methods [38]. Elute DNA in a low-EDTA TE buffer or nuclease-free water and quantify using a fluorescence-based method suitable for low-concentration DNA.
BEAMing Assay Workflow
The core BEAMing process involves several steps to partition and amplify individual DNA molecules for digital quantification [1].
- Primer Design and Probe Selection: Design PCR primers and hydrolysis (TaqMan) probes to amplify a short amplicon (typically 50-100 bp) spanning the mutation of interest. The mutant and wild-type probes must be differentially labeled with fluorophores (e.g., FAM for mutant, VIC for wild-type).
- Water-in-Oil Emulsion PCR:
- Prepare the PCR Mix: Combine purified cfDNA with primers, probes, DNA polymerase, and a population of magnetic beads whose surfaces are coated with oligonucleotides complementary to the primer sequences.
- Create the Emulsion: Vigorously mix the aqueous PCR reaction with oil and surfactants to generate a stable water-in-oil emulsion. This creates millions of microscopic aqueous compartments, each ideally containing a single DNA template and a single bead [1].
- Amplify the DNA: Perform PCR amplification. Within each compartment, the DNA template is amplified and clonally attached to the surface of the single bead present.
- Emulsion Breakage and Bead Recovery: After amplification, break the emulsion using a destabilizing solvent. Recover the beads by centrifugation and wash them thoroughly to remove oil and excess reagents.
- Flow Cytometry Hybridization and Analysis:
- Hybridize with Probes: Incubate the beads with fluorescently labeled probes specific to the wild-type and mutant sequences. This is a critical hybridization step that occurs after amplification.
- Analyze by Flow Cytometry: Resuspend the beads and analyze them on a flow cytometer. The beads pass through the laser interrogation point individually. Beads that carried a wild-type template will hybridize only to the wild-type probe, beads that carried a mutant template will hybridize only to the mutant probe, and beads that carried no template will not fluoresce.
Table 1: Key Reagents and Materials for BEAMing Assay
| Item | Function | Specification/Notes |
|---|---|---|
| Streck cfDNA BCT | Blood collection | Preserves cfDNA, prevents gDNA release from white blood cells [38]. |
| QIAamp Circulating Nucleic Acid Kit | cfDNA extraction | Silica-membrane technology for high yield [38]. |
| Magnetic Beads | Template immobilization | 1-5 µm diameter, covalently linked to oligonucleotide primers. |
| TaqMan Probes | Mutation detection | FAM-labeled mutant probe, VIC-labeled wild-type probe. |
| Emulsion Oil & Surfactants | Compartmentalization | Forms stable water-in-oil microreactors for digital PCR [1]. |
| Hot-Start DNA Polymerase | DNA Amplification | Reduces non-specific amplification during emulsion setup. |
Data Analysis and Mutation Quantification
The analysis of flow cytometry data from a BEAMing assay involves a clear gating strategy to identify and count the different bead populations [39].
- Create a Bead Gate: On a Forward Scatter (FSC) vs. Side Scatter (SSC) dot plot, draw a gate around the bead population to exclude any debris or aggregates [39].
- Create a Single-Bead Gate: Plot FSC-Area vs FSC-Height to gate on single beads and exclude doublets.
- Analyze Fluorescence: From the single-bead gate, create a two-parameter dot plot of Fluorescence 1 (FAM, mutant probe) vs. Fluorescence 2 (VIC, wild-type probe). Four distinct bead populations should be visible:
- FAM-positive/VIC-negative: Beads carrying the mutant sequence.
- VIC-positive/FAM-negative: Beads carrying the wild-type sequence.
- Double-negative: Beads that did not carry a DNA template (PCR-negative).
- Double-positive: Beads that carried both sequences (rare, can indicate mixed templates or errors).
- Calculate Mutant Allele Frequency (MAF): Count the number of beads in the mutant and wild-type populations. Calculate the MAF using the formula: MAF (%) = [Number of Mutant Beads / (Number of Mutant Beads + Number of Wild-type Beads)] × 100
BEAMing Flow Cytometry Gating Strategy
Table 2: Flow Cytometry Configuration for BEAMing Analysis
| Parameter | Setting | Purpose |
|---|---|---|
| Detector | FSC | Triggering; detect particles based on size [39]. |
| Laser | 488 nm blue | Excitation of FITC (FAM) and PE (VIC). |
| Fluorescence Detector 1 | FITC (530/30 nm) | Detect FAM signal from mutant probes. |
| Fluorescence Detector 2 | PE (575/26 nm) | Detect VIC signal from wild-type probes. |
| Threshold | FSC | Ignore sub-bead-size debris. |
| Events to Record | ≥ 100,000 | Ensure sufficient beads for statistical power. |
Troubleshooting and Technical Considerations
- Low Bead Recovery After Emulsion Breakage: Ensure the emulsion is stable during PCR by optimizing the oil-to-aqueous phase ratio and vortexing thoroughly. Use fresh surfactants.
- High Background Fluorescence: Titrate the hybridization probe concentration to find the optimal signal-to-noise ratio. Increase the stringency of post-hybridization washes.
- Unexpected Double-Positive Beads: This can be due to non-specific probe hybridization, cross-talk between fluorescence channels, or the presence of beads carrying both sequences (e.g., from droplet coalescence). Check compensation on the flow cytometer and ensure emulsion stability.
- Poor Assay Sensitivity: The analytical sensitivity of BEAMing is exceptionally high, capable of detecting mutant alleles at frequencies as low as 0.001% to 0.1% [37] [38]. If sensitivity is inadequate, ensure high-quality cfDNA input, optimize PCR efficiency, and analyze a larger number of beads to survey a greater number of genome equivalents.
In the era of precision oncology, the analysis of circulating tumor DNA (ctDNA) via liquid biopsy has emerged as a pivotal tool for guiding therapeutic decisions in metastatic colorectal cancer (mCRC). RAS mutation status (encompassing KRAS and NRAS) serves as a critical predictive biomarker for resistance to anti-epidermal growth factor receptor (EGFR) monoclonal antibodies, such as cetuximab and panitumumab [5] [40]. Testing for these mutations is therefore essential to ensure that these targeted therapies are only administered to patients with RAS wild-type tumors, who are likely to benefit [40].
BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) represents a highly sensitive digital PCR-based methodology for detecting and quantifying rare mutant DNA sequences, such as RAS mutations, from a patient's plasma [5]. This application note details the implementation of BEAMing-based RAS mutation testing within a clinical research setting, providing a standardized protocol and contextualizing its performance against other methodologies.
Clinical and Technical Background
RAS Mutations as a Predictive Biomarker
Comprehensive RAS analysis extends beyond the traditional KRAS exon 2 to include mutations in KRAS exons 3 and 4, and NRAS exons 2, 3, and 4. Approximately 55.9% of mCRC patients harbor a mutation in one of these RAS genes, rendering them unsuitable for anti-EGFR therapy [40]. The clinical imperative for accurate testing is underscored by data showing that patients with RAS wild-type tumors experience a statistically significant improvement in overall survival when treated with EGFR inhibitors combined with chemotherapy, compared to chemotherapy alone (26.0 versus 20.2 months) [40].
BEAMing Technology Fundamentals
BEAMing is a sophisticated technique that transforms individual DNA molecules into magnetic beads for analysis. The process involves compartmentalizing single DNA molecules from a plasma-derived ctDNA sample into water-in-oil microemulsions, along with PCR reagents. Within each microreactor, a clonal amplification reaction occurs, generating thousands of copies of the original molecule bound to a single bead. Subsequently, these beads are analyzed via flow cytometry using allele-specific fluorescent probes to differentiate and quantify mutant and wild-type DNA molecules. This process allows BEAMing to detect mutant alleles with a high sensitivity, at a level as low as 0.01% mutant allele frequency [5].
Performance Characteristics and Validation
The OncoBEAM RAS CRC assay is designed to detect 34 mutations across codons 12, 13, 59, 61, 117, and 146 of the KRAS and NRAS genes [5]. Multiple studies have validated its performance against standard tissue-based testing.
Table 1: Concordance Studies between BEAMing and Tissue-Based RAS Testing
| Study / Cohort | Sample Size (n) | Concordance Rate | Sensitivity | Specificity |
|---|---|---|---|---|
| Multi-Center European Evaluation [5] | 238 | 93.3% | 92.6% | 94.0% |
| ColoBEAM Study (2025) [41] | 202 | 83.2% | 77.3% | 94.3% |
| ColoBEAM (Chemotherapy-naive pts) [41] | Subgroup | - | 86.1% | 91.3% |
| ColoBEAM (Liver mets) [41] | Subgroup | - | 88.6% | 89.7% |
Table 2: Comparison of ctDNA Detection Methodologies
| Method of Detection | Example Techniques | Detection Limit (% ctDNA) | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Digital PCR | BEAMing, Droplet Digital PCR (ddPCR) | ~0.01% | Very high sensitivity; rapid turnaround; cost-effective for limited mutations. | Limited to a small number of pre-defined mutations. |
| Targeted Deep Sequencing (NGS) | SafeSeqS, TAm-Seq, CAPP-Seq | ~0.01–2.0% | Broader coverage of genes/mutations; high sensitivity. | Can require complex bioinformatics; longer turnaround time. |
| Whole Genome Sequencing (NGS) | Digital Karyotyping, PARE | ~1% | Hypothesis-free; captures all variant types. | Lower sensitivity; higher cost; computationally intensive. |
| Whole Exome Sequencing (NGS) | Various Panels | ~5% | Focus on protein-coding regions. | Lower sensitivity; not ideal for liquid biopsy. |
Experimental Protocol: OncoBEAM RAS Testing
Pre-Analytical Phase: Sample Collection and Processing
Materials:
- K2EDTA or Streck Cell-Free DNA blood collection tubes.
- Refrigerated centrifuge.
- DNA Micro Kit (Qiagen) or equivalent.
Workflow:
- Blood Collection: Draw a minimum of 10 ml of whole blood into K2EDTA tubes. Invert gently 8-10 times to mix.
- Plasma Separation: Process samples within one hour of collection.
- Centrifuge at 820 × g for 10 minutes at 4°C to separate plasma from cellular components.
- Transfer the supernatant (plasma) to a fresh microcentrifuge tube without disturbing the buffy coat.
- Perform a second, higher-speed centrifugation at 16,000 × g for 10 minutes at 4°C to pellet any remaining cells and debris.
- Plasma Storage: Aliquot the clarified plasma and store at -80°C until DNA extraction.
- ctDNA Extraction: Isolate ctDNA from 1-4 ml of plasma using the DNA Micro Kit, following the manufacturer's instructions. Elute in a small volume (e.g., 20-50 µl) of provided elution buffer.
- DNA Quantification: Measure the concentration of double-stranded DNA using a fluorescence-based assay (e.g., Qubit dsDNA HS Assay). Spectrophotometric methods are not recommended due to low concentration and potential for contaminant interference.
Analytical Phase: BEAMing PCR and Mutation Detection
Research Reagent Solutions:
Table 3: Essential Reagents for BEAMing RAS Testing
| Reagent / Material | Function | Example / Specification |
|---|---|---|
| Plasma cfDNA | Analytical substrate | Isolated from patient plasma, volume ≥1mL equivalent |
| HotStart High-Fidelity DNA Polymerase | Initial target amplification | Phusion HotStart (NEB) |
| Magnetic Streptavidin Beads | Solid-phase PCR support | MyOne Streptavidin C1 Beads (Invitrogen) |
| Emulsification Oil/Detergent Mix | Microreactor formation | ABIL WE09, Mineral Oil, TegoSoft DEC |
| Biotinylated Capture Oligonucleotides | Bead functionalization | 5'-dual biotin-T-Spacer18-tcccgcgaaattaatacgac-3' |
| Allele-Specific Fluorescent Probes | Mutation detection & quantification | FAM- and HEX-labeled probes for WT/mutant RAS |
| Flow Cytometer | Bead population analysis | Capable of detecting FITC/HEX channels |
Workflow:
- Initial PCR Amplification:
- Perform initial PCR in eight separate 25 µl reactions to amplify the RAS gene regions of interest from the extracted ctDNA.
- Use HotStart high-fidelity DNA polymerase and primers designed for the specific RAS codons.
- Cycling Conditions: 98°C for 30s; 35 cycles of (98°C for 10s, 57°C for 10s, 72°C for 10s); final extension at 72°C.
Emulsion PCR (BEAMing):
- Pool the initial PCR products and use a small amount (e.g., 18 pg) as template for the emulsion PCR.
- Prepare a PCR mix containing the template, Platinum Taq DNA polymerase, dNTPs, MgCl₂, primers, and ~6×10^7 magnetic streptavidin beads coated with a capture oligonucleotide.
- Create a water-in-oil emulsion by vigorously shaking the PCR mix with an oil/emulsifier mixture. This generates millions of microreactors, each ideally containing a single bead and a single DNA molecule.
- Perform emulsion PCR with a specialized cycling program to amplify the DNA molecules onto the beads within their individual compartments.
Bead Recovery and Hybridization:
- Break the emulsions using a buffer containing Triton-X-100 and SDS.
- Recover the beads by centrifugation and wash thoroughly.
- Denature the DNA on the beads with NaOH to create single-stranded templates.
- Hybridize the beads with a mix of fluorescently labeled probes that are complementary to either the wild-type or specific mutant sequences.
Flow Cytometry and Analysis:
- Analyze the beads using a flow cytometer.
- Beads that hybridize with a wild-type probe will fluoresce in one channel (e.g., HEX), while beads that hybridize with a mutant probe will fluoresce in another (e.g., FAM). Beads that do not hybridize or that hybridize with both are excluded.
- The result is calculated as the ratio of mutant beads to the total number of wild-type plus mutant beads, providing a quantitative measure of the mutant allele fraction in the original sample.
RAS Signaling Pathway and Therapeutic Implications
The RAS family of proteins (including KRAS, NRAS, and HRAS) are small GTPases that act as critical molecular switches in intracellular signaling networks. In normal physiology, they cycle between an active GTP-bound state and an inactive GDP-bound state, tightly regulated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). Oncogenic mutations in RAS (commonly at codons 12, 13, and 61) lock the protein in its active GTP-bound state, leading to constitutive, ligand-independent signaling [42].
This persistent activation drives oncogenesis through multiple downstream effector pathways. The primary pathway is the RAF-MEK-ERK (MAPK) cascade, which promotes cell proliferation and survival. Additionally, activated RAS engages the PI3K-AKT-mTOR pathway, further enhancing cell survival and growth. In mCRC, this constitutive signaling downstream of the EGFR renders anti-EGFR monoclonal antibodies therapeutically ineffective [40]. Therefore, identifying these mutations is essential for excluding patients from anti-EGFR therapy.
BEAMing technology provides a highly sensitive, specific, and quantitative method for detecting RAS mutations in the ctDNA of patients with mCRC. Its high concordance with standard tissue testing and ability to provide rapid turnaround make it a robust tool for clinical research and a valuable adjunct to tissue-based genotyping. The implementation of this protocol enables researchers to accurately identify patients who are unlikely to benefit from anti-EGFR therapy, thereby optimizing patient selection for targeted treatments and advancing personalized medicine in colorectal cancer.
BEAMing (Beads, Emulsion, Amplification, and Magnetics) represents a highly sensitive digital PCR-based methodology for detecting and quantifying circulating tumor DNA (ctDNA) in liquid biopsies. This technology enables the identification of specific EGFR mutations in non-small cell lung cancer (NSCLC) patients with exceptional sensitivity, capable of detecting mutant alleles at frequencies as low as 0.01% against a background of wild-type DNA [43]. The clinical significance of EGFR mutation detection lies in its critical role in guiding targeted therapy decisions, with mutations in exons 18-21 predicting response to tyrosine kinase inhibitors (TKIs) such as gefitinib, erlotinib, afatinib, and osimertinib [44].
The technological foundation of BEAMing integrates emulsion PCR with flow cytometry to achieve single-molecule resolution. This approach allows for the absolute quantification of mutant EGFR alleles without the need for standard curves, providing superior sensitivity compared to conventional sequencing methods or real-time PCR [3]. When applied to EGFR mutation detection in NSCLC, BEAMing demonstrates remarkable concordance with tissue-based testing, with reported agreement rates of 90-100% across different exons when compared with standard qPCR methods [3].
BEAMing addresses a critical need in precision oncology by enabling non-invasive genotyping and serial monitoring of tumor genomics throughout treatment. This capability is particularly valuable in advanced NSCLC, where tumor heterogeneity and evolving resistance mechanisms necessitate repeated genomic assessment [1]. The technology's performance characteristics make it suitable for multiple clinical applications, including initial therapy selection, monitoring of treatment response, and early detection of resistance mutations such as T790M [3].
Performance Characteristics and Validation Data
Analytical Sensitivity and Specificity
BEAMing technology demonstrates exceptional performance characteristics for EGFR mutation detection in ctDNA. Validation studies conducted across multiple institutions have established the methodology's reliability through direct comparison with established tissue-based testing methods [3].
Table 1: BEAMing Performance Metrics for EGFR Mutation Detection
| Parameter | Exon 19 | Exon 20 | Exon 21 (L858R) | Exon 21 (L861Q) |
|---|---|---|---|---|
| Concordance with EMR-qPCR (%) | 98.8 | 98.9 | 95.5 | - |
| Concordance with Diatech qPCR (%) | 90.0 | 100 | 96.0 | 98.0 |
| Sensitivity | >90% | >95% | >90% | >95% |
| Specificity | >95% | >95% | >95% | >95% |
| Mutation Frequency Detection Limit | 0.01% | 0.01% | 0.01% | 0.01% |
The analytical sensitivity of BEAMing was rigorously validated through controlled experiments using DNA from mutant cell lines (H1975 and PC9) diluted in wild-type DNA (A549 background). These studies confirmed the technology's ability to reliably detect mutant alleles at frequencies as low as 0.01%, which is significantly lower than the detection limits of conventional sequencing methods (typically 5-10%) and comparable to other digital PCR platforms [3].
Comparison with Alternative Detection Platforms
BEAMing occupies a distinct position in the ecosystem of ctDNA detection technologies, balancing high sensitivity with the ability to interrogate multiple mutations simultaneously.
Table 2: Comparison of EGFR Mutation Detection Platforms
| Technology | Sensitivity | Multiplexing Capability | Turnaround Time | Key Applications |
|---|---|---|---|---|
| BEAMing | 0.01% | Moderate | 2-3 days | Treatment monitoring, resistance detection |
| ddPCR | 0.01-0.1% | Low | 1-2 days | Targeted mutation tracking |
| NGS (CAPP-Seq) | 0.1% | High | 5-7 days | Comprehensive profiling, novel mutation discovery |
| ARMS-PCR | 1-5% | Low | 1-2 days | Initial therapy selection |
| Sanger Sequencing | 10-20% | Low | 3-5 days | Historical standard |
When compared to next-generation sequencing (NGS) approaches, BEAMing offers superior sensitivity for detecting known mutations but lacks the ability to identify novel or unexpected variants without prior knowledge [4]. This characteristic makes BEAMing particularly well-suited for monitoring known EGFR mutations during treatment, where the genetic targets are well-defined and high sensitivity is required to detect emerging resistance mutations early [1] [3].
Detailed BEAMing Protocol for EGFR Mutation Detection
Pre-analytical Sample Processing
Proper sample collection and processing are critical for successful EGFR mutation detection using BEAMing technology. Adherence to standardized protocols minimizes pre-analytical variables that could compromise assay sensitivity [38] [43].
Blood Collection and Plasma Separation:
- Collect 10 mL of peripheral blood into EDTA-containing tubes or specialized cell-free DNA blood collection tubes (e.g., Streck, PAXgene) [38].
- Process samples within 2-6 hours of collection when using EDTA tubes. Tubes with preservatives allow storage for up to 7 days at room temperature [38].
- Centrifuge samples at 820 × g for 10 minutes at room temperature to separate plasma from cellular components.
- Transfer the supernatant to a fresh tube and perform a second centrifugation at 16,000 × g for 10 minutes to remove remaining cellular debris [3].
- Aliquot cleared plasma and store at -80°C if not processing immediately. Avoid more than three freeze-thaw cycles [38].
ctDNA Extraction:
- Extract ctDNA from 1 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) or equivalent [3].
- Elute DNA in a volume of 20-50 μL of TE buffer or nuclease-free water.
- Quantify DNA concentration using a spectrophotometer (e.g., Nanodrop ND1000). Typical yields range from 5-50 ng/mL of plasma, depending on tumor burden [3].
BEAMing PCR Workflow
The core BEAMing methodology involves several sequential steps that transform individual DNA molecules into bead-bound amplicons for subsequent analysis.
Initial PCR Amplification:
- Set up eight separate 25 μL PCR reactions, each containing:
- Template DNA from 250 μL of plasma
- 1× Phusion High-Fidelity PCR Buffer
- 1.5 U HotStart Phusion Polymerase
- 0.2 μM of each primer (EGFR exon-specific)
- 0.25 mM each dNTP
- 0.5 mM MgCl₂
- Cycling conditions:
- 98°C for 30 seconds
- 35 cycles of: 98°C for 10 seconds, 57°C for 10 seconds, 72°C for 10 seconds
- Final extension: 72°C for 5 minutes [3]
- Pool PCR products from all eight reactions and quantify using spectrophotometry.
Emulsion PCR and Mutation Detection:
- Prepare 150 μL PCR mixture containing:
- 18 pg of template DNA from the initial PCR
- 40 U Platinum Taq DNA Polymerase
- 1× PCR Buffer
- 0.2 mM dNTPs
- 5 mM MgCl₂
- 0.05 μM Tag1 primer
- 8 μM Tag2 primer
- ~6×10⁷ magnetic streptavidin beads coated with Tag1 oligonucleotide
- Create a water-in-oil emulsion by adding 600 μL of oil/emulsifier mixture (7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC) and shaking in a TissueLyser for 10 seconds at 15 Hz, then 7 seconds at 17 Hz [3].
- Perform emulsion PCR with the following conditions:
- 94°C for 2 minutes
- 3 cycles of: 94°C for 10 seconds, 68°C for 45 seconds, 70°C for 75 seconds
- 3 cycles of: 94°C for 10 seconds, 65°C for 45 seconds, 70°C for 75 seconds
- 3 cycles of: 94°C for 10 seconds, 62°C for 45 seconds, 70°C for 75 seconds
- 50 cycles of: 94°C for 10 seconds, 57°C for 45 seconds, 70°C for 75 seconds
- Break emulsions by adding 150 μL of breaking buffer (10 mM Tris-HCl pH 7.5, 1% Triton-X-100, 1% SDS, 100 mM NaCl, 1 mM EDTA) and mixing at 20 Hz for 20 seconds.
- Recover beads by centrifugation at 3,200 × g for 2 minutes and remove the oil phase.
- Denature DNA on beads with 0.1 M NaOH for 5 minutes.
- Perform allele-specific hybridization with fluorescently labeled probes complementary to mutant and wild-type sequences (15-18 nt in length) [3].
- Analyze beads using flow cytometry (e.g., FACSArray III) to quantify mutant and wild-type populations.
EGFR Signaling Pathway and Mutation Impact
Research Reagent Solutions and Materials
Successful implementation of BEAMing for EGFR mutation detection requires specific reagents and instrumentation optimized for this methodology.
Table 3: Essential Research Reagents for BEAMing EGFR Detection
| Reagent/Instrument | Specification | Function in Protocol | Commercial Examples |
|---|---|---|---|
| Blood Collection Tubes | Cell-free DNA preservative tubes | Maintains sample integrity during transport | Streck cfDNA BCT, PAXgene Blood ccfDNA |
| DNA Extraction Kit | Silica membrane-based extraction | Isolves high-quality ctDNA from plasma | QIAamp Circulating Nucleic Acid Kit (Qiagen) |
| High-Fidelity Polymerase | Hot-start, high-fidelity enzyme | Initial target amplification with minimal errors | Phusion HotStart Flex (NEB) |
| Magnetic Beads | Streptavidin-coated, 1μm diameter | Solid support for emulsion PCR | Dynabeads MyOne Streptavidin C1 |
| Emulsion Reagents | Oil-surfactant mixture | Creates stable water-in-oil microreactors | ABIL WE09, TegoSoft DEC, Mineral oil |
| Detection Probes | Fluorescently labeled oligonucleotides | Mutant vs. wild-type allele discrimination | FAM-/HEX-labeled probes (15-18 nt) |
| Flow Cytometer | 2-laser capability | Quantifies mutant and wild-type bead populations | FACSArray III, MACSQuant Analyzer |
Implementation Considerations and Troubleshooting
Quality Control Measures
Implementation of BEAMing technology for EGFR mutation detection requires rigorous quality control procedures to ensure reliable results:
- Sample Adequacy Controls: Include extraction blanks to monitor contamination and positive controls using DNA from mutant cell lines (e.g., PC9 for exon 19 deletions, H1975 for L858R) diluted in wild-type DNA (A549) [3].
- Assay Performance Monitoring: Establish threshold values for minimum bead count (typically >100,000 beads per sample) and minimum input DNA quantity.
- Interpretation Criteria: Define threshold for mutant allele calling based on background signal in negative controls, typically >3 standard deviations above background.
Troubleshooting Common Issues
- Low Bead Recovery: Optimize emulsion stability by ensuring proper oil:aqueous phase ratio and vigorous mixing. Verify bead coating efficiency.
- High Background Signal: Increase hybridization stringency by optimizing temperature and salt concentration. Verify probe specificity and purity.
- Poor Sensitivity: Ensure rapid sample processing to prevent DNA degradation. Verify plasma is completely free of cellular contamination through rigorous centrifugation.
- Inconsistent Results Between Replicates: Standardize emulsion formation using mechanical homogenization rather than vortexing. Ensure consistent bead counting during flow cytometry analysis.
BEAMing technology provides a robust, highly sensitive platform for detecting EGFR mutations in ctDNA from NSCLC patients. The methodology offers significant advantages for monitoring treatment response and detecting resistance mutations during targeted therapy. With concordance rates exceeding 90% compared to tissue-based testing and sensitivity for mutant allele detection down to 0.01%, BEAMing represents a powerful tool for liquid biopsy applications in precision oncology [3].
Future developments in BEAMing technology are likely to focus on increasing multiplexing capabilities to simultaneously detect a broader spectrum of EGFR mutations and resistance mechanisms. Integration with other analytical approaches, such as fragmentomic analysis or methylation profiling, may further enhance the clinical utility of BEAMing for comprehensive tumor genotyping [4]. As standardization improves and regulatory frameworks evolve, BEAMing and related digital PCR technologies are positioned to become integral components of personalized treatment pathways for NSCLC patients, enabling more dynamic and minimally invasive assessment of tumor genomics throughout the disease course.
Monitoring Treatment Response and Resistance Mechanisms
BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) represents a highly sensitive and quantitative approach for analyzing circulating tumor DNA (ctDNA), enabling researchers and clinicians to monitor tumor dynamics and treatment response with high precision [19] [3]. This technique combines emulsion PCR with flow cytometry to detect and quantify specific somatic mutations present in ctDNA, which constitutes only a small fraction (sometimes less than 0.01%) of total cell-free DNA in circulation [19]. In the context of precision oncology, BEAMing facilitates real-time, non-invasive assessment of tumor burden, minimal residual disease (MRD), and emerging resistance mutations during therapy [2] [14]. This application note provides detailed protocols and analytical frameworks for implementing BEAMing-based ctDNA analysis in cancer research and drug development, with a specific focus on monitoring treatment response and resistance mechanisms.
BEAMing technology enables the digital detection of rare mutant DNA fragments against a background of wild-type DNA through a multi-step process that achieves exceptional sensitivity and specificity [3]. The fundamental principle involves converting single DNA molecules into magnetic beads coated with thousands of copies of the original template, creating a visually detectable and quantifiable signal via flow cytometry [19]. This approach allows for precise quantification of mutant allele frequencies, with demonstrated clinical utility in tracking tumor dynamics during therapy [19] [3].
The exceptional sensitivity of BEAMing (capable of detecting mutant alleles at frequencies as low as 0.01%) stems from its unique workflow: individual DNA molecules are separated into water-in-oil emulsion droplets along with magnetic beads, facilitating millions of parallel PCR reactions in isolated compartments [3]. This compartmentalization prevents cross-contamination and enables clonal amplification of single DNA fragments. Subsequent hybridization with mutation-specific fluorescent probes allows for precise enumeration of mutant and wild-type alleles using flow cytometry [19] [3].
Table 1: Key Analytical Performance Characteristics of BEAMing Technology
| Performance Parameter | Specification | Experimental Demonstration |
|---|---|---|
| Detection Sensitivity | ≤0.01% mutant allele frequency | Detection of EGFR mutations in NSCLC patient plasma [3] |
| Dynamic Range | 0.01% to 100% mutant allele frequency | Quantification of APC and KRAS mutations in colorectal cancer [19] |
| Sample Input | DNA from 250-500 μL plasma per reaction | BEAMing protocol for EGFR mutation detection [3] |
| Concordance with Tissue | 90-100% for various mutations | 90% for exon 19, 100% for exon 20, 96% for L858R in EGFR [3] |
| Half-life Measurement | Capable of measuring ctDNA kinetics | Estimated half-life of 114 minutes post-surgical resection [19] |
BEAMing Workflow and Experimental Protocol
Sample Collection and Processing
Proper sample collection and processing are critical for obtaining reliable BEAMing results. The following protocol has been optimized for plasma collection and cell-free DNA isolation:
Blood Collection: Collect peripheral blood (10-20 mL) in EDTA-containing tubes or specialized cell-free DNA BCT tubes (Streck) to preserve sample integrity [45]. Process samples within 1-2 hours of collection to prevent leukocyte lysis and contamination with genomic DNA.
Plasma Separation: Perform two-step centrifugation:
Cell-free DNA Isolation: Extract cfDNA from 1-4 mL plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) according to manufacturer's instructions with modified proteinase K incubation (extend to 1 hour) [45]. Elute DNA in 70-140 μL of AVE buffer.
DNA Quantification: Measure DNA concentration using fluorometric methods (e.g., Qubit) or spectrophotometry (Nanodrop ND1000) [3]. Typical yields range from 5-100 ng/mL plasma, depending on tumor burden and disease stage.
BEAMing Assay Procedure
The core BEAMing protocol involves multiple steps that require meticulous optimization:
Initial Amplification: Perform high-fidelity PCR to amplify target regions containing mutations of interest:
- Reaction setup: 25 μL reactions containing template DNA (from 250 μL plasma), 5× Phusion High-Fidelity PCR Buffer, 1.5 U HotStart Phusion polymerase, 0.2 μM of each primer, 0.25 mM dNTPs, and 0.5 mM MgCl₂ [3].
- Cycling conditions: 98°C for 30s; 35 cycles of (98°C for 10s, primer-specific annealing temperature for 10s, 72°C for 10s); final extension at 72°C for 5-10 minutes [3].
- Pool and quantify amplification products using spectrophotometry.
Emulsion PCR Setup: Prepare water-in-oil emulsion to compartmentalize individual DNA molecules and beads:
- PCR mixture: 150 μL containing 18 pg template DNA, 40 U Platinum Taq DNA polymerase, 1× PCR buffer, 0.2 mM dNTPs, 5 mM MgCl₂, 0.05 μM Tag1 primer, 8 μM Tag2 primer, and ~6×10⁷ magnetic streptavidin beads coated with Tag1 oligonucleotide [3].
- Create emulsion by adding 600 μL oil/emulsifier mixture (7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC) and shaking in a TissueLyser for 10s at 15 Hz followed by 7s at 17 Hz [3].
- Verify emulsion quality microscopically (40× magnification) to ensure proper bead distribution.
Emulsion PCR Amplification:
- Thermal cycling: 94°C for 2 minutes; 3 cycles of (94°C for 10s, 68°C for 45s, 70°C for 75s); 3 cycles of (94°C for 10s, 65°C for 45s, 70°C for 75s); 3 cycles of (94°C for 10s, 62°C for 45s, 70°C for 75s); 50 cycles of (94°C for 10s, 57°C for 45s, 70°C for 75s) [3].
Emulsion Breaking and Bead Recovery:
- Add 150 μL breaking buffer (10 mM Tris-HCl pH 7.5, 1% Triton-X-100, 1% SDS, 100 mM NaCl, 1 mM EDTA) to each well and mix with TissueLyser at 20 Hz for 20s [3].
- Centrifuge at 3,200 × g for 2 minutes and remove oil phase.
- Repeat breaking step twice, then wash beads with 150 μL wash buffer (20 mM Tris-HCl pH 8.4, 50 mM KCl).
- Denature DNA with 0.1 M NaOH for 5 minutes, then wash and resuspend in wash buffer.
Mutation Detection by Hybridization:
- Hybridize beads with fluorescently labeled probes complementary to mutant (e.g., FAM-labeled) and wild-type (e.g., PE-labeled) sequences (15-18 nt length) [3].
- Analyze beads using flow cytometry (e.g., FACSArray or CyFlow Cube) to enumerate mutant and wild-type populations.
- Calculate mutant allele frequency as (mutant beads / [mutant + wild-type beads]) × 100%.
Research Reagent Solutions
Table 2: Essential Research Reagents for BEAMing Assays
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Blood Collection Tubes | Cell-free DNA BCT tubes (Streck) | Preserves blood sample integrity during transport and storage [45] |
| Nucleic Acid Isolation | QIAamp Circulating Nucleic Acid Kit (Qiagen) | Isolves cell-free DNA from plasma samples [45] [3] |
| Polymerase Enzymes | HotStart Phusion Polymerase (NEB), Platinum Taq (Invitrogen) | High-fidelity amplification and emulsion PCR [3] |
| Magnetic Beads | Streptavidin-coated magnetic beads (MyOne, Invitrogen) | Solid support for DNA amplification and detection [3] |
| Emulsion Components | ABIL WE09, Mineral oil, TegoSoft DEC | Creates water-in-oil emulsion for compartmentalized PCR [3] |
| Detection Probes | Fluorescently labeled allele-specific oligos (FAM/PE) | Differentiates mutant and wild-type alleles during flow analysis [3] |
| Control Materials | Genomic DNA from cell lines (e.g., PC9, H1975, A549) | Assay validation and sensitivity determination [3] |
Applications in Treatment Response Monitoring
BEAMing technology enables precise quantification of ctDNA dynamics during cancer therapy, providing researchers with a powerful tool for assessing treatment efficacy and detecting emerging resistance. The applications span multiple cancer types and therapeutic modalities.
Monitoring Surgical Response and Minimal Residual Disease
In colorectal cancer patients undergoing surgical resection, BEAMing has demonstrated exceptional utility in monitoring treatment response and detecting minimal residual disease (MRD). Studies have shown that ctDNA levels measured by BEAMing drop precipitously following complete tumor resection, with a median decrease of 99.0% observed within days after surgery [19]. The half-life of ctDNA post-resection was estimated at approximately 114 minutes, highlighting the dynamic nature of this biomarker [19]. In cases where resection was incomplete, ctDNA levels persisted or increased, enabling real-time assessment of surgical efficacy [19].
Longitudinal monitoring of ctDNA after surgery can detect molecular recurrence months before clinical or radiographic evidence of disease. In colorectal cancer, ctDNA-based MRD detection has been shown to provide a lead time of 2-15 months compared to conventional imaging [45]. This early warning system enables researchers to study window-of-opportunity interventions and assess novel adjuvant therapies in defined high-risk populations.
Targeted Therapy Response Assessment
BEAMing technology provides critical insights into response patterns during targeted therapy, particularly for tumors driven by oncogenic mutations. In non-small cell lung cancer (NSCLC) with EGFR mutations, BEAMing enables quantitative monitoring of mutant allele frequencies during EGFR tyrosine kinase inhibitor (TKI) therapy [3]. Declining ctDNA levels correlate with radiographic response and often precede size changes measurable by RECIST criteria [2] [14].
The high sensitivity of BEAMing also facilitates early detection of resistance mechanisms. For example, the emergence of EGFR T790M mutations can be identified in plasma weeks to months before clinical progression, enabling researchers to study resistance mechanisms and assess the efficacy of next-generation inhibitors [14] [3]. This application is particularly valuable for studying sequential targeted therapies and combination approaches designed to overcome or prevent resistance.
Table 3: ctDNA Dynamics During Cancer Therapy Measured by BEAMing
| Clinical Scenario | ctDNA Response Pattern | Research Implications |
|---|---|---|
| Complete Surgical Resection | Rapid decline (>99%) within 24-48 hours; half-life ~114 minutes [19] | Biomarker for assessing surgical efficacy; baseline for MRD monitoring |
| Effective Systemic Therapy | Significant decrease in mutant allele frequency; often precedes radiographic response [2] [14] | Early endpoint for drug efficacy studies; correlation with tumor burden |
| Incomplete Resection/Residual Disease | Persistent or rising ctDNA levels post-treatment [19] | Identification of non-responders for treatment intensification studies |
| Acquired Resistance | Emergence of new resistance mutations (e.g., T790M) during ongoing therapy [14] [3] | Studying resistance mechanisms; evaluating sequential targeted therapies |
| Molecular Relapse | Detectable ctDNA months before clinical recurrence [45] | Window for studying salvage therapies; understanding tumor dormancy |
Data Analysis and Interpretation
Quantitative Analysis of BEAMing Data
Proper analysis of BEAMing data requires specialized approaches to distinguish true signal from background noise and derive meaningful biological insights:
Mutant Allele Frequency Calculation:
- Calculate mutant allele frequency (MAF) as: MAF = (Number of mutant beads) / (Number of mutant beads + Number of wild-type beads) × 100% [3].
- Establish background signal using negative controls (normal plasma or wild-type only samples).
- Apply threshold for positive detection (typically 3 standard deviations above background).
Absolute Quantification:
Statistical Considerations:
- Account for sampling variability, especially at very low mutant frequencies (<0.1%).
- Utilize Poisson statistics for digital quantification in low-abundance samples.
- Establish significance thresholds for changes in mutant allele frequency (typically >2-fold change considered significant).
Integration with Complementary Biomarkers
BEAMing data should be interpreted in the context of complementary biomarkers and clinical parameters:
Imaging Correlation: Compare ctDNA dynamics with radiographic assessment (RECIST criteria) to validate molecular response [2]. Note that ctDNA changes often precede radiographic changes.
Protein Biomarkers: Correlate with conventional serum tumor markers (e.g., CEA in colorectal cancer) [19] [45]. ctDNA typically shows superior sensitivity and specificity for monitoring tumor dynamics.
Resistance Mutation Patterns: Monitor multiple mutations simultaneously to understand clonal evolution during therapy [14]. The emergence of subclonal resistance mutations can inform combination therapy strategies.
Troubleshooting and Technical Considerations
Successful implementation of BEAMing assays requires attention to several technical challenges:
Low ctDNA Abundance: For early-stage cancers or low-shedding tumors, consider increasing plasma input volume (up to 4 mL) and implementing fragment size selection to enrich for ctDNA [14]. The use of unique molecular identifiers (UMIs) can help distinguish true mutations from amplification artifacts [2].
Assay Sensitivity Optimization: Regularly validate assay sensitivity using dilution series of mutant DNA in wild-type background [3]. Establish limit of detection (LOD) and limit of quantification (LOQ) for each mutation assay.
Sample Quality Control: Monitor sample integrity through DNA fragment size analysis. ctDNA typically shows a characteristic fragmentation pattern (~90-150 bp) that differs from high-molecular-weight genomic DNA [14].
Multiple Mutation Tracking: For comprehensive resistance monitoring, develop parallel assays for common resistance mutations in the relevant cancer type. This enables tracking of polyclonal resistance mechanisms.
BEAMing technology provides researchers and drug developers with a highly sensitive and quantitative platform for monitoring treatment response and resistance mechanisms through ctDNA analysis. The protocols and applications detailed in this document enable real-time assessment of tumor dynamics, detection of minimal residual disease, and identification of emerging resistance mutations during therapy. As precision oncology continues to evolve, BEAMing-based ctDNA analysis offers unprecedented opportunities to study tumor evolution under therapeutic pressure and develop more effective, adaptive treatment strategies. The integration of this technology into translational research protocols will accelerate drug development and enhance our understanding of treatment response and resistance across diverse cancer types.
Minimal Residual Disease (MRD) Detection Strategies
Circulating tumor DNA (ctDNA) has emerged as a transformative biomarker for Minimal Residual Disease (MRD) detection in solid tumors. MRD refers to the presence of residual tumor cells after curative-intent treatment that evade standard radiographic detection but can lead to future relapse [46]. ctDNA is a fraction of cell-free DNA (cfDNA) shed into the bloodstream by tumor cells through apoptosis, necrosis, and active secretion, carrying the comprehensive genetic and epigenetic landscape of the parent tumor [46] [47]. The analysis of ctDNA for MRD detection represents a paradigm shift in oncology, enabling the identification of molecular relapse months or even years before clinical or radiographic evidence emerges [46]. This application note details the detection strategies, methodologies, and protocols for ctDNA-based MRD analysis within the broader context of BEAMing technology research.
Biology and Technical Challenges of ctDNA
Fundamental Characteristics
ctDNA possesses distinct biological properties that inform detection methodologies. These fragments are typically shorter than healthy cfDNA, with a mean length of approximately 143 base pairs compared to 166 base pairs for normal cfDNA [46]. The concentration of ctDNA in blood is influenced by tumor type, stage, burden, and location, with the fraction of ctDNA in total cfDNA ranging from <0.05% to 90% [46]. A critical feature is its short half-life of 16 minutes to 2.5 hours, allowing ctDNA to provide a "real-time" snapshot of tumor dynamics [46].
Technical Challenges in MRD Detection
Accurate MRD detection presents significant technical challenges due to the extremely low concentration of ctDNA in peripheral blood following curative-intent therapy, where variant allele frequencies (VAF) can be <0.01% [46]. This necessitates highly sensitive and specific platforms capable of distinguishing rare tumor-derived mutations from background wild-type DNA and accounting for biological confounders such as clonal hematopoiesis of indeterminate potential (CHIP) [46].
MRD Detection Strategies and Methodologies
Strategic Approaches
Two primary strategies dominate ctDNA-based MRD detection: tumor-informed and tumor-agnostic approaches [46] [48].
Table 1: Comparison of MRD Detection Strategies
| Feature | Tumor-Informed Approach | Tumor-Agnostic Approach |
|---|---|---|
| Principle | Customized panel based on prior knowledge of tumor mutational profile | Fixed panel independent of tumor tissue analysis |
| Workflow | 1. Tumor tissue sequencing (WES/WGS)2. Personalized panel design3. Plasma tracking of patient-specific mutations | Direct plasma analysis using pre-selected gene panels |
| Sensitivity | High (can track multiple patient-specific mutations) | Moderate to high |
| Specificity | High (reduces false positives from CHIP) | Moderate |
| Turnaround Time | Longer (requires tissue sequencing and custom design) | Shorter |
| Tumor Uniqueness | 55.7-81.5% of genes are patient-specific [48] | Limited to pre-designed content |
Detection Technologies
Multiple technological platforms have been developed to address the sensitivity requirements for MRD detection:
- PCR-based Methods: Including droplet digital PCR (ddPCR) and BEAMing technology (Beads, Emulsions, Amplification, and Magnetics), reliably detecting known genomic alterations with sensitivity down to VAF of 0.01% [46] [5].
- Next-Generation Sequencing (NGS) Approaches:
BEAMing Technology for ctDNA MRD Detection
Principles of BEAMing
BEAMing (Beads, Emulsions, Amplification, and Magnetics) represents an advanced emulsion PCR-based digital detection platform that enables absolute quantification of rare mutant DNA molecules [5]. The technology transforms individual DNA molecules into magnetic beads coupled with thousands of identical DNA copies through compartmentalized water-in-oil microemulsions, followed by flow cytometry analysis to differentiate mutant from wild-type sequences [5].
BEAMing Workflow
BEAMing Experimental Protocol
Pre-Analytical Phase
- Blood Collection: Draw blood into K2/K3-EDTA tubes or cell preservation tubes. Invert 8-10 times gently to mix additives [47].
- Plasma Separation: Process within 4-6 hours for EDTA tubes (up to 5-7 days for cell preservation tubes). Perform two-step centrifugation:
- First centrifugation: 800-1,600×g at 4°C for 10 minutes
- Second centrifugation: 14,000-16,000×g at 4°C for 10 minutes [47]
- Plasma QC: Visually inspect for hemolysis (orange/red discoloration), icterus (yellowish/greenish), or lipemia (opaque) [47].
- Storage: Store plasma at -80°C for long-term preservation. Avoid multiple freeze-thaw cycles [47].
BEAMing Analytical Protocol
- cfDNA Extraction: Extract cfDNA from plasma using silica membrane columns or magnetic beads. Quantify using fluorometry [5].
- Primer Design: Design allele-specific primers for mutations identified through prior tumor sequencing (tumor-informed) or hotspot mutations (tumor-agnostic).
- Emulsion Preparation:
- Create water-in-oil emulsion with reaction droplets containing:
- 1-2 DNA molecules
- PCR reagents
- Magnetic beads with complementary oligonucleotides
- Create water-in-oil emulsion with reaction droplets containing:
- Emulsion PCR:
- Thermal cycling conditions:
- Initial denaturation: 95°C for 5 min
- 40-50 cycles of: 95°C for 30s, 58-62°C for 30s, 72°C for 45s
- Final extension: 72°C for 7 min
- Thermal cycling conditions:
- Bead Recovery: Break emulsion and recover beads using magnetic separation.
- Mutation Detection:
- Hybridize with fluorescence-labeled mutation-specific probes
- Analyze by flow cytometry
- Mutant beads fluoresce differentially from wild-type beads
- Quantification: Calculate mutant allele frequency using formula:
- % Mutation = (Mutant beads / Total beads) × 100 [5]
Performance Characteristics and Validation
Analytical Performance
BEAMing technology demonstrates a detection limit of 0.01% mutant allele frequency, enabling identification of rare ctDNA molecules in background wild-type DNA [5]. The technology has been clinically validated for RAS mutation testing in colorectal cancer, showing 93.3% overall concordance with standard tissue testing [5].
Table 2: Performance Characteristics of MRD Detection Methods
| Technology | Detection Limit | Advantages | Limitations |
|---|---|---|---|
| BEAMing | 0.01% | High sensitivity for known mutations; absolute quantification | Limited to known mutations |
| ddPCR | 0.01% | High sensitivity; easy implementation | Limited multiplexing capability |
| Hybrid Capture NGS | 0.01%-0.1% | Broad coverage; discovery capability | Higher input requirements; complex bioinformatics |
| Amplicon NGS | 0.02%-0.1% | High sensitivity; flexible panel design | PCR artifacts; limited genomic coverage |
Clinical Validation in Solid Tumors
In lung cancer, a meta-analysis of 16 studies (1,251 patients) demonstrated that ctDNA MRD detection has high specificity (0.86-0.95) but moderate sensitivity (0.41-0.76) for predicting recurrence [50]. The landmark strategy (single timepoint after treatment) showed higher specificity but lower sensitivity compared to surveillance strategy (longitudinal monitoring) [50]. The tumor-informed approach demonstrates superior performance, with the MEDAL study showing better sensitivity and predictive value for disease-free survival compared to tumor-agnostic fixed panels [48].
Research Reagent Solutions
Table 3: Essential Research Reagents for BEAMing MRD Detection
| Reagent/Material | Function | Specifications |
|---|---|---|
| Cell-Free DNA Blood Collection Tubes | Preserves blood cell integrity during storage/storage | Streck, PAXgene, or similar cell-stabilizing tubes |
| Magnetic Beads with Oligonucleotides | Solid support for DNA amplification | Streptavidin-coated beads with biotinylated primers |
| Emulsion Oil Phase | Creates microcompartments for digital PCR | Surfactant-containing mineral or silicon oils |
| Mutation-Specific Fluorescent Probes | Detect mutant vs. wild-type sequences | FAM, VIC, or other fluorophore-labeled hybridization probes |
| Allele-Specific Primers | Amplify target mutations with high specificity | Designed with 3'-end mismatch for allele discrimination |
| DNA Extraction Kits | Isolve high-quality cfDNA from plasma | Silica membrane or magnetic bead-based systems |
| Fluorometric Quantification Kits | Accurately measure cfDNA concentration | Qubit dsDNA HS Assay or similar |
MRD Clinical Decision Pathway
BEAMing technology provides a highly sensitive and specific platform for ctDNA-based MRD detection in solid tumors, with significant implications for clinical research and drug development. The tumor-informed approach enhances detection performance by leveraging patient-specific mutational profiles, though tumor-agnostic methods offer practical advantages in certain settings. As MRD detection technologies continue to evolve, standardization of pre-analytical procedures, analytical validation, and clinical interpretation will be critical for advancing their application in therapeutic development and personalized cancer management.
Optimizing BEAMing Assays: Addressing Technical Challenges and Limitations
Pre-analytical Variables and Sample Quality Control
The analytical reliability of circulating tumor DNA (ctDNA) analysis using BEAMing (beads, emulsion, amplification, and magnetics) technology is fundamentally dependent on pre-analytical sample quality. ctDNA molecules represent a minute fraction of total cell-free DNA (cfDNA) in circulation, sometimes constituting <0.1% of total cfDNA, creating significant challenges for reliable detection [14]. Pre-analytical variables—those factors affecting samples from collection through processing—directly impact the integrity, quantity, and quality of ctDNA, thereby influencing downstream analytical results and clinical interpretations [51]. This application note provides detailed protocols for standardizing pre-analytical procedures to ensure sample quality for BEAMing-based ctDNA analysis in cancer research and drug development.
Critical Pre-analytical Variables in ctDNA Analysis
Biological and Physiological Considerations
Multiple biological and physiological factors introduce significant intra- and inter-individual variability in cfDNA characteristics, potentially confounding ctDNA analysis results. These variables must be considered during patient preparation and data interpretation [51].
Table 1: Biological and Physiological Variables Affecting ctDNA Analysis
| Variable Category | Specific Factors | Impact on cfDNA/ctDNA |
|---|---|---|
| Demographic Factors | Age, gender | Baseline cfDNA levels may vary |
| Lifestyle Factors | Diet, exercise | Acute exercise may temporarily elevate cfDNA |
| Physiological States | Pregnancy, menstruation, obesity | Altered cfDNA levels and composition |
| Pathological Conditions | Inflammation, infection, diabetes | Increased background cfDNA from non-tumor cells |
| Medical Interventions | Surgery, chemotherapy, radiotherapy | Massive cfDNA release following tissue damage |
| Cancer Biology | Tumor type, stage, vascularity | Impacts ctDNA shedding rates and fragment size |
Research indicates that cfDNA originates through various mechanisms including apoptosis (producing characteristic 160-180 bp fragments), necrosis (yielding larger >10,000 bp fragments), and active release from living cells (fragments 150-6,000 bp) [51]. Understanding these biological variables is essential for distinguishing tumor-derived DNA from background cfDNA in BEAMing analysis.
Blood Collection and Sample Handling
Standardized blood collection procedures are critical for maintaining cfDNA integrity and preventing genomic DNA contamination from hematopoietic cell lysis.
Recommended Materials
Table 2: Essential Research Reagents for Blood Collection and Processing
| Item | Specification | Function | Example Products |
|---|---|---|---|
| Blood Collection Tubes | cfDNA-stabilizing tubes | Preserves nucleic acid integrity, prevents cellular lysis | Streck cfDNA BCT, PAXgene Blood ccfDNA Tubes |
| Centrifuge | Swing-bucket rotor capable of 1600×g and 16,000×g | Sequential plasma separation | Refrigerated centrifuges |
| Plasma Storage Tubes | Low DNA binding | Prevents cfDNA adsorption to tube walls | LoBind tubes |
Detailed Blood Collection Protocol
Venipuncture: Collect peripheral blood using a standard venipuncture technique with a 21-gauge or larger needle to minimize shear forces that could lyse blood cells.
Tube Filling: Draw a minimum of 10 mL blood into cfDNA-stabilizing tubes, ensuring correct fill volume to maintain proper preservative-to-blood ratio.
Tube Mixing: Gently invert tubes 8-10 times immediately after collection to ensure complete mixing with preservatives.
Transportation: Maintain samples at ambient temperature (18-25°C) and transport to the laboratory within 24 hours of collection [36].
Temporary Storage: If processing cannot occur within 24 hours, store samples at 4°C for a maximum of 72 hours. Avoid freeze-thaw cycles of whole blood.
Plasma Processing and cfDNA Extraction
The plasma processing methodology significantly impacts cfDNA yield, purity, and fragment size distribution, all critical parameters for BEAMing analysis.
Two-Step Centrifugation Protocol
First Centrifugation Step:
- Conditions: 1600×g for 10 minutes at 4°C
- Outcome: Separation of plasma from cellular components
- Post-centrifugation: Carefully transfer supernatant to a new tube, avoiding disturbance of the buffy coat layer which contains white blood cells
Second Centrifugation Step:
- Conditions: 16,000×g for 10 minutes at 4°C
- Outcome: Removal of any remaining cellular debris and platelets
- Post-centrifugation: Transfer cleared plasma to fresh LoBind tubes for immediate extraction or storage [36]
Plasma Storage: Aliquot processed plasma into working volumes to avoid repeated freeze-thaw cycles. Store at -80°C until cfDNA extraction.
cfDNA Extraction and Quantification
Extraction Method: Use commercial cfDNA extraction kits specifically designed for low-abundance DNA recovery. Extract from 2-4 mL plasma following manufacturer's protocols [36].
Quantification: Utilize fluorescence-based methods (e.g., Qubit dsDNA HS Assay) for accurate quantification of low-concentration DNA. Avoid spectrophotometric methods which lack sensitivity and specificity for cfDNA.
Quality Assessment: Analyze cfDNA fragment size distribution using microfluidic capillary electrophoresis (e.g., Agilent TapeStation 4200 with Cell-Free DNA ScreenTape). Expect a peak at approximately 166 bp representing mononucleosomal DNA [36].
Sample Processing Workflow for ctDNA Analysis
Quality Control Checkpoints and Acceptance Criteria
Implementing rigorous quality control checkpoints throughout the pre-analytical phase is essential for generating reliable BEAMing data.
Table 3: Quality Control Checkpoints and Acceptance Criteria
| QC Checkpoint | Parameter | Acceptance Criteria | Corrective Action |
|---|---|---|---|
| Post-Collection | Time-to-processing | ≤24 hours (ambient) | Document deviation; note potential gDNA contamination |
| Post-Centrifugation | Sample appearance | Clear plasma, no hemolysis | Discard heavily hemolyzed samples |
| cfDNA Extraction | Yield | >0.1 ng/μL from 4 mL plasma | Concentrate if low yield; repeat extraction if below LOD |
| cfDNA Quality | Fragment size | Peak ~166 bp | Size selection may be required if high molecular weight DNA present |
| Pre-BEAMing | cfDNA fraction | >10% tumor DNA for optimal detection | Note limited sensitivity if below threshold |
For BEAMing technology specifically, which relies on precise amplification and detection of rare mutations, the following additional QC parameters are recommended:
Wild-Type DNA Assessment: Evaluate background levels of wild-type DNA which can dilute mutant alleles and reduce detection sensitivity.
Inhibition Testing: Include control reactions to detect PCR inhibitors that may affect emulsion amplification efficiency.
Input DNA Consistency: Standardize input DNA amounts across samples to ensure comparable detection limits.
Impact of Pre-analytical Variables on BEAMing Results
Variations in pre-analytical procedures can introduce significant artifacts in BEAMing analysis:
Cell Lysis During Collection: Improper collection or handling can cause white blood cell lysis, increasing background wild-type DNA and reducing mutant allele detection sensitivity [51].
Extended Storage Times: Delays in processing can reduce ctDNA yield and increase fragment size due to release of high molecular weight DNA from lysed blood cells.
Incomplete Centrifugation: Residual cells or platelets in plasma can contaminate cfDNA extracts with genomic DNA, altering the mutant-to-wild-type allele ratio.
Suboptimal Extraction Methods: Inefficient recovery of short DNA fragments can preferentially deplete ctDNA, which tends to be shorter than non-malignant cfDNA [14].
Pre-analytical Issues and Their Impacts on BEAMing Analysis
Standardization of pre-analytical procedures is fundamental to generating reliable, reproducible ctDNA data using BEAMing technology. The comprehensive protocols outlined in this application note address the most critical variables in sample collection, processing, and quality control. Implementation of these standardized procedures across research laboratories and clinical trials will enhance data comparability, improve assay sensitivity, and ultimately strengthen the validity of research conclusions in ctDNA analysis for drug development. As the field advances, continued refinement of pre-analytical standards will be necessary to accommodate emerging technologies and expanding applications of liquid biopsy in precision oncology.
Strategies for Low Frequency Mutation Detection (<0.1%)
The analysis of circulating tumor DNA (ctDNA) has emerged as a powerful, minimally invasive tool for cancer genotyping, treatment monitoring, and the detection of minimal residual disease (MRD) [1] [9]. A significant challenge in this field is the reliable detection of low-frequency mutations, often present at allele frequencies below 0.1%, against a background of wild-type DNA [5] [4]. This technical hurdle is central to advancing liquid biopsy applications, particularly for early cancer detection and monitoring treatment response. BEAMing technology represents one of the most sensitive approaches for this purpose. This document outlines the principles, protocols, and key applications of BEAMing for detecting mutations below the 0.1% threshold, providing a structured guide for researchers and drug development professionals.
BEAMing (Beads, Emulsion, Amplification, and Magnetics) is a digital PCR-based method that combines emulsion PCR with flow cytometry to achieve ultra-sensitive detection of rare mutant alleles [5]. Its core principle involves physically separating individual DNA molecules such that each can be amplified and analyzed in isolation, thereby overcoming the limitations of traditional bulk PCR.
The process allows for the detection of a single mutant DNA molecule among 10,000 wild-type molecules, achieving a detection sensitivity as low as 0.01% mutant allele frequency [5] [52]. This makes it particularly suited for analyzing ctDNA, where tumor-derived DNA can constitute less than 0.1% of the total cell-free DNA (cfDNA) in plasma, especially in patients with early-stage disease or low tumor burden [53] [9].
Table 1: Key Characteristics of BEAMing Technology
| Feature | Description | Performance/Implication |
|---|---|---|
| Basic Principle | Emulsion-based digital PCR combined with flow cytometry | Physical separation of DNA molecules for individual analysis |
| Detection Limit | Can detect 1 mutant in a background of 10,000 wild-type molecules [5] [52] | ~0.01% mutant allele frequency |
| Analytical Sensitivity | High for pre-defined hotspot mutations [5] | Ideal for tracking known mutations in ctDNA |
| Analytical Specificity | High; mutations are confirmed by specific hybridization probes [5] | Low false-positive rate |
| Concordance with Tissue | Demonstrated high concordance (e.g., 93.3% for RAS in mCRC) [5] | Validated for clinical decision-making |
Experimental Workflow and Protocol
The successful application of BEAMing for low-frequency mutation detection relies on a meticulously controlled workflow, from blood collection to final data analysis. The following protocol is compiled from established methods in colorectal cancer and glioma studies [5] [53] [52].
Pre-Analytical Phase: Sample Collection and Plasma Isolation
The integrity of pre-analytical steps is critical for accurate low-frequency mutation detection.
- Blood Collection: Collect 10-20 mL of peripheral blood into cell-stabilizing blood collection tubes (BCTs), such as Streck cell-free DNA BCT tubes [53] [52]. These tubes contain preservatives that prevent leukocyte lysis and the release of wild-type genomic DNA, which would dilute the mutant allele fraction.
- Plasma Processing: Process samples within the validated stability window of the BCTs (e.g., up to 7 days for Streck tubes at room temperature). Perform a double centrifugation protocol [53]:
- First Spin: 800–1600 × g for 10 minutes at 4°C to separate plasma from blood cells.
- Second Spin: Transfer the supernatant to a new tube and centrifuge at 16,000 × g for 10 minutes to remove any remaining cellular debris.
- Plasma Storage: Aliquot the clarified plasma and store at -80°C until cfDNA extraction to avoid freeze-thaw cycles that can degrade DNA [53].
cfDNA Extraction and BEAMing Assay
The core of the protocol involves isolating cfDNA and performing the BEAMing reaction.
- cfDNA Extraction: Extract cfDNA from 2-5 mL of plasma using commercially available kits (e.g., QIAamp Circulating Nucleic Acid Kit). Elute DNA in a low-volume elution buffer (e.g., 50-100 µL) to maximize concentration.
- BEAMing Reaction:
- Primer Design: Design PCR primers specific to the genomic region containing the hotspot mutation of interest (e.g., KRAS G12/G13, IDH1 R132H).
- Emulsion PCR: The extracted cfDNA is mixed with PCR reagents and magnetic beads. This mixture is vigorously vortexed with oil to create a water-in-oil emulsion, forming millions of microreactors. Each microreactor ideally contains a single bead and no more than one DNA molecule.
- Amplification: The emulsion is subjected to PCR. If a DNA molecule is present in a microreactor, it amplifies and binds to the bead.
- Emulsion Breaking: After PCR, the emulsion is broken, and the beads are recovered.
- Hybridization: The beads are incubated with fluorescently labeled probes specific for the wild-type and mutant sequences.
- Flow Cytometry Analysis: Beads are analyzed by flow cytometry. Beads that hybridize with the mutant probe are counted as mutant, while those that hybridize only with the wild-type probe are counted as wild-type. The mutant allele frequency is calculated as (Number of mutant beads / Total number of beads) × 100%.
The following diagram illustrates the core BEAMing workflow.
Critical Considerations for Low-Frequency Detection
- Avoiding Contamination: Use dedicated pre- and post-PCR areas. Include negative controls (no-template and wild-type DNA) in every run to monitor for cross-contamination.
- Addressing Clonal Hematopoiesis (CHIP): Be aware that mutations detected in plasma may originate from clonal hematopoiesis of indeterminate potential (CHIP) rather than the tumor, which can lead to false-positive results [1]. Correlation with clinical context is essential.
- Data Interpretation: Set a limit of detection (LOD) and limit of blank (LOB) using control samples. A mutation is typically called positive when its frequency is above the statistically defined LOD and is reproducibly detected.
Research Reagent Solutions
A successful BEAMing assay depends on specific, high-quality reagents. The table below details essential materials and their functions.
Table 2: Essential Research Reagents for BEAMing Protocols
| Reagent/Material | Function | Specific Examples & Notes |
|---|---|---|
| Cell-Stabilizing BCTs | Prevents leukocyte lysis during blood transport/storage, preserving the wild-type DNA background and ctDNA integrity. | Streck Cell-Free DNA BCTs [52]; PAXgene Blood ccfDNA Tubes [53]. |
| cfDNA Extraction Kit | Isolves short-fragment, low-concentration cfDNA from plasma with high efficiency and purity. | QIAamp Circulating Nucleic Acid Kit; other silica-membrane based kits. |
| BEAMing Beads | Serve as solid support for the amplification of single DNA molecules in emulsion microreactors. | Streptavidin-coated magnetic beads (if using biotinylated primers). |
| Hotspot-Specific Primers & Probes | Amplify the target region and allow for specific discrimination between wild-type and mutant sequences. | Designed for mutations in genes like KRAS, NRAS, IDH1, EGFR [5] [52]. |
| Fluorophore-Labeled Probes | Enable detection and quantification of mutant and wild-type alleles via flow cytometry. | Typically, a different fluorophore (e.g., FAM, VIC) for mutant vs. wild-type probes. |
| Emulsion Oil & Surfactants | Create and stabilize the water-in-oil emulsion, forming millions of independent PCR microreactors. | Specialized emulsion PCR oil and surfactant solutions. |
Applications in Cancer Research
BEAMing technology has been successfully applied in various oncology research contexts, demonstrating its value for sensitive ctDNA analysis.
- Guidance for Targeted Therapy: In metastatic colorectal cancer (mCRC), the OncoBEAM RAS assay is used to identify patients with RAS mutations who are unlikely to benefit from anti-EGFR therapy [5]. The high concordance (93.3%) with standard tissue testing underscores its reliability for clinical research.
- Monitoring Treatment Response: The short half-life of ctDNA (16 minutes to 2.5 hours) makes it an ideal biomarker for real-time monitoring of tumor dynamics [53]. BEAMing can be used to track changes in mutant allele frequency during therapy, providing an early readout of drug efficacy or emergence of resistance.
- Challenging Tumor Types: BEAMing has shown promise in detecting mutations in cancers where ctDNA shedding is low, such as gliomas. One study detected IDH1 mutations in the plasma of 50% of patients with IDH1-mutant gliomas, a notable achievement given the blood-brain barrier [52].
Comparison with Other Detection Methods
BEAMing is one of several highly sensitive platforms for mutation detection. The table below compares it with other common techniques.
Table 3: Comparison of Low-Frequency Mutation Detection Methods
| Method | Detection Limit | Advantages | Limitations |
|---|---|---|---|
| BEAMing | ~0.01% [5] | Very high sensitivity for known hotspots; high specificity due to probe hybridization. | Limited to pre-defined mutations; requires specialized workflow. |
| Droplet Digital PCR (ddPCR) | ~0.001%-0.01% [54] | High sensitivity; easier to implement than BEAMing; absolute quantification. | Best suited for a limited number of known mutations per reaction. |
| Next-Generation Sequencing (NGS) | ~0.1%-2.0% (varies by protocol) [5] | Interrogates a broad range of genes and mutation types simultaneously; discovery-based. | Generally less sensitive than digital PCR methods; higher cost and longer turnaround for targeted applications. |
The following diagram outlines the decision-making process for selecting a suitable detection method based on research goals.
BEAMing technology provides a robust and highly sensitive framework for detecting low-frequency mutations down to 0.01% in ctDNA, addressing a core need in modern cancer research and drug development. Its application in identifying actionable mutations, monitoring MRD, and tracking treatment response offers a powerful, minimally invasive complement to traditional tissue biopsy. Adherence to standardized pre-analytical and analytical protocols, as detailed in this document, is paramount for generating reliable and reproducible data. As the field of liquid biopsy continues to evolve, BEAMing remains a cornerstone technology for pushing the boundaries of detecting and quantifying rare tumor-derived variants.
Addressing Tumor Heterogeneity and Clonal Evolution
BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) represents a highly sensitive digital PCR-based method for analyzing circulating tumor DNA (ctDNA) in liquid biopsies. This approach is particularly suited for addressing the critical challenges of tumor heterogeneity and clonal evolution in oncology research and drug development. By enabling the precise detection and quantification of rare tumor-specific mutations from a patient's blood, BEAMing facilitates non-invasive genomic profiling that captures the spatial and temporal genomic diversity of malignancies [1] [4]. As tumors evolve under therapeutic pressure, selecting for resistant clones, BEAMing provides a dynamic window into these processes, allowing researchers to monitor acquired resistance mutations that often precede clinical evidence of disease progression [14] [2].
The clinical significance of this technology lies in its ability to generate real-time, actionable genetic data when traditional tissue biopsies are impractical or fail to represent the full heterogeneity of the disease [55]. For researchers and drug development professionals, BEAMing offers a robust platform for identifying actionable mutations, assessing minimal residual disease (MRD), and tracking treatment response through longitudinal monitoring [1] [2].
Key Applications in Oncology Research
Overcoming Technical Challenges
BEAMing technology addresses several limitations of conventional sequencing approaches when applied to ctDNA analysis. The table below summarizes the key technical challenges and how BEAMing provides solutions:
Table 1: Technical Challenges and BEAMing Solutions in ctDNA Analysis
| Technical Challenge | BEAMing Solution | Research Implication |
|---|---|---|
| Low abundance of ctDNA in early-stage disease or MRD [14] | Digital quantification through emulsion PCR enables detection of mutant alleles at frequencies as low as 0.01% [55] | Enables studies of early-stage cancer dynamics and MRD assessment |
| Tumor heterogeneity [1] | Single-molecule amplification allows parallel detection of multiple mutations from same sample | Captures subclonal diversity not visible in tissue biopsies |
| Need for rapid, targeted mutation detection [4] | Focused panels for known mutations with rapid turnaround time | Ideal for longitudinal therapy response studies and resistance monitoring |
| Differentiation of true mutations from sequencing artifacts [2] | Emulsion-based compartmentalization reduces background noise | Improves specificity for low-frequency variant detection |
Monitoring Clonal Evolution
BEAMing technology provides researchers with a powerful tool for tracking the dynamics of clonal populations during treatment. The high sensitivity of BEAMing allows detection of emerging resistant subclones often weeks or months before clinical or radiographic progression becomes evident [14] [2]. In EGFR-mutant non-small cell lung cancer (NSCLC), for instance, BEAMing can monitor for the emergence of the T790M resistance mutation, guiding timely switches to third-generation EGFR inhibitors [14]. Similarly, in colorectal cancer, BEAMing enables tracking of KRAS mutation dynamics, while in breast cancer, it can monitor ESR1 mutations developing under aromatase inhibitor therapy [4] [2].
Figure 1: BEAMing Technology in Monitoring Therapy Resistance
Experimental Protocols for BEAMing Analysis
Sample Collection and Processing Protocol
Principle: Proper pre-analytical sample handling is critical for accurate BEAMing analysis due to the low abundance and fragility of ctDNA [4].
Materials:
- Streck Cell-Free DNA Blood Collection Tubes or K2EDTA tubes
- Plasma separation tubes
- QIAamp Circulating Nucleic Acid Kit (Qiagen) or similar
- Magnetic stand for 1.5 mL tubes
- TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0)
Procedure:
- Blood Collection: Collect 10 mL peripheral blood into cell-free DNA stabilization tubes. Invert gently 8-10 times for mixing.
- Plasma Separation: Centrifuge at 1600 × g for 20 minutes at room temperature within 2 hours of collection.
- Secondary Centrifugation: Transfer supernatant to fresh tube. Centrifuge at 16,000 × g for 10 minutes at 4°C to remove residual cells.
- Plasma Storage: Aliquot cleared plasma and store at -80°C until DNA extraction.
- cfDNA Extraction: Use commercial circulating nucleic acid kit according to manufacturer's instructions.
- DNA Quantification: Quantify cfDNA using fluorometric methods (e.g., Qubit dsDNA HS Assay).
- Quality Assessment: Verify fragment size distribution (expected peak ~166 bp) using Bioanalyzer or TapeStation.
Technical Notes:
- Process samples within 6 hours if using K2EDTA tubes, or within 72 hours if using stabilization tubes.
- Avoid repeated freeze-thaw cycles of plasma and extracted cfDNA.
- Include a negative control ( plasma from healthy donor) with each extraction batch.
BEAMing Assay Protocol for EGFR Mutation Detection
Principle: This protocol adapts the methodology from Mirikar et al. (2025) for detecting EGFR mutations in NSCLC patients [55].
Materials:
- BEAMing kit for EGFR mutations (commercial or custom)
- Magnetic beads coated with streptavidin
- PCR reagents: dNTPs, polymerase, buffer
- Emulsion oil phase (mineral oil with surfactants)
- Flow cytometer with sorting capability
- Biotinylated primers for EGFR exons 18-21
- Probes for wild-type and mutant alleles (differentially labeled)
Procedure:
- Primer Extension:
- Prepare 25 μL reaction containing: 10 ng cfDNA, 1× PCR buffer, 200 μM dNTPs, 0.5 μM biotinylated forward primer, 0.5 μM reverse primer, and 1 U DNA polymerase.
- Thermal cycling: 95°C for 5 min; 35 cycles of 95°C for 30 s, 60°C for 30 s, 72°C for 30 s; final extension 72°C for 7 min.
Emulsion Preparation:
- Mix amplification products with 10^7 streptavidin-coated magnetic beads in aqueous phase.
- Add to oil phase (4:1 ratio) and vortex vigorously for 10 minutes to create water-in-oil emulsion.
Emulsion PCR:
- Transfer emulsion to PCR tubes or plates.
- Thermal cycling: 95°C for 5 min; 50 cycles of 95°C for 30 s, 60°C for 30 s, 72°C for 30 s.
Bead Recovery and Purification:
- Break emulsion by adding 500 μL isopropanol per 100 μL emulsion. Vortex briefly.
- Pellet beads using magnetic stand. Discard supernatant.
- Wash beads twice with 500 μL TE buffer.
Hybridization and Detection:
- Resuspend beads in hybridization buffer containing fluorescently labeled probes for wild-type and mutant alleles.
- Incubate at 45°C for 30 minutes.
- Wash beads to remove unbound probes.
- Analyze by flow cytometry to count beads bound to wild-type vs. mutant probes.
Calculation:
- Mutant Allele Frequency (%) = (Number of mutant beads / Total number of beads) × 100
Figure 2: BEAMing Assay Workflow for ctDNA Analysis
Research Reagent Solutions
The following table details essential reagents and materials required for implementing BEAMing technology in ctDNA analysis research:
Table 2: Essential Research Reagents for BEAMing-Based ctDNA Analysis
| Reagent/Material | Function | Example Products |
|---|---|---|
| cfDNA Extraction Kit | Isolation of high-quality cell-free DNA from plasma samples | QIAamp Circulating Nucleic Acid Kit, MagMAX Cell-Free DNA Isolation Kit |
| Biotinylated Primers | Target-specific amplification with bead attachment capability | Custom-designed primers for hotspots in EGFR, KRAS, BRAF, PIK3CA |
| Streptavidin-Coated Magnetic Beads | Solid support for PCR amplification and subsequent analysis | Dynabeads MyOne Streptavidin C1, MagPrep Streptavidin Beads |
| Emulsion Oil Phase | Creation of water-in-oil compartments for digital PCR | Bio-Rad Droplet Generation Oil, Thermo Fisher Emulsion Oil |
| High-Fidelity DNA Polymerase | Accurate amplification with minimal errors | Q5 High-Fidelity DNA Polymerase, Platinum SuperFi II DNA Polymerase |
| Fluorescently Labeled Probes | Detection of wild-type and mutant alleles | FAM and VIC-labeled TaqMan probes, Cy3 and Cy5-labeled hybridization probes |
| Magnetic Separation Rack | Bead purification and washing | DynaMag Magnet, MagnaRack |
| Flow Cytometer | Detection and quantification of bead-bound alleles | BD FACSymphony, Beckman Coulter CytoFLEX |
Performance Data and Validation
Analytical Validation of BEAMing Technology
The performance of BEAMing technology has been extensively validated in clinical research settings. The table below summarizes key performance metrics from recent studies:
Table 3: Performance Metrics of BEAMing Technology in ctDNA Analysis
| Parameter | Performance Metric | Experimental Details |
|---|---|---|
| Concordance with Tissue Biopsy | 98.8% for exon 19, 98.9% for exon 20, 95.5% for exon 21 (EGFR) | Comparison with real-time qPCR in 100 NSCLC patients [55] |
| Limit of Detection | Capable of detecting mutant alleles at frequencies of 0.01%-0.1% | Dependent on input DNA quantity and specific mutation assayed [14] [55] |
| Analytical Sensitivity | 96% detection rate in early-stage breast cancer at baseline | SV-based ctDNA assay with median VAF of 0.15% [14] |
| Dynamic Range | Linear quantification from 0.01% to 100% mutant allele frequency | Digital nature enables accurate quantification across wide range [4] |
| Precision | High reproducibility with CV < 10% for replicate samples | Inter-assay and intra-assay validation data [55] |
Comparison with Alternative Methodologies
BEAMing technology occupies a distinct niche in the landscape of ctDNA analysis methods, offering a balance of sensitivity, specificity, and throughput suitable for tracking tumor heterogeneity:
Table 4: Method Comparison for ctDNA Heterogeneity Analysis
| Method | Optimal Use Case | Sensitivity | Throughput | Advantages for Heterogeneity Studies |
|---|---|---|---|---|
| BEAMing | Targeted mutation tracking in longitudinal studies | 0.01%-0.1% | Medium | Absolute quantification, low input requirement, minimal artifacts [55] [4] |
| ddPCR | Validation of known mutations | 0.01%-0.1% | Medium | Rapid turnaround, cost-effective for few targets [14] [2] |
| NGS Panels | Unbiased discovery of novel mutations | 1%-5% (standard); 0.1%-1% (error-corrected) | High | Comprehensive coverage, ability to detect structural variants [1] [14] |
| Structural Variant-Based Assays | MRD detection in fusion-driven cancers | <0.01% for personalized assays | Low to Medium | High specificity, low background in normal DNA [14] |
Implementation Considerations
Integration in Drug Development Workflows
For drug development professionals, BEAMing technology offers valuable applications throughout the therapeutic development pipeline. In early-phase clinical trials, BEAMing can identify patient subgroups most likely to respond to targeted therapies based on their mutational profile [1]. During late-phase trials, it enables monitoring of emerging resistance mechanisms, providing insights into drug durability and guiding combination therapy strategies [2]. The technology's ability to quantitatively track mutant allele frequencies over time makes it particularly valuable for pharmacodynamic studies and determining optimal biological dosing [4].
Limitations and Alternative Approaches
While BEAMing provides exceptional sensitivity for detecting known mutations, researchers should consider its limitations. The technology is primarily suited for targeted analysis of pre-defined mutations rather than discovery of novel alterations [4]. Additionally, challenges remain in detecting certain genomic alterations such as copy number variations and gene fusions via BEAMing alone [1]. For comprehensive profiling of heterogeneous tumors, researchers may consider complementary approaches including structural variant-based assays, fragmentomics analysis, or methylation profiling to capture the full spectrum of molecular alterations [14]. Recent advances in multimodal analysis that combine mutation detection with epigenetic features have demonstrated improved sensitivity for detecting residual disease and monitoring clonal evolution [14] [2].
Limitations in Early-Stage Cancer and Low-Shedding Tumors
Circulating tumor DNA (ctDNA) consists of fragmented DNA molecules released into the bloodstream by apoptotic or necrotic tumor cells, carrying tumor-specific genetic alterations [56] [9]. This liquid biopsy component has emerged as a promising tool for non-invasive cancer detection, monitoring treatment response, and identifying resistance mutations [57] [58]. BEAMing technology (Beads, Emulsions, Amplification, and Magnetics) represents a highly sensitive platform for ctDNA analysis, combining emulsion PCR with flow cytometry to detect and quantify rare mutant DNA sequences against a background of wild-type DNA [5].
The fundamental challenge in analyzing ctDNA lies in its extremely low concentration in blood, particularly in early-stage cancers and low-shedding tumors [21] [57]. ctDNA typically represents only 0.1% to 1.0% of total cell-free DNA (cfDNA) in plasma, creating a significant technical hurdle for reliable detection [9]. This limitation becomes especially critical in minimal residual disease (MRD) monitoring and early cancer detection, where ctDNA levels can fall below 0.01% [5] [21]. Understanding these limitations is essential for researchers and drug development professionals seeking to implement BEAMing technology in clinical studies and diagnostic applications.
Quantitative Performance Limitations
Detection Sensitivity Across Cancer Stages
The sensitivity of ctDNA detection using BEAMing technology is highly dependent on tumor stage and volume. In stage IV metastatic cancers, ctDNA is detectable in nearly 100% of cases, while early-stage malignancies show significantly lower detection rates of approximately 50% [5]. This stark difference highlights the fundamental challenge of applying BEAMing technology to early-stage disease and low-shedding tumors.
Table 1: ctDNA Detection Performance Across Tumor Stages and Sizes
| Tumor Stage | Tumor Size | Detection Rate | ctDNA Fraction | Key Limitations |
|---|---|---|---|---|
| Early Stage (I/II) | <1 cm diameter | ~50% [5] | Often <0.1% [21] | Insufficient for tumors <5 mm [56] |
| Early Stage (I/II) | 1-2 cm diameter | 59-71% [56] | 0.1%-0.5% [21] | High false-negative rates [56] |
| Advanced Stage (IV) | >2 cm diameter | ~100% [5] | 0.5%-10% [5] | Good detection but limited early intervention value |
| Post-Treatment (MRD) | Microscopic | 50-80% (varies) [21] | 0.01%-0.1% [21] | Requires ultra-deep sequencing |
Analytical Performance Metrics
BEAMing technology demonstrates variable performance characteristics depending on the application and tumor context. While the technology can achieve detection limits as low as 0.01% variant allele frequency (VAF) under ideal conditions, real-world performance in early-stage cancers is considerably less sensitive [5].
Table 2: Analytical Performance of BEAMing Technology for ctDNA Analysis
| Performance Parameter | Reported Range | Factors Influencing Performance | Clinical Implications |
|---|---|---|---|
| Limit of Detection (LOD) | 0.01%-0.5% VAF [5] [21] | Input DNA quantity, sequencing depth, tumor shedding | 0.5% LOD detects only ~50% of alterations [21] |
| Sensitivity in Early Cancer | 69%-98% [56] | Tumor size, location, vascularity | False negatives limit screening utility [56] |
| Specificity | ~99% [56] | Assay design, error correction, background mutation | Low false-positive rate enables clinical use [5] |
| Concordance with Tissue | 91.8%-93.3% [5] | Tumor heterogeneity, timing of collection | High concordance in metastatic setting [5] |
| Input DNA Requirements | 60 ng recommended [21] | Blood volume, cfDNA concentration, extraction efficiency | Low cfDNA yield limits sensitivity [21] |
Protocol for Assessing BEAMing Technology Limitations
Experimental Workflow for Sensitivity Determination
Title: BEAMing ctDNA Analysis Workflow
Step-by-Step Methodology
Sample Collection and Processing
- Collect 10-20 mL whole blood into cell-stabilization tubes (e.g., Streck, EDTA)
- Process within 2-6 hours of collection to prevent leukocytic DNA contamination [59]
- Perform double centrifugation: 1,600×g for 10 min followed by 16,000×g for 10 min to obtain platelet-poor plasma
- Aliquot and store at -80°C until DNA extraction
cfDNA Extraction and Quantification
- Extract cfDNA from 4-8 mL plasma using silica-membrane technology
- Quantify using fluorometric methods (e.g., Qubit dsDNA HS Assay)
- Assess DNA fragmentation profile using Bioanalyzer or TapeStation
- Critical Step: Calculate haploid genome equivalents (GEs) using the formula: GEs = (DNA mass in ng × 6.022×10²³) / (average fragment length × 647×10⁹) [21]
BEAMing Assay Implementation
- Design primers targeting tumor-specific mutations (e.g., KRAS, NRAS, EGFR)
- Perform emulsion PCR to compartmentalize individual DNA molecules
- Incorporate unique molecular identifiers (UMIs) to distinguish true mutations from PCR errors [21]
- Use flow cytometry to detect and quantify mutant-bearing beads
Sensitivity Determination for Low-Shedding Tumors
- Prepare dilution series of mutant DNA in wild-type DNA (1% to 0.001%)
- Process alongside clinical samples to establish limit of detection (LOD)
- Calculate LOD using 20 replicates of wild-type DNA to determine 95% detection level
- Validation: Include pre-characterized samples with known VAF (0.1%, 0.5%, 1%)
Data Analysis and Quality Control
Technical Limitations and Optimization Strategies
Key Limitations in Early-Stage Detection
The application of BEAMing technology to early-stage cancers and low-shedding tumors faces several fundamental technical constraints that impact assay performance and clinical utility.
Biological Limitations: Early-stage tumors (<1 cm diameter) release minimal ctDNA into circulation, often resulting in concentrations below the detection threshold of current BEAMing assays [56]. Tumor location and vascularity significantly impact ctDNA shedding, with pancreatic, prostate, and renal cancers typically demonstrating lower shedding rates compared to colorectal, bladder, and ovarian cancers [5]. The short half-life of ctDNA (approximately 114 minutes) provides opportunities for real-time monitoring but necessitates careful timing of sample collection [56].
Technical Barriers: The stochastic limitation of detecting rare mutant molecules in small blood volumes presents a fundamental challenge. A 10 mL blood draw from a patient with early-stage lung cancer might yield only ~8,000 haploid genome equivalents, with a mere 8 mutant molecules present at 0.1% VAF, making detection statistically improbable [21]. Pre-analytical variables including blood collection tubes, processing time, centrifugation protocols, and DNA extraction methods significantly impact assay sensitivity and reproducibility [59].
Analytical Challenges: Achieving 99% detection probability for variants at 0.1% VAF requires approximately 10,000× sequencing depth, which remains prohibitively expensive for routine clinical implementation [21]. Error rates in PCR amplification and sequencing can generate false-positive signals that mimic true low-frequency variants, necessitating sophisticated error-correction algorithms [21].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for BEAMing ctDNA Analysis
| Reagent/Category | Specific Examples | Function | Considerations for Low-Shedding Tumors |
|---|---|---|---|
| Blood Collection Tubes | Streck Cell-Free DNA BCT, EDTA tubes | Preserves blood sample integrity | Streck tubes allow longer processing windows (up to 72h) [59] |
| DNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit, MagMAX Cell-Free DNA Isolation Kit | Isolates cfDNA from plasma | Silica-membrane methods provide higher yield for low-concentration samples [59] |
| PCR Components | Hot Start polymerase, dNTPs, UMIs | Amplifies target sequences | UMI incorporation essential for error correction [21] |
| Reference Materials | Seraseq ctDNA Reference Materials, Horizon Multiplex I | Assay validation and quality control | Critical for establishing LOD in low-VAF range [59] |
| Bead Systems | Magnetic beads with specific surface chemistry | Compartmentalization of reactions | Coating affects emulsion stability and hybridization efficiency [5] |
Pathway to Clinical Utility in Challenging Scenarios
Decision Framework for Application
Title: Clinical Application Decision Pathway
Future Directions and Optimization Strategies
Several emerging approaches show promise for addressing the current limitations of BEAMing technology in early-stage and low-shedding tumors. Integrating multimodal analysis combining ctDNA mutation detection with epigenetic markers such as methylation patterns and fragmentomics may enhance sensitivity for low-shedding tumors [9]. Technical refinements including improved emulsion stability, more efficient DNA capture, and advanced error-correction algorithms could lower the detection limit from the current 0.5% to 0.1%, potentially increasing alteration detection from 50% to approximately 80% [21].
Standardization of pre-analytical variables through implementation of rigorous quality control assays enables development of robust workflows that facilitate the implementation of ctDNA analysis into clinical routine [59]. For minimal residual disease monitoring, tumor-informed approaches that track multiple patient-specific mutations show enhanced sensitivity compared to fixed-panel designs [57].
While BEAMing technology faces significant limitations in early-stage cancer and low-shedding tumors, systematic optimization of experimental protocols and careful interpretation of results within clinical context can maximize its utility in research and drug development settings. Researchers should consider these limitations when designing studies and applying BEAMing technology to ensure appropriate implementation and interpretation of results.
Multiplex Assay Design for Multiple Mutation Detection
The analysis of circulating tumor DNA (ctDNA) via liquid biopsy has emerged as a major minimally invasive biomarker in oncology, enabling molecular profiling, treatment monitoring, and minimal residual disease (MRD) detection [12] [60]. Detecting multiple mutations in parallel is crucial for overcoming tumor heterogeneity and achieving the high sensitivity required for MRD assessment, where ctDNA concentrations can be exceptionally low [60]. Multiplex assay design is fundamental to this process, allowing simultaneous assessment of numerous genomic alterations from limited ctDNA samples. This application note details the design and implementation of multiplex assays within the context of BEAMing technology for ctDNA analysis, providing structured protocols and performance data to guide researchers and drug development professionals.
Background and Clinical Significance
Circulating tumor DNA consists of short, tumor-derived DNA fragments present in the bloodstream, representing the entire tumor genome and its (sub)clones [12]. Its concentration correlates with tumor burden and cancer stage, and its short half-life (16 minutes to 2.5 hours) makes it valuable for real-time treatment monitoring [12]. In metastatic settings, ctDNA analysis identifies actionable mutations to guide targeted therapies. In MRD detection after curative-intent treatment, identifying the small volume of remaining tumor cells requires extremely sensitive methods to detect low variant allele frequencies (VAFs) often below 0.1% [60].
Multiplexed detection is essential for MRD because tracking multiple genomic alterations in parallel minimizes sampling error and increases the likelihood of detecting ctDNA [60]. Both tumor-informed (requiring prior tissue sequencing) and tumor-naïve (using predefined mutation panels) approaches benefit from multiplexing. Tumor-informed assays offer enhanced sensitivity by reducing background noise, while tumor-naïve assays provide faster turnaround and simpler logistics [12] [60]. BEAMing technology represents a highly sensitive approach for multiplex mutation detection in this context.
BEAMing (Beads, Emulsion, Amplification, and Magnetics) is a sensitive, PCR-based digital detection technology. Its core principle involves compartmentalizing individual DNA molecules with magnetic beads in water-in-oil emulsions for parallel amplification, followed by flow cytometry to detect and quantify mutant alleles.
Key Principles and Workflow
The following diagram illustrates the core BEAMing process for detecting mutant DNA molecules amidst a background of wild-type sequences:
Comparison with Other Detection Platforms
Selecting an appropriate ctDNA detection platform requires careful consideration of performance characteristics. The table below summarizes a comparative study of four platforms for KRAS mutation detection in metastatic colorectal cancer (mCRC) patient plasma and synthetic reference samples [61].
Table 1: Platform Comparison for KRAS Mutation Detection in Plasma cfDNA
| Platform | Technology | Sensitivity in mCRC Patients | Breadth of KRAS Targets | Maximum Sample Throughput | Total Annual Costs |
|---|---|---|---|---|---|
| BEAMing | Digital PCR (bead-based) | High (detected more mutations) | Moderate | Moderate | Highest |
| ddPCR | Digital PCR (droplet-based) | High (detected more mutations) | Moderate (G12/G13 screening) | High | Low |
| Idylla | Real-time PCR (cartridge-based) | Lower | Limited (predefined panel) | Low | Lowest |
| COBAS z480 | Real-time PCR (plate-based) | Lower | Limited (predefined panel) | High | Moderate |
This comparative study demonstrated that BEAMing and ddPCR detected more KRAS mutations among mCRC patients than Idylla and COBAS z480, highlighting their superior sensitivity for multiplex detection in challenging samples [61].
Essential Research Reagent Solutions
Successful implementation of BEAMing and related multiplex assays requires specific, high-quality reagents. The following table details key materials and their functions in the experimental workflow.
Table 2: Essential Research Reagents for BEAMing-based ctDNA Analysis
| Research Reagent | Function in Assay | Key Characteristics |
|---|---|---|
| Cell-free DNA BCT Tubes | Blood collection and plasma stabilization | Prevents cfDNA degradation and release of genomic DNA from blood cells during shipment and storage [61]. |
| Magnetic Streptavidin Beads | Solid support for PCR amplification | Beads serve as mobile solid support for clonal amplification of individual DNA molecules in emulsion microreactors [61]. |
| Biotinylated Primers | Target amplification and bead attachment | Designed for mutation hotspots; biotin group enables binding to streptavidin-coated beads for compartmentalized amplification [61]. |
| Emulsion Oil & Surfactants | Creation of water-in-oil microreactors | Forms stable emulsion compartments, each containing a single bead and DNA template for isolated PCR reactions [61]. |
| Mutation-Specific Fluorescent Probes | Detection of amplified mutant sequences | Fluorescently-labeled probes hybridize specifically to amplified mutant sequences on beads for detection by flow cytometry [61]. |
| Wild-Type-Specific Fluorescent Probes | Detection of amplified wild-type sequences | Differently labeled probes identify beads containing amplified wild-type sequences, enabling digital counting and ratio calculation [61]. |
| Fragmented Wild-Type gDNA | Assay control and standardization | Used in synthetic reference samples to establish assay sensitivity, specificity, and limit of detection by spiking with mutant sequences [61]. |
Detailed Experimental Protocol
Sample Collection and cfDNA Isolation
Materials:
- Cell-free DNA BCT tubes (Streck)
- QIAsymphony Circulating DNA Kit (Qiagen) or similar
- Microcentrifuge
Procedure:
- Blood Collection: Collect patient blood into 10 mL Cell-free DNA BCT tubes. Invert gently 8-10 times for mixing [61].
- Plasma Separation: Process tubes within 48 hours of collection.
- Centrifuge at 1700 × g for 10 minutes at room temperature.
- Transfer supernatant to a fresh tube without disturbing the buffy coat.
- Centrifuge at 20,000 × g for 10 minutes at room temperature.
- Transfer cell-free plasma to a new tube, avoiding any pellet [61].
- cfDNA Isolation: Isolate cfDNA from 4 mL plasma using the QIAsymphony Circulating DNA Kit according to manufacturer's instructions, eluting in 60 μL elution buffer [61].
- Quantity and Quality Control: Measure cfDNA concentration using a fluorometric method (e.g., Qubit). Analyze fragment size distribution using a BioAnalyzer High Sensitivity DNA kit to confirm expected cfDNA profile (peak ~160-170 bp) [61].
BEAMing Assay for Multiplex Mutation Detection
Materials:
- Magnetic streptavidin beads
- Biotinylated PCR primers for target mutations
- Emulsion oil and surfactants
- Mutation-specific and wild-type-specific fluorescent probes
- Thermal cycler
- Flow cytometer
Procedure:
- Primer-Bead Preparation:
- Incubate biotinylated primers with magnetic streptavidin beads according to manufacturer's instructions to create primer-bound beads.
- Wash beads to remove unbound primers.
Emulsion PCR Setup:
- Prepare PCR master mix containing:
- 10-50 ng isolated cfDNA
- Primer-bound beads
- PCR reagents (polymerase, dNTPs, buffer)
- Probe mixture
- Create a water-in-oil emulsion by vigorously mixing the aqueous PCR mix with emulsion oil using a homogenizer. Each aqueous microdroplet should contain an average of less than one bead and one DNA molecule [61].
- Prepare PCR master mix containing:
Emulsion PCR Amplification:
- Run the following thermocycling profile:
- Initial denaturation: 95°C for 5 min
- 45-50 cycles of:
- Denaturation: 95°C for 30 sec
- Annealing: 55-60°C for 30 sec
- Extension: 72°C for 30 sec
- Final extension: 72°C for 5 min
- Maintain emulsion stability throughout thermal cycling.
- Run the following thermocycling profile:
Emulsion Breaking and Recovery:
- Break the emulsion by adding a breaking solution and gentle mixing.
- Recover the beads by magnetic separation and wash thoroughly to remove oil and contaminants.
Fluorescent Hybridization:
- Hybridize beads with mutation-specific and wild-type-specific fluorescent probes.
- Use different fluorophores for mutant and wild-type probes (e.g., FAM for mutant, VIC for wild-type).
- Incubate at precise hybridization temperature to ensure allele-specific binding.
Flow Cytometry Analysis:
- Analyze beads using a flow cytometer equipped with appropriate lasers and filters.
- Count at least 1,000,000 beads per sample for reliable VAF quantification.
- Distinguish four bead populations: mutant-only, wild-type-only, double-positive (potential hybridisation errors), and negative.
Data Analysis and VAF Calculation:
- Calculate Variant Allele Frequency using the formula: VAF = (Number of mutant beads) / (Number of mutant beads + Number of wild-type beads) × 100%
- Apply background correction based on negative control samples.
The following workflow summarizes the key stages of the BEAMing protocol from sample to result:
Quality Control and Validation
Synthetic Reference Samples:
- Prepare fragmented wild-type genomic DNA using enzymatic fragmentation (e.g., dsDNA Fragmentase) or sonication [61].
- Spike with synthetic DNA fragments containing known KRAS mutations (e.g., G12A, G12C, G13D) at defined mutant allele frequencies (e.g., 0.50%, 0.04%, 0.02%) [61].
- Use these constructed references to validate assay sensitivity, specificity, and limit of detection across multiple replicates.
Controls:
- Include a wild-type only control (0% VAF) in each run to determine background signal and set thresholds for positivity [61].
- Use no-template controls to monitor for contamination.
Troubleshooting Guide
Table 3: Common BEAMing Assay Issues and Solutions
| Problem | Potential Cause | Solution |
|---|---|---|
| Low bead count | Emulsion instability; inefficient breaking | Optimize oil-to-aqueous phase ratio; ensure proper breaking solution preparation and mixing [61]. |
| High background signal | Non-specific probe binding; insufficient washing | Optimize hybridization temperature and time; increase wash stringency and number of washes. |
| Poor separation between mutant and wild-type populations | Suboptimal probe design; cross-hybridization | Redesign probes for better specificity; validate probe performance with control samples. |
| Low mutant recovery | Inefficient primer binding; PCR inhibition | Check primer design and concentration; add inhibition removal steps to DNA extraction. |
| Inconsistent results between replicates | Emulsion heterogeneity; pipetting errors | Standardize emulsion preparation protocol; use positive displacement pipettes for viscous solutions. |
Applications in Cancer Research and Drug Development
BEAMing technology enables multiple applications in oncology research:
- MRD Detection and Recurrence Monitoring: Tracking patient-specific mutations after curative-intent therapy provides a highly sensitive method for detecting molecular relapse before radiographic evidence [60].
- Therapy Response Monitoring: Serial ctDNA analysis during treatment can reveal emerging resistance mutations (e.g., KRAS mutations in colorectal cancer during EGFR inhibitor therapy), enabling timely treatment adaptation [12].
- Clinical Trial Biomarker Development: Utilizing BEAMing as a companion diagnostic in clinical trials can help identify patient subgroups most likely to respond to targeted therapies based on their mutation profile [60].
The high sensitivity and multiplexing capacity of BEAMing make it particularly valuable for these applications, especially when tracking multiple mutations across different genes to comprehensively monitor tumor evolution and therapy resistance.
Reducing Background Noise and False Positives
In the field of liquid biopsy, BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) has emerged as a highly sensitive approach for detecting circulating tumor DNA (ctDNA). This method enables the quantification of rare mutant DNA fragments present in blood plasma against a substantial background of wild-type DNA. A significant challenge in this sensitive detection is the prevalence of biological background noise and the risk of false positive results, which can directly impact clinical interpretation. This application note details the primary sources of this noise and provides validated protocols to enhance the specificity of BEAMing assays in ctDNA analysis.
The accurate detection of ctDNA is confounded by several technical and biological factors that introduce noise into the system. The table below summarizes the primary sources and their impact on BEAMing assays.
Table 1: Major Sources of Background Noise and False Positives in ctDNA Analysis
| Noise Source | Type | Impact on BEAMing Assay | Potential Consequence |
|---|---|---|---|
| Clonal Hematopoiesis (CH) | Biological | Somatic mutations in blood cells are detected in cell-free DNA (cfDNA) and misattributed to tumor origin [12]. | False positive results; inappropriate therapy decisions. |
| Sequencing Errors | Technical | Base misincorporation during amplification or sequencing mimics true low-frequency variants [12]. | Overestimation of variant allele frequency (VAF). |
| Low Abundance of ctDNA | Biological/Preamplification | The signal from mutant DNA fragments can be obscured by the high background of wild-type DNA, especially in early-stage disease [19]. | Reduced sensitivity; failure to detect true variants. |
Among biological confounders, clonal hematopoiesis (CH) is particularly notable. CH is part of the normal aging process and involves the accumulation of hematopoietic clones with somatic mutations in cancer-associated genes [12]. When these mutations are detected in plasma cfDNA without a matched tumor tissue sample for comparison, they can be misinterpreted as tumor-derived, leading to false positives [12]. This risk is especially pronounced in tumor-naïve (agnostic) testing approaches [12].
Strategies for Noise Reduction and Specificity Enhancement
Wet-Lab Optimizations
Refinements in the core BEAMing protocol are critical for suppressing noise.
- Utilization of High-Fidelity Polymerases: Employing DNA polymerases with proofreading activity (e.g., HotStart Phusion polymerase) during the initial amplification steps significantly reduces errors introduced by amplification, which is a major source of technical noise [3].
- Emulsion PCR for Physical Segregation: The core of BEAMing involves performing PCR within millions of individual water-in-oil emulsion droplets, each containing a single magnetic bead and ideally a single DNA molecule. This confines amplification, preventing cross-contamination and allowing for precise digital quantification [19] [3].
- Rigorous Pre-Analytical Controls: Standardizing blood sample processing is crucial. This includes plasma separation via double centrifugation (e.g., 820 × g for 10 min, followed by 16,000 × g for 10 min) to remove cellular debris and prevent contamination by genomic DNA from lysed blood cells [3].
In Silico and Analytical Filtration Methods
Post-sequencing, bioinformatic and analytical strategies are employed to enhance specificity.
- Paired White Blood Cell (WBC) Sequencing: The most effective strategy to mitigate clonal hematopoiesis is the parallel sequencing of matched white blood cells. Mutations identified in both cfDNA and WBCs can be filtered out as CH-derived, significantly reducing false positives [12].
- Unique Molecular Identifiers (UMIs): While more common in NGS, the principle of tagging individual DNA molecules with barcodes prior to amplification can be adapted. This allows for bioinformatic correction of PCR and sequencing errors by grouping and comparing reads originating from the same original molecule [62].
Table 2: Key Research Reagent Solutions for BEAMing Assays
| Reagent / Material | Function / Application | Key Characteristics |
|---|---|---|
| Streptavidin Magnetic Beads | Solid support for emulsion PCR; capture of biotinylated amplicons [3]. | Uniform size; high biotin-binding capacity. |
| High-Fidelity DNA Polymerase | Initial target amplification from plasma DNA template [3]. | Proofreading (3'→5' exonuclease) activity for high accuracy. |
| Emulsion Oil/Emulsifier Mixture | Generation of stable water-in-oil microreactors for compartmentalized PCR [3]. | Stable emulsion formation; compatible with PCR reagents. |
| Allele-Specific Fluorescent Probes | Hybridization-based detection of wild-type vs. mutant sequences on beads [3]. | High specificity; distinct fluorophores for multiplexing. |
Experimental Protocol: BEAMing with Noise Reduction
This protocol is adapted from studies on EGFR mutation detection in NSCLC and colorectal cancer monitoring [19] [3].
Workflow Overview:
Sample Preparation and DNA Extraction
- Blood Collection and Plasma Separation: Collect peripheral blood into EDTA tubes. Process within one hour of draw.
- Centrifuge at 820 × g for 10 minutes at 4°C to separate plasma.
- Transfer the supernatant plasma to a fresh tube and centrifuge at 16,000 × g for 10 minutes to pellet any remaining cellular debris [3].
- Aliquot and store the cleared plasma at -80°C until DNA extraction.
- cfDNA Extraction: Isolate cfDNA from 1-4 mL of plasma using a silica-membrane based kit (e.g., QIAamp DNA Micro Kit, Qiagen) according to the manufacturer's instructions. Elute in a low-volume elution buffer (e.g., 20-50 µL) [3].
- Parallel WBC DNA Extraction: Isolate genomic DNA from the initial blood cell pellet using a standard gDNA extraction kit. This will be used for subsequent CH filtering.
BEAMing PCR and Detection
- Initial Target Amplification: Perform the first PCR to amplify the genomic region of interest from plasma cfDNA.
- Reaction Setup: Set up multiple 25 µL reactions per sample. Each reaction should contain template DNA from a defined volume of plasma (e.g., 250 µL plasma equivalent), high-fidelity buffer, 1.5 U of HotStart Phusion polymerase, 0.2 µM of each gene-specific primer, 0.25 mM dNTPs, and 0.5 mM MgCl₂ [3].
- Thermal Cycling: 98°C for 30 s; 35 cycles of (98°C for 10 s, [Primer-Specific Tm, e.g., 57°C] for 10 s, 72°C for 10 s); final extension at 72°C for 5 min [3].
- Pool PCR products from the same sample and quantify.
- Emulsion PCR (BEAMing)
- Bead Preparation: Use ~6×10⁷ magnetic streptavidin beads coated with a 5'-biotinylated capture oligonucleotide [3].
- Create Emulsion: Combine the amplified DNA template, beads, PCR reagents, and Taq polymerase with an oil/emulsifier mixture (e.g., 7% ABIL WE 09, 20% mineral oil, 73% TegoSoft DEC). Shake vigorously on a TissueLyser to create a stable microemulsion where each aqueous compartment contains, on average, less than one bead and one DNA molecule [3].
- Emulsion PCR: Dispense the emulsion into a PCR plate and run the amplification program with a touchdown or constant annealing temperature protocol [3].
- Bead Recovery and Hybridization
- Break Emulsion: Add a breaking buffer (e.g., containing Triton-X-100 and SDS) to the reacted emulsion and mix to coalesce the aqueous phases. Pellet the beads by centrifugation [3].
- Denature DNA: Treat beads with 0.1 M NaOH to denature the double-stranded DNA, leaving single-stranded DNA bound to the beads via the biotin-streptavidin linkage [3].
- Allele-Specific Hybridization: Incubate the beads with fluorescently labeled probes complementary to the wild-type and mutant sequences. Use probes of 15-18 nt length, designed for different mutations (e.g., for EGFR L858R, exon 19 del) [3].
- Flow Cytometry and Quantification
- Analyze the beads using a flow cytometer (e.g., FACSAria III) to distinguish between beads bound to mutant probes, wild-type probes, or both.
- The fraction of mutant DNA fragments is calculated as (number of mutant beads) / (number of mutant + wild-type beads). The absolute concentration of mutant fragments can be determined by multiplying this fraction by the total concentration of DNA fragments of the gene, as determined by a separate quantitative PCR assay [19].
Data Analysis and CH Filtering Workflow
- Cross-Referencing with WBC Sequencing Data: For any mutation identified in the plasma cfDNA via BEAMing, cross-reference it with the sequencing data obtained from the patient's matched WBC-DNA.
- Variant Classification: Mutations found in both cfDNA and WBC-DNA are classified as originating from clonal hematopoiesis and are filtered out from the final tumor-associated variant list [12]. Only mutations found in cfDNA but absent in WBC-DNA are reported as tumor-derived ctDNA.
BEAMing technology provides a powerful and highly sensitive platform for the digital detection of ctDNA. The reliability of its results, however, is critically dependent on systematic efforts to reduce background noise and false positives. By integrating the outlined wet-lab optimizations—particularly the mandatory inclusion of paired white blood cell sequencing for CH filtering—researchers and clinical developers can significantly improve the specificity and clinical utility of their BEAMing assays, enabling more accurate tumor genotyping, disease monitoring, and treatment decision-making.
Quality Assurance and Standardization Protocols
BEAMing (Beads, Emulsions, Amplification, and Magnetics) represents a highly sensitive digital PCR-based methodology for detecting and quantifying circulating tumor DNA (ctDNA) in blood samples from cancer patients. This technology addresses the fundamental challenge of identifying rare mutant DNA fragments within a vast excess of wild-type DNA, enabling detection sensitivity as low as 0.01% mutant allele frequency [5] [63]. As a liquid biopsy approach, BEAMing enables non-invasive assessment of tumor-associated mutations, monitoring of treatment response, and identification of emerging resistance mechanisms without the need for repeated tissue biopsies [5] [2].
The clinical validity of BEAMing technology has been extensively demonstrated in colorectal cancer, where OncoBEAM RAS testing shows 93.3% overall concordance with standard tissue testing methods [5]. This high concordance, combined with rapid turnaround times, positions BEAMing as a valuable tool for molecular profiling in advanced cancer patients, particularly when tissue samples are unavailable or insufficient for comprehensive genomic analysis [5]. The quality assurance and standardization protocols outlined in this document provide a framework for implementing BEAMing technology in clinical research settings, ensuring reliable, reproducible results across laboratories and over time.
Quality Assurance Parameters and Performance Standards
Analytical Performance Metrics
Robust quality assurance for BEAMing technology requires strict adherence to predefined performance metrics across multiple parameters. These standards ensure the technology meets the sensitivity and specificity requirements necessary for reliable ctDNA analysis in clinical research contexts.
Table 1: Key Analytical Performance Metrics for BEAMing Technology
| Parameter | Performance Standard | Validation Method |
|---|---|---|
| Sensitivity | Detection of mutant alleles at 0.01% allele frequency [5] | Dilution series of mutant DNA in wild-type background |
| Specificity | >99% agreement with known wild-type samples [63] | Analysis of healthy donor samples and confirmed wild-type controls |
| Concordance with Tissue | 91.8-93.3% for RAS testing in colorectal cancer [5] | Comparison against standard tissue testing results |
| Precision | <10% coefficient of variation for replicate samples | Intra-assay and inter-assay replication studies |
| Dynamic Range | Linear quantification from 0.01% to 100% mutant allele frequency [63] | Analysis of samples with known mutation percentages |
Pre-analytical Quality Controls
Pre-analytical variables significantly impact BEAMing assay performance and must be standardized to ensure result reliability. Blood collection protocols should utilize specialized blood collection tubes (BCTs) with cell-stabilizing preservatives, such as Streck cfDNA tubes, PAXgene Blood ccfDNA tubes (Qiagen), or similar products, which maintain ctDNA stability for up to 3-7 days at room temperature [38] [25]. Plasma separation should follow a standardized dual-centrifugation protocol: initial low-speed centrifugation (800-1,900 × g for 10 minutes) to pellet blood cells, followed by high-speed centrifugation (12,000-16,000 × g for 10 minutes) to remove remaining cellular debris [38] [25]. Plasma should be aliquoted and stored at -80°C to preserve ctDNA integrity, with freeze-thaw cycles minimized to prevent nucleic acid degradation [38].
Standardized BEAMing Workflow Protocol
Sample Preparation and DNA Extraction
The initial phase of the BEAMing protocol focuses on obtaining high-quality ctDNA from patient blood samples while minimizing contamination with genomic DNA from blood cells.
Blood Collection and Processing: Collect peripheral blood (2 × 10 mL recommended for single-analyte testing) into appropriate BCTs using butterfly needles to minimize hemolysis [38]. Process samples within the stability window for the chosen collection tube (within 2-6 hours for EDTA tubes; up to 7 days for specialized BCTs) [38] [25]. Perform plasma separation using the standardized dual-centrifugation protocol and store aliquots at -80°C until DNA extraction.
ctDNA Extraction: Extract ctDNA using silica membrane-based columns (e.g., QIAamp Circulating Nucleic Acid Kit) or magnetic bead-based systems (e.g., QIAamp MinElute ccfDNA Mini Kit) [38] [25]. Silica membrane methods generally yield more ctDNA, while magnetic bead systems offer advantages for automated processing and recovery of smaller DNA fragments [25]. Quantify extracted DNA using fluorometric methods and assess fragment size distribution to confirm typical ctDNA characteristics (peak ~170 bp) [2].
BEAMing Process Steps
The core BEAMing technology involves partitioning individual DNA molecules into water-in-oil emulsions for clonal amplification, followed by flow cytometric analysis to detect and quantify mutations.
Pre-Amplification: Perform limited-cycle (typically 15-25 cycles) conventional PCR to amplify target regions of interest using primers incorporating known tag sequences [63]. This step enriches for target sequences while introducing sequence tags necessary for subsequent emulsion PCR.
Emulsion PCR Preparation: Incubate pre-amplified DNA products with streptavidin-coated magnetic beads that have been conjugated to biotinylated primers complementary to the sequence tags [63]. Mix the bead-DNA complex with PCR reagents and oil to create water-in-oil microemulsions, with droplet size calibrated to contain an average of one bead and one DNA template molecule per droplet [64] [63].
Emulsion PCR Amplification: Perform PCR amplification within the emulsion droplets, resulting in clonal amplification where each bead becomes coated with thousands of copies of a single DNA molecule [63]. Break the emulsions after amplification and recover the beads using magnetic separation [64].
Mutation Detection: Hybridize bead-bound DNA with mutation-specific fluorescent probes designed to distinguish wild-type from mutant sequences [63]. Use flow cytometry to quantify the ratio of mutant to wild-type beads, enabling digital quantification of the original mutant DNA fraction in the patient sample [63].
The BEAMing technology workflow illustrating the multi-step process from blood collection to digital quantification of mutant DNA fragments.
Research Reagent Solutions and Essential Materials
Implementation of BEAMing technology requires specific high-quality reagents and materials to ensure optimal assay performance and minimize technical variability.
Table 2: Essential Research Reagents for BEAMing Technology
| Reagent/Material | Function | Recommendation |
|---|---|---|
| High-Fidelity DNA Polymerase | PCR amplification with minimal errors | Phusion High-Fidelity DNA Polymerase (50-fold lower error rate than Taq) [63] |
| Specialized Blood Collection Tubes | Preserve ctDNA integrity during transport | Streck cfDNA BCT, PAXgene Blood ccfDNA tubes [38] [25] |
| Silica-Membrane Extraction Kits | High-yield ctDNA isolation | QIAamp Circulating Nucleic Acid Kit (Qiagen) [38] [25] |
| Magnetic Beads | Solid support for emulsion PCR | Streptavidin-coated paramagnetic beads (1-2μm diameter) [64] [63] |
| Mutation-Specific Probes | Detect mutant vs. wild-type sequences | Fluorescently-labeled hybridization probes with appropriate quenching [63] |
| Emulsion Oil Phase | Create water-in-oil microreactors | Surfactant-stabilized oil formulations [64] |
Applications in Clinical Research and Drug Development
BEAMing technology provides valuable applications across multiple domains of cancer research and therapeutic development, enabled by its sensitive quantification of ctDNA.
Treatment Response Monitoring: BEAMing enables real-time assessment of tumor dynamics during therapy through serial ctDNA measurements. Research demonstrates that ctDNA levels decrease rapidly following effective surgical resection (99% median decrease) with an estimated half-life of approximately 114 minutes [19]. In patients undergoing systemic therapy, rising ctDNA levels often indicate emerging treatment resistance or disease progression before radiographic evidence [2].
Minimal Residual Disease Detection: The exceptional sensitivity of BEAMing technology enables detection of minimal residual disease following curative-intent therapy. Studies show that ctDNA positivity after completion of therapy predicts recurrence with high accuracy, potentially identifying patients who might benefit from additional treatment interventions [2].
Clinical Trial Enrichment: BEAMing technology facilitates patient stratification for clinical trials by non-invasively identifying specific mutations required for eligibility [63]. This approach was successfully implemented in metastatic breast cancer research, where BEAMing detected PIK3CA mutations in ctDNA with 100% concordance with tumor tissue results [63].
Troubleshooting and Technical Optimization
Successful implementation of BEAMing technology requires attention to potential technical challenges and systematic optimization of key parameters.
Reduced Sensitivity: If mutant detection sensitivity declines below the 0.01% threshold, verify emulsion quality and droplet size distribution to ensure proper compartmentalization [63]. Assess DNA polymerase fidelity and consider implementing unique molecular identifiers (UMIs) to distinguish true mutations from amplification artifacts [2].
High Background Signal: Excessive background fluorescence in flow cytometry can result from non-specific probe hybridization. Optimize hybridization stringency conditions and include appropriate negative controls (wild-type only samples) to establish background thresholds [63].
Low Bead Recovery: Inefficient bead recovery after emulsion breaking reduces counting statistics and assay precision. Ensure proper functioning of magnetic separation equipment and optimize surfactant concentrations in the breaking solution [64].
Standardization of BEAMing technology across research laboratories requires meticulous attention to pre-analytical variables, reagent quality, and technical execution. The protocols outlined herein provide a foundation for implementing this powerful ctDNA analysis platform, enabling reliable detection and quantification of tumor-specific mutations in blood samples from cancer patients.
Troubleshooting Common Technical Failures
Beads, Emulsion, Amplification, and Magnetics (BEAMing) is a highly sensitive digital PCR-based technology that enables the detection and quantification of rare mutant DNA sequences, such as circulating tumor DNA (ctDNA), within a vast background of wild-type DNA from plasma. This technology is pivotal for applications in oncology, including cancer diagnosis, monitoring treatment response, and detecting minimal residual disease (MRD), as it can identify specific mutations with high precision [4] [3]. The core principle involves converting single DNA molecules into magnetic beads bound with thousands of copies of the original sequence, which are then analyzed via flow cytometry to distinguish mutant from wild-type alleles [3]. Despite its power, the BEAMing workflow is technically complex, and its success is susceptible to failures at multiple stages, from sample collection to data analysis. This document outlines common technical failures encountered during BEAMing for ctDNA analysis and provides detailed troubleshooting protocols to ensure robust and reliable results for researchers and drug development professionals.
Common Technical Failures and Troubleshooting Protocols
The successful application of BEAMing technology relies on meticulous attention to pre-analytical, analytical, and post-analytical variables. The following sections detail specific failure points, their underlying causes, and step-by-step corrective actions.
Pre-Analytical Stage: Sample Collection and Plasma Preparation
The integrity of ctDNA is paramount and can be compromised before analysis even begins.
- Failure Point: Degradation of ctDNA and contamination by genomic DNA from lysed blood cells.
- Root Cause: Improper blood collection, handling, or storage conditions can lead to cell lysis, releasing a large amount of wild-type genomic DNA that dilutes the already scarce ctDNA, lowering the mutant allele frequency below the detection limit [36].
- Troubleshooting Protocol:
- Collection: Draw blood into cfDNA-stabilizing tubes (e.g., Streck cfDNA BCT) and invert gently 8-10 times to mix the preservative. Do not use EDTA tubes for prolonged storage before processing [36].
- Processing Time: Centrifuge blood samples within 1-2 hours of collection. If using stabilizing tubes, process within 24-48 hours as per manufacturer's guidelines [36].
- Centrifugation: Perform a two-step centrifugation protocol.
- Storage: Aliquot the clarified plasma to avoid freeze-thaw cycles and store at -80°C until DNA extraction.
Analytical Stage: Emulsion PCR and BEAMing Reaction
The core BEAMing reaction is sensitive to reagent quality and reaction conditions.
- Failure Point: Low emulsion stability or poor PCR efficiency, leading to a low yield of bead-bound amplicons.
- Root Cause: Inconsistent emulsion formation due to improper oil-to-aqueous phase ratios, suboptimal PCR cycling conditions, or poor-quality enzymes and reagents.
- Troubleshooting Protocol:
- Emulsion Formation:
- Ensure the oil/emulsifier mixture is prepared correctly (e.g., 7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC) [3].
- Use a TissueLyser or similar homogenizer for consistent and vigorous shaking to create a stable microemulsion. Shake for 10 seconds at 15 Hz, followed by 7 seconds at 17 Hz [3].
- Visual Inspection: Check the emulsion under an inverted microscope at 40x magnification. A successful emulsion will show uniform, aqueous compartments dispersed in the oil phase [3].
- PCR Amplification:
- Use a high-fidelity, hot-start DNA polymerase (e.g., HotStart Phusion polymerase) to minimize non-specific amplification [3].
- Verify the purity and concentration of primers and template DNA using a spectrophotometer (e.g., Nanodrop) [3].
- Adhere strictly to the PCR cycling conditions, which typically involve an initial denaturation at 98°C, followed by 35 cycles of denaturation (98°C), annealing (57-68°C, gradient optimized), and extension (72°C) [3].
- Emulsion Formation:
Post-Analytical Stage: Bead Recovery and Flow Cytometry
The final stages are critical for accurate quantification of mutant alleles.
- Failure Point: High background noise, non-specific signals, or low bead count during flow cytometry analysis.
- Root Cause: Incomplete breaking of the emulsion, inefficient washing of beads, or degradation of fluorescently labeled probes.
- Troubleshooting Protocol:
- Emulsion Breaking:
- Use a breaking buffer containing Triton-X-100 and SDS to effectively disrupt the emulsion.
- Ensure thorough mixing with a TissueLyser at 20 Hz for 20 seconds to recover all magnetic beads [3].
- Bead Washing:
- Perform multiple washes with wash buffer (e.g., 20 mM Tris-HCl, pH 8.4, 50 mM KCl) to remove excess reagents, surfactants, and unincorporated probes [3].
- Use a magnetic separator to efficiently pellet and wash the beads between steps.
- Allele-Specific Hybridization:
- Design fluorescently labeled probes (15-18 nt) complementary to both mutant and wild-type sequences with high specificity [3].
- Include stringent washing steps after hybridization to remove mismatched probes.
- Protect fluorescent probes from light to prevent photobleaching.
- Emulsion Breaking:
Quantitative Performance Data and Validation
Robust validation is required to define the operational limits of the BEAMing assay. The following tables summarize key performance metrics.
Table 1: Sensitivity and Specificity of BEAMing PCR for EGFR Mutation Detection [3]
| Parameter | Exon 19 | Exon 20 | Exon 21 (L858R) | Exon 21 (L861Q) |
|---|---|---|---|---|
| Concordance with EMR-qPCR (%) | 98.8 | 98.9 | 95.5 | N/A |
| Concordance with Diatech qPCR (%) | 90.0 | 100 | 96.0 | 98.0 |
| Cohen's Kappa | Significant (p < 0.001) | Significant (p < 0.001) | Significant (p < 0.001) | Significant (p < 0.001) |
Table 2: Key Reagent Solutions for BEAMing RT-PCR
| Research Reagent | Function / Role in Experiment | Example / Specification |
|---|---|---|
| cfDNA Stabilizing Tubes | Preserves blood sample integrity by preventing white blood cell lysis and cfDNA degradation during transport and storage. | Streck cfDNA BCT [36] |
| High-Fidelity DNA Polymerase | Amplifies target DNA sequences from plasma with high accuracy and minimal errors during the initial PCR step. | HotStart Phusion Polymerase (NEB) [3] |
| Magnetic Streptavidin Beads | Serve as solid support for PCR amplification; bind biotinylated primers to isolate and quantify individual DNA molecules. | MyOne Streptavidin C1 Beads (Invitrogen) [3] |
| Emulsifier/Oil Mixture | Creates stable water-in-oil microreactors for digital PCR, enabling single-molecule amplification on beads. | ABIL WE09, Mineral Oil, TegoSoft DEC [3] |
| Fluorescently Labeled Probes | Enable allele-specific detection and differentiation of mutant vs. wild-type sequences via flow cytometry. | 15-18 nt probes, dual-color FACS system [3] |
Workflow and Pathway Diagrams
The following diagrams illustrate the core BEAMing workflow and the critical emulsion formation process.
Diagram 1: BEAMing Workflow. The process from plasma isolation to mutant allele quantification.
Diagram 2: Emulsion Formation. Critical step creating microreactors for single-molecule PCR.
BEAMing Performance: Validation Studies and Technology Comparisons
Analytical validation is a critical prerequisite for the clinical application of any liquid biopsy method, establishing that an test accurately and reliably measures the intended analyte. For BEAMing (Beads, Emulsions, Amplification, and Magnetics) technology used in circulating tumor DNA (ctDNA) analysis, this process quantitatively demonstrates that the assay can detect tumor-specific mutations with high sensitivity, specificity, and reproducibility. BEAMing technology addresses the fundamental challenge of identifying rare mutant DNA molecules within a vast excess of wild-type circulating cell-free DNA (cfDNA) by combining emulsion PCR with flow cytometry to digitally detect and quantify specific mutations. This technical foundation enables BEAMing to detect mutated ctDNA at allele frequencies as low as 0.01%, making it one of the most sensitive platforms available for liquid biopsy applications [5]. The clinical imperative for such high sensitivity is clear: studies have shown that ctDNA can constitute less than 0.025% of total cfDNA in patients with early-stage disease, and plasma from patients with small tumor burden often contains vanishingly low amounts of ctDNA, sometimes less than 1-100 copies per 1 mL of plasma [38]. This protocol outlines the comprehensive analytical validation of BEAMing assays for ctDNA analysis, providing researchers and drug development professionals with standardized methods to establish assay performance characteristics essential for clinical research and diagnostic applications.
Performance Characteristics of BEAMing Technology
Sensitivity and Specificity Profiles
Sensitivity and specificity form the cornerstone of analytical validation, defining an assay's ability to correctly identify true positive and true negative samples, respectively. For BEAMing-based ctDNA analysis, sensitivity is defined as the lowest allele frequency at which a mutation can be reliably detected, while specificity reflects the assay's capacity to distinguish true mutations from background artifacts. In head-to-head comparisons, BEAMing technology has demonstrated a limit of detection (LOD) of 0.01% mutant allele frequency, enabling identification of rare mutant molecules in patient plasma [5]. Clinical studies utilizing molecular amplification pools (MAPs) for error-reduction in ctDNA sequencing—a methodological approach related to BEAMing—have reported sensitivity of 98.5% and specificity of 98.9% when validated against droplet digital PCR (ddPCR) as a reference method [65].
The exceptional sensitivity of BEAMing stems from its technical design: the platform partitions individual DNA molecules into water-in-oil microemulsions, where each molecule is amplified in isolation and subsequently bound to magnetic beads. This compartmentalization creates a "digital" readout where each bead corresponds to a single starting DNA molecule, allowing precise quantification of mutant and wild-type alleles through flow cytometry [5]. This approach overcomes the limitations of conventional PCR, where rare mutant sequences can be obscured by the amplification background of abundant wild-type DNA.
Table 1: Analytical Sensitivity and Specificity of BEAMing Technology in Clinical Studies
| Study | Cancer Type | Number of Samples | Sensitivity | Specificity | Concordance with Tissue |
|---|---|---|---|---|---|
| Bettegowda et al. [5] | Multiple solid tumors | Not specified | 0.01% LOD (mutant allele frequency) | Not specified | Not specified |
| Thierry et al. [5] | mCRC | 106 | 92.6% (positive percent agreement) | 94% (negative percent agreement) | 91.8% |
| Clinical Evaluation [5] | mCRC | 238 | Not specified | Not specified | 93.3% |
| MAPs Sequencing [65] | Lung cancer | 356 | 98.5% | 98.9% | Similar to ddPCR (0.1% AF) |
Reproducibility and Concordance Data
Reproducibility, encompassing both inter-laboratory and intra-assay consistency, represents another critical metric for analytical validation. For ctDNA assays, reproducibility remains particularly challenging for samples with ultra-low ctDNA content, highlighting the importance of interlaboratory harmonization of testing procedures [38]. BEAMing technology has demonstrated strong concordance with standard tissue-based testing methods across multiple studies. A large performance evaluation across six European centers reported 93.3% overall concordance between OncoBEAM RAS CRC testing and standard-of-care tissue testing for 238 metastatic colorectal cancer (mCRC) patients [5]. In this evaluation, plasma RAS mutations were detected in 112 of 121 KRAS mutant cases identified by tissue-based testing (92.6% positive percent agreement), while no RAS mutations were found in 110 of 117 tissue-negative cases (94% negative percent agreement) [5].
The reproducibility of BEAMing is facilitated by its standardized workflow and quantitative output format. Unlike sequencing-based methods that may require complex bioinformatic pipelines for variant calling, BEAMing provides direct digital quantification of mutant alleles through flow cytometry, reducing potential sources of technical variability. Furthermore, the technology's ability to assess hundreds of millions of individual DNA molecules with standard laboratory equipment contributes to its robust performance across different testing environments [5].
Experimental Protocols
Pre-Analytical Phase: Blood Collection and Plasma Processing
The pre-analytical phase is critically important for ctDNA analysis, as improper specimen collection or handling can significantly impact assay performance through contamination with genomic DNA from lysed blood cells.
Table 2: Blood Collection and Plasma Processing Protocols for BEAMing ctDNA Analysis
| Procedure Step | Recommendation | Technical Notes | References |
|---|---|---|---|
| Blood Collection | Use butterfly needles; avoid thin needles/prolonged tourniquet | 2×10 mL blood for single-analyte LB; larger volumes for MRD | [38] |
| Collection Tubes | EDTA tubes (process within 2-6 hours at 4°C) or cell-stabilizing BCTs (e.g., Streck, PAXgene) | BCTs allow room temperature storage/transport for up to 7 days | [38] |
| Centrifugation | Double centrifugation: 1st: 380-3,000g for 10min (RT); 2nd: 12,000-20,000g for 10min (4°C) | Single low-speed centrifugation for PEG-mediated enrichment | [38] |
| Plasma Storage | At -80°C | 10 years for mutation detection; 9 months for quantitative analysis | [38] |
| ctDNA Extraction | Silica membrane columns (e.g., QIAamp Circulating Nucleic Acid Kit) or magnetic beads | Silica membranes yield more ctDNA than magnetic beads | [38] |
BEAMing Assay Protocol
The BEAMing protocol transforms the challenge of detecting rare mutations into a digital detection problem through compartmentalized amplification.
Step 1: DNA Extraction and Quantification
- Extract ctDNA from plasma using a validated method (e.g., silica membrane columns).
- Quantify total cfDNA using fluorometric methods (e.g., Qubit dsDNA HS Assay).
- Verify DNA fragment size distribution (expected peak ~166 bp) using bioanalyzer or tape station.
Step 2: Target Amplification and Primer Design
- Design PCR primers to amplify regions containing mutations of interest (e.g., KRAS codons 12, 13, 59, 61, 117, 146; NRAS homologous regions).
- For the OncoBEAM RAS CRC assay, design primers to detect 34 mutations across KRAS and NRAS oncogenes.
- Perform initial amplification of target regions from ctDNA using biotinylated primers.
Step 3: Emulsion PCR Preparation
- Prepare water-in-oil emulsions containing PCR reagents, magnetic beads with streptavidin coating, and amplified DNA fragments.
- Optimize emulsion conditions to ensure most droplets contain either one DNA molecule or no DNA molecules (Poisson distribution).
- The emulsion system creates millions of individual microreactors where each bead captures a single DNA molecule.
Step 4: Emulsion PCR Amplification
- Perform PCR amplification within the emulsion droplets.
- Each bead captures amplicons from a single DNA molecule, creating clonal populations on each bead.
- After amplification, break the emulsion and purify the beads containing amplified DNA.
Step 5: Mutation Detection by Hybridization
- Incubate beads with fluorescently labeled probes specific for wild-type and mutant sequences.
- Use different fluorophores for wild-type and mutant probes to enable discrimination.
- Include appropriate controls: known wild-type samples, mutant controls at various allele frequencies (e.g., 0.01%, 0.1%, 1%), and no-template controls.
Step 6: Flow Cytometry Analysis
- Analyze beads using flow cytometry to quantify the ratio of mutant to wild-type beads.
- Establish gating strategies to distinguish mutant-positive, wild-type-positive, and negative beads.
- Calculate mutant allele frequency as: (number of mutant beads)/(number of mutant + wild-type beads) × 100.
Step 7: Data Analysis and Quality Control
- Implement quality control metrics including minimum bead count (typically >100,000 beads per reaction), threshold for background signal, and concordance with expected control values.
- For the OncoBEAM RAS CRC test, validate against established standards to ensure detection of mutations at 0.01% allele frequency with high specificity [5].
BEAMing Workflow: Sample to Result
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for BEAMing ctDNA Analysis
| Reagent/Category | Function | Specific Examples |
|---|---|---|
| Blood Collection Tubes with Stabilizers | Preserve blood cell integrity during transport/storage; prevent wild-type gDNA release | cfDNA BCT (Streck), PAXgene Blood ccfDNA (Qiagen), Roche cfDNA tubes [38] |
| Nucleic Acid Extraction Kits | Isolate high-quality ctDNA from plasma; minimize fragmentation | QIAamp Circulating Nucleic Acid Kit (silica membrane), Maxwell RSC ccfDNA (magnetic beads) [38] |
| BEAMing Reaction Components | Enable compartmentalized amplification and detection | Streptavidin-coated magnetic beads, biotinylated primers, emulsion oils, fluorescent hybridization probes [5] |
| Reference Standards | Validate assay performance; establish LOD/LOL | Horizon Discovery multiplex reference standards, Seraseq ctDNA controls [5] |
| qPCR/ddPCR Reagents | Orthogonal validation; absolute quantification | Bio-Rad ddPCR supermix, EGFR/RAS mutation assays [65] |
Methodological Visualizations
Analytical Validation Framework
Discussion and Technical Considerations
Addressing Pre-Analytical Variables
The impact of pre-analytical variables on BEAMing assay performance cannot be overstated. Physiological and pathological factors significantly influence ctDNA levels and quality. Chronic or acute diseases (e.g., diabetes, kidney disease, inflammation), surgical trauma (causing transient ccfDNA increases for weeks), and even circadian dynamics (higher ctDNA levels at night) can affect ctDNA measurements [38]. Furthermore, approaches to stimulate transient ctDNA release before blood collection—including irradiation (ctDNA spike 6-24 hours post-procedure), ultrasound (sonobiopsy for brain tumors), and mechanical stress (mammography for breast cancer)—have shown promise for enhancing detection sensitivity [38]. These factors must be standardized and reported to ensure reproducible results across studies and clinical implementations.
Comparison with Alternative Methodologies
BEAMing occupies a distinct position in the landscape of ctDNA detection technologies, offering a different balance of sensitivity, multiplexing capability, and workflow complexity compared to other methods.
Table 4: Comparison of BEAMing with Other ctDNA Detection Technologies
| Method | Detection Limit | Advantages | Limitations |
|---|---|---|---|
| BEAMing | 0.01% | High sensitivity; digital quantification; clinical validation | Limited to known mutations; moderate throughput |
| ddPCR | 0.01% | Absolute quantification; high sensitivity; easy implementation | Limited multiplexing; targeted approach only |
| Targeted NGS (CAPP-Seq, Safe-Seq) | 0.01-2.0% | Broader mutation screening; discovery capability | Higher cost; complex bioinformatics; personalization needed |
| Whole Exome/Genome Sequencing | 1-5% | Comprehensive; no prior knowledge needed | Low sensitivity; high cost; extensive data analysis |
BEAMing technology demonstrates particular strength in clinical applications where high sensitivity for a defined set of mutations is required, such as expanded RAS testing in colorectal cancer or EGFR mutation testing in non-small cell lung cancer [5]. The technology's high concordance with tissue-based testing (93.3% in multi-center studies) and ability to detect emerging resistance mutations make it particularly valuable for monitoring treatment response and disease evolution [5] [2].
Reproducibility Challenges and Solutions
Despite its robust performance, several factors can affect the reproducibility of BEAMing assays, particularly in multi-center studies. The reproducibility of ctDNA-based liquid biopsy assays remains insufficient for samples with ultra-low ctDNA content, highlighting the need for interlaboratory harmonization of testing procedures [38]. Several approaches can enhance reproducibility:
- Standardized blood collection protocols using validated blood collection tubes
- Implementation of reference standards at various allele frequencies to monitor assay performance
- Harmonized DNA extraction methods to maximize yield and minimize fragmentation
- Rigorous quality control metrics including minimum bead counts and background mutation thresholds
- Blinded validation studies across multiple sites to identify and address sources of variability
These measures are particularly important when implementing BEAMing for minimal residual disease (MRD) detection, where ctDNA levels are minimal and technical variability can significantly impact clinical interpretation.
Liquid biopsy, particularly the analysis of circulating tumor DNA (ctDNA), has emerged as a transformative, minimally invasive approach for cancer genomic profiling. The BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) represents one of the most clinically validated platforms for ctDNA analysis, demonstrating remarkable sensitivity in detecting tumor-specific mutations. This application note synthesizes evidence from major studies evaluating the concordance between BEAMing-based ctDNA analysis and traditional tissue biopsy, providing researchers and drug development professionals with critical insights for implementing this technology in precision oncology workflows.
Clinical Concordance Evidence Across Cancer Types
Extensive clinical validation studies have established that BEAMing technology demonstrates high concordance with standard tissue biopsy across multiple cancer types, positioning it as a reliable alternative when tissue is unavailable or insufficient.
Table 1: Concordance Rates of BEAMing ctDNA Analysis Versus Tissue Biopsy Across Cancer Types
| Cancer Type | Study Cohort Size | Key Genes Analyzed | Overall Concordance | Positive Percent Agreement | Negative Percent Agreement | Citation |
|---|---|---|---|---|---|---|
| Colorectal Cancer | 238 patients | KRAS, NRAS (34 mutations) | 93.3% | 92.6% | 94.0% | [5] |
| Early-Stage Breast Cancer | 246 patients | TP53, PIK3CA, AKT1, ERBB2 | 25.6% | 29.6% (detection rate) | 81.7% (tissue detection rate) | [66] |
| Advanced Biliary Tract Cancer | 102 patients | FGFR2, IDH1, ERBB2, PIK3CA | 84.8% sensitivity | 79.4% PPV | N/R | [67] |
The variable concordance in early-stage breast cancer (25.6%) highlights a fundamental biological constraint: tumors with lower burden and earlier disease stages shed less ctDNA into circulation, making detection more challenging. This limitation is less pronounced in advanced cancers, where tumor burden is higher and ctDNA is more abundant [66].
BEAMing Technology: Principles and Workflow
BEAMing addresses the technical challenge of identifying rare DNA molecules by combining emulsion PCR with flow cytometry to achieve high-resolution detection of mutant DNA sequences amidst a predominance of wild-type DNA.
Core Technological Principles
BEAMing technology enables the detection of ctDNA mutations at frequencies as low as 0.01%, far surpassing the sensitivity of conventional PCR methods [5]. This exceptional sensitivity is achieved through:
- Compartmentalized Amplification: Individual DNA molecules are separated into water-in-oil microemulsions, with each droplet containing approximately one DNA template and one magnetic bead, ensuring clonal amplification without cross-contamination [64].
- Sequence-Specific Detection: Amplified products bound to magnetic beads are analyzed via flow cytometry using mutation-specific fluorescent probes, allowing precise discrimination between wild-type and mutant sequences [5].
- Digital Quantification: Each bead corresponds to a single DNA molecule from the original sample, enabling absolute quantification of mutant allele frequency in the circulation [64].
Detailed Experimental Protocol
Protocol: BEAMing-Based ctDNA Analysis for RAS Mutations in Colorectal Cancer
Based on the OncoBEAM RAS CRC CE-IVD platform
Step 1: Plasma Collection and cfDNA Extraction
- Collect whole blood in EDTA-containing tubes. Process within 2 hours of collection to prevent leukocyte lysis and contamination with genomic DNA.
- Perform double centrifugation: first at 1,600×g for 10 minutes, then transfer supernatant and centrifuge at 16,000×g for 10 minutes at 4°C to remove residual cells [66].
- Extract cfDNA from 4-5 mL plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen). Elute in 50-100 µL AVE buffer. Quantify using Qubit dsDNA HS Assay [66].
Step 2: BEAMing Reaction Setup
- Prepare first-round PCR master mix containing:
- 20 ng cfDNA (minimum input)
- RAS-specific primers (covering KRAS/NRAS codons 12, 13, 59, 61, 117, 146)
- DNA polymerase with proofreading capability
- dNTPs, MgCl₂, and reaction buffer
- Partition the PCR reaction into water-in-oil microemulsions using vigorous vortexing with oil-surfactant mixture. Each aqueous compartment should contain approximately one DNA template and one streptavidin-coated magnetic bead (1.0 µm diameter) conjugated with biotinylated PCR primers [64].
Step 3: Emulsion PCR Amplification
- Perform thermal cycling as follows:
- Initial denaturation: 95°C for 5 minutes
- 45-50 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: 58-60°C for 45 seconds
- Extension: 72°C for 60 seconds
- Final extension: 72°C for 7 minutes
- Break emulsions using butanol or direct disruption method. Recover beads using magnetic separation [64].
Step 4: Hybridization and Flow Cytometry
- Incubate beads with mutation-specific fluorescent probes for KRAS and NRAS mutations.
- Use wild-type-specific probes with different fluorophores to distinguish mutant and wild-type populations.
- Analyze using flow cytometry with a minimum of 1,000,000 beads per sample to ensure adequate detection sensitivity [5].
- Set threshold for mutant allele calling at 0.01% of total beads analyzed.
Step 5: Data Analysis and Interpretation
- Calculate mutant allele frequency (MAF) as: (Mutant beads / Total beads) × 100
- For clinical reporting, establish a validated cutoff (typically 0.02-0.05%) considering background signal and assay noise.
- Generate comprehensive report including detected mutations, MAF, and confidence metrics [5].
Research Reagent Solutions
Table 2: Essential Research Reagents for BEAMing ctDNA Analysis
| Reagent/Kit | Function | Specification | |
|---|---|---|---|
| QIAamp Circulating Nucleic Acid Kit | cfDNA extraction from plasma | Optimized for low-abundance nucleic acids; includes carrier RNA to improve yield | [66] |
| OncoBEAM RAS CRC Panel | Mutation detection | Covers 34 mutations in KRAS/NRAS hotspots | [5] |
| Streptavidin-Coated Magnetic Beads | Solid support for amplification | 1.0 µm diameter; functionalized with streptavidin for biotin binding | [64] |
| Biotinylated Primer Sets | Target amplification | Specific for RAS gene regions of interest | [64] |
| Mutation-Specific Fluorescent Probes | Detection | Dual-color system for mutant/wild-type discrimination | [5] |
| Emulsion Oil/Detergent Mix | Microreactor formation | Creates stable water-in-oil emulsions for compartmentalized PCR | [64] |
BEAMing Workflow and Signaling Pathways
The following diagram illustrates the complete BEAMing workflow from sample collection to mutation detection:
Diagram 1: BEAMing Workflow for ctDNA Analysis. The process transforms liquid biopsy samples into digitally quantifiable mutation data through compartmentalized amplification.
The RAS signaling pathway analysis is crucial for understanding the clinical significance of mutations detected via BEAMing in colorectal cancer:
Diagram 2: RAS Signaling Pathway and Therapeutic Implications. BEAMing detection of RAS mutations identifies patients who will not respond to anti-EGFR therapies, guiding treatment selection.
BEAMing technology provides a robust, clinically validated methodology for ctDNA analysis that demonstrates high concordance with tissue biopsy, particularly in advanced cancers. The exceptional sensitivity of BEAMing (0.01% detection limit) enables reliable detection of rare mutant alleles in circulation, supporting its utility for treatment selection, response monitoring, and resistance mutation detection. While concordance remains highest in advanced malignancies with substantial tumor shedding, ongoing technical refinements continue to expand applications across cancer stages and types. For researchers and drug development professionals, BEAMing represents a powerful tool for non-invasive genomic profiling that can complement or, in appropriate clinical contexts, substitute for traditional tissue-based approaches.
Head-to-Head Comparison with Droplet Digital PCR (ddPCR)
Circulating tumor DNA (ctDNA) analysis has emerged as a cornerstone of liquid biopsy, enabling non-invasive assessment of tumor genetics to guide clinical decision-making in oncology. Among the various digital PCR platforms available, two of the most prominent and clinically validated technologies are BEAMing (Beads, Emulsion, Amplification, and Magnetics) and Droplet Digital PCR (ddPCR). This application note provides a detailed, evidence-based comparison of these two platforms, focusing on their analytical performance, methodological considerations, and applicability in clinical research settings. As ctDNA assays become increasingly integrated into drug development pipelines and clinical trial protocols, understanding the nuanced differences between these technologies is paramount for researchers and drug development professionals seeking to implement robust liquid biopsy workflows.
Performance Comparison: Analytical Concordance and Discordance
Multiple studies have demonstrated strong overall concordance between BEAMing and ddPCR for ctDNA analysis. A landmark 2019 study by O'Leary et al. directly compared both technologies using 363 baseline plasma samples from patients with advanced breast cancer in the phase 3 PALOMA-3 trial [20].
Table 1: Mutation Detection Concordance Between BEAMing and ddPCR
| Gene | BEAMing Detection Rate | ddPCR Detection Rate | Agreement (κ statistic) | Discordancy Rate |
|---|---|---|---|---|
| ESR1 | 24.2% (88/363) | 25.3% (92/363) | κ = 0.91 (95% CI, 0.85-0.95) | 3.9% |
| PIK3CA | 26.2% (95/363) | 22.9% (83/363) | κ = 0.87 (95% CI, 0.81-0.93) | 5.0% |
The high κ values indicate almost perfect agreement between the two technologies, suggesting sufficient reproducibility for clinical use [20]. This robust concordance is particularly notable given that these assays were performed in different laboratories under standardized conditions.
Despite strong overall agreement, discordant results do occur and primarily stem from biological and technical factors:
- Stochastic sampling effects: The majority of discordant calls occur at allele frequencies <1%, where sampling statistics play a significant role [20]
- Mutation-specific differences: Higher rates of discordancy have been observed for less common mutations (P = 0.019) [20]
- Platform-specific sensitivity: A 2023 comparison study of digital PCR platforms found differential sensitivity in mutation detection, with solid dPCR demonstrating 100% detection of EGFR mutations compared to 58.8% for ddPCR [68]
Technical Workflows and Methodological Considerations
BEAMing Technology Workflow
BEAMing combines emulsion PCR with flow cytometry to detect and quantify rare mutant DNA fragments [5]. The process involves several sophisticated steps:
Diagram 1: BEAMing Workflow for ctDNA Analysis
The BEAMing process begins with plasma isolation from peripheral blood, optimally collected in cell-stabilizing tubes (e.g., Streck cfDNA BCT tubes) to prevent white blood cell lysis and preserve ctDNA integrity [69]. Following cfDNA extraction, the core BEAMing process involves:
- Compartmentalization: Individual DNA molecules are compartmentalized into water-in-oil microemulsions along with magnetic beads [5]
- Emulsion PCR: Each DNA molecule is amplified within its microreactor, creating thousands of identical copies bound to a single bead [5]
- Hybridization: The beads are subsequently hybridized with fluorescent probes specific for mutant or wild-type sequences [5]
- Enumeration: Flow cytometry quantifies the ratio of mutant to wild-type beads, enabling precise quantification of mutant allele frequency [5]
This workflow allows BEAMing to achieve exceptional sensitivity, reliably detecting mutant alleles at frequencies as low as 0.01% (1 mutant in 10,000 wild-type alleles) [5] [52].
ddPCR Technology Workflow
Droplet Digital PCR employs a different partitioning strategy to achieve single-molecule amplification:
Diagram 2: ddPCR Workflow for ctDNA Analysis
The ddPCR workflow involves:
- Droplet Generation: The PCR reaction mixture is partitioned into approximately 20,000 nanoliter-sized water-in-oil droplets [20]
- Endpoint PCR: Each droplet undergoes thermal cycling to amplify target sequences
- Droplet Reading: A droplet reader analyzes the fluorescence of each droplet to classify it as positive (mutant), positive (wild-type), or negative
- Poisson Correction: Statistical analysis applies Poisson correction to account for the possibility of multiple target molecules per droplet, enabling absolute quantification without standard curves
Pre-analytical Considerations for Both Platforms
Both technologies share critical pre-analytical requirements that significantly impact assay performance:
- Blood Collection: Blood should be drawn using large gauge diameter needles (≤21 G) into K₂EDTA or cell-stabilizing tubes (e.g., Streck cfDNA collection tubes) [69]
- Processing Time: Plasma isolation should occur within 4-6 hours for K₂EDTA tubes or within 2-7 days for cell-stabilizing tubes [69]
- Centrifugation Protocol: Sequential centrifugation at progressively increasing speeds (800–1,600 g) at 4°C is recommended for optimal plasma separation [69]
- Plasma vs Serum: Analysis should be performed on plasma rather than serum due to lower background wild-type DNA concentration in plasma [5]
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for BEAMing and ddPCR
| Reagent Category | Specific Examples | Function | Technical Considerations |
|---|---|---|---|
| Blood Collection Tubes | Streck cfDNA BCT tubes, K₂EDTA tubes | Preserves ctDNA integrity, prevents white blood cell lysis | Streck tubes allow longer processing windows (2-7 days); K₂EDTA requires processing within 4-6 hours [69] [52] |
| DNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit, other commercial cfDNA kits | Isolves high-purity cfDNA from plasma | Tailor purification method to upstream pre-analytical and downstream analysis [69] |
| PCR Reagents | Target-specific primers and probes, polymerase master mixes | Amplifies target DNA sequences | BEAMing requires specialized reagents for emulsion formation and bead coupling [5] |
| Reference Standards | Commercial ctDNA reference standards, synthetic DNA controls | Validates assay performance, determines sensitivity and specificity | Essential for establishing limit of detection (LOD), particularly for low-frequency mutations [20] |
| Bioinformatic Tools | Unique Molecular Identifier (UMI) analysis, Poisson correction algorithms | Analyzes raw data, corrects for technical artifacts | Molecular barcoding helps distinguish true mutations from sequencing/PCR errors [69] |
Application-Specific Performance Considerations
Clinical Utility in Different Cancer Types
Both platforms have demonstrated utility across multiple solid tumors, though performance characteristics vary by application:
Colorectal Cancer: BEAMing has been extensively validated for RAS mutation testing in colorectal cancer, with the OncoBEAM RAS CRC assay showing 93.3% overall concordance with standard tissue testing [5]. The assay detects 34 mutations in codons 12, 13, 59, 61, 117, and 146 of KRAS and NRAS genes [5].
Gliomas: BEAMing has shown promise in detecting IDH1 mutations in gliomas, where ctDNA levels are typically very low due to the blood-brain barrier. One study demonstrated 100% specificity but modest (50%) sensitivity in detecting IDH1 mutations in plasma, with false negatives primarily occurring when no tumor progression was evident by MRI [52].
Treatment Response Monitoring: Both technologies effectively monitor tumor dynamics during therapy. BEAMing has been used to track ctDNA levels in patients with colorectal cancer undergoing surgery or chemotherapy, with studies showing a rapid decrease (median 99.0%) in ctDNA following complete tumor resection and an estimated ctDNA half-life of 114 minutes [19].
Technical Comparison Table
Table 3: Technical Comparison of BEAMing and ddPCR Platforms
| Parameter | BEAMing | Droplet Digital PCR |
|---|---|---|
| Sensitivity | ≤0.01% mutant allele frequency [5] [52] | ~0.01% mutant allele frequency [20] |
| Partitioning Method | Water-in-oil emulsions with magnetic beads [5] | Water-in-oil droplets [20] |
| Detection Method | Flow cytometry with fluorescent hybridization probes [5] | Fluorescence detection of individual droplets [20] |
| Multiplexing Capacity | Limited by flow cytometry fluorescence channels [5] | Limited by available fluorescence channels [2] |
| Throughput | Moderate | Moderate to high |
| Sample Volume Requirements | 5-10 ml for metastatic setting; up to 60 ml for MRD detection [69] | Typically 2-5 ml plasma [68] |
| Tumor-Informed Approach | Required for optimal sensitivity [69] | Can be used with or without tumor tissue analysis [2] |
| Absolute Quantification | Yes, via bead counting [5] | Yes, via Poisson statistics [20] |
| Clinical Validation | Extensive for RAS in CRC, ESR1/PIK3CA in breast cancer [20] [5] | Extensive for multiple applications [20] [2] |
BEAMing and ddPCR represent two highly sensitive digital PCR technologies with demonstrated utility in ctDNA analysis for clinical research and drug development. Both platforms show strong concordance for mutation detection in most clinical scenarios, with discordance primarily occurring at very low allele frequencies (<1%) due to stochastic sampling effects. The choice between platforms depends on specific research requirements, including needed sensitivity, multiplexing capabilities, throughput needs, and available infrastructure. BEAMing offers exceptional sensitivity for predefined hotspot mutations and has been extensively clinically validated, particularly for RAS testing in colorectal cancer. ddPCR provides a more flexible platform for various mutation detection applications without requiring tumor tissue information. As ctDNA analysis continues to evolve and integrate into clinical trial designs and precision oncology workflows, understanding these technological nuances enables researchers to select the optimal platform for their specific application.
Performance Against Next-Generation Sequencing Platforms
Circulating tumor DNA (ctDNA) analysis has emerged as a cornerstone of precision oncology, enabling non-invasive tumor genotyping, therapy selection, and treatment monitoring [70] [2]. Among the technologies developed for ctDNA analysis, BEAMing technology represents a highly sensitive approach that combines digital PCR principles with flow cytometry to detect rare tumor-derived mutations in blood [70]. As next-generation sequencing (NGS) platforms increasingly dominate the liquid biopsy landscape, understanding the performance characteristics of BEAMing relative to various NGS approaches becomes essential for researchers and drug development professionals selecting appropriate analytical tools for specific applications.
BEAMing (Beads, Emulsion, Amplification, and Magnetics) technology achieves its high sensitivity through a process that converts single DNA molecules into magnetic beads coated with thousands of copies of the original DNA, enabling precise enumeration of mutant alleles via flow cytometry [70]. This technology offers a detection sensitivity of up to 0.02% variant allele frequency (VAF), making it particularly valuable for applications requiring identification of rare variants in limited sample volumes [70]. The following application note provides a comprehensive performance comparison between BEAMing technology and contemporary NGS platforms, along with detailed experimental protocols for their evaluation in ctDNA analysis.
Technology Comparison
The analytical performance of BEAMing technology and NGS platforms varies significantly across multiple parameters, influencing their suitability for different research applications.
Table 1: Performance Comparison of BEAMing vs. NGS Platforms for ctDNA Analysis
| Parameter | BEAMing Technology | Targeted NGS Panels | Whole Genome Sequencing | Structural Variant-Based NGS |
|---|---|---|---|---|
| Sensitivity (Limit of Detection) | 0.02% VAF [70] | ~0.1% VAF (conventional); <0.01% VAF (error-corrected) [14] [2] | >1% VAF (limited by sequencing depth) [70] | <0.01% VAF (parts-per-million sensitivity) [14] |
| Target Approach | Targeted (known mutations) [70] | Targeted (gene panels) or untargeted [70] | Untargeted (genome-wide) [70] | Personalized (tumor-informed) [14] |
| Multiplexing Capability | Limited (few mutations per reaction) [70] | High (dozens to hundreds of genes) [70] [2] | Comprehensive (entire genome) [70] | Moderate (patient-specific breakpoints) [14] |
| Throughput | Medium | High | Low to Medium | Medium |
| Primary Applications | Therapy selection, resistance monitoring, quantitative tracking of specific mutations [70] [2] | Comprehensive profiling, mutation discovery, tumor heterogeneity assessment [70] [2] | Discovery research, copy number alteration analysis [70] | Minimal residual disease, early-stage cancer detection [14] |
| Input Requirements | 4-5 mL plasma (5-10 ng/mL ctDNA) [70] | 4-5 mL plasma (5-10 ng/mL ctDNA) [70] | Higher input requirements | 4-5 mL plasma (5-10 ng/mL ctDNA) [70] |
| Turnaround Time | Rapid (hours to 1 day) [70] | Moderate to Long (3-7 days) [70] | Long (1-2 weeks) [70] | Moderate to Long (5-10 days) [14] |
The performance data reveals a fundamental trade-off between breadth of genomic interrogation and detection sensitivity. BEAMing technology excels in scenarios requiring ultra-sensitive detection of pre-defined mutations, particularly for monitoring treatment response and emerging resistance mechanisms [70] [2]. In advanced lung squamous cell carcinoma, for example, quantitative ctDNA dynamics using targeted approaches have successfully identified molecular responders to immunochemotherapy, with significant differences in progression-free survival (HR = 0.19, p < 0.001) and overall survival (HR = 0.24, p < 0.001) [16].
In contrast, NGS platforms offer more comprehensive genomic coverage, enabling detection of novel alterations and better characterization of tumor heterogeneity [70] [2]. Error-corrected NGS methods have closed the sensitivity gap with BEAMing, achieving detection limits below 0.01% VAF through techniques like unique molecular identifiers (UMIs), duplex sequencing, and concatenating original duplex for error correction (CODEC) [14] [2]. Structural variant-based NGS assays represent a particularly promising approach, detecting tumor-specific chromosomal rearrangements with parts-per-million sensitivity in early-stage breast cancer [14].
Experimental Protocols
BEAMing Technology Workflow Protocol
Sample Preparation and DNA Extraction
Principle: Optimal recovery of ctDNA requires careful sample handling to prevent contamination and degradation. ctDNA fragments typically range from 70-200 base pairs, with a half-life of 16 minutes to 2.5 hours in circulation [70].
Procedure:
- Blood Collection: Collect 10 mL of whole blood into EDTA or specialized cell-free DNA blood collection tubes [70] [71].
- Plasma Separation: Centrifuge at 800-1600 × g for 10 minutes at 4°C within 2 hours of collection. Transfer supernatant to a fresh tube and perform a second centrifugation at 16,000 × g for 10 minutes to remove residual cells [71].
- cfDNA Extraction: Use commercial cfDNA extraction kits following manufacturer's protocols. Elute in 20-50 μL of low-EDTA TE buffer or molecular grade water.
- Quality Control: Quantify cfDNA using fluorometric methods (e.g., Qubit). Expect yields of 5-10 ng/mL of plasma from cancer patients [70]. Assess fragment size distribution using Bioanalyzer or TapeStation.
BEAMing Reaction Setup
Principle: BEAMing converts single DNA molecules into magnetic beads coated with thousands of copies of the original DNA through emulsion PCR, enabling digital enumeration of mutant alleles [70].
Procedure:
- Primer Design: Design biotinylated primers specific to mutations of interest. For the biotin-streptavidin complex formation, ensure 5' biotin modification [70].
- Emulsion PCR:
- Prepare PCR mixture containing: 10-20 ng cfDNA, biotinylated primers, dNTPs, polymerase, and magnetic beads coated with streptavidin.
- Create water-in-oil emulsion by vigorous vortexing or using microfluidic devices, generating ~10,000 droplets per microliter.
- Perform thermal cycling: Initial denaturation at 95°C for 2 min; 40-50 cycles of 95°C for 15 sec, 60°C for 30 sec, 72°C for 30 sec; final extension at 72°C for 5 min.
- Emulsion Breaking: Recover beads by breaking emulsion using detergent or alcohol-based methods. Wash beads thoroughly to remove oil and contaminants.
- Hybridization: Incubate beads with fluorescently-labeled probes specific to wild-type and mutant sequences. Use different fluorophores for discrimination.
- Flow Cytometry Analysis: Analyze beads using flow cytometer with appropriate laser and filter settings. Count at least 1,000,000 beads per sample to achieve 0.02% sensitivity [70].
- Data Analysis: Calculate mutant allele frequency as (mutant beads / total beads) × 100%.
NGS-Based ctDNA Analysis Protocol
Library Preparation and Target Enrichment
Principle: NGS library preparation for ctDNA requires special consideration for low-input, fragmented DNA, with incorporation of unique molecular identifiers (UMIs) to distinguish true mutations from PCR/sequencing errors [2] [71].
Procedure:
- Library Construction:
- Use commercial ctDNA-specific library preparation kits.
- Repair DNA ends, add dA-tails, and ligate adapters containing UMIs.
- Use 10-30 ng of input cfDNA, with minimum amplification cycles (4-8 cycles) to maintain library complexity.
- Target Enrichment:
- For hybrid capture-based panels: Hybridize with biotinylated probes covering regions of interest (50-200 genes), capture with streptavidin beads, and wash stringently.
- For amplicon-based panels: Use two-step PCR with target-specific primers in first PCR and indexing primers in second PCR.
- Library QC: Quantify using qPCR or fragment analyzer. Pool libraries at equimolar ratios.
Sequencing and Data Analysis
Procedure:
- Sequencing: Load pooled libraries onto NGS platform (Illumina, Ion Torrent). Aim for minimum 10,000x raw coverage for 0.1% VAF detection, with higher coverage (50,000-100,000x) for lower VAF detection [14] [71].
- Bioinformatic Processing:
- Demultiplex reads and trim adapter sequences.
- Group reads by UMI families and generate consensus sequences to reduce sequencing errors.
- Align to reference genome (GRCh38) using optimized aligners (BWA-MEM, NovoAlign).
- Call variants using ctDNA-optimized callers (MuTect, VarScan2) with minimum supporting reads of 3-5 for low-frequency variants.
- Apply filters for mapping quality, base quality, and strand bias.
- Annotate variants using databases (COSMIC, dbSNP, ClinVar).
Analytical Validation Protocol
Principle: Comprehensive validation ensures reliable detection of low-frequency variants, addressing unique challenges of ctDNA analysis including low analyte concentration and potential interference from clonal hematopoiesis [72] [71].
Procedure:
- Limit of Detection (LOD) Study:
- Prepare reference materials with known VAF (0.01%-5%) using cell line DNA or synthetic DNA fragments.
- Test each VAF level with 5-8 replicates across multiple days.
- Determine LOD as the lowest VAF with ≥95% detection rate.
Precision Studies:
- Assess repeatability (within-run) and reproducibility (between-run, between-operator, between-instrument) using low-VAF samples (0.1%, 0.5%, 1%).
- Include at least 3 replicates per condition.
Specificity Studies:
- Evaluate analytical specificity using samples with known clonal hematopoiesis mutations.
- Test cross-reactivity with common germline polymorphisms.
Accuracy/Concordance Studies:
- Compare results with orthogonal methods (digital PCR, orthogonal NGS).
- Assess tissue-plasma concordance when matched samples available.
Table 2: Validation Performance Criteria Based on BLOODPAC Recommendations [72] [71]
| Validation Parameter | Acceptance Criteria | BEAMing Performance | NGS Performance |
|---|---|---|---|
| Limit of Detection | ≤0.5% VAF for most applications; <0.1% for MRD | 0.02% VAF [70] | 0.01-0.1% VAF (method dependent) [14] |
| Repeatability | CV <20% at LOD | Typically <15% | Typically <20% |
| Reproducibility | >95% concordance between replicates | >98% | >95% |
| Specificity | >99% for variant calling | >99.9% | >99% (with UMI) |
| Input DNA Linear Range | 1-50 ng | 1-100 ng | 1-100 ng |
| Tissue-Concordance | >80% for detectable variants | >85% | 70-95% (study dependent) |
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of BEAMing and NGS ctDNA analysis requires carefully selected reagents and materials optimized for low-input, high-sensitivity applications.
Table 3: Essential Research Reagents for BEAMing and NGS ctDNA Analysis
| Reagent/Material | Function | BEAMing Application | NGS Application |
|---|---|---|---|
| Cell-Free DNA Blood Collection Tubes | Stabilize nucleated cells and prevent genomic DNA contamination | Essential for pre-analytical standardization [72] [71] | Essential for pre-analytical standardization [72] [71] |
| cfDNA Extraction Kits | Isolation of short-fragment DNA from plasma | Critical for optimal recovery of 70-200 bp fragments [70] | Critical for optimal recovery of 70-200 bp fragments [70] |
| Biotinylated Primers | Target-specific amplification with bead attachment | Essential for emulsion PCR and bead capture [70] | Used in hybrid capture panels |
| Streptavidin-Coated Magnetic Beads | Solid support for PCR amplification and probe hybridization | Core component for BEAMing reaction [70] | Used in hybrid capture enrichment |
| Fluorophore-Linked Probes | Mutation detection and quantification | Essential for flow cytometry detection (wild-type vs. mutant) [70] | N/A |
| Unique Molecular Identifiers (UMIs) | Tagging individual DNA molecules for error correction | Not typically used | Essential for distinguishing true mutations from artifacts [2] |
| Hybrid Capture Probes | Target enrichment for sequencing | N/A | Essential for focused genomic regions |
| High-Fidelity Polymerase | Accurate amplification with low error rates | Critical for mutation detection specificity | Critical for library amplification |
| Reference Standard Materials | Assay validation and quality control | Essential for LOD studies [72] [71] | Essential for LOD studies [72] [71] |
Applications in Drug Development
Therapy Selection and Resistance Monitoring
BEAMing technology provides distinct advantages in clinical trial contexts requiring rapid turnaround for known biomarkers. In metastatic colorectal cancer, BEAMing-based KRAS mutation detection has demonstrated utility for patient stratification to anti-EGFR therapies [70]. Similarly, in non-small cell lung cancer, BEAMing enables monitoring of EGFR T790M resistance mutations, informing treatment transitions to third-generation EGFR inhibitors without repeated tissue sampling [2].
The quantitative nature of BEAMing makes it particularly valuable for tracking dynamic changes in mutation burden during treatment. Studies across multiple solid tumors have established that early changes in ctDNA levels (within 2-4 weeks of treatment initiation) correlate strongly with subsequent radiographic response and survival outcomes [2] [16]. In advanced lung squamous cell carcinoma, quantitative ctDNA dynamics using a novel MinerVa-Delta algorithm successfully identified molecular responders to immunochemotherapy, with significant differences in progression-free survival (HR = 0.19, p < 0.001) and overall survival (HR = 0.24, p < 0.001) [16].
Minimal Residual Disease Assessment
For MRD detection, structural variant-based NGS approaches have demonstrated exceptional sensitivity, detecting ctDNA in 96% of early-stage breast cancer patients at baseline with median VAF of 0.15%, including 10% of patients with VAF <0.01% [14]. These approaches leverage patient-specific chromosomal rearrangements, which are essentially unique to the tumor and not present in healthy cells, enabling highly specific detection.
BEAMing technology can be adapted for MRD applications when tumor-specific mutations are known, though it requires prior tumor sequencing to identify appropriate targets. In colorectal cancer, BEAMing-based MRD assessment after curative resection has identified patients at high recurrence risk up to 1 year before clinical evidence of relapse [70]. Similarly, in lymphoma, ctDNA-based MRD assays have proven more sensitive than standard PET or CT imaging for predicting relapse and guiding immunochemotherapy [14].
BEAMing technology maintains a crucial niche in the ctDNA analysis landscape, offering unparalleled sensitivity for quantifying known mutations when ultra-low detection limits are required. Its performance characteristics make it particularly valuable for applications in therapy monitoring, resistance mutation detection, and clinical trial biomarker assessment where specific mutations are targeted. Meanwhile, NGS platforms provide comprehensive genomic profiling capabilities that are essential for discovery research and heterogeneous tumors, with advancing error-correction methodologies continually improving their sensitivity thresholds.
The choice between BEAMing and NGS platforms ultimately depends on the specific research question, with factors including required sensitivity, number of targets, sample availability, and turnaround time influencing technology selection. As ctDNA analysis continues to evolve, both technologies will play complementary roles in advancing precision oncology and drug development, enabling non-invasive assessment of tumor dynamics and treatment response.
Regulatory Status and Clinical Guidelines Adoption
BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) represents a sophisticated digital PCR platform for analyzing circulating tumor DNA (ctDNA), offering exceptional sensitivity for detecting rare tumor-derived DNA molecules in patient blood. This technology addresses a critical need in precision oncology by enabling non-invasive tumor genotyping, treatment response monitoring, and minimal residual disease (MRD) detection. The clinical adoption of BEAMing is guided by evolving regulatory frameworks and professional guidelines that establish standards for analytical validation and clinical implementation. As a bead-based emulsion polymerase chain reaction method, BEAMing achieves single-molecule sensitivity by compartmentalizing DNA fragments into water-in-oil microemulsions, converting each target DNA molecule into a magnetic bead containing thousands of identical copies, and detecting mutations via flow cytometry with fluorescently labeled probes. This technical foundation supports its growing incorporation into clinical practice for cancer management, particularly in settings requiring high sensitivity mutation detection.
Current Regulatory Landscape
The regulatory status for BEAMing-based ctDNA tests varies globally, with several key milestones achieved for clinical implementation.
Regulatory Approvals and Clearances
CE Marking: The OncoBEAM RAS CRC test has received CE Mark approval in the European Union for expanded RAS mutation testing in colorectal cancer patients, enabling its clinical use [5]. This test detects 34 mutations in KRAS and NRAS genes, providing comprehensive mutation profiling to guide anti-EGFR therapy decisions.
FDA Recognition: While BEAMing technology itself is not yet directly approved by the U.S. Food and Drug Administration (FDA), the regulatory agency has published guidance supporting the use of ctDNA as a biomarker in drug development contexts [73]. The FDA recognizes ctDNA as a valid marker for enriching clinical trial populations and using ctDNA dynamics as an early indicator of drug activity.
Laboratory-Developed Tests (LDTs): BEAMing platforms are implemented as LDTs in certified clinical laboratories, operating under Clinical Laboratory Improvement Amendments (CLIA) regulations that establish quality standards for laboratory testing [5].
Table 1: Global Regulatory Status of BEAMing ctDNA Tests
| Region | Regulatory Status | Key Approved/Validated Tests | Primary Clinical Applications |
|---|---|---|---|
| European Union | CE Mark obtained | OncoBEAM RAS CRC test | RAS mutation testing in metastatic colorectal cancer [5] |
| United States | Implemented as LDTs; FDA guidance for drug development | Various laboratory-developed BEAMing assays | Mutation detection in advanced cancers; clinical trial endpoints [73] |
| International | Variable adoption; often relies on central laboratories | OncoBEAM platform across cancer types | NSCLC, colorectal cancer genotyping [3] |
Professional Guideline Recommendations
Clinical practice guidelines have begun incorporating ctDNA testing, with specific implications for BEAMing technology:
Lung Cancer Applications: BEAMing demonstrates high concordance with tissue-based testing for EGFR mutations in non-small cell lung cancer (NSCLC), with studies showing 98.8%, 98.9%, and 95.5% concordance for exons 19, 20, and 21, respectively, when compared to standard qPCR methods [3].
Colorectal Cancer Guidelines: Professional oncology guidelines acknowledge the utility of ctDNA for RAS testing when tissue is unavailable, with BEAMing showing 93.3% overall concordance with standard tissue testing in metastatic colorectal cancer [5].
Pre-analytical Standards: Recent guidelines specify requirements for blood collection tubes (EDTA or cell preservation tubes), processing timelines (within 4-6 hours for EDTA tubes), and plasma preparation protocols (two-step centrifugation at 800-1,600×g followed by 14,000-16,000×g) to ensure ctDNA analysis quality [47].
Analytical Validation and Performance Standards
BEAMing technology undergoes rigorous validation to establish performance characteristics for clinical implementation.
Sensitivity and Specificity Metrics
The exceptional sensitivity of BEAMing enables detection of mutant alleles at frequencies as low as 0.01% [5], making it particularly suitable for ctDNA analysis where tumor DNA represents a small fraction of total cell-free DNA. Validation studies demonstrate:
- High Concordance with Tissue: In colorectal cancer, BEAMing exhibits 92.6% positive percent agreement and 94% negative percent agreement with tissue-based RAS testing [5].
- Robust Mutation Detection: For NSCLC EGFR testing, BEAMing compared favorably with qPCR methods, achieving 90-100% gene-level concordance across different exons [3].
- Quantification Capability: BEAMing provides precise mutant allele frequency quantification, enabling monitoring of tumor dynamics and treatment response [5].
Table 2: Performance Characteristics of BEAMing ctDNA Analysis
| Performance Parameter | BEAMing Performance | Comparative Method | Clinical Context |
|---|---|---|---|
| Limit of Detection | 0.01% mutant allele frequency [5] | Conventional PCR (1-5%) | Detection of rare mutant molecules in blood |
| Concordance with Tissue | 93.3% overall for RAS (CRC) [5] | Standard tissue testing | Metastatic colorectal cancer |
| EGFR Exon 19 Concordance | 98.8% [3] | Real-time qPCR | NSCLC targeted therapy selection |
| EGFR Exon 21 Concordance | 95.5% [3] | Real-time qPCR | NSCLC targeted therapy selection |
| Turnaround Time | Rapid (days) | Tissue sequencing (weeks) | Timely treatment decisions |
Quality Control Considerations
Implementation of BEAMing requires strict quality control measures:
- Sample Adequacy: Assessment of total cell-free DNA concentration and fragmentation patterns to ensure sample quality [47].
- Control Materials: Use of reference standards and cell line DNA with known mutation status to validate each run [3].
- Result Interpretation: Establishment of cutoff values for mutant allele frequency reporting, typically above the 0.01% limit of detection [5].
Experimental Protocol: BEAMing ctDNA Analysis for EGFR Mutations in NSCLC
This protocol details the application of BEAMing technology for detecting EGFR mutations in non-small cell lung cancer patients, based on published methodology [3].
Pre-analytical Phase: Blood Collection and Plasma Processing
- Blood Collection: Draw 10 mL peripheral blood into K2- or K3-EDTA tubes. Invert tubes 8-10 times immediately after collection to mix blood with anticoagulant [47].
- * Plasma Separation*: Process samples within 1 hour of collection. Perform first centrifugation at 820×g for 10 minutes at room temperature. Transfer supernatant to fresh tube without disturbing buffy coat. Perform second centrifugation at 16,000×g for 10 minutes to remove remaining cellular debris [3].
- * Plasma Storage*: Aliquot cleared plasma and store at -80°C until DNA extraction. Avoid repeated freeze-thaw cycles.
- ctDNA Extraction: Extract ctDNA from 1 mL plasma using silica column-based kits (e.g., QIAamp DNA Micro Kit, Qiagen). Elute in low-EDTA TE buffer or nuclease-free water. Quantify DNA using fluorometric methods (e.g., Qubit) rather than UV spectrophotometry, which is less accurate for low-concentration samples.
BEAMing PCR Assay Procedure
Primary PCR Amplification:
- Set up eight separate 25 μL PCR reactions per sample containing:
- Template DNA from 250 μL plasma equivalent
- 5× Phusion High-Fidelity PCR Buffer
- 1.5 U HotStart Phusion DNA Polymerase
- 0.2 μM forward and reverse primers (EGFR exons 19, 20, 21)
- 0.25 mM each dNTP
- 0.5 mM MgCl₂
- Cycling conditions: 98°C for 30s; 35 cycles of: 98°C for 10s, 57°C for 10s, 72°C for 10s; final extension at 72°C for 5 minutes [3].
- Pool PCR products from all eight reactions and quantify using fluorometry.
- Set up eight separate 25 μL PCR reactions per sample containing:
Emulsion PCR (BEAMing):
- Prepare 150 μL PCR mixture containing:
- 18 pg pooled primary PCR product
- 40 U Platinum Taq DNA Polymerase
- 1× PCR Buffer
- 0.2 mM dNTPs
- 5 mM MgCl₂
- 0.05 μM Tag1 primer
- 8 μM Tag2 primer
- ~6×10⁷ magnetic streptavidin beads coated with Tag1 oligonucleotide
- Create microemulsion by adding 600 μL oil/emulsifier mixture (7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC) and one 5 mm steel bead to PCR mixture. Shake in TissueLyser at 15 Hz for 10s, then 17 Hz for 7s.
- Verify emulsion quality under microscope (40× magnification) to ensure proper bead compartmentalization.
- Perform emulsion PCR with following cycling conditions: 94°C for 2min; 3 cycles of 94°C for 10s, 68°C for 45s, 70°C for 75s; 3 cycles of 94°C for 10s, 65°C for 45s, 70°C for 75s; 3 cycles of 94°C for 10s, 62°C for 45s, 70°C for 75s; 50 cycles of 94°C for 10s, 57°C for 45s, 70°C for 75s [3].
- Prepare 150 μL PCR mixture containing:
Emulsion Breaking and Bead Recovery:
- Add 150 μL breaking buffer (10 mM Tris-HCl pH 7.5, 1% Triton X-100, 1% SDS, 100 mM NaCl, 1 mM EDTA) to each well.
- Shake at 20 Hz for 20s in TissueLyser to break emulsions.
- Recover beads by centrifugation at 3,200×g for 2 minutes, remove oil phase.
- Repeat breaking step twice, then wash beads with 150 μL wash buffer (20 mM Tris-HCl pH 8.4, 50 mM KCl).
- Denature DNA with 0.1 M NaOH for 5 minutes, then wash again with wash buffer.
Mutation Detection by Hybridization:
- Resuspend beads in 150 μL hybridization buffer containing fluorescently labeled probes complementary to wild-type and mutant sequences (15-18 nt length).
- Hybridize at appropriate temperature based on probe characteristics.
- Analyze beads using flow cytometry to distinguish mutant and wild-type populations based on fluorescence signals.
Data Analysis and Interpretation
- Calculate mutant allele frequency as percentage of mutant beads relative to total beads (mutant + wild-type).
- Establish threshold for positive detection based on validation data (typically >0.01% mutant allele frequency).
- Compare with clinical cutoffs established for specific mutations (e.g., T790M, L858R, exon 19 deletions).
Signaling Pathways and Workflow Diagrams
Figure 1: BEAMing ctDNA Analysis Workflow. The complete process from blood collection to result interpretation, highlighting key steps in pre-analytical, analytical, and post-analytical phases.
Figure 2: EGFR Signaling Pathway and Clinical Application. (Left) Oncogenic EGFR signaling driven by mutations in tyrosine kinase domain, activating downstream survival pathways. (Right) Clinical implementation of BEAMing for EGFR mutation detection and TKI therapy monitoring.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for BEAMing ctDNA Analysis
| Reagent/Category | Specific Examples | Function in BEAMing Protocol |
|---|---|---|
| Blood Collection Tubes | K2/K3-EDTA tubes; Cell preservation tubes (Streck, PAXgene) | Prevent coagulation and preserve cell-free DNA by stabilizing blood cells [47] |
| DNA Extraction Kits | QIAamp DNA Micro Kit (Qiagen); Magnetic bead-based systems | Isolation of high-quality ctDNA from plasma with efficient recovery of short fragments [3] |
| High-Fidelity DNA Polymerases | Phusion Hot Start Polymerase (NEB); Platinum Taq DNA Polymerase (Invitrogen) | Accurate primary PCR amplification; emulsion PCR with thermostable enzyme [3] |
| Magnetic Beads | Streptavidin-coated magnetic beads (MyOne, Invitrogen) | Solid support for DNA amplification and separation of mutant/wild-type molecules [3] |
| Emulsion Components | ABIL WE 09; TegoSoft DEC; Mineral oil | Create stable water-in-oil microemulsions for compartmentalized PCR [3] |
| Allele-Specific Probes | Fluorescently labeled oligonucleotides (15-18 nt) | Differentiation of mutant and wild-type sequences through hybridization [3] |
| Reference Standards | DNA from cell lines (PC9, H1975, A549); Synthetic DNA controls | Assay validation, sensitivity determination, and quality control [3] |
The integration of BEAMing technology into clinical practice requires careful consideration of regulatory, technical, and validation parameters. Current guidelines support its use for mutation detection in advanced cancers where tissue is limited, with growing evidence for MRD monitoring applications. The high sensitivity and quantitative nature of BEAMing position it as a valuable tool for precision oncology, particularly in guiding targeted therapy decisions and monitoring treatment resistance. As regulatory frameworks evolve and clinical evidence expands, BEAMing methodology is poised for increased adoption across molecular pathology laboratories, ultimately enhancing non-invasive cancer management through robust ctDNA analysis.
Multicenter Studies and Real-World Performance Data
Beads, Emulsion, Amplification, and Magnetics (BEAMing) is a highly sensitive digital PCR technology that enables the detection and quantification of rare circulating tumor DNA (ctDNA) molecules in patient blood samples. This technology addresses the fundamental challenge of identifying tumor-derived DNA fragments that often constitute less than 0.1% of total cell-free DNA in plasma, making it particularly valuable for clinical applications where non-invasive monitoring is critical [5]. BEAMing technology functions by partitioning individual DNA molecules into water-in-oil microemulsions, where each droplet contains a single magnetic bead that captures amplified DNA copies. Subsequent flow cytometry analysis differentiates mutant from wild-type sequences using fluorescent probes, allowing precise enumeration even at extremely low variant allele frequencies [5] [52]. In the context of advancing precision oncology, BEAMing represents a significant methodological innovation for liquid biopsy applications, providing clinicians and researchers with a tool for rapid genotyping without invasive tissue sampling.
Analytical Performance: Sensitivity and Reproducibility
Technical Foundations and Detection Limits
BEAMing technology demonstrates exceptional analytical sensitivity, reliably detecting mutated ctDNA molecules at frequencies as low as 0.01% (1 mutant allele in 10,000 wild-type alleles) [5]. This high sensitivity stems from its unique workflow that combines emulsion PCR with flow cytometric detection. The process begins with the attachment of single DNA molecules to magnetic beads, followed by amplification within individual emulsion droplets. This compartmentalization ensures that amplified products remain attached to their original beads, maintaining the one-to-one relationship between starting molecules and detected signals [5]. The resulting beads are then analyzed via flow cytometry using allele-specific fluorescent probes, enabling precise discrimination and quantification of mutant alleles. This approach significantly reduces background noise and enhances the detection of rare variants, making it particularly suitable for monitoring minimal residual disease and early treatment response assessment [52].
When compared to other ctDNA detection platforms in multicenter evaluations, BEAMing consistently demonstrates robust performance. A comprehensive evaluation of nine ctDNA assays revealed that while all test assays were generally comparable or superior to the benchmark assay, factors such as ctDNA extraction efficiency, input quantity, and variant type significantly impacted performance [13]. BEAMing maintained high sensitivity even at medium cfDNA inputs (20-50 ng), with performance variations becoming more pronounced at lower inputs (<20 ng) [13]. This technical reliability across different sample conditions makes BEAMing particularly valuable for real-world clinical applications where sample quality and quantity may vary.
Comparative Method Performance
Table 1: Comparison of BEAMing with Other ctDNA Detection Technologies
| Method | Detection Limit (% ctDNA) | Key Advantages | Key Limitations |
|---|---|---|---|
| BEAMing | 0.01% | High sensitivity, quantitative, clinically validated | Limited to known mutations, complex workflow |
| ddPCR | ~0.01% | High sensitivity, easy workflow | Limited multiplexing capability |
| Targeted NGS | 0.01-2.0% | Broad mutation coverage, relatively inexpensive | Requires assay personalization |
| Whole Genome Sequencing | ~1% | Comprehensive coverage without personalization | Expensive, low sensitivity |
| Whole Exome Sequencing | ~5% | Focus on coding regions | Expensive, limited sensitivity [5] |
Clinical Validation: Multicenter Concordance Studies
Colorectal Cancer Applications
The clinical validity of BEAMing technology has been extensively demonstrated through multiple prospective multicenter studies, particularly in metastatic colorectal cancer (mCRC) where RAS mutation status determines eligibility for anti-EGFR therapies. A landmark real-world performance comparison across 10 hospital laboratories in Spain evaluated 236 mCRC patients and found an overall agreement of 89% (210/236 patients; kappa = 0.770) between plasma-based OncoBEAM RAS testing and standard tissue-based analysis [74]. After resolving discordant cases through additional testing, the final concordance reached 92% (217/236 patients; kappa = 0.853), demonstrating the reliability of BEAMing for clinical decision-making [74].
Notably, the study identified specific clinical factors influencing concordance rates. Patients with exclusively lung metastases showed lower concordance (68.8%), suggesting potentially reduced ctDNA shedding from pulmonary metastases compared to hepatic lesions (94.5-94.8% concordance) [74]. This finding highlights the importance of considering tumor location when interpreting liquid biopsy results. Additionally, patients who had undergone primary tumor resection before blood collection showed lower concordance (87.4% vs. 95.7% in unresected patients, P=0.033), possibly reflecting reduced overall tumor burden and consequently lower ctDNA levels [74].
Cross-Cancer Application Evidence
Beyond colorectal cancer, BEAMing technology has demonstrated utility across multiple tumor types. In gliomas, where liquid biopsy applications are particularly challenging due to the blood-brain barrier, BEAMing successfully detected IDH1 mutations in plasma with 100% specificity, though with modest sensitivity (50%) [52]. The study surveyed six patients with IDH1-mutant tumors and detected the same mutations in plasma in three cases, all of which were collected either at diagnosis or during progressive disease [52]. The high false-negative rate (86%) underscores the particular challenges of ctDNA detection in CNS malignancies but simultaneously confirms BEAMing's specificity when mutations are detected [52].
Table 2: Multicenter Performance Data for BEAMing RAS Testing in mCRC
| Study Parameter | Vassiliou et al. (2017) | Vidal et al. (2018) |
|---|---|---|
| Number of Patients | 238 | 236 |
| Number of Centers | 6 European centers | 10 Spanish hospitals |
| Overall Concordance | 93.3% | 89% (92% after discordance resolution) |
| Positive Percent Agreement | 92.6% | Not specified |
| Negative Percent Agreement | 94.0% | Not specified |
| RAS Mutation Rate in Tissue | 51% | 55.5% |
| RAS Mutation Rate in Plasma | 50% | 51.3% [5] [74] |
Experimental Protocols and Workflows
Sample Collection and Processing
The BEAMing protocol begins with proper sample collection and handling, which are critical for reliable results. Blood samples should be collected in specialized tubes containing preservatives, such as Streck cell-free DNA BCT tubes or EDTA tubes, with 10mL typically drawn per collection [74] [52]. Plasma separation must occur within specific timeframes to prevent genomic DNA contamination from blood cell lysis. The process involves two centrifugation steps: first at 1600-2000 × g for 10-20 minutes to separate plasma from blood cells, followed by a second centrifugation at 16,000 × g for 10 minutes to remove remaining cellular debris [75]. The processed plasma should be stored at -80°C if DNA extraction cannot be performed immediately, with freeze-thaw cycles minimized to prevent DNA fragmentation.
Cell-free DNA extraction employs commercial kits optimized for short fragment recovery, with quality control performed using fluorometric methods rather than spectrophotometry due to the low concentrations typically obtained [75]. The extracted cfDNA should demonstrate a characteristic fragment size distribution peaking at approximately 166 base pairs, confirming the preservation of the typical cfDNA profile. DNA quantity and quality assessments are critical, as input amounts directly impact assay sensitivity; studies indicate that inputs below 20 ng may compromise variant detection, particularly at low allele frequencies [13].
BEAMing Wet-Lab Methodology
The core BEAMing process involves several sequential steps after DNA extraction. First, target regions are amplified using conventional PCR with primers designed for specific mutations of interest (e.g., RAS mutations in colorectal cancer or IDH1 mutations in gliomas) [5] [52]. The amplified products are then subjected to emulsion PCR, where individual DNA molecules are compartmentalized with magnetic beads in water-in-oil microemulsions. Within each droplet, PCR amplification occurs with the DNA products becoming covalently linked to the beads, creating clonal populations where each bead carries thousands of copies of a single original DNA molecule [5].
Following emulsion breakdown, the beads are hybridized with fluorescent probes specific for wild-type or mutant sequences. Flow cytometry then enumerates the beads, distinguishing mutant from wild-type populations based on fluorescence signals [5] [52]. The mutant allele fraction is calculated as the ratio of mutant beads to the total beads (mutant + wild-type), providing precise quantification of the mutation burden in the original sample. This digital counting approach provides absolute quantification without the need for standard curves and offers a linear dynamic range across multiple orders of magnitude [5].
BEAMing Workflow: From Sample to Result
Bioinformatics and Data Analysis
The analytical phase of BEAMing relies on robust flow cytometry data interpretation. Beads are classified into four populations based on fluorescence signals: wild-type only (single fluorescence), mutant only (different single fluorescence), double-positive (both fluorescences, indicating potential hybrid artifacts), and double-negative (no fluorescence) [5]. The mutant allele frequency is calculated as the percentage of mutant beads relative to the total bead count excluding double-positive and double-negative populations. Thresholds for positivity must be established using appropriate controls, with typical limits of detection validated at 0.01% for many applications [5].
Quality control parameters include minimum bead counts (typically >10,000 beads per sample), background signal assessment in negative controls, and concordance with expected values in positive controls [5] [52]. For clinical applications, results are often reported as positive, negative, or indeterminate, with specific thresholds for mutant allele fractions determined through extensive clinical validation studies. In the case of the OncoBEAM RAS CRC assay, a cutoff of 0.01% mutant allele fraction is typically employed, though clinical interpretation may consider higher thresholds for therapeutic decision-making [5] [74].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for BEAMing Experiments
| Reagent/Material | Function | Specification Notes |
|---|---|---|
| Cell-Free DNA Collection Tubes | Preserves blood sample integrity | Streck Cell-Free DNA BCT or similar; prevents genomic DNA contamination |
| Magnetic Beads | Solid support for DNA amplification | Streptavidin-coated beads for biotinylated oligonucleotide capture |
| Emulsion Oil | Creates reaction compartments | Forms stable water-in-oil microemulsions for partitioned PCR |
| Allele-Specific Fluorescent Probes | Mutant vs. wild-type discrimination | FAM and VIC-labeled probes for flow cytometry detection |
| PCR Primers | Target sequence amplification | Designed for specific hotspot mutations with appropriate modification |
| Hybridization Buffers | Facilitates probe binding | Optimized for specific hybridization conditions |
| Reference Control Materials | Assay validation and QC | Synthetic oligonucleotides with known mutations; reference plasma samples [5] [74] [52] |
Real-World Evidence and Clinical Utility
Real-world evidence continues to demonstrate the clinical utility of BEAMing technology across diverse settings. In clinical practice, BEAMing-based liquid biopsy has addressed significant gaps in cancer care, particularly when tissue samples are unavailable, insufficient, or difficult to obtain [5]. The rapid turnaround time associated with blood-based testing enables quicker treatment decisions compared to tissue sequencing, which often requires complex logistics and extended processing times [5] [74]. This advantage is particularly critical in advanced cancer patients with rapidly progressing disease, where timely intervention is essential.
Longitudinal monitoring applications represent another area where BEAMing technology provides unique clinical value. The high sensitivity and quantitative nature of the technology make it suitable for tracking mutation burden over time, allowing clinicians to monitor treatment response and emerging resistance [5] [76]. In colorectal cancer, studies have demonstrated that post-operative ctDNA detection using BEAMing and other sensitive technologies strongly predicts recurrence with hazard ratios exceeding 30.0, providing a significantly more accurate prognostic assessment than conventional carcinoembryonic antigen testing [76]. This prognostic capability enables more personalized adjuvant therapy decisions and surveillance intensity modulation.
Despite its demonstrated utility, real-world implementation of BEAMing faces challenges, including biological factors such as variable ctDNA shedding across tumor types and locations [74] [52]. The technology's limitation to known mutations also necessitates prior knowledge of the mutational landscape, unlike comprehensive next-generation sequencing approaches that can detect novel alterations [5] [21]. Nevertheless, for applications focused on specific hotspot mutations in genes like RAS, IDH1, and EGFR, BEAMing remains a robust and clinically validated solution with extensive real-world evidence supporting its utility in modern oncology practice.
Clinical Decision Pathway for BEAMing Testing
Cost-Benefit Analysis and Implementation Considerations
BEAMing (Beads, Emulsion, Amplification, and Magnetics) technology represents a highly sensitive digital PCR platform for circulating tumor DNA (ctDNA) analysis, enabling detection of rare mutant DNA molecules at frequencies as low as 0.01% (approximately 1 mutant allele in 10,000 wild-type alleles) [5] [52]. This sensitivity makes it particularly valuable for liquid biopsy applications in oncology, where ctDNA often constitutes a minute fraction of total cell-free DNA (cfDNA), especially in early-stage cancers and minimal residual disease (MRD) monitoring [7] [38]. The technology combines emulsion PCR with flow cytometry to detect and quantify somatic mutations in plasma, addressing critical needs in personalized cancer management [5].
BEAMing's clinical utility is well-established in multiple cancer types. In metastatic colorectal cancer (mCRC), the OncoBEAM RAS CRC test demonstrates 93.3% overall concordance with standard tissue testing, enabling rapid turnaround for anti-EGFR therapy decisions [5]. Similarly, in gliomas, BEAMing successfully detects IDH1 mutations in plasma with 100% clinical specificity, despite the challenges posed by the blood-brain barrier [52]. As liquid biopsies gain prominence for cancer diagnosis, treatment monitoring, and MRD detection, understanding the economic and implementation aspects of BEAMing technology becomes crucial for laboratories and healthcare systems [1] [7].
Cost Analysis of ctDNA Testing
The economic evaluation of ctDNA testing, including BEAMing technology, reveals substantial cost variability influenced by testing platform, methodology, and operational context. A comprehensive micro-costing framework developed for Dutch clinical practice demonstrates that total expenses per sample range from €168 to €7,638 ($199 to $9,124), highlighting the significant impact of technological choices and testing volumes [77].
Table 1: Cost Components of ctDNA Testing
| Cost Category | Specific Considerations | Impact on Total Cost |
|---|---|---|
| Platform & Methodology | PCR-based vs. NGS-based platforms; targeted vs. comprehensive panels | Major driver; NGS panels typically higher cost |
| Testing Volume | Scale of operations; batch processing efficiency | Higher volumes generally reduce cost per sample |
| Personnel | Technical expertise required for operation and analysis | Significant variable across healthcare settings |
| Reagents & Consumables | Specialized tubes, extraction kits, amplification reagents | Substantial recurring expense |
| Equipment & Overhead | Instrument acquisition, maintenance, facility costs | Fixed costs distributed across testing volume |
| Quality Control | Standardization, validation, failure rates | Essential for reliability; adds to overall expense |
This cost structure sensitivity means that laboratories must carefully consider their specific use cases. For instance, in stage II colon cancer, economic models indicate that ctDNA-guided adjuvant chemotherapy selection becomes cost-effective only when test costs fall below €1,500, assuming current Dutch clinical practice parameters [78]. The balance between test performance characteristics (sensitivity, specificity) and cost is particularly important for population-level applications such as MRD detection and early-stage cancer screening [78] [38].
Benefit Considerations in Clinical Applications
Clinical Utility and Health Economic Impact
The primary benefits of BEAMing and similar ctDNA technologies stem from their ability to guide precision oncology decisions non-invasively. In metastatic colorectal cancer, BEAMing enables expanded RAS testing with rapid turnaround times, addressing critical gaps in care when tissue samples are unavailable, inadequate, or difficult to obtain [5]. This facilitates appropriate patient selection for anti-EGFR therapies, potentially improving clinical outcomes while avoiding ineffective treatments in mutation-positive patients.
In the adjuvant setting for stage II colon cancer, ctDNA testing offers the potential to optimize chemotherapy allocation. Current models project that combining ctDNA status with existing clinicopathological features (pT4, pMMR) could reduce recurrences by approximately 3.6% while increasing quality-adjusted life years (QALYs) by +0.038 compared to guideline-based selection alone [78]. This translates to improved population health outcomes through both better identification of high-risk patients who would benefit from adjuvant therapy and avoidance of unnecessary treatment toxicity in low-risk patients.
Operational and Clinical Workflow Benefits
Beyond direct clinical impact, BEAMing technology offers significant operational advantages:
- Minimally Invasive Monitoring: Enables repeated assessments to track treatment response and resistance development without repeated tissue biopsies [1] [7]
- Capturing Tumor Heterogeneity: Provides a more comprehensive mutational profile than single-site tissue biopsies by reflecting contributions from multiple tumor sites [1] [79]
- Rapid Turnaround Time: Facilitates timely clinical decisions, particularly important when tissue processing delays could compromise treatment initiation [5]
- Longitudinal Disease Monitoring: Allows dynamic assessment of tumor evolution during therapy, enabling earlier intervention in case of molecular progression [5] [7]
Technical Protocols and Implementation Framework
BEAMing Technology Workflow Protocol
The BEAMing methodology transforms rare DNA mutation detection into a digital counting process through several optimized steps:
1. Blood Collection and Plasma Processing
- Collect 2×10 mL peripheral blood in cell-stabilizing tubes (e.g., Streck cfDNA BCT) [38] [52]
- Process within 2-6 hours if using EDTA tubes; up to 7 days with specialized preservative tubes [7] [38]
- Perform two-step centrifugation: initial at 1,200-1,600× g for 10 min followed by high-speed centrifugation at 12,000-20,000× g for 10 min at 4°C [7] [38]
- Aliquot plasma and store at -80°C until DNA extraction to prevent degradation [7]
2. ctDNA Extraction
- Use silica membrane-based columns (e.g., QIAamp Circulating Nucleic Acid Kit) or magnetic bead-based systems [38]
- Automated extraction platforms provide more consistent yields and reduce manual variability [7]
3. Emulsion PCR (BEAMing Process)
- Partition individual DNA molecules into water-in-oil microemulsions with PCR reagents and magnetic beads [5]
- Each compartment contains approximately one DNA molecule and one magnetic bead [5]
- Perform PCR amplification within each microemulsion, creating clonal amplifications bound to beads [5]
4. Hybridization and Flow Cytometry
- Hybridize beads with fluorescent probes specific for wild-type and mutant sequences [5] [52]
- Analyze using flow cytometry to count mutant and wild-type beads [5]
- Calculate mutant allele frequency based on ratio of mutant to total beads [5]
Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for BEAMing Experiments
| Reagent/Material | Function | Implementation Notes |
|---|---|---|
| Cell-Stabilizing Blood Collection Tubes (Streck, PAXgene) | Preserve blood cell integrity during transport/storage | Enable room temperature storage for up to 7 days; critical for multi-site trials [7] [38] |
| Silica-Membrane ctDNA Extraction Kits | Isolate high-purity ctDNA from plasma | Provide superior yields vs. magnetic bead methods; minimize wild-type DNA contamination [38] |
| Emulsion PCR Reagents | Partition and amplify individual DNA molecules | Require precise oil-surfactant mixtures for stable microemulsion formation [5] |
| Mutation-Specific Fluorescent Probes | Detect wild-type vs. mutant sequences | Design for high specificity; multiplexing possible for multiple mutations [5] [52] |
| Magnetic Beads with Primer Linkage | Support clonal amplification and detection | Surface chemistry critical for efficient DNA binding and amplification [5] |
| Quality Control Standards | Validate assay performance and sensitivity | Include wild-type controls, mutation standards at various VAFs [59] |
Implementation Challenges and Solutions
Technical and Analytical Considerations
Successful BEAMing implementation requires addressing several technical challenges:
Pre-analytical Variability: Blood collection and processing protocols significantly impact ctDNA quality and quantity. Standardized protocols using specialized collection tubes that prevent leukocyte lysis are essential for reliable results, particularly in multi-center studies [7] [38]. Plasma should be separated from blood cells within 2-6 hours when using conventional EDTA tubes, though specialized tubes extend this window to 3-7 days [38].
Assay Sensitivity Limitations: While BEAMing achieves exceptional sensitivity for point mutations (0.01%), it has reduced performance for detecting copy number alterations and gene fusions [1]. The technology is ideally suited for hotspot mutation detection but may require complementary approaches for comprehensive genomic profiling.
Clonal Hematopoiesis Interference: Mutations derived from clonal hematopoiesis of indeterminate potential (CHIP) can cause false-positive results [1]. This challenge necessitates careful interpretation of mutation profiles, particularly when mutations commonly associated with CHIP are detected without corresponding tissue confirmation.
Clinical Integration and Validation
Implementing BEAMing technology in clinical practice requires rigorous validation and integration strategies:
Analytical Validation: Laboratories must establish performance characteristics for each mutation included in clinical panels, determining limits of detection, analytical specificity, and precision using well-characterized reference materials [59]. This includes validation of the entire workflow from blood collection through final analysis.
Clinical Concordance Studies: Before implementation, BEAMing results should be compared with standard tissue testing in the intended-use population. For RAS testing in mCRC, studies demonstrate >90% concordance between plasma and tissue results, providing confidence in liquid biopsy results when tissue is unavailable [5].
Interpretation Frameworks: Developing clinical reports that appropriately communicate technical limitations and clinical implications is essential. This includes specifying detected mutations, variant allele frequencies, and limitations related to assay sensitivity and potential CHIP contributions [1] [7].
BEAMing technology for ctDNA analysis represents a powerful tool in precision oncology, offering significant clinical benefits through non-invasive mutation detection and monitoring. The cost-benefit analysis supports its implementation particularly in scenarios where tissue biopsies are challenging or repeated monitoring is necessary. Successful integration requires careful attention to pre-analytical variables, analytical validation, and interpretation frameworks that account for both technical and biological limitations.
Future directions for BEAMing technology include expanding mutation panels, further improving sensitivity for MRD detection, and demonstrating cost-effectiveness in broader clinical applications. As evidence accumulates from ongoing clinical trials, particularly in the adjuvant setting for various solid tumors, the role of BEAMing in routine cancer management is likely to expand, potentially transforming approaches to cancer diagnosis, treatment selection, and monitoring.
The analysis of circulating tumor DNA (ctDNA) using BEAMing technology (Beads, Emulsion, Amplification, and Magnetics) represents a highly sensitive and quantitative approach for detecting specific tumor-derived mutations in patient blood samples [1] [4]. However, tumor biology is complex, and a single analyte often provides an incomplete picture of disease status. The future of liquid biopsy lies in integrating multiple modalities to create a more comprehensive diagnostic profile. Combining BEAMing with other liquid biopsy analytes and technologies addresses inherent limitations of single-analyte approaches and enables a more robust assessment of cancer presence, evolution, and treatment response [80] [81].
This multi-analyte approach leverages the complementary strengths of different biomarkers: while BEAMing excels at quantifying specific mutations with high sensitivity, other components like methylation patterns, fragmentomics, and protein markers provide additional layers of information about tumor origin, burden, and heterogeneity [4] [2]. Such integration is particularly crucial for applications in early cancer detection, minimal residual disease (MRD) monitoring, and guiding precision oncology interventions, where sensitivity and specificity must be maximized [9] [81].
Key Liquid Biopsy Modalities for Integration
Circulating Tumor DNA (ctDNA) Methylation Analysis
DNA methylation is an epigenetic modification that regulates gene expression, and cancer cells exhibit distinct methylation patterns that can be detected in ctDNA [4]. Unlike somatic mutations analyzed by BEAMing, methylation changes are highly prevalent and tissue-specific, making them valuable markers for determining cancer origin and detecting early-stage disease [81].
- Complementary Value to BEAMing: While BEAMing targets a predefined set of mutations, methylation profiling can screen for cancer presence without prior knowledge of the tumor's genetic landscape. This is particularly valuable for early detection and screening in asymptomatic populations [81].
- Integration Protocol: A sequential workflow can be implemented where a broad, methylation-based screen identifies potential cancer cases, followed by BEAMing assays to quantify specific, actionable mutations for targeted therapy selection [4] [81].
Circulating Tumor Cells (CTCs)
CTCs are intact tumor cells shed into the bloodstream from primary or metastatic sites. They provide a complete cellular snapshot of the tumor, including DNA, RNA, proteins, and functional characteristics [80] [9].
- Complementary Value to BEAMing: BEAMing analyzes freely circulating DNA fragments, while CTCs offer the opportunity for whole-genome and transcriptome analysis of viable tumor cells. This allows for the study of gene expression, signaling pathways, and functional mechanisms of drug resistance that are not accessible through ctDNA alone [80] [9].
- Integration Protocol: After isolating CTCs from blood samples using systems like the FDA-cleared CellSearch platform or microfluidic devices, cells can be enumerated and then subjected to single-cell or pooled RNA/DNA sequencing. BEAMing can be performed in parallel on plasma from the same blood draw to correlate mutation abundance with the presence of viable tumor cells [9].
Extracellular Vesicles (EVs) and Tumor-Educated Platelets (TEPs)
EVs, including exosomes, are lipid-bilayer particles released by cells that carry proteins, nucleic acids (DNA, RNA, miRNA), and lipids from their cell of origin. TEPs are platelets that have been altered by interactions with tumors, absorbing tumor-derived biomolecules and reflecting the tumor's RNA and protein profile [80].
- Complementary Value to BEAMing: EVs and TEPs provide a rich source of RNA and protein biomarkers. They can offer information on gene fusions, alternative splicing, and signaling pathway activation, complementing the DNA-based mutational profile from BEAMing [80].
- Integration Protocol: EVs are typically isolated from plasma via ultracentrifugation, density gradient centrifugation, or size-exclusion chromatography. TEPs are isolated through platelet-specific purification. Subsequent RNA and protein analysis from these components can be correlated with BEAMing-derived ctDNA mutation data for a multi-omic view of the tumor [80].
Fragmentomics and End Motif Analysis
Fragmentomics involves the study of the size, distribution, and end sequences of cell-free DNA fragments. Tumor-derived ctDNA has been shown to have characteristic fragmentation patterns and end motifs that differ from those of non-tumor cfDNA [4] [2].
- Complementary Value to BEAMing: Fragmentomic analysis can help distinguish tumor-derived signals from non-tumor background, thereby improving the specificity of liquid biopsy assays. It can be applied genome-wide without the need for tumor-specific markers, enhancing the detection of cancers with low ctDNA shedding [4] [2].
- Integration Protocol: Following cfDNA extraction, a portion of the sample can be subjected to low-coverage whole-genome sequencing (WGS) for fragmentome analysis (e.g., using the DELFI approach). This data can be combined with the targeted, high-sensitivity mutation data from BEAMing to increase overall assay performance for early detection [4].
Table 1: Comparison of Key Liquid Biopsy Modalities for Integration with BEAMing
| Modality | Biomarker Type | Key Information Provided | Primary Integration Benefit |
|---|---|---|---|
| ctDNA Methylation | Epigenetic | Tissue of origin, early carcinogenesis | Expands detection capability without prior knowledge of tumor mutations |
| Circulating Tumor Cells (CTCs) | Cellular | Whole genome/transcriptome of viable tumor cells, functional studies | Provides information on living tumor cell biology and heterogeneity |
| Extracellular Vesicles (EVs) | Particulate (RNA, protein) | Gene fusions, RNA expression, protein biomarkers | Enables multi-analyte (RNA, protein) analysis from the same source |
| Fragmentomics | Physical DNA traits | DNA packaging & cleavage patterns, tumor vs. non-tumor origin | Improves specificity and can detect low-shedding tumors |
Experimental Protocols for Integrated Analysis
Protocol 1: Combined ctDNA Mutation and Methylation Analysis
This protocol details a sequential workflow for integrating BEAMing-based mutation detection with methylation analysis to enhance the detection of esophageal cancer [81].
Sample Collection and Processing:
- Collect patient blood in cell-stabilizing tubes (e.g., Streck Cell-Free DNA BCT).
- Process within 6 hours: centrifuge at 1600 × g for 20 min to separate plasma.
- Perform a second high-speed centrifugation at 16,000 × g for 10 min to remove residual cells.
- Extract cfDNA from plasma using a silica-membrane or magnetic bead-based kit. Elute in a low-EDTA TE buffer.
Parallel Analysis:
- Methylation Analysis:
- Treat a portion of cfDNA with sodium bisulfite using a commercial conversion kit.
- Perform methylation-specific PCR (qMSP) or NGS on a panel of genes hypermethylated in the target cancer (e.g., SEPTIN9, TFPI2 for esophageal cancer) [81].
- BEAMing Analysis:
- Use the remaining cfDNA for BEAMing assay.
- Design BEAMing primers to target known driver mutations (e.g., TP53, KRAS) identified in the tumor type of interest [1] [2].
- Follow standard BEAMing workflow: emulsion PCR on magnetic beads, hybridization with fluorescent probes, and flow cytometry analysis to count mutant and wild-type DNA beads [4].
- Methylation Analysis:
Data Integration:
- A positive result from either assay triggers a more detailed investigation.
- Quantify the correlation between methylation signal intensity and mutant allele frequency from BEAMing to assess tumor burden and clonal heterogeneity.
Protocol 2: Multi-Modal MRD Detection in Colorectal Cancer
This protocol outlines a strategy for detecting Minimal Residual Disease (MRD) in colorectal cancer patients post-surgery by integrating fragmentomics with tumor-informed BEAMing [82] [2].
Tumor Tissue Analysis (Tumor-Informed Approach):
- Sequence the resected primary tumor (via WES or a targeted panel) to identify 10-16 patient-specific somatic mutations.
- Select the most suitable mutations (e.g., clonal, not associated with CHIP) for designing personalized BEAMing assays.
Longitudinal Plasma Analysis:
- Collect plasma samples pre-surgery, 4 weeks post-surgery, and then every 3-6 months for monitoring.
- Extract cfDNA from each time point.
- Fragmentomic Analysis:
- Perform low-pass WGS (~0.1x coverage) on a portion of cfDNA.
- Use bioinformatics tools (e.g., DELFI) to analyze genome-wide fragmentation profiles and derive a "tumor fraction" estimate [4].
- BEAMing Analysis:
- Perform the personalized BEAMing assay on the same cfDNA sample to quantify the presence of tumor-specific mutations with high sensitivity (~0.01%) [2].
Data Integration and MRD Calling:
- MRD positivity is defined by a confirmed detection of tumor-derived signal in either assay.
- The fragmentomic score provides an orthogonal, tumor-agnostic validation of the BEAMing result, increasing confidence in the MRD call, especially in cases with low variant allele frequency.
The following workflow diagram illustrates the multi-modal MRD detection protocol:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Kits for Multi-Modal Liquid Biopsy Research
| Reagent / Kit | Primary Function | Role in Integrated Workflow |
|---|---|---|
| Cell-Free DNA Blood Collection Tubes (e.g., Streck BCT, PAXgene) | Stabilizes nucleated blood cells to prevent genomic DNA contamination during shipment/storage. | Ensures pre-analytical quality for all downstream analyses (BEAMing, fragmentomics, methylation) [4]. |
| Magnetic Beads (Streptavidin-Coated) | Solid support for PCR amplification and probe hybridization in BEAMing. | Core component of the BEAMing assay to physically separate and count mutant DNA molecules [4]. |
| Sodium Bisulfite Conversion Kit | Deaminates unmethylated cytosine to uracil, allowing methylation status to be read as a C-to-T sequence change. | Essential pre-processing step for DNA methylation analysis via sequencing or PCR [81]. |
| Unique Molecular Identifier (UMI) Adapters | Short random nucleotide sequences ligated to each DNA fragment prior to PCR amplification. | Tags original DNA molecules to correct for PCR errors and duplicates in NGS-based fragmentomics and methylation studies, improving quantification [2]. |
| EV Isolation Kits (e.g., ultracentrifugation, precipitation) | Isolate extracellular vesicles from plasma or other biofluids based on size, density, or surface markers. | Enables downstream RNA and protein analysis from EVs, complementing DNA-based assays [80]. |
| CTC Enrichment Kits (e.g., immunomagnetic, microfluidic) | Enrich circulating tumor cells from whole blood using antibodies against surface markers (e.g., EpCAM). | Allows for isolation of viable tumor cells for culture or genomic analysis, providing a layer of cellular information [9]. |
Quantitative Performance of Integrated Approaches
Integrating multiple liquid biopsy modalities consistently demonstrates enhanced performance compared to single-analyte approaches. The table below summarizes key performance metrics from recent studies, highlighting the additive value of a multi-modal strategy.
Table 3: Performance Metrics of Single vs. Multi-Modal Liquid Biopsy Approaches
| Application / Cancer Type | Single-Modality (e.g., BEAMing only) | Integrated Multi-Modal Approach | Key Finding / Improvement |
|---|---|---|---|
| Early Cancer Detection [4] [81] | Sensitivity: ~50-70% (Specificity ~95%) | Sensitivity: >90% (Specificity ~95%) | Combining mutation (BEAMing) and methylation analysis significantly increases sensitivity for stage I/II tumors. |
| MRD Detection in Colorectal Cancer [82] [2] | Lead Time: 3-5 months (BEAMing) | Lead Time: 5-8 months (BEAMing + Fragmentomics) | Multi-modal detection predicts radiographic recurrence with a significantly longer lead time. |
| Therapy Response Monitoring in NSCLC [1] [2] | Accuracy: ~85% (ctDNA clearance) | Accuracy: >95% (ctDNA + EV miRNA) | Adding EV-derived miRNA profiles improves accuracy in predicting long-term treatment response. |
| Tumor Origin Determination [80] [81] | Limited capability (Mutation profile only) | Accuracy: ~85-90% (Methylation + Fragmentomics) | Methylation and fragmentomic patterns are highly tissue-specific, enabling prediction of cancer origin in cases of unknown primary. |
The integration of BEAMing technology with other liquid biopsy modalities represents a paradigm shift in cancer diagnostics and monitoring. By combining the high sensitivity of BEAMing for known mutations with the broad, tumor-agnostic screening power of methylation analysis, fragmentomics, and EV/CTC analysis, researchers and clinicians can obtain a more holistic and actionable view of a patient's disease [80] [2] [81].
Future developments will focus on standardizing these complex workflows, validating them in large-scale prospective clinical trials, and leveraging machine learning to optimally fuse the multi-modal data streams into a single, clinically reportable result [1] [9]. As these technologies mature and become more accessible, integrated liquid biopsy panels are poised to become the standard of care in precision oncology, enabling earlier detection, more precise monitoring, and more personalized treatment strategies for cancer patients.
Conclusion
BEAMing technology has established itself as a robust, highly sensitive platform for ctDNA analysis with demonstrated clinical utility across multiple cancer types. The technology's ability to detect mutations at frequencies as low as 0.01% and provide precise quantification makes it particularly valuable for treatment monitoring, resistance mutation detection, and minimal residual disease assessment. While BEAMing shows excellent concordance with both tissue biopsy and alternative digital PCR methods, successful implementation requires careful attention to pre-analytical variables and understanding of its limitations in early-stage disease. Future directions include expanded multiplexing capabilities, integration with next-generation sequencing for broader mutation profiling, and development of standardized protocols for clinical trial implementation. As precision oncology continues to evolve, BEAMing remains a critical tool for researchers and drug development professionals seeking to translate liquid biopsy applications into improved patient outcomes.
References
- 1. Clinical applications of cell-free DNA-based liquid biopsy ... [https://www.sciencedirect.com/science/article/pii/S1936523325002505]
- 2. Circulating tumor DNA to monitor treatment response in ... [https://www.nature.com/articles/s41698-025-00876-y]
- 3. Comparative analysis of EGFR mutations in circulating ... [https://www.nature.com/articles/s41598-025-85160-6]
- 4. A Review of Circulating Tumor DNA (ctDNA) and the Liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12032849/]
- 5. Incorporating BEAMing technology as a liquid biopsy into ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5834030/]
- 6. Recent advances in ctDNA detection using electrochemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11448507/]
- 7. Implementing ctDNA Analysis in the Clinic: Challenges and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7693855/]
- 8. A clinician's handbook for using ctDNA throughout the patient ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-022-01551-7]
- 9. Liquid biopsy in cancer: current status, challenges and ... [https://www.nature.com/articles/s41392-024-02021-w]
- 10. Tracing the history of clinical practice of liquid biopsy [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1574736/full]
- 11. The Use of ctDNA for BRAF Mutation Testing in Routine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8833667/]
- 12. Circulating Tumor DNA detection in cancer - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12450317/]
- 13. Analytical evaluation of circulating tumor DNA sequencing ... [https://www.nature.com/articles/s41598-024-54361-w]
- 14. Monitoring and Assessment of Circulating Tumor DNA in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550271/]
- 15. Implementation of Circulating Tumor DNA (ctDNA) Testing ... [https://www.sciencedirect.com/science/article/pii/S2950195425000359]
- 16. Quantitative and Dynamic ctDNA as a Biomarker of ... [https://www.sciencedirect.com/science/article/pii/S1556086425007518]
- 17. A comparative study on ctDNA and tumor DNA mutations in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10691057/]
- 18. Potential Applications of Circulating Tumor DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6689482/]
- 19. Circulating mutant DNA to assess tumor dynamics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2820391/]
- 20. Comparison of BEAMing and Droplet Digital PCR for Circulating Tumor DNA Analysis - PubMed [https://pubmed.ncbi.nlm.nih.gov/31551314/]
- 21. Real-World Technical Hurdles of ctDNA NGS Analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12564474/]
- 22. Circulating tumour DNA assays in focus: highlights from ... [https://www.nature.com/articles/s44276-025-00181-y]
- 23. Advances in CTC and ctDNA detection techniques [https://breast-cancer-research.biomedcentral.com/articles/10.1186/s13058-025-02024-7]
- 24. The prospects and limitations of liquid biopsy utilization for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11937291/]
- 25. Circulating tumor DNA laboratory processes and clinical ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1520733/full]
- 26. Improvement of the sensitivity of circulating tumor DNA- ... [https://www.explorationpub.com/Journals/etat/Article/1002333]
- 27. Circulating tumor DNA laboratory processes and clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12119280/]
- 28. Circulating tumor DNA in cancer diagnosis, monitoring, and ... [https://jenci.springeropen.com/articles/10.1186/s43046-022-00109-4]
- 29. Pre-Analytical Evaluation of Streck Cell-Free DNA Blood ... [https://www.mdpi.com/2075-4418/13/7/1288]
- 30. Clinical Practice Guidelines for Pre-Analytical Procedures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8548242/]
- 31. Investigation of appropriate pre-analytical procedure for ... [https://www.oncotarget.com/article/25881/text/]
- 32. Emulsion PCR: Techniques and Applications [https://pubmed.ncbi.nlm.nih.gov/26843044/]
- 33. BEAMing [https://en.wikipedia.org/wiki/BEAMing]
- 34. Review Miniaturising the laboratory in emulsion droplets [https://www.sciencedirect.com/science/article/abs/pii/S0167779906001582]
- 35. A novel emulsion PCR method coupled with fluorescence ... [https://www.nature.com/articles/s41598-018-36981-1]
- 36. Implementation of circulating tumor DNA (ctDNA) testing in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12356032/]
- 37. Revolution of Circulating Tumor DNA [https://pmc.ncbi.nlm.nih.gov/articles/PMC12192262/]
- 38. Improvement of the sensitivity of circulating tumor DNA-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12332530/]
- 39. How to Interpret Flow Cytometry Data [https://www.fortislife.com/resources/antibody-resources/how-to-interpret-flow-cytometry-data]
- 40. RAS testing in metastatic colorectal cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC4830882/]
- 41. Final results of the multicentric ColoBEAM study [https://pubmed.ncbi.nlm.nih.gov/41235357/]
- 42. Profiling Colorectal Cancer in the Landscape Personalized ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8122648/]
- 43. status of Current in precision oncology for hepatocellular... ctDNA [https://jeccr.biomedcentral.com/articles/10.1186/s13046-021-01940-8]
- 44. Emerging biomarkers for early detection of lung cancer [https://link.springer.com/article/10.1007/s44272-025-00046-y]
- 45. Liquid profiling of circulating tumor DNA in colorectal cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC9120900/]
- 46. liquid remnants of solid tumors treated with curative intent... Detecting [https://pmc.ncbi.nlm.nih.gov/articles/PMC10152452/]
- 47. Clinical Practice Guideline for Blood-based Circulating Tumor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10813828/]
- 48. ( Minimal ) in Solid Tumors: Residual ... Disease MRD Detection [https://www.linkedin.com/pulse/minimal-residual-disease-mrd-solid-tumors-detection-strategy-rfdmc]
- 49. TruSight Oncology 500 ctDNA v2 Documentation [https://support.illumina.com/sequencing/sequencing_kits/trusight-oncology-500-ctDNA-v2/documentation.html]
- 50. Accuracy of minimal by circulating tumor... residual disease detection [https://bmcmedicine.biomedcentral.com/articles/10.1186/s12916-023-02849-z]
- 51. The impact of preanalytical variables on the analysis of cell ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1385041/full]
- 52. Detection of IDH1 Mutations in Plasma Using BEAMing ... [https://www.mdpi.com/2072-6694/14/12/2891]
- 53. Circulating tumor DNA in colorectal cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC11961841/]
- 54. Revolution of Circulating Tumor DNA: From Bench ... [https://www.mdpi.com/1467-3045/47/6/428]
- 55. Comparative analysis of EGFR mutations in circulating ... [https://pubmed.ncbi.nlm.nih.gov/39775010/]
- 56. Exploring Circulating Tumor DNA (CtDNA) and Its Role in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10516512/]
- 57. Circulating tumor DNA: current challenges for clinical utility [https://www.jci.org/articles/view/154941]
- 58. Techniques of using circulating tumor DNA as a liquid ... [https://www.sciencedirect.com/science/article/pii/S2001037018301958]
- 59. Considerations and quality controls when analyzing cell ... [https://www.sciencedirect.com/science/article/pii/S2214753518300184]
- 60. Commercial ctDNA for minimal residual disease detection of... assays [https://pmc.ncbi.nlm.nih.gov/articles/PMC9016631/]
- 61. Performance of four platforms for KRAS mutation detection in plasma... [https://www.nature.com/articles/s41598-020-64822-7?error=cookies_not_supported]
- 62. Real-World Technical Hurdles of ctDNA NGS Analysis [https://www.mdpi.com/2079-9721/13/10/312]
- 63. BEAMing for Cancer [https://www.genengnews.com/insights/beaming-for-cancer/]
- 64. US20130040300A9 - Methods for Beaming [https://patents.google.com/patent/US20130040300A9/en]
- 65. Sensitivity, specificity, and accuracy of a liquid biopsy ... [https://www.nature.com/articles/s41598-021-89592-8]
- 66. Concordance of Targeted Sequencing from Circulating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10526508/]
- 67. Concordance of ctDNA and tissue genomic profiling in ... [https://www.sciencedirect.com/science/article/pii/S0168827824026412]
- 68. Comparison of digital PCR systems for the analysis ... [https://www.sciencedirect.com/science/article/pii/S0009898123000414]
- 69. ctDNA applications and integration in colorectal cancer [https://www.nature.com/articles/s41571-020-0392-0]
- 70. Clinical Circulating Tumor DNA Testing for Precision Oncology [https://pmc.ncbi.nlm.nih.gov/articles/PMC10101787/]
- 71. Generic Protocols for the Analytical Validation of Next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7462123/]
- 72. Understanding BLOODPAC's Analytical Validation Protocols [https://www.bloodpac.org/bloodpac-blog/understanding-bloodpacs-analytical-validation-protocols-for-ctdna-assays]
- 73. The emerging clinical utility of ctDNA to guide adjuvant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12173074/]
- 74. Prospective multicenter real-world RAS mutation ... [https://www.nature.com/articles/s41416-018-0293-5]
- 75. Circulating Tumor DNA (ctDNA) Sequencing [https://microbenotes.com/circulating-tumor-dna-ctdna-sequencing/]
- 76. Clinical trial and real-world evidence of circulating tumor ... [https://www.sciencedirect.com/science/article/pii/S2949820124000547]
- 77. A Micro-Costing Framework for Circulating Tumor DNA ... [https://www.sciencedirect.com/science/article/pii/S1525157822003099]
- 78. Early evaluation of the effectiveness and cost-effectiveness of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11339739/]
- 79. Clinical Utility of ctDNA Analysis in Lung Cancer—A Review [https://www.mdpi.com/2543-6031/93/3/17]
- 80. Liquid biopsy – a narrative review with an update on current ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12333414/]
- 81. Multi-analyte liquid biopsy approaches for early detection ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1622984/full]
- 82. Research trends and hotspots of circulating tumor DNA in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12116327/]