A Comprehensive Guide to Building and Validating Pharmacophore Models for Virtual Screening in Drug Discovery
This article provides a detailed, step-by-step guide for researchers and drug development professionals on constructing, applying, and validating pharmacophore models for virtual screening.
A Comprehensive Guide to Building and Validating Pharmacophore Models for Virtual Screening in Drug Discovery
Abstract
This article provides a detailed, step-by-step guide for researchers and drug development professionals on constructing, applying, and validating pharmacophore models for virtual screening. It covers the foundational concepts of pharmacophores, compares structure-based and ligand-based modeling methodologies, and outlines best practices for model refinement and troubleshooting. Furthermore, the guide explores rigorous validation techniques, including the use of ROC curves and enrichment factors, and compares the performance of pharmacophore-based screening with docking-based methods. The content is designed to equip scientists with the practical knowledge needed to effectively implement this powerful computer-aided drug discovery technique to reduce time and costs in lead compound identification.
Understanding Pharmacophores: The Essential Framework for Molecular Recognition
The pharmacophore concept stands as one of the most enduring and fruitful paradigms in medicinal chemistry and computer-aided drug design. As an abstract representation of molecular interactions, it provides the foundational framework for understanding structure-activity relationships and enables the rational design of therapeutic compounds. In modern drug discovery, pharmacophore models serve as essential tools for virtual screening, de novo design, and lead optimization, dramatically reducing the time and cost associated with bringing new drugs to market. The evolution of this concept from a qualitative notion to a quantitatively precise definition mirrors the advancement of drug discovery itself, transitioning from observational chemistry to computationally-driven molecular design. This technical guide explores the pharmacophore's historical origins, its formal IUPAC definition, and its practical application in contemporary virtual screening workflows, providing researchers with both theoretical foundation and methodological protocols for implementing pharmacophore-based strategies in drug development projects.
Historical Evolution of the Pharmacophore Concept
The conceptual foundation of the pharmacophore emerged long before the term itself was formally coined. In 1909, Paul Ehrlich introduced the foundational idea by describing a "molecular framework that carries the essential features responsible for a drug's biological activity" [1]. This initial conceptualization established the principle that specific molecular features, rather than the entire molecular structure, mediate biological activity. Around the same period, Emil Fischer's "lock and key" hypothesis provided a complementary physical model for understanding the stereochemical complementarity between ligands and their biological targets [2].
The term "pharmacophore" was popularized significantly later by Lemont Kier in 1967 and appeared in a published work in 1971 [3]. Kier's work represented a critical step toward formalizing the concept, moving from vague notions of important functional groups to a more systematic understanding of essential molecular features. F. W. Shueler also contributed to this conceptual evolution, employing the expression "pharmacophoric moiety" in his 1960s publications, which closely aligned with the modern pharmacophore understanding [3].
Throughout the late 20th century, computational advances transformed the pharmacophore from a theoretical concept to an actionable tool in drug discovery. The development of automated pharmacophore generation algorithms in the 1990s and early 2000s enabled researchers to systematically extract common chemical features from sets of active molecules and create predictive models for virtual screening [1]. This period marked the transition of pharmacophores from descriptive frameworks to prescriptive tools that could actively guide drug design decisions and compound prioritization.
The Modern IUPAC Definition and Core Principles
The International Union of Pure and Applied Chemistry established the current formal definition of a pharmacophore as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [4]. This precise definition, endorsed in 2015 as part of IUPAC's Recommendations on computational drug design terminology, carries several fundamental implications for modern drug discovery.
The definition emphasizes that pharmacophores represent abstract features rather than specific chemical structures or functional groups. This abstraction is crucial for enabling "scaffold hopping" – the identification of structurally diverse compounds that share the same pattern of essential interactions [2] [5]. By focusing on the spatial arrangement of chemical features rather than atomic connectivity, pharmacophore models can identify novel chemotypes that would be missed by similarity-based screening approaches.
A second critical aspect of the IUPAC definition is its emphasis on supramolecular interactions, positioning the pharmacophore as an interface concept that encompasses both ligand properties and complementary target features. This conceptual framework acknowledges that pharmacophores exist not as intrinsic molecular properties alone, but as relational attributes defined through interaction with biological targets [4] [1]. The definition also establishes the direct link between the pharmacophore and biological response, making explicit that a valid pharmacophore model must account for the structural determinants of efficacy, not merely binding.
Table 1: Essential Pharmacophore Features and Their Interaction Types
| Feature Type | Geometric Representation | Complementary Feature | Interaction Type | Structural Examples |
|---|---|---|---|---|
| Hydrogen Bond Acceptor (HBA) | Vector or Sphere | Hydrogen Bond Donor | Hydrogen Bonding | Amines, Carboxylates, Ketones |
| Hydrogen Bond Donor (HBD) | Vector or Sphere | Hydrogen Bond Acceptor | Hydrogen Bonding | Amines, Amides, Alcoholes |
| Aromatic (AR) | Plane or Sphere | Aromatic, Positive Ionizable | π-Stacking, Cation-π | Any Aromatic Ring |
| Positive Ionizable (PI) | Sphere | Negative Ionizable, Aromatic | Ionic, Cation-π | Ammonium Ions, Metal Cations |
| Negative Ionizable (NI) | Sphere | Positive Ionizable | Ionic | Carboxylates, Phosphates |
| Hydrophobic (H) | Sphere | Hydrophobic | Hydrophobic Contact | Alkyl Groups, Alicycles, Halogens |
Methodological Approaches to Pharmacophore Modeling
Structure-Based Pharmacophore Modeling
Structure-based pharmacophore modeling derives interaction features directly from the three-dimensional structure of a macromolecular target or a target-ligand complex. This approach requires high-quality structural data, typically from X-ray crystallography, NMR spectroscopy, or increasingly, from computationally-predicted structures using tools like AlphaFold2 [2]. The methodology involves systematic analysis of the binding site to identify key interaction points and their spatial relationships, which are then translated into pharmacophore features.
The standard workflow for structure-based pharmacophore development begins with protein preparation, which includes adding hydrogen atoms, assigning proper protonation states, and correcting any structural deficiencies in the experimental coordinates [2]. The subsequent binding site detection step identifies the physiologically relevant cavity, which can be guided by experimental data on known ligands or computed using algorithms like GRID or LUDI that analyze geometric, energetic, and evolutionary constraints [2]. The core feature generation process then identifies potential interaction points by probing the binding site with functional groups or by analyzing existing protein-ligand complexes to determine conserved interactions. The final feature selection step distills the most essential features to create a selective yet sufficiently general model [2].
When a co-crystallized ligand is present, the pharmacophore features can be placed more accurately based on observed interactions, and exclusion volumes can be incorporated to represent spatial constraints of the binding pocket [2]. The resulting models typically exhibit high specificity and can effectively guide virtual screening even for structurally novel compounds.
Ligand-Based Pharmacophore Modeling
Ligand-based pharmacophore modeling approaches generate hypotheses based on the structural and physicochemical properties of known active compounds, without requiring direct knowledge of the target structure. This method is particularly valuable when the macromolecular target is poorly characterized structurally but sufficient ligand activity data is available. The fundamental premise is that compounds sharing similar biological activities likely interact with the same target through common molecular features arranged in a conserved spatial orientation [6].
The ligand-based workflow initiates with training set selection, requiring a carefully curated set of structurally diverse molecules with measured activities against the target of interest. Ideally, this set should include both active and inactive compounds to enhance model discriminative power [3]. The subsequent conformational analysis generates representative low-energy conformations for each molecule, often using algorithms like poling or Monte Carlo methods to ensure adequate coverage of conformational space [6]. The critical molecular superimposition step aligns compounds to maximize overlap of putative pharmacophore features, employing either point-based methods (minimizing RMSD between corresponding features) or property-based approaches (optimizing overlap of molecular interaction fields) [6].
Following alignment, the feature extraction process identifies conserved chemical features across the aligned set, balancing generality with specificity to create a model with appropriate discriminative power [6]. Finally, model validation assesses the ability of the pharmacophore hypothesis to correctly classify active and inactive compounds, with iterative refinement to improve predictive performance [3].
Table 2: Common Software Tools for Pharmacophore Modeling
| Software Package | Approach | Key Algorithms | Primary Applications |
|---|---|---|---|
| Catalyst (Accelrys) | Ligand-based | Hip-Hop, HypoGen | Virtual Screening, 3D-QSAR |
| DISCO | Ligand-based | Clique Detection | Feature Pattern Recognition |
| GASP | Ligand-based | Genetic Algorithm | Molecular Alignment |
| Phase | Ligand-based | Scoring & Matching | Virtual Screening, QSAR |
| LigandScout | Structure-based | Interaction Mapping | Structure-based Design |
| MOE | Both | Pharmacophore Query | Virtual Screening |
Experimental Protocols and Implementation
Protocol for Structure-Based Pharmacophore Generation
Objective: To create a structure-based pharmacophore model from a protein-ligand complex structure for virtual screening applications.
Required Materials and Software:
- Protein Data Bank structure of target with bound ligand
- Molecular modeling software (e.g., LigandScout, MOE, Discovery Studio)
- Protein preparation tools (e.g., Schrodinger Protein Preparation Wizard, MOE QuickPrep)
- Virtual screening database (e.g., ZINC, ChEMBL)
Methodology:
- Structure Retrieval and Preparation: Download the high-resolution (preferably <2.5 Å) crystal structure of the target protein in complex with a high-affinity ligand from the Protein Data Bank. Remove all non-essential water molecules, cofactors, and alternate conformations. Add hydrogen atoms appropriate for physiological pH (7.4) and optimize their orientations using molecular mechanics force fields.
Binding Site Analysis: Define the binding site using the coordinates of the cocrystallized ligand, expanding by 5-10 Å to include all residues potentially involved in ligand recognition. Analyze conserved interactions (hydrogen bonds, hydrophobic contacts, ionic interactions) between the ligand and binding site residues.
Feature Identification and Mapping: Identify key interaction features directly observed in the crystal structure. Map hydrogen bond donors/acceptors, hydrophobic regions, charged/ionizable groups, and aromatic rings. Define feature tolerances based on the observed geometry and potential for isosteric replacement.
Exclusion Volume Placement: Incorporate exclusion volumes to represent steric constraints of the binding pocket, placing spheres at positions occupied by protein atoms that would clash with potential ligands.
Model Validation: Validate the initial model by screening a small set of known active and inactive compounds. Adjust feature definitions and tolerances to maximize enrichment of active compounds while minimizing false positives.
This protocol typically requires 2-3 days for a trained computational chemist, with the majority of time spent on careful structure preparation and iterative model validation [2].
Protocol for Ligand-Based Pharmacophore Generation
Objective: To develop a quantitative pharmacophore model from a set of known active and inactive compounds without structural information about the biological target.
Required Materials and Software:
- Set of 20-50 compounds with known biological activities (IC50, Ki, or EC50 values)
- Conformational analysis software (e.g., Catalyst, OMEGA)
- Pharmacophore generation suite (e.g., Catalyst/HypoGen, Phase)
- Diverse set of inactive compounds for model validation
Methodology:
- Training Set Compilation: Compile a structurally diverse set of 16-24 compounds spanning at least 4 orders of magnitude in activity values. Include both highly active and moderately active compounds to ensure the model captures essential versus ancillary features.
Conformational Space Exploration: Generate comprehensive conformational ensembles for each compound using the "best conformer generation" method or poling algorithm to ensure coverage of potential bioactive conformations. Maintain an energy threshold of 10-20 kcal/mol above the global minimum to include relevant excited states.
Pharmacophore Hypothesis Generation: Using automated algorithms (e.g., HypoGen), generate multiple pharmacophore hypotheses that correlate feature composition and spatial arrangement with biological activity. The algorithm typically employs a subtractive approach that eliminates features common to inactive compounds.
Statistical Validation: Evaluate hypotheses based on correlation coefficients, cost analysis, and root mean square deviation. Select the hypothesis with the lowest total cost value and highest predictive index for further validation.
Test Set Prediction: Challenge the selected model with a test set of 10-20 compounds not included in the training set. Calculate correlation between predicted and experimental activities and assess the model's scaffold-hopping capability by examining its performance across diverse chemical classes.
This ligand-based protocol typically requires 3-5 days, with computational time heavily dependent on the size and flexibility of the training set compounds [6].
Applications in Virtual Screening and Drug Discovery
Pharmacophore models serve as powerful filters in virtual screening workflows, enabling efficient prioritization of candidate compounds from large chemical databases. By encoding the essential steric and electronic features required for biological activity, pharmacophore queries can rapidly eliminate compounds lacking critical interaction elements while identifying novel chemotypes that fulfill the interaction pattern [2] [1]. This approach is particularly valuable for screening massive databases like ZINC, which contains hundreds of millions of commercially available compounds.
In a recent application targeting monoamine oxidase inhibitors, researchers combined pharmacophore-based virtual screening with machine learning to accelerate the identification of novel chemotypes [7]. The pharmacophore model served as an initial filter to reduce the chemical space before applying more computationally intensive docking studies, demonstrating the efficiency of this hierarchical approach. The study identified 24 synthesized compounds with MAO-A inhibitory activity, validating the predictive capability of the method [7].
Another innovative application involves the use of ensemble pharmacophores to address flexibility in both ligands and targets. In the discovery of novel tubulin inhibitors, researchers generated multiple pharmacophore representations based on different X-ray structures of tubulin-ligand complexes [8]. This ensemble approach captured the inherent plasticity of the colchicine binding site and enabled the identification of novel diaryl tetrazole compounds with potent antiproliferative activity, demonstrating the value of dynamic pharmacophore representations for flexible targets.
Recent Advances and Future Perspectives
The field of pharmacophore modeling continues to evolve through integration with emerging computational methodologies. Machine learning approaches are now being employed to accelerate pharmacophore-based virtual screening, with models trained to predict docking scores without performing explicit molecular docking calculations [7]. These hybrid approaches can achieve speed improvements of up to 1000-fold compared to traditional docking-based virtual screening while maintaining comparable predictive accuracy [7].
Another significant advancement involves the incorporation of pharmacophore constraints directly into generative molecular design. Frameworks like DiffPharm utilize diffusion models to generate novel molecular structures that explicitly satisfy 3D pharmacophore constraints [9]. This inverse design approach represents a paradigm shift from screening existing compounds to actively creating molecules tailored to specific interaction patterns, potentially dramatically expanding accessible chemical space for drug discovery.
The increasing availability of high-quality protein structures from both experimental determination and computational prediction (e.g., AlphaFold2) is expanding opportunities for structure-based pharmacophore approaches [2]. As these structural models continue to improve in accuracy and coverage, pharmacophore modeling will likely play an increasingly central role in target-based drug discovery, particularly for understudied proteins emerging from genomic studies.
Future developments are expected to focus on dynamic pharmacophore models that explicitly incorporate protein flexibility, solvation effects, and allosteric mechanisms. These advanced models will provide more realistic representations of molecular recognition events, potentially improving the success rates of virtual screening campaigns and reducing attrition in later stages of drug development.
Table 3: Key Research Reagent Solutions for Pharmacophore Modeling
| Resource Category | Specific Tools/Services | Key Functionality | Application Context |
|---|---|---|---|
| Structural Databases | RCSB Protein Data Bank (PDB) | Source of experimental 3D structures | Structure-based pharmacophore generation |
| Compound Databases | ZINC, ChEMBL | Libraries of screening compounds | Virtual screening, training set creation |
| Software Platforms | Catalyst, LigandScout, MOE | Automated pharmacophore generation | Model development and screening |
| Conformational Analysis | OMEGA, Catalyst ConFirm | Generation of bioactive conformations | Ligand-based model preparation |
| Validation Tools | ROC Curves, Enrichment Factors | Assessment of model performance | Model selection and optimization |
| Machine Learning Integration | Scikit-learn, DeepChem | Docking score prediction | Accelerated virtual screening |
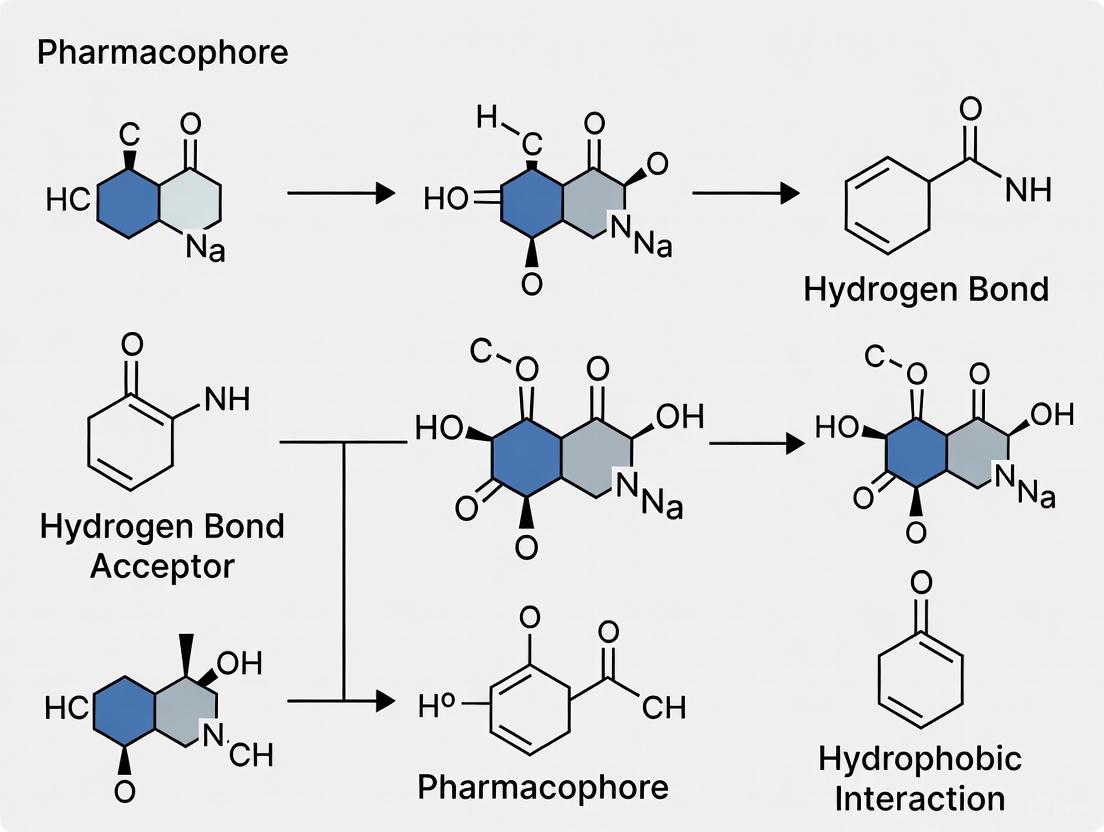
A pharmacophore is defined as the ensemble of steric and electronic features that are necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or block) its biological response [10] [11]. This abstract representation captures the essential three-dimensional arrangement of molecular features shared by ligands that exhibit similar biological activity against a given target, independent of their underlying chemical scaffold [10]. The concept originated with Paul Ehrlich in the late 19th century and was formalized by Schueler in 1960, evolving into a cornerstone of modern rational drug design [10] [11]. In practical terms, pharmacophore models shift the focus from specific atoms and bonds to the fundamental chemical functionalities required for molecular recognition and binding, enabling the identification of structurally diverse compounds that share a common biological activity profile [11].
Core Pharmacophoric Features and Their Characteristics
The activity of a pharmacophore is governed by a set of core physicochemical features and their precise spatial arrangement. These features represent the key functional elements that enable a ligand to form stable complexes with its biological target through complementary interactions [12] [11]. The most critical features include hydrogen bond donors and acceptors, hydrophobic areas, and ionizable groups, each contributing distinct energetic and steric properties to the binding event.
Table 1: Core Pharmacophoric Features and Their Properties
| Feature Type | Atomic/Groups Involved | Interaction Type | Spatial Representation | Common Tolerances |
|---|---|---|---|---|
| Hydrogen Bond Donor (HBD) | N-H, O-H | Electrostatic, directed | Vector (Donor Point + Projection) | Distance: ±1.0–1.5 Å [10] |
| Hydrogen Bond Acceptor (HBA) | O, N (with lone pairs) | Electrostatic, directed | Vector (Acceptor Point + Projection) | Distance: ±1.0–1.5 Å [10] |
| Hydrophobic Area (H) | Alkyl chains, aromatic rings | Van der Waals, entropic (desolvation) | Spherical centroid or volume | Sphere radius: 4–6 Å [10] |
| Positive Ionizable (PI) | Protonated amines (pKa 7-10) | Strong electrostatic (salt bridge) | Spherical point | pKa range: 7-10 at pH 7.4 [10] |
| Negative Ionizable (NI) | Carboxylates, phosphates (pKa 3-5) | Strong electrostatic (salt bridge) | Spherical point | pKa range: 3-5 at pH 7.4 [10] |
| Aromatic Ring (AR) | Phenyl, heterocyclic rings | Cation-π, π-π stacking | Planar ring with centroid and normal vector | Geometric, planar |
Table 2: Quantitative Electronic and Physical Descriptors for Feature Optimization
| Feature Type | Charge Descriptor (Partial Charge | q | ) | Lipophilicity (logP) Context | Geometric Descriptors |
|---|---|---|---|---|---|
| HBD / HBA | > 0.2 e (electron charge units) [10] | Not primary driver | Inter-feature distances, angles, vectors [10] | ||
| Hydrophobic Area | Not primary driver | Optimal logP 2–5 for permeability [10] | Volume, surface area | ||
| Ionizable Groups | Full charge (pKa-dependent) [10] | Can impact solubility/permeability | Spherical point with tolerance | ||
| Aromatic Ring | π-electron density | Can increase logP | Centroid, plane, normal vector |
Hydrogen Bond Donors and Acceptors
Hydrogen bond donors are features containing a hydrogen atom bonded to an electronegative atom (like oxygen or nitrogen), which can be donated to form a favorable electrostatic interaction with a hydrogen bond acceptor on the target [11]. Hydrogen bond acceptors are atoms, typically oxygen or nitrogen with available lone pairs, that can accept a hydrogen bond from a donor [11]. In pharmacophore models, these are often represented as vector features to capture the directionality of the interaction, which is crucial for binding affinity and specificity [10]. The directionality arises from the optimal linear geometry of the D-H···A interaction (where D is the donor and A is the acceptor). Typical tolerance for the distance between donor and acceptor features in a model is ±1.0–1.5 Å to account for conformational flexibility [10].
Hydrophobic Areas
Hydrophobic areas are regions of the ligand that are non-polar and favor van der Waals interactions with complementary non-polar surfaces in the target's binding pocket [12] [11]. The burial of these groups upon ligand binding is energetically favorable primarily due to the desolvation effect—the release of ordered water molecules from the hydrophobic surfaces into the bulk solvent [10]. In pharmacophore models, these features are typically represented as spherical centroids or volumes encompassing atoms in alkyl chains or the faces of aromatic rings [10]. The size of these volumes, often with radii of 4–6 Å, helps define the extent of the hydrophobic interaction required for activity [10].
Ionizable Groups
Ionizable groups are functional groups that can carry a formal positive or negative charge at physiological pH (approximately 7.4), such as basic amines (positive ionizable) or acidic carboxylates (negative ionizable) [11]. These features enable strong, long-range electrostatic interactions and salt bridges with oppositely charged residues in the protein target (e.g., aspartate, glutamate, lysine, arginine) [10]. The inclusion of these features in a pharmacophore model is dependent on the group's protonation state. For instance, a basic group with a pKa between 7 and 10 is expected to be protonated and positively charged at pH 7.4, while an acidic group with a pKa between 3 and 5 is expected to be deprotonated and negatively charged [10].
Experimental and Computational Protocols for Feature Identification
The accurate identification and placement of pharmacophoric features rely on well-established computational protocols. The following methodologies detail the process for both structure-based and ligand-based approaches.
Structure-Based Pharmacophore Modeling Protocol
This protocol is used when a 3D structure of the target protein (often with a bound ligand) is available, typically from the Protein Data Bank (PDB) [11].
Protein Preparation:
- Source: Obtain the 3D structure from the PDB. If an experimental structure is unavailable, use computational techniques like homology modeling (e.g., MODELLER) or machine learning-based methods (e.g., AlphaFold2) [11].
- Preparation Steps:
- Add hydrogen atoms, which are often missing in X-ray structures.
- Assign correct protonation states to residues (e.g., His, Asp, Glu) based on their local environment using tools like PROPKA.
- Repair any missing residues or side-chain atoms.
- Perform energy minimization to relieve steric clashes.
Binding Site Detection:
- If the structure is a protein-ligand complex, the binding site is defined by the co-crystallized ligand.
- For apo structures, use computational tools like GRID (which uses molecular interaction fields) or LUDI (which uses geometric rules and knowledge-based distributions) to identify potential binding pockets [11].
Feature Generation and Selection:
- Analyze the protein-ligand interactions (or the protein's interaction potential in an apo structure) to generate a map of potential interaction points.
- Key interactions (e.g., a hydrogen bond with a key catalytic residue, a salt bridge with a charged residue, or hydrophobic contact with a conserved residue) are translated into corresponding pharmacophore features (HBA, HBD, H, PI, NI) [13] [11].
- Select only the essential features that are critical for binding affinity and specificity. This can be done by:
- Removing features that do not contribute significantly to the calculated binding energy.
- Identifying conserved interactions across multiple protein-ligand complexes.
- Incorporating information from site-directed mutagenesis studies.
- Add exclusion volumes (XVOL) to represent the shape of the binding pocket and steric restrictions, preventing clashes with the protein [11].
Ligand-Based Pharmacophore Modeling Protocol
This protocol is used when a set of known active ligands is available, but the 3D structure of the target is unknown [12] [11].
Ligand Dataset Curation:
- Assemble a set of 3-10 structurally diverse molecules that are confirmed to be active against the target.
- Include a set of known inactive compounds if available, to help identify features that discriminate between active and inactive molecules.
Conformational Analysis:
- For each active ligand, generate an ensemble of low-energy 3D conformers. This is critical because the pharmacophore must be based on the bioactive conformation.
- Use methods such as systematic search, Monte Carlo sampling, or molecular dynamics simulations to explore the conformational space [12].
Molecular Alignment and Hypothesis Generation:
- Use common feature alignment or flexible alignment algorithms to superimpose the conformers of the active ligands [12].
- The goal is to find a 3D alignment where key chemical features from all active molecules overlap. This common set of overlapping features forms the initial pharmacophore hypothesis [10] [12].
- The principle of superposition is the cornerstone of this approach, assuming that active molecules share a common spatial arrangement of interaction points [10].
Feature Identification and Model Refinement:
- Identify the essential features consistently present across all active ligands and absent in inactives. Statistical methods or iterative refinement are used to exclude non-essential (auxiliary) features that may modulate affinity but are not critical for binding [10].
- Define the spatial constraints, including distances, angles, and tolerances between the selected features. Tolerances (e.g., distance ±1.5 Å) account for small deviations in the binding mode and conformational flexibility [10].
Application in Virtual Screening: A Case Study
The primary application of a validated pharmacophore model is in virtual screening (VS) of large compound libraries to identify novel hit compounds [12]. The following case study illustrates a complete protocol.
Case Study: Identification of SARS-CoV-2 PLpro Inhibitors from Marine Natural Products [13]
- Objective: Discover novel inhibitors of the SARS-CoV-2 papain-like protease (PLpro) by screening the Comprehensive Marine Natural Product Database (CMNPD).
- Pharmacophore Model: A structure-based model was developed using a potent co-crystallized inhibitor. The final model contained 9 pharmacophoric features representing interactions with five key binding sites on PLpro [13].
- Virtual Screening Protocol:
- Pharmacophore Screening: The 9-feature model was used as a 3D query to screen the CMNPD, yielding 66 initial hits that matched the pharmacophore.
- Molecular Weight Filter: The hit library was downsized by applying a filter for molecular weight ≤ 500 g/mol, resulting in 50 candidates for docking.
- Comparative Molecular Docking: The 50 hits were docked into the PLpro binding site using two different docking engines, AutoDock and AutoDock Vina. Using multiple engines relieves disparities in their search and scoring functions.
- Consensus Scoring: Docking results from both programs were compared. The compound CMNPD28766 (aspergillipeptide F) was ranked in the top 1% by both engines and also had the highest pharmacophore-fit score (75.916), identifying it as the best candidate [13].
- Validation with Molecular Dynamics (MD): The stability of the aspergillipeptide F-PLpro complex was confirmed through MD simulations, which quantified Cα-atom movements and showed a stable conformation with a low free energy of binding [13].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Essential Software Tools for Pharmacophore Modeling and Virtual Screening
| Tool Name | Type/Availability | Primary Function in Workflow | Key Capabilities |
|---|---|---|---|
| Discovery Studio | Commercial Package | End-to-end model development & screening | Comprehensive suite for structure/ligand-based modeling, docking, and ADMET prediction [12]. |
| MOE (Molecular Operating Environment) | Commercial Package | End-to-end model development & screening | Integrated software for structure-based design, pharmacophore modeling, QSAR, and simulation [12]. |
| LigandScout | Commercial Package | Advanced structure-based modeling | Creates pharmacophores from PDB complexes; performs virtual screening with exclusion volumes [12]. |
| Pharmer | Open-Source Tool | Pharmacophore screening | Efficient search of large chemical databases for molecules matching a 3D pharmacophore query [12]. |
| AutoDock / Vina | Free Tool | Molecular Docking | Predicts bound conformations and scores ligand-receptor interactions [14] [13]. |
| RDKit | Open-Source Tool | Cheminformatics & Feature Identification | Provides fundamental cheminformatics functions, including feature detection and fingerprinting, used in many pipelines [15]. |
| GROMACS / AMBER | Free/Commercial Tool | Molecular Dynamics (MD) | Validates binding stability and calculates free energy of binding post-screening [14] [13]. |
| PharmaGist | Open-Source Tool | Ligand-based modeling | Aligns multiple flexible ligands to generate shared pharmacophore hypotheses [12]. |
In structure-based drug discovery, the binding site and the pharmacophore represent two complementary perspectives for understanding and exploiting drug-target interactions. The binding site is a physically defined location on a protein where a ligand binds, characterized by specific amino acid residues and structural features that facilitate molecular recognition [16]. In contrast, a pharmacophore provides an abstract description of the steric and electronic features that are necessary for molecular recognition and triggering (or blocking) a biological response [2]. It represents a functional pattern rather than a concrete structural entity.
The fundamental relationship between these concepts is that the pharmacophore effectively translates the physical properties of a binding site into a set of chemical features that a ligand must possess to bind effectively. While the binding site constitutes the "lock," the pharmacophore describes the essential characteristics of the "key" that can operate it [2]. This whitepaper explores both concepts in detail, provides methodologies for their investigation, and demonstrates how integrating knowledge of binding sites with pharmacophore modeling creates powerful frameworks for virtual screening in drug discovery research.
The Binding Site: The Protein's Interaction Landscape
Definition and Key Characteristics
A binding site is typically a buried cavity or surface cleft on a protein that possesses specific chemical and structural properties complementary to its ligand. These sites are not static; their flexibility and dynamics are crucial for function, as they can adopt an ensemble of conformers depending on the binding partner and environment [16]. In enzymes, binding sites are often active sites where chemical reactions occur, while in transporters and receptors, they facilitate binding that triggers conformational changes [16].
Key characteristics of binding sites include:
- Specific spatial arrangements of amino acid residues that form interaction points
- Complementary chemical properties (hydrophobicity, charge distribution, hydrogen bonding capability)
- Structural adaptability to accommodate different ligands
- Energetically favorable regions for molecular interactions
Experimental and Computational Approaches for Binding Site Identification
Table 1: Experimental Methods for Binding Site Characterization
| Method | Key Principle | Resolution | Key Applications |
|---|---|---|---|
| X-ray Crystallography | Analysis of electron density in protein crystals | Atomic (~1-2 Å) | High-resolution structure determination of protein-ligand complexes |
| Cryo-Electron Microscopy | Electron scattering from frozen hydrated samples | Near-atomic (2-4 Å) | Structure determination of large complexes and membrane proteins |
| NMR Spectroscopy | Analysis of nuclear magnetic moments in solution | Atomic | Studying protein dynamics and binding in solution |
| Cysteine Scanning Mutagenesis | Systematic mutation of residues to cysteine and testing reactivity | Residue-level | Mapping functional residues in binding sites [16] |
Table 2: Computational Methods for Binding Site Detection and Analysis
| Method | Key Principle | Tools/Examples | Strengths |
|---|---|---|---|
| Pocket Detection | Geometric analysis of protein surface to identify cavities | GRID, LUDI [2] | Fast identification of potential binding pockets |
| Molecular Dynamics (MD) Simulations | Sampling protein flexibility and conformational changes | GROMACS, AMBER | Accounts for protein flexibility and solvation effects [17] |
| Small Molecule Mapping | Probing surfaces with molecular fragments to find favorable positions | MCSS, FTMap [16] | Identifies "sticky" regions that preferentially bind molecular fragments |
| Binding Site Prediction Servers | Machine learning and evolutionary conservation | ConSurf, DeepSite | Identifies functionally important regions |
Core Principles and Feature Definitions
A pharmacophore represents the essential molecular features a compound must possess to achieve optimal interactions with a specific biological target. According to the IUPAC definition, it is "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2]. This abstract representation enables the identification of structurally diverse compounds that share key interaction capabilities.
Table 3: Fundamental Pharmacophore Features and Their Chemical Significance
| Feature Type | Chemical Group Examples | Role in Molecular Recognition | Target Complement |
|---|---|---|---|
| Hydrogen Bond Acceptor (HBA) | Carbonyl oxygen, Nitrile nitrogen, Ether oxygen | Forms hydrogen bonds with donor groups | Backbone NH, Ser/Thr/Tyr OH |
| Hydrogen Bond Donor (HBD) | Amine, Amide NH, Hydroxyl | Forms hydrogen bonds with acceptor groups | Backbone C=O, Asp/Glu COO- |
| Hydrophobic (H) | Alkyl chains, Aromatic rings | Drives desolvation and van der Waals interactions | Leu, Ile, Val, Phe side chains |
| Positive Ionizable (PI) | Primary amine, Guanidino | Forms salt bridges and charge-charge interactions | Asp, Glu carboxylate |
| Negative Ionizable (NI) | Carboxylate, Phosphate | Forms salt bridges and charge-charge interactions | Arg, Lys ammonium groups |
| Aromatic (AR) | Phenyl, Pyridine, Heterocycles | Enables π-π and cation-π interactions | Phe, Tyr, Trp, His side chains |
Pharmacophore Representation and Fingerprints
Pharmacophores can be encoded as molecular fingerprints for efficient virtual screening and machine learning applications. The ErG (Extended Reduced Graph) fingerprint represents a 2D pharmacophore fingerprint that captures detailed properties required for target interaction [18]. Similarly, atom-pair based 2D pharmacophore fingerprints represent all atom-atom pharmacophore feature pairs along with their topological distances, creating histograms for each feature pair type [19]. These representations enable rapid similarity comparison between molecules and facilitate scaffold hopping by focusing on interaction capabilities rather than specific structural elements.
Methodological Approaches: Building Pharmacophore Models
Structure-Based Pharmacophore Modeling
Structure-based pharmacophore modeling derives pharmacophore features directly from the 3D structure of a protein target, typically from protein-ligand complexes. This approach provides a complementary map of the interaction potential within a binding site [2].
Experimental Protocol: Structure-Based Pharmacophore Modeling
Step 1: Protein Structure Preparation
- Obtain 3D structure from PDB or via homology modeling (AlphaFold2)
- Add hydrogen atoms, assign protonation states, and optimize hydrogen bonding networks
- Remove crystallographic artifacts and correct missing residues
- Energy minimization to relieve steric clashes
Step 2: Binding Site Identification and Analysis
- Define binding site using co-crystallized ligand or computational detection tools (GRID, LUDI)
- Analyze key interacting residues and their properties
- Critical Tip: Include some endogenous molecules in screening libraries as validation for docking parameters [16]
Step 3: Pharmacophore Feature Generation
- Use protein-ligand complex to identify key interaction points
- Map interaction features (hydrogen bonds, hydrophobic contacts, ionic interactions)
- Generate exclusion volumes to represent steric constraints of the binding pocket
Step 4: Feature Selection and Model Validation
- Select features most critical for binding affinity
- Validate model using known active and inactive compounds
- Optimize model based on enrichment performance
Structure-Based Pharmacophore Modeling Workflow
Ligand-Based Pharmacophore Modeling
When protein structural information is unavailable, ligand-based approaches can develop pharmacophore models using the structural and activity information of known ligands. The QPhAR (Quantitative Pharmacophore Activity Relationship) method represents a novel approach that constructs quantitative pharmacophore models from molecular datasets, enabling activity prediction based on pharmacophore alignment to a consensus model [20].
Experimental Protocol: Ligand-Based Pharmacophore Modeling with QPhAR
Step 1: Dataset Curation and Conformation Generation
- Collect compounds with known biological activities (IC50, Ki values)
- Generate representative 3D conformations for each compound
- Apply energy minimization and conformational sampling
Step 2: Pharmacophore Perception and Alignment
- Identify pharmacophore features for each compound
- Generate a consensus (merged) pharmacophore from all training samples
- Align individual pharmacophores to the consensus model
Step 3: Model Training and Validation
- Use relative position information to the merged pharmacophore as model input
- Apply machine learning to derive quantitative relationship with biological activities
- Validate model using cross-validation and external test sets
Step 4: Virtual Screening and Hit Prioritization
- Apply model to screen compound libraries
- Rank hits by predicted activity values
- Select candidates for experimental validation
Ligand-Based QPhAR Modeling Workflow
Comparative Performance: Pharmacophore vs. Docking-Based Virtual Screening
Table 4: Benchmark Comparison of Virtual Screening Approaches Across Eight Protein Targets [21] [22]
| Target | PBVS Enrichment | DBVS Enrichment (DOCK) | DBVS Enrichment (GOLD) | DBVS Enrichment (Glide) | Performance Advantage |
|---|---|---|---|---|---|
| ACE | High | Moderate | Moderate | Moderate | PBVS Superior |
| AChE | High | Low | Low | Moderate | PBVS Superior |
| AR | High | Low | Low | Low | PBVS Superior |
| DacA | High | Moderate | Moderate | Moderate | PBVS Superior |
| DHFR | High | Moderate | Moderate | High | PBVS Superior |
| ERα | High | Moderate | Moderate | Moderate | PBVS Superior |
| HIV-pr | Moderate | High | Moderate | Moderate | DBVS Superior |
| TK | High | Moderate | Moderate | Moderate | PBVS Superior |
| Average Hit Rate (Top 2%) | 32.5% | 12.8% | 14.2% | 18.6% | PBVS Superior |
| Average Hit Rate (Top 5%) | 45.2% | 24.6% | 26.3% | 29.8% | PBVS Superior |
The benchmark study demonstrated that pharmacophore-based virtual screening (PBVS) significantly outperformed docking-based virtual screening (DBVS) in retrieving active compounds from databases for most targets [21] [22]. Of the sixteen sets of virtual screens conducted, PBVS achieved higher enrichment factors in fourteen cases compared to DBVS methods. This performance advantage was particularly evident when considering the early enrichment of hit lists, with PBVS achieving an average hit rate of 32.5% in the top 2% of ranked compounds compared to 12.8-18.6% for docking methods [21].
Integrated Applications and Advanced Approaches
Machine Learning and Pharmacophore Integration
Recent advances have integrated pharmacophore modeling with machine learning to enhance predictive performance. The ErG pharmacophore fingerprint has been successfully used in multi-class classification models to predict E3 ligase binding selectivity, achieving 93.8% accuracy in assigning binders to their correct E3 ligase targets [18]. This approach enables rational design of targeted protein degraders by predicting the probability of compounds binding to different E3 ligases.
TransPharmer represents another innovative integration, combining pharmacophore-informed generative models with GPT-based frameworks for de novo molecule generation [23]. This approach maintains crucial non-bond interactions with receptors while producing structurally distinct compounds, effectively enabling scaffold hopping in drug discovery.
Emerging Trends: Residue-Based Pharmacophores for Protein Therapeutics
As therapeutic modalities expand beyond small molecules to include protein-based drugs, residue-based pharmacophore approaches have emerged to study protein-protein interactions [17]. These methods employ molecular dynamics simulations to account for solvation, conformational flexibility, and entropic effects, providing better approximation of free energy of binding. Applications include identifying receptor-ligand partners, engineering protein interfaces for selectivity, and designing therapeutic antibodies [17].
Table 5: Key Software Tools for Pharmacophore Modeling and Virtual Screening
| Tool/Resource | Type | Key Function | Application Context |
|---|---|---|---|
| LigandScout [21] | Software | Structure-based pharmacophore generation from protein-ligand complexes | Virtual screening, feature identification |
| Catalyst/Hypogen [20] | Software | Ligand-based pharmacophore modeling and quantitative activity prediction | QSAR, lead optimization |
| QPhAR [20] [24] | Algorithm | Quantitative pharmacophore model construction from molecular datasets | Activity prediction, model validation |
| PMapper [19] | Command-line Tool | Pharmacophore fingerprint generation | Molecular similarity, machine learning |
| GRID [2] | Software | Molecular interaction field calculation | Binding site detection, interaction analysis |
| GRAIL [17] | Method | Grid-based pharmacophore representation with MD simulation | Protein-protein interaction studies |
| ErG Fingerprint [18] | Descriptor | 2D pharmacophore fingerprint for machine learning | Binding specificity prediction, library design |
| TransPharmer [23] | Generative Model | Pharmacophore-informed molecule generation | De novo design, scaffold hopping |
The binding site and pharmacophore represent complementary perspectives in drug discovery - the former defining the physical interaction landscape on the protein, while the latter abstracts the essential chemical features required for molecular recognition. Integrated approaches that leverage structural knowledge of binding sites to inform pharmacophore model development create powerful frameworks for virtual screening. Benchmark studies demonstrate that pharmacophore-based methods frequently outperform docking-based approaches in enrichment performance, particularly in early retrieval of active compounds. Emerging trends integrating pharmacophore concepts with machine learning and generative models promise to further accelerate the discovery of novel bioactive ligands with improved structural diversity and therapeutic potential.
{# The Critical Role of Pharmacophore Models in Modern Computer-Aided Drug Discovery (CADD)
A pharmacophore is an abstract description of the structural features of a compound that are essential for its biological activity [25]. According to the International Union of Pure and Applied Chemistry (IUPAC), it is defined as "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2]. This conceptual framework, first introduced by Paul Ehrlich in 1909 and later refined by Emil Fisher's "Lock & Key" principle, has evolved into a sophisticated tool that forms the backbone of many modern computer-aided drug discovery (CADD) workflows [2] [26].
Pharmacophore modeling represents a successful and expanded area of computational drug design that enables researchers to move beyond specific atomic structures to focus on the essential chemical functionalities required for molecular recognition [25]. By schematically illustrating the essential components of molecular recognition, pharmacophores provide a powerful approach for representing and identifying active molecules in both two and three dimensions [25]. The core principle underlying pharmacophore modeling is that molecules sharing common chemical functionalities in a similar spatial arrangement are likely to exhibit biological activity toward the same target [2]. This abstraction makes pharmacophores particularly valuable for identifying structurally diverse compounds that interact with the same biological target—a process known as scaffold hopping [20].
Pharmacophore Modeling Approaches
The generation of pharmacophore models can be accomplished through two primary computational approaches, each with distinct requirements and applications. The choice between these methods depends on the available data, computational resources, and the specific objectives of the drug discovery project [2].
Structure-Based Pharmacophore Modeling
Structure-based pharmacophore modeling relies on the three-dimensional structural information of the macromolecular target, typically obtained from X-ray crystallography, NMR spectroscopy, or computational methods such as homology modeling [2]. With the advent of advanced structure prediction tools like ALPHAFOLD2, this approach has become increasingly accessible even when experimental structures are unavailable [2].
Table 1: Key Steps in Structure-Based Pharmacophore Modeling
| Step | Description | Tools & Methods |
|---|---|---|
| Protein Preparation | Critical evaluation and optimization of the target structure, including protonation states, hydrogen atom placement, and correction of structural errors. | Molecular mechanics force fields, energy minimization [2] |
| Ligand-Binding Site Detection | Identification of the key region where ligands interact with the protein target. | GRID (molecular interaction fields), LUDI (geometric rules), manual analysis from co-crystallized ligands [2] |
| Feature Generation | Mapping potential interaction points in the binding site to define complementary pharmacophore features. | Analysis of protein-ligand complexes or apo structures [2] |
| Feature Selection | Selection of the most relevant features essential for bioactivity to create the final pharmacophore hypothesis. | Conservation analysis, energy contribution assessment, spatial constraints [2] |
The quality of the input structure directly influences the quality of the resulting pharmacophore model [2]. When a protein-ligand complex structure is available, pharmacophore feature generation can be achieved more accurately by analyzing the specific interactions between the ligand functional groups and the target protein [2]. Exclusion volumes can be added to represent spatial restrictions from the binding site shape, creating a more selective model [2].
Ligand-Based Pharmacophore Modeling
In the absence of a macromolecular target structure, ligand-based pharmacophore modeling provides a powerful alternative by deriving common chemical features from a set of known active ligands [2]. This approach is based on the fundamental premise that compounds binding to the same biological target likely share essential molecular features necessary for binding and activity [26].
The ligand-based approach involves analyzing the three-dimensional structures of multiple active compounds to identify shared pharmacophore features and their spatial relationships [2]. This process typically requires the generation of multiple conformations for each compound to account for flexibility and ensure coverage of the bioactive conformation [26]. Advanced algorithms then align these conformations and extract common chemical features that define the pharmacophore model [26].
Quantitative Structure-Activity Relationship (QSAR) or Quantitative Structure-Property Relationship (QSPR) modeling can be integrated with ligand-based pharmacophore approaches to create predictive models that correlate pharmacophore features with biological activity levels [2]. The recently developed QPhAR (Quantitative Pharmacophore Activity Relationship) method represents a significant advancement in this area, enabling the construction of robust quantitative models that can generalize to underrepresented or missing molecular features in the training set by leveraging pharmacophoric interaction patterns [20].
Applications in Virtual Screening and Drug Discovery
Pharmacophore models serve as versatile tools throughout the drug discovery pipeline, with virtual screening representing one of their most prominent applications.
Virtual Screening
Pharmacophore-based virtual screening involves using pharmacophore queries to search large chemical databases and identify compounds that match the essential feature arrangement [2]. This approach significantly enriches screening libraries with compounds that have a higher probability of biological activity, thereby improving hit rates while reducing costs compared to traditional high-throughput screening [27].
Advanced tools like Pharmer have revolutionized pharmacophore search capabilities by introducing novel computational approaches that scale with query complexity rather than database size [27]. Pharmer employs innovative data structures like the KDB-tree and Bloom fingerprints to enable exact pharmacophore searches of millions of compounds in minutes—more than an order of magnitude faster than previous technologies [27].
Table 2: Pharmacophore-Based Virtual Screening Tools and Applications
| Tool/Method | Approach | Key Features | Applications |
|---|---|---|---|
| Pharmer | Alignment-based search using spatial indexing | KDB-tree data structure, Bloom fingerprints, exact search | High-throughput screening of large databases [27] |
| QPhAR Workflow | Quantitative pharmacophore activity relationship | Machine learning integration, automated feature optimization | Activity prediction, hit prioritization [24] |
| ML-Accelerated Screening | Ensemble machine learning models | Docking score prediction, 1000x speed increase | Rapid identification of MAO inhibitors [7] |
| Structure-Based Screening | Protein structure-derived queries | Exclusion volumes, interaction complementarity | Target-focused screening [2] |
Integration with Other CADD Methods
Pharmacophore approaches are frequently combined with other computational techniques to enhance their effectiveness and accuracy. The integration of pharmacophore modeling with molecular docking simulations represents a particularly powerful combination [25]. In this hybrid approach, pharmacophore models can pre-filter compound libraries to reduce the number of candidates for more computationally intensive docking studies, or alternatively, docking results can inform the development of more refined pharmacophore models [25].
Machine learning techniques have opened new frontiers in pharmacophore-based drug discovery. Recent advances include the development of ensemble ML models that can predict docking scores without performing time-consuming molecular docking procedures, achieving a 1000-fold acceleration in binding energy predictions [7]. These models learn from docking results, allowing researchers to choose their preferred docking software while bypassing the limitations of insufficient experimental activity data [7].
Advanced Protocols and Recent Advances
Automated Pharmacophore Optimization with QPhAR
The QPhAR algorithm represents a significant advancement in automated pharmacophore modeling by addressing the traditional limitation of manual, expert-dependent refinement [24]. This method automates the selection of features that drive pharmacophore model quality using structure-activity relationship (SAR) information extracted from validated QPhAR models [24].
The QPhAR workflow begins with dataset preparation and splitting into training and test sets, followed by QPhAR model generation using the training set molecules [24]. The model is validated through cross-validation and leave-one-out analysis before the refined pharmacophore is automatically generated from the model [24]. This pharmacophore is then used for virtual screening, with hits ranked by their QPhAR-predicted activity values [24].
Studies have demonstrated that QPhAR-based refined pharmacophores outperform traditional baseline pharmacophores (generated from the most active compounds) on composite scoring metrics, showing particular utility in predicting hERG liability and other ADMET properties [24].
Machine Learning-Accelerated Virtual Screening Protocol
A recent innovative protocol for monoamine oxidase (MAO) inhibitor discovery demonstrates the power of integrating machine learning with pharmacophore-based screening [7]:
- Activity Data Collection: MAO-A and MAO-B ligands with corresponding activity data are obtained from the ChEMBL database [7].
- Docking Score Calculation: Smina docking software is used to calculate docking scores for the compounds [7].
- Machine Learning Model Training: Multiple types of molecular fingerprints and descriptors are used to construct ensemble models that predict docking scores [7].
- Pharmacophore-Constrained Screening: The ZINC database is screened using pharmacophoric constraints [7].
- Synthesis and Testing: Top-ranked compounds are synthesized and evaluated for biological activity [7].
This approach achieved a 1000-fold acceleration in binding energy predictions compared to classical docking-based screening and successfully identified novel MAO-A inhibitors with percentage efficiency indices close to known drugs at the lowest tested concentrations [7].
Table 3: Key Research Reagents and Computational Tools for Pharmacophore Modeling
| Resource Type | Examples | Function/Purpose |
|---|---|---|
| Software Platforms | MOE (Molecular Operating Environment), Discovery Studio, PHASE | Comprehensive suites for pharmacophore modeling, visualization, and screening [28] [20] |
| Open-Source Tools | Pharmer | Efficient pharmacophore search using spatial indexing and Bloom filters [27] |
| Protein Databases | RCSB Protein Data Bank (PDB) | Source of experimental 3D protein structures for structure-based modeling [2] |
| Compound Libraries | ZINC, ChEMBL | Collections of screening compounds with structural and activity data [7] |
| Conformation Generators | iConfGen, Monte Carlo methods | Generation of 3D molecular conformations for flexible alignment [26] [20] |
| Machine Learning Libraries | Scikit-learn, TensorFlow, PyTorch | Implementation of QSAR and docking score prediction models [7] [20] |
Workflow Visualization
Diagram 1: Comprehensive Workflow for Pharmacophore-Based Drug Discovery. This diagram illustrates the integrated approach combining structure-based and ligand-based methods with machine learning advancements.
Pharmacophore modeling has evolved into an indispensable component of modern computer-aided drug discovery, providing a powerful abstract representation of the essential features required for molecular recognition [25]. The integration of structure-based and ligand-based approaches, combined with recent advances in machine learning and automated optimization algorithms, has significantly enhanced the accuracy and efficiency of pharmacophore methods [24] [7].
As drug discovery faces increasing pressures to reduce costs and development timelines, pharmacophore-based strategies offer robust solutions for enriching screening libraries, identifying novel chemotypes through scaffold hopping, and predicting ADMET properties [25] [20]. The continued development of quantitative pharmacophore methods, efficient search algorithms like Pharmer, and ML-accelerated screening protocols promises to further expand the role of pharmacophores in rational drug design [24] [7] [27].
For researchers embarking on pharmacophore-based virtual screening, the key success factors include careful selection of the modeling approach based on available data, rigorous validation of models, and leveraging the growing ecosystem of computational tools and databases. By adhering to these principles and incorporating the latest methodological advances, scientists can fully harness the power of pharmacophore models to accelerate the discovery of novel therapeutic agents.
A Step-by-Step Guide to Structure-Based and Ligand-Based Pharmacophore Modeling
Structure-based pharmacophore modeling is a foundational technique in modern computer-aided drug discovery. This method abstracts the essential steric and electronic features from a three-dimensional protein structure that are necessary for optimal supramolecular interactions with a ligand, enabling the virtual screening of compound libraries to identify novel drug candidates [2]. The profound advantage of this approach lies in its independence from known active ligands; it requires only the 3D structure of the target protein, either from experimental methods or computational prediction, to derive a model that defines the spatial and functional constraints a molecule must satisfy to bind effectively [2].
The reliability of the input protein structure is paramount, as it directly influences the quality and predictive power of the resulting pharmacophore model. Traditionally, researchers have relied on experimental structures from the Protein Data Bank (PDB), often in complex with a ligand. However, the rapid advancement of deep learning-based protein structure prediction tools, most notably AlphaFold2 (AF2), has provided researchers with highly accurate models for nearly every protein encoded by the human genome, dramatically expanding the scope of targets accessible to structure-based methods [29]. This guide provides a comprehensive technical framework for building and validating structure-based pharmacophore models, leveraging both PDB and AF2 structures to drive virtual screening campaigns.
Core Concepts and Workflow
Definition of a Pharmacophore
The International Union of Pure and Applied Chemistry (IUPAC) defines a pharmacophore as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2]. In practical terms, a pharmacophore model represents these key chemical functionalities as geometric entities—such as spheres, planes, and vectors—that define the allowed spatial coordinates for interactions.
The most critical pharmacophoric features include [2]:
- Hydrogen Bond Acceptor (HBA)
- Hydrogen Bond Donor (HBD)
- Hydrophobic (H) area
- Positively Ionizable (PI) group
- Negatively Ionizable (NI) group
- Aromatic (AR) ring
To accurately represent the physical constraints of the binding site, exclusion volumes (XVOL) are often added to the model. These volumes define regions in space that are occupied by the protein and into which a ligand cannot penetrate, thus shaping the steric boundaries of potential drug molecules [2].
The following diagram illustrates the complete, integrated workflow for structure-based pharmacophore modeling, encompassing both traditional PDB and modern AlphaFold2-derived structures.
Input Structure Preparation and Analysis
Sourcing and Preparing Experimental PDB Structures
When using a structure from the PDB, the initial preparation phase is critical for generating a reliable pharmacophore model. The workflow involves several key steps [2]:
- Structure Quality Assessment: Begin by evaluating the resolution, R-value, and free R-value of the structure. For instance, a high-quality structure like FAK1 in complex with inhibitor P4N (PDB ID: 6YOJ) has a resolution of 1.36 Å, which is excellent for modeling [30].
- Completeness Check: Identify and model any missing residues or loops. For the 6YOJ structure, residues 570–583 and 687–689 were missing and were successfully modeled using tools like MODELLER, selecting the model with the lowest zDOPE score [30].
- Structure Preparation: Add hydrogen atoms, assign correct protonation states to residues (e.g., for Asp, Glu, His), and perform energy minimization to relieve steric clashes and ensure proper geometry [2].
Table 1: Key Protein Preparation Steps and Tools
| Step | Description | Common Tools/Software |
|---|---|---|
| Quality Assessment | Evaluate resolution, R-factors, and completeness | PDB Validation Reports, MolProbity |
| Missing Residue Modeling | Fill in gaps in the protein sequence | MODELLER, Swiss-Model, Rosetta |
| Hydrogen Addition & Optimization | Add H atoms and optimize side-chain rotamers | MOE, Schrödinger Protein Preparation Wizard, UCSF Chimera |
| Protonation State Assignment | Determine correct charges for acidic/basic residues | PROPKA, H++ server |
| Energy Minimization | Relax the structure to remove steric clashes | GROMACS, AMBER, OpenMM |
Utilizing and Validating AlphaFold2 Models
For targets without experimental structures, AlphaFold2 (AF2) provides a powerful alternative. However, specific considerations must be addressed [29]:
- Model Quality Metrics: The primary metric for assessing AF2 model confidence is the pLDDT score, which is per-residue estimate of confidence on a scale from 0-100. Residues with pLDDT > 90 are considered high accuracy, while those with pLDDT < 70 may have low reliability, particularly in flexible loops or disordered regions. Global model quality can be further assessed using QMEAN Z-scores and MolProbity scores for stereochemical quality [29].
- Binding Site Analysis: Pay close attention to the pLDDT scores of residues forming the binding pocket. If confidence is low in this critical region, the utility of the model for pharmacophore generation is compromised.
- Handling Missing Components: AF2 predicts protein structures without ligands, ions, or cofactors. The AlphaFill tool can be used to transplant these missing components from experimentally determined structures into AF2 models [29].
- Addressing Conformational Bias: Standard AF2 models may be biased toward dominant conformational states present in the training data. For example, most kinase structures in the PDB are in the DFG-in state, leading AF2 to preferentially predict this state. The Multi-State Modeling (MSM) protocol overcomes this by providing state-specific templates to AF2, enabling the prediction of diverse conformational states (e.g., DFG-in vs. DFG-out for kinases) crucial for discovering different inhibitor types [31].
Table 2: Key AlphaFold2 Model Validation Metrics
| Metric | Target Value | Interpretation |
|---|---|---|
| pLDDT (per-residue) | > 70 (Good), > 90 (High) | Measures local confidence; crucial for binding site residues. |
| RMSD (Global/Backbone) | < 2.0 Å | Compares overall fold to a reference (experimental) structure. |
| MolProbity Score | < 2.0 | Combined measure of stereochemical quality (lower is better). |
| Ramachandran Favored (%) | > 90% | Percentage of residues in favored regions of phi/psi space. |
| QMEAN Z-Score | Around 0 | Global model quality score relative to high-resolution structures. |
Pharmacophore Model Generation
Binding Site Identification
The first step in model generation is the precise identification of the ligand-binding site [2]. This can be achieved through:
- Analysis of Holo-Structures: If a protein-ligand complex is available, the binding site is defined by the coordinates of the bound ligand.
- Computational Prediction: For apo-structures, tools like GRID or LUDI can be employed. GRID uses molecular interaction fields (MIFs) with different chemical probes to identify energetically favorable binding regions, while LUDI applies geometric rules derived from known protein-ligand interactions to predict potential binding sites [2].
Feature Generation and Selection
Once the binding site is defined, the software identifies potential pharmacophore features that complement the protein's functional groups [2]:
- Interaction Analysis: The binding site is analyzed for residues capable of forming hydrogen bonds, hydrophobic contacts, ionic interactions, etc.
- Feature Mapping: Tools like the Pharmit server can automatically process a protein-ligand complex (e.g., FAK1-P4N) and detect critical pharmacophoric features involved in the interaction. Initial runs often generate many features, requiring careful selection [30].
- Feature Selection: The initial model containing many features must be refined to include only the essential ones. This can be done by removing features that do not contribute significantly to binding energy, identifying conserved interactions across multiple complex structures, or incorporating key functional residues from sequence analysis [2]. Exclusion volumes are added to represent the steric boundaries of the pocket.
Model Validation and Virtual Screening
Validation Using Known Actives and Decoys
Before deploying a model for screening, its predictive power must be statistically validated [30]. This process involves:
- Dataset Curation: A set of known active compounds and a set of chemically similar but presumed inactive molecules (decoys) are required. Databases like DUD-E (Directory of Useful Decoys: Enhanced) provide pre-compiled sets for many targets [30].
- Screening and Metrics Calculation: The pharmacophore model is used to screen the validation library. Standard statistical metrics are then calculated to evaluate performance.
Table 3: Key Statistical Metrics for Pharmacophore Model Validation
| Metric | Formula | Interpretation |
|---|---|---|
| Sensitivity (Recall) | (True Positives / All Actives) × 100 | The model's ability to identify known active compounds. |
| Specificity | (True Negatives / All Decoys) × 100 | The model's ability to reject inactive decoy compounds. |
| Enrichment Factor (EF) | (Hitssₜₐᵣgₑₜ / Nₜₐᵣgₑₜ) / (Hitssₜₒₜₐₗ / Nₜₒₜₐₗ) | Measures how much more likely the model is to find actives compared to random selection. |
| Goodness of Hit (GH) | Complex formula combining sensitivity and specificity. | A composite score; a value of 0.7-0.8 indicates an excellent model. |
The model with the best combined metrics (e.g., high sensitivity, specificity, and GH score) is selected for the final virtual screening [30].
Virtual Screening and Hit Identification
The validated pharmacophore model serves as a query to search large chemical databases such as ZINC, ChEMBL, or in-house collections [30] [27]. This process, known as pharmacophore-based virtual screening, can be performed using tools like Pharmit or Pharmer. Pharmer uses efficient data structures (KDB-trees) and search algorithms to rapidly screen millions of compounds by aligning their conformers to the query pharmacophore [27]. The output is a list of candidate "hit" molecules that match the pharmacophore hypothesis. These hits are typically prioritized further by assessing their drug-likeness, predicting their ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties, and subjecting them to more precise molecular docking studies [30].
Table 4: Essential Tools and Resources for Structure-Based Pharmacophore Modeling
| Category | Tool/Resource | Primary Function | Key Features |
|---|---|---|---|
| Protein Structure Databases | Protein Data Bank (PDB) | Repository for experimental 3D structures of proteins and nucleic acids. | Provides structures solved by X-ray, Cryo-EM, and NMR. |
| Protein Structure Prediction | AlphaFold Protein Structure Database | Repository of pre-computed AlphaFold2 models for a vast range of proteomes. | Offers easy access to AF2 models with per-residue confidence scores (pLDDT). |
| Protein Preparation & Analysis | UCSF Chimera, MODELLER | Molecular modeling and visualization; homology modeling of missing loops/regions. | Used for adding hydrogens, energy minimization, and filling missing residues. |
| Binding Site Detection | GRID, LUDI | Identifies potential ligand-binding pockets on a protein structure. | GRID uses interaction energy calculations; LUDI uses geometric rules. |
| Pharmacophore Modeling & Screening | Pharmit, Pharmer | Web-based and standalone tools for creating pharmacophore models and screening compound libraries. | Pharmit is a web server for interactive screening; Pharmer is optimized for high-speed searches of large databases [30] [27]. |
| Chemical Databases | ZINC, DUD-E | Publicly accessible databases of commercially available compounds (ZINC) and sets of actives/decoys for validation (DUD-E). | Essential for both validation and finding potential hit molecules [30]. |
Structure-based pharmacophore modeling represents a powerful strategy for initiating drug discovery campaigns, particularly for targets with limited chemical starting points. The integration of highly accurate AlphaFold2 models has significantly expanded the universe of druggable targets, while methodologies like Multi-State Modeling help overcome historical conformational biases. By rigorously preparing the input structure—whether from the PDB or AF2—generating and validating the model with robust statistical measures, and leveraging efficient screening tools, researchers can confidently employ this methodology to identify novel and diverse chemical matter. This guide provides a foundational technical framework for scientists to apply these principles effectively in their virtual screening research.
In the landscape of computer-aided drug design (CADD), pharmacophore modeling represents a foundational approach for identifying novel therapeutic agents by capturing the essential steric and electronic features responsible for biological activity. The International Union of Pure and Applied Chemistry (IUPAC) defines a pharmacophore as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [32]. In practical terms, a pharmacophore is not a real molecule or functional group association, but rather an abstract concept that describes the common steric and electrostatic complementarities of bioactive compounds with their target [32]. Ligand-based pharmacophore modeling specifically addresses scenarios where the three-dimensional structure of the biological target is unknown or unavailable, relying instead on the structural information and biological activity data of known ligands to infer the critical features necessary for binding and activity [2] [33].
This approach operates on the fundamental principle that compounds sharing common chemical functionalities in a similar spatial arrangement are likely to exhibit biological activity toward the same target [2]. By distilling the essential molecular recognition elements from a set of active compounds, researchers can create three-dimensional queries to screen large chemical databases efficiently, identify novel chemotypes with potential activity (a process known as scaffold hopping), and guide lead optimization efforts [2] [32]. The effectiveness of ligand-based pharmacophore modeling has been demonstrated across various therapeutic areas, including oncology [34], infectious diseases [35], central nervous system disorders [7], and cardiovascular diseases [36], establishing it as a versatile and valuable tool in modern drug discovery.
Theoretical Foundations and Key Concepts
Fundamental Pharmacophore Features
Pharmacophore models represent molecular interactions through simplified chemical feature types that capture the essential interactions between a ligand and its biological target. The most significant pharmacophoric feature types include [2]:
- Hydrogen Bond Acceptors (HBA): Atoms or regions that can accept hydrogen bonds, typically oxygen or nitrogen atoms with lone electron pairs.
- Hydrogen Bond Donors (HBD): Atoms or groups that can donate hydrogen bonds, usually featuring a hydrogen atom bonded to an electronegative atom (O-H, N-H).
- Hydrophobic Areas (H): Non-polar regions of the molecule that participate in van der Waals interactions with complementary hydrophobic regions of the target.
- Positively and Negatively Ionizable Groups (PI/NI): Functional groups that can carry formal positive or negative charges under physiological conditions, enabling electrostatic interactions.
- Aromatic Rings (AR): Planar, conjugated ring systems that can engage in π-π stacking or cation-π interactions.
These features are represented in three-dimensional space as geometric entities such as points, spheres, vectors, and planes, with tolerance radii accounting for some spatial flexibility [2] [32]. Additional shape constraints or exclusion volumes can be incorporated to represent the steric boundaries of the binding pocket and improve model selectivity [2].
Comparison with Structure-Based Approaches
Ligand-based and structure-based approaches represent complementary strategies for pharmacophore model development, each with distinct advantages and limitations:
Table 1: Comparison of Ligand-Based and Structure-Based Pharmacophore Modeling Approaches
| Aspect | Ligand-Based Approach | Structure-Based Approach |
|---|---|---|
| Prerequisite Data | Known active ligands with biological activity data | 3D structure of the target (from X-ray, NMR, or homology modeling) |
| Feature Identification | Derived from common patterns among active compounds | Derived from complementarity to binding site features |
| Best Application | Targets with unknown 3D structure but known active ligands | Targets with available 3D structure, especially with bound ligands |
| Advantages | Does not require target structure; Can incorporate extensive SAR data | Can identify novel interaction points not present in known ligands |
| Limitations | Limited by diversity and quality of known active compounds | Dependent on quality and relevance of the protein structure |
The choice between these approaches depends largely on data availability, data quality, computational resources, and the intended application of the generated models [2]. In many modern drug discovery campaigns, these approaches are used synergistically to leverage their respective strengths.
Methodological Workflow
The development of a ligand-based pharmacophore model follows a systematic workflow that transforms a set of known active compounds into a validated three-dimensional query for virtual screening. The complete process is visualized in Figure 1 below:
Figure 1. Comprehensive Workflow for Ligand-Based Pharmacophore Modeling. The process begins with data collection and progresses through training set selection, model generation, and rigorous validation before application in virtual screening.
Training Set Selection and Preparation
The initial and arguably most critical step in ligand-based pharmacophore modeling is the careful selection of training set compounds. The training set should include molecules with the following characteristics [34] [36]:
- A wide range of biological activities (typically spanning 3-4 orders of magnitude in IC₅₀ or Kᵢ values)
- Structural diversity to avoid bias toward specific chemical scaffolds
- High-quality biological data obtained from consistent assay conditions
- Representative examples of active, moderately active, and inactive compounds to establish structure-activity relationships
A well-designed training set ensures the generated model can distinguish between active and inactive compounds and possesses predictive capability for novel chemotypes. For instance, in a study targeting DNA Topoisomerase I inhibitors, the training set included 29 camptothecin derivatives with IC₅₀ values ranging from 0.003 μM to 11.4 μM, categorized into most active (<0.1 μM), active (0.1-1.0 μM), moderately active (1.0-10.0 μM), and inactive (>10.0 μM) groups [34].
Once selected, compounds undergo structure preparation including [34] [36]:
- 2D to 3D structure conversion
- Hydrogen addition and geometry optimization using force fields (e.g., CHARMM)
- Energy minimization using algorithms like steepest descent and conjugate gradient
- Generation of multiple conformations to represent accessible conformational space
Pharmacophore Feature Mapping and Model Generation
With prepared training set compounds, the next step involves identifying potential pharmacophoric features and generating hypothesis models. The feature mapping process identifies the chemical features present in the training set using predefined definitions (often encoded as SMARTS patterns) [37]. The 3D QSAR pharmacophore generation methodology then constructs hypotheses that best correlate the spatial arrangement of these features with biological activity [34] [36].
The HypoGen algorithm, implemented in software such as Discovery Studio, employs a three-phase process for model generation [36]:
- Constructive Phase: Identifies hypotheses common to the most active compounds by examining all possible pharmacophore configurations for the conformations of the most active compounds.
- Subtractive Phase: Removes pharmacophore configurations that are also present in the least active molecules (typically defined as compounds with activity 3.5 orders of magnitude less than the most active compound).
- Optimization Phase: Improves the hypothesis score through simulated annealing, varying features and locations to optimize activity prediction.
This process generates multiple pharmacophore hypotheses that are evaluated based on cost parameters, correlation coefficients, and predictive capability.
Conformational Sampling and Molecular Representation
A crucial aspect of ligand-based pharmacophore modeling is accounting for molecular flexibility, as small molecules typically exist in multiple conformations in solution. Most implementations address this by generating conformational ensembles for each compound, often within a defined energy window (e.g., 20 kcal/mol above the global minimum) [36]. The Poling algorithm or other diversity-based methods are employed to ensure comprehensive coverage of accessible conformational space while avoiding redundant conformers [36].
For 3D pharmacophore representation, novel approaches have been developed that utilize canonical pharmacophore signatures based on quadruplets of features. This representation encodes both the content and topology of pharmacophores through a Morgan-like algorithm applied to complete graphs of pharmacophore features, with binned distances between features enabling fuzzy matching [37]. Special handling of stereoconfiguration ensures distinction between pharmacophores with different spatial organization of features [37].
Experimental Protocols and Validation Strategies
Detailed Protocol for 3D QSAR Pharmacophore Generation
Based on published methodologies for various targets [34] [36], the following protocol provides a detailed workflow for generating and validating 3D QSAR pharmacophore models:
Compound Preparation and Conformation Generation
- Draw 2D structures using chemical drawing software (e.g., ChemDraw, ChemSketch)
- Convert to 3D structures and add hydrogen atoms
- Perform geometry optimization using force fields (e.g., CHARMM) with smart minimizer executing 2000 steps of steepest descent followed by conjugate gradient algorithms (convergence gradient: 0.001 kcal/mol)
- Generate multiple conformers using a poling algorithm with these parameters:
- Maximum conformations: 255 per compound
- Energy threshold: 20 kcal/mol above global minimum
- Root mean square deviation (RMSD) threshold: 0.5 Å for conformation diversity
Pharmacophore Model Generation using HypoGen Algorithm
- Perform feature mapping to identify relevant pharmacophore features (HBA, HBD, HY-AL, HY-AR, RA)
- Set uncertainty value to 2.0 (meaning the actual activity is assumed to be within a range that is twice the reported value)
- Define minimum inter-feature distance to 2.0 Å
- Run HypoGen algorithm to generate top 10 pharmacophore hypotheses
- Evaluate hypotheses based on cost values: total cost, fixed cost, null cost, and configuration cost
Statistical Validation of Models
- Calculate correlation coefficient between experimental and estimated activities
- Assess root mean square deviation (RMSD) of predictions
- Analyze cost differences: (null cost - total cost) > 70 indicates >90% statistical significance
- Perform Fischer randomization test (95% or 99% confidence level) to ensure model not generated by chance
- Execute leave-one-out validation to assess model robustness
Model Validation Techniques
Rigorous validation is essential to establish the predictive power and reliability of pharmacophore models. Multiple validation strategies should be employed [38] [36]:
Test Set Prediction: Use a separate set of compounds (not included in training) to evaluate the model's ability to predict external data. A good model should show a high correlation between experimental and predicted activities for the test set [34].
Fischer Randomization: Generate random pharmacophore models by scrambling activity data to establish statistical significance. At 95% confidence level, 19 random spreadsheets should be generated; if the original hypothesis has a lower cost than all randomized ones, the model is significant at the 95% level [36].
Leave-One-Out Validation: Iteratively remove one compound from the training set, regenerate the model, and predict the omitted compound's activity. This assesses the model's dependence on any single compound.
ROC Analysis and Enrichment Factors: For virtual screening applications, evaluate model performance using Receiver Operating Characteristic (ROC) curves and calculate enrichment factors (EF) to quantify the model's ability to prioritize active compounds over inactive ones [38]. AUC values of 0.7-0.8 indicate good performance, while values >0.8 represent excellent performance [38].
Table 2: Quantitative Validation Metrics for Pharmacophore Models
| Validation Method | Optimal Values | Interpretation |
|---|---|---|
| Correlation Coefficient (r) | >0.9 | Strong correlation between predicted and experimental activities |
| Cost Difference | >70 | >90% statistical significance |
| RMSD | <1.0 | Low error in activity prediction |
| Fischer Randomization | 95% or 99% confidence | Model not generated by chance |
| ROC AUC | 0.7-0.8 (good), >0.8 (excellent) | Discrimination ability between active and inactive compounds |
Virtual Screening Applications
Screening Workflow and Hit Identification
Validated pharmacophore models serve as powerful 3D queries for virtual screening of large compound databases to identify novel hit compounds. The screening process typically follows a multi-step workflow [34] [32]:
- Database Preparation: Convert screening database (e.g., ZINC, ChEMBL) into searchable 3D format with pre-computed conformations
- Pharmacophore Screening: Use the validated model as a 3D query to identify matching compounds
- Multi-Stage Filtration:
- Lipinski's Rule of Five: Filter for drug-like properties (molecular weight ≤500, LogP ≤5, HBD ≤5, HBA ≤10)
- SMART Filtration: Remove compounds with undesirable functional groups or reactive motifs
- Activity Estimation: Retain compounds with predicted activity below a threshold (e.g., <1.0 μM)
- Molecular Docking: Further evaluate filtered hits using molecular docking to assess binding mode and complementarity with the target
- ADMET Profiling: Predict absorption, distribution, metabolism, excretion, and toxicity properties to prioritize leads with favorable pharmacokinetic and safety profiles
This comprehensive approach was successfully demonstrated in a study identifying Topoisomerase I inhibitors, where screening of 1,087,724 drug-like molecules from the ZINC database ultimately yielded three potential hit molecules (ZINC68997780, ZINC15018994, and ZINC38550809) with stable binding confirmed through molecular dynamics simulation [34].
Performance Optimization and Pre-filtering Strategies
To enhance screening efficiency with large compound databases, several pre-filtering strategies are employed [32]:
- Feature-count Matching: Quick elimination of compounds lacking the necessary pharmacophore feature types
- Pharmacophore Fingerprints: Binary representations encoding presence/absence of specific pharmacophoric patterns for rapid similarity searching
- Descriptor-based Similarity: Fast similarity calculations using molecular descriptors to identify structurally similar compounds
- Shape-based Pre-screening: Rapid shape comparison to eliminate compounds with incompatible steric properties
These pre-filtering strategies can dramatically reduce the number of compounds requiring computationally expensive 3D alignment, improving screening throughput by several orders of magnitude while maintaining sensitivity for true active compounds [32].
Successful implementation of ligand-based pharmacophore modeling requires access to specialized software tools, compound databases, and computational resources. The following table summarizes key components of the research toolkit:
Table 3: Essential Resources for Ligand-Based Pharmacophore Modeling Research
| Resource Category | Specific Tools/Databases | Key Functionality |
|---|---|---|
| Commercial Software | Discovery Studio (Accelrys), MOE (Chemical Computing Group), Phase (Schrödinger), LigandScout (Inte:Ligand) | Comprehensive pharmacophore modeling, virtual screening, and analysis platforms |
| Open-Source Tools | PharmaGist, USRCAT, Pharmer, pmapper | Free alternatives for specific pharmacophore tasks with varying capabilities |
| Compound Databases | ZINC, ChEMBL, PubChem, DrugBank | Sources of chemical structures and bioactivity data for training sets and virtual screening |
| Conformation Generators | CONFIRM, Omega, RDKit Conformer Generation | Generation of representative conformational ensembles for flexible matching |
| Validation Tools | DUD-E server, ROC analysis utilities, Statistical packages | Decoy generation and model validation capabilities |
The selection of appropriate tools depends on research objectives, available resources, and specific requirements of the drug discovery project. Commercial platforms typically offer integrated workflows and user-friendly interfaces, while open-source tools provide flexibility and customization options for specialized applications [37] [32].
Ligand-based pharmacophore modeling represents a powerful and well-established approach in computer-aided drug design, particularly valuable when structural information about the biological target is limited. By systematically extracting common chemical features from known active compounds and arranging them in three-dimensional space, this methodology captures the essential elements required for molecular recognition and biological activity. The strength of this approach lies in its ability to integrate structural and activity data to create predictive models that can guide virtual screening, scaffold hopping, and lead optimization efforts.
As drug discovery faces increasing challenges with novel and difficult targets, ligand-based pharmacophore modeling continues to evolve through integration with machine learning methods [7], novel pharmacophore representation schemes [37], and enhanced virtual screening algorithms. When implemented following rigorous validation protocols and applied as part of a comprehensive drug discovery workflow, this methodology significantly accelerates the identification of novel chemical starting points for therapeutic development, ultimately contributing to more efficient and successful drug discovery campaigns.
A pharmacophore is defined by the International Union of Pure and Applied Chemistry (IUPAC) as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2]. This abstract concept provides a powerful framework for understanding molecular recognition and is extensively applied in computer-aided drug design (CADD). Pharmacophore modeling serves as a critical tool in virtual screening by reducing the complexity of molecular interactions to a set of essential features, enabling the efficient identification of novel hit compounds from extensive chemical libraries [2] [39]. The robustness of pharmacophore models stems from their focus on chemical functionalities rather than specific molecular scaffolds, facilitating the identification of structurally diverse compounds with similar biological activity [2].
The development of a pharmacophore model represents a foundational step in structure-based drug design, creating a template that encapsulates the key interactions necessary for a ligand to bind effectively to its target protein. This approach is particularly valuable when handling large compound databases, as it enables rapid filtering based on essential chemical features before applying more computationally intensive methods like molecular docking [39]. The effectiveness of pharmacophore modeling has been demonstrated across numerous therapeutic areas, from cancer research targeting proteins like XIAP [40] to metabolic disorders focusing on enzymes like ketohexokinase-C [14] and viral infections targeting viral proteases [13]. This whitepaper provides a comprehensive technical guide to the core steps in pharmacophore model development—protein preparation, binding site detection, and feature selection—framed within the context of virtual screening research.
Protein Preparation: The Critical Foundation
The initial and arguably most crucial step in structure-based pharmacophore modeling is the preparation of the protein structure. The quality of the input protein structure directly determines the reliability of the resulting pharmacophore model, as any deficiencies in the structural data will propagate through the entire modeling process [2].
Source and Quality Assessment of Protein Structures
The primary source for protein structures is the RCSB Protein Data Bank (PDB), which contains thousands of high-resolution structures solved primarily through X-ray crystallography or NMR spectroscopy [2]. When selecting a structure, priority should be given to the following characteristics: high resolution (preferably <2.0 Å), completeness of the structure in the binding site region, presence of a relevant co-crystallized ligand, and minimal mutations or missing residues in critical regions [2] [40]. In cases where experimental structures are unavailable, computational techniques such as homology modeling or cutting-edge tools like AlphaFold2 can generate reliable 3D models [2]. A critical evaluation of the input structure is essential before proceeding, including assessment of stereochemical parameters through Ramachandran plots and verification of overall structural integrity [2].
Preparation Workflow and Technical Considerations
The protein preparation workflow involves several standardized steps to ensure the structure is optimized for computational analysis. The initial step involves adding hydrogen atoms, which are typically not resolved in X-ray crystal structures [2]. This is followed by assignment of protonation states for amino acid residues, which should reflect physiological conditions and may require specialized tools for predicting pKa values of specific residues like histidines, glutamic acid, and aspartic acid [2] [40]. The structure should also be checked for missing heavy atoms or side chains, which may need to be modeled computationally. Additionally, non-protein components such as water molecules, ions, and cofactors must be critically evaluated—some tightly bound waters may participate in crucial hydrogen-bonding networks and should be retained, while others may be removed to simplify the model [2]. The final preparation step typically involves energy minimization to relieve steric clashes and optimize the geometry of the added atoms while preserving the overall protein fold [2].
Table 1: Key Steps in Protein Preparation
| Step | Description | Tools/Methods | Critical Considerations |
|---|---|---|---|
| Structure Sourcing | Obtain 3D structure from PDB or computational modeling | RCSB PDB, AlphaFold2, homology modeling | Resolution, completeness, relevance of co-crystallized ligand |
| Hydrogen Addition | Add and optimize hydrogen atoms | Molecular modeling software | Correct protonation states at physiological pH |
| Missing Components | Address missing residues/side chains | Modeler, Prime, MODELLER | Particularly critical in binding site regions |
| Water/Co-factor处理 | Evaluate non-protein components | Interaction analysis, energy calculations | Retain functionally important waters |
| Energy Minimization | Relieve steric clashes | Molecular mechanics force fields | Preserve crystal structure integrity |
Binding Site Detection and Characterization
Accurate identification and characterization of the ligand-binding site is the next critical step in structure-based pharmacophore modeling, as it defines the spatial context for all subsequent feature generation [2].
Binding Site Identification Methods
Binding site detection can be approached through multiple methodologies. When the protein structure contains a co-crystallized ligand, the binding site is often defined as the residues within a specific radius (typically 5-10 Å) of the bound ligand [40]. In the absence of a ligand, computational tools can predict potential binding pockets based on various properties. GRID is a grid-based method that uses different molecular probes to sample protein surfaces and identify regions with energetically favorable interactions [2]. LUDI applies knowledge-based rules derived from distributions of non-bonded contacts in experimental structures to predict interaction sites [2]. Other approaches include geometric methods that identify surface cavities and concavities, and energy-based methods that evaluate interaction potentials across the protein surface [41]. Many modern tools combine multiple approaches to improve prediction accuracy.
Binding Site Analysis and Characterization
Once identified, the binding site requires detailed characterization to understand its properties and potential interaction capabilities. This involves mapping the chemical environment, including hydrophobic patches, hydrogen-bonding capabilities (donors and acceptors), charged regions, and aromatic clusters [2]. The shape and volume of the binding site should be analyzed, as this information can be incorporated into the pharmacophore model as exclusion volumes to represent sterically forbidden regions [2] [41]. If multiple protein-ligand complexes are available, analysis of conserved interactions can help identify critical features that should be prioritized in the pharmacophore model [2]. For targets with known active compounds, mutagenesis data can provide experimental validation of important residues [2].
Table 2: Binding Site Detection Methods and Applications
| Method Type | Representative Tools | Underlying Principle | Strengths | Limitations |
|---|---|---|---|---|
| Geometry-based | POCKET, PocketPicker | Identifies surface cavities and pockets | Fast computation | May miss cryptic sites |
| Energy-based | GRID, Q-SiteFinder | Molecular interaction energy calculations | Accounts for chemical properties | Computationally more intensive |
| Knowledge-based | LUDI | Statistical analysis of known structures | Leverages experimental data | Dependent on database completeness |
| Template-based | SiteMap, AutoLigand | Comparison to known binding sites | High accuracy for similar targets | Limited to well-characterized folds |
| Data-driven/ML | P2Rank, DeepPocket | Machine learning algorithms | Improving accuracy with more data | Training set dependencies |
Pharmacophore Feature Selection and Model Generation
The core of pharmacophore modeling involves identifying and selecting the key chemical features from the prepared protein structure and binding site that are essential for molecular recognition and biological activity [2].
Fundamental Pharmacophore Features
The most essential pharmacophore features include hydrogen bond acceptors (HBA) and donors (HBD), which are represented as vectors indicating the direction of hydrogen bond formation [2]. Hydrophobic features (H) represent areas favorable for hydrophobic interactions and are typically depicted as spheres in 3D space [39]. Charged features include positive ionizable (PI) and negative ionizable (NI) groups that participate in electrostatic interactions or salt bridges [2]. Aromatic features (AR) capture potential π-π stacking or cation-π interactions [39]. Exclusion volumes (XVOL) are incorporated to represent steric constraints from the protein structure, ensuring that generated ligands do not clash with the binding site [2] [41]. Modern pharmacophore modeling tools like LigandScout can automatically identify these features from protein-ligand complexes by analyzing interaction patterns [40].
Feature Selection and Hypothesis Generation
The initial feature detection typically generates more features than necessary, requiring a careful selection process to create a refined pharmacophore hypothesis. This selection can be guided by analyzing interaction energy contributions, where features that contribute significantly to binding energy are prioritized [2]. If multiple protein-ligand structures are available, identifying conserved interactions across different complexes helps select biologically relevant features [2]. Information from sequence alignments or genetic variation studies can highlight functionally critical residues [2]. Spatial constraints from the receptor structure can also guide feature selection to ensure geometric compatibility [2]. The complexity of the final model should balance comprehensiveness with practicality—too many features may make the model overly specific, while too few may reduce its discriminative power [2] [39].
Advanced Shape-Based Features
Recent advances in pharmacophore modeling have incorporated shape-based features to improve screening accuracy. Negative image-based (NIB) models use the shape of the binding cavity itself as a key feature, creating a pseudo-ligand that represents the optimal steric fit [41]. Tools like O-LAP generate shape-focused pharmacophore models by clustering overlapping atoms from docked active ligands to create cavity-filling models that better represent the binding site geometry [41]. These approaches can significantly enhance virtual screening enrichment by incorporating explicit shape complementarity into the feature set [41].
Integrated Workflow and Experimental Validation
The development of a robust pharmacophore model requires systematic integration of the previously described steps, followed by rigorous validation to ensure predictive capability.
Comprehensive Workflow Integration
A typical structure-based pharmacophore modeling workflow integrates all stages from initial protein preparation to final model generation. The process begins with protein structure acquisition and preparation, followed by binding site detection and analysis [2] [40]. The subsequent feature identification phase maps all potential interaction points within the binding site, which are then refined through strategic feature selection to create the pharmacophore hypothesis [2]. This hypothesis can be further optimized using known active compounds to improve its discriminative power [41]. The entire process is iterative, with model validation often leading to refinements in feature selection or binding site definition.
Diagram Title: Pharmacophore Modeling Workflow
Model Validation Strategies
Validation is essential to confirm the pharmacophore model's ability to distinguish active from inactive compounds [39]. The most common method uses receiver operating characteristic (ROC) curves and area under the curve (AUC) values to quantify model performance [40]. The early enrichment factor (EF), particularly EF1%, measures the model's ability to identify true actives in the top percent of screened compounds [40]. Some methods use decoy sets with known actives to test the model's retrieval capability [40]. Additionally, the model can be tested against known inactive compounds to verify it does not incorrectly identify them as hits [39].
Integration with Virtual Screening
Validated pharmacophore models are deployed as filters in virtual screening campaigns to rapidly reduce large compound libraries to manageable sizes [2] [39]. The pharmacophore model serves as a query to search databases, with compounds matching the feature arrangement progressing to more computationally intensive methods like molecular docking [2] [13]. This hierarchical approach optimizes computational resources by applying rapid pharmacophore screening before precise docking calculations [42] [43]. Successful applications of this strategy have identified novel inhibitors for various targets, including XIAP for cancer therapy [40] and ketohexokinase-C for metabolic disorders [14].
Table 3: Essential Resources for Structure-Based Pharmacophore Modeling
| Resource Category | Specific Tools/Services | Primary Function | Key Applications in Workflow |
|---|---|---|---|
| Protein Databases | RCSB PDB, AlphaFold Protein Structure Database | Source of 3D protein structures | Initial protein structure acquisition |
| Protein Preparation | Schrödinger Protein Preparation Wizard, MOE, BIOVIA Discovery Studio | Structure optimization and refinement | Hydrogen addition, protonation state assignment, energy minimization |
| Binding Site Detection | GRID, LUDI, SiteMap, P2Rank | Identification and analysis of binding pockets | Binding site characterization and mapping |
| Pharmacophore Modeling | LigandScout, MOE, PHASE, O-LAP | Feature identification and model generation | Pharmacophore feature selection and hypothesis generation |
| Virtual Screening Platforms | ZINC Database, Enamine REAL, Schrodinger Maestro | Compound libraries and screening environments | Model validation and virtual screening applications |
| Validation Tools | DUDE, DUD-E | Decoy sets for model validation | Performance assessment through enrichment calculations |
The development of robust pharmacophore models through meticulous protein preparation, accurate binding site detection, and strategic feature selection represents a powerful methodology in modern drug discovery. When properly validated and integrated into virtual screening workflows, these models significantly accelerate the identification of novel bioactive compounds by efficiently navigating vast chemical spaces. As computational methods continue to advance, incorporating techniques like machine learning [43] and molecular dynamics [44] [45], pharmacophore modeling remains an indispensable tool for researchers seeking to bridge structural biology and therapeutic development. The systematic approach outlined in this whitepaper provides a framework for developing pharmacophore models that balance molecular complexity with practical screening utility, offering researchers a strategic advantage in the challenging landscape of drug discovery.
In computer-aided drug discovery, a pharmacophore model abstractly represents the spatial and electronic functional features necessary for a molecule to interact with its biological target [2] [39]. These features include hydrogen bond donors (HBD) and acceptors (HBA), hydrophobic areas (H), positively and negatively ionizable groups (PI/NI), and aromatic rings (AR) [2]. The core challenge, however, lies in the dynamic nature of small molecules. Ligands are not rigid; they exist as ensembles of interconverting three-dimensional structures. Therefore, identifying a molecule's bioactive conformation—the specific 3D structure it adopts when bound to the target—is a critical prerequisite for constructing a meaningful pharmacophore model and for the subsequent success of virtual screening campaigns [46].
This guide provides an in-depth technical overview of the methods and best practices for handling ligand flexibility through conformational analysis and generating bioactive conformers, specifically within the workflow of building a pharmacophore model for virtual screening.
Theoretical Foundation: Why Ligand Flexibility Matters
The concept of the bioactive conformation is intrinsically linked to the pharmacophore. A pharmacophore model is, by definition, a three-dimensional arrangement of steric and electronic features [2]. If the conformer used to build or screen against this model does not represent the true binding pose, the model's ability to identify active compounds is severely compromised. The primary goal of conformational analysis in this context is to ensure that the conformational ensemble generated for a ligand includes, or can be used to deduce, this bioactive state.
Conformational flexibility arises from the rotation around single bonds, leading to different torsional angles and, consequently, distinct three-dimensional shapes with potentially different energies. The challenge is to sample this conformational space efficiently, balancing computational cost with the need to cover the relevant low-energy states that a ligand is likely to populate in solution and upon binding.
Methodologies for Conformational Analysis
Several computational strategies have been developed to generate conformational ensembles, each with its own strengths and optimal use cases.
Knowledge-Based and Systematic Search Methods
Knowledge-based methods, such as those implemented in ConfGen, apply empirically derived heuristics and rules about preferred torsional angles to rapidly generate a set of low-energy, diverse conformers [46]. These methods are highly efficient for generating a manageable number of plausible conformations for ligand-based virtual screening.
Systematic search methods, while sometimes computationally expensive, exhaustively explore all possible rotatable bonds by incrementing torsion angles through a defined range (e.g., every 120 degrees for sp³ carbon atoms). This approach ensures comprehensive coverage but can lead to a combinatorial explosion for highly flexible molecules.
Stochastic and Simulation-Based Methods
Stochastic methods, like Monte Carlo algorithms, randomly change torsion angles to explore the conformational landscape. This approach is less likely to be trapped in local minima and can be effective for complex molecules, though it may require many steps to ensure adequate coverage.
Simulation-based methods, primarily Molecular Dynamics (MD) simulations, model the physical movement of atoms over time. MD is particularly valuable for studying the time-dependent behavior of molecules and capturing the influence of solvation. However, standard MD simulations may not efficiently cross high energy barriers, potentially limiting the sampling of diverse conformations on typical computational timescales.
Table 1: Comparison of Conformational Search Methodologies
| Method | Key Principle | Advantages | Limitations | Suitable for |
|---|---|---|---|---|
| Knowledge-Based | Pre-defined torsional libraries & rules | High speed, computationally efficient | May miss unusual conformations | High-throughput virtual screening [46] |
| Systematic Search | Exhaustive torsion scanning | Comprehensive coverage | Combinatorial explosion for flexible molecules | Small to medium-sized molecules |
| Stochastic | Random changes to torsions | Good escape from local minima | Can be inefficient; unpredictable | Complex, macrocyclic molecules |
| Simulation-Based (MD) | Newtonian mechanics over time | Includes solvation & dynamics | Computationally intensive; poor barrier crossing | Refinement & stability assessment [47] |
Generating the Bioactive Conformer
Generating a diverse set of conformers is only the first step. The next, more critical step is to identify which of these conformers represents the bioactive state.
Ligand-Based Pharmacophore Modeling
When the structure of the biological target is unknown, the bioactive conformation must be inferred from a set of known active ligands. Ligand-based pharmacophore modeling aligns multiple active compounds in 3D space to identify their common chemical features, and the alignment process inherently implies a bioactive-like conformation for each molecule [2] [48].
A standard protocol, as demonstrated in a study on MMP-9 inhibitors, involves:
- Data Set Curation: A diverse set of active ligands with known biological activity (e.g., IC50) is collected. Activities are often converted to pIC50 (-logIC50) for modeling [48].
- Conformer Generation: A comprehensive conformational ensemble is generated for each ligand. For instance, the Phase module uses distance-dependent dielectric for solvation treatment during this process [48].
- Pharmacophore Hypothesis Generation: The software identifies common pharmacophoric features and their spatial arrangements across the active ligands. A hypothesis is typically a set of features like donor, acceptor, hydrophobic, and aromatic rings (e.g., the
DDHRRmodel) [48]. - Model Validation: The model is validated using statistical parameters (e.g., R², Q²) and its ability to predict the activity of a test set of compounds not used in model development [48] [49].
The conformers selected by this process for the active ligands are considered reasonable approximations of their bioactive conformations.
Structure-Based Pharmacophore Modeling
When an experimental 3D structure of the target (e.g., from X-ray crystallography) is available, a more direct approach can be used. A structure-based pharmacophore is built by analyzing the interaction points within the protein's binding site [2]. A ligand's bioactive conformer can be generated by docking it into this site.
The workflow generally includes:
- Protein Preparation: The protein structure is prepared by adding hydrogen atoms, assigning correct protonation states, and optimizing the structure [2].
- Binding Site Definition: The ligand-binding site is identified, often based on the location of a co-crystallized ligand or using computational tools like GRID [2].
- Molecular Docking: Ligands are flexibly docked into the binding site, generating multiple poses. The top-ranked pose from docking software like PLANTS is frequently used as the bioactive conformer for downstream pharmacophore modeling or validation [41].
- Pharmacophore Feature Generation: Essential chemical features are derived from the protein-ligand interaction pattern, and exclusion volumes can be added to represent the shape of the binding pocket [2].
Emerging AI-Powered Approaches
Recent advances in deep learning are creating new paradigms for handling ligand flexibility and pharmacophore mapping. DiffPhore is a knowledge-guided diffusion model that generates 3D ligand conformations "on-the-fly" to maximally map onto a given pharmacophore model [50]. It leverages large datasets of 3D ligand-pharmacophore pairs to learn the mapping relationships, outperforming traditional methods in predicting binding conformations [50].
Another approach, PGMG (Pharmacophore-Guided deep learning approach for bioactive Molecule Generation), uses pharmacophore hypotheses as input to generate novel bioactive molecules directly, introducing a latent variable to handle the complex many-to-many mapping between pharmacophores and molecules [15].
Experimental Protocol: A Practical Workflow
Below is a detailed, step-by-step protocol for generating and validating bioactive conformers within a pharmacophore modeling project, synthesizing methodologies from the cited literature.
Step 1: Ligand Preparation and Conformer Generation
- Input: 2D structures (e.g., SMILES strings) of known active ligands.
- Software: Use a tool like ConfGen [46] or the LigPrep/ConfGen suite.
- Parameters:
- Forcefield: OPLS3e [48].
- Dielectric constant: Use a distance-dependent dielectric for implicit solvation [48].
- Energy window: Retain conformers within a specified energy threshold (e.g., 10 kcal/mol) from the global minimum.
- Maximum number of conformers per ligand: This can be optimized based on ligand flexibility, but presets in ConfGen offer a good balance of speed and accuracy [46].
- Output: A multi-conformer 3D structure file for each ligand.
Step 2: Pharmacophore Model Development and Conformer Selection
- Software: PHASE module [48].
- Procedures:
- Activity Data: Assign activity values (e.g., pIC50) and define activity thresholds (e.g., pIC50 > 8.3 as active, <5.5 as inactive) [48].
- Training/Test Set: Divide the data set into a training set (~70%) for model development and a test set (~30%) for validation [48].
- Feature Identification: Define the pharmacophoric features (HBD, HBA, H, AR, etc.) on the training set ligands.
- Hypothesis Generation: Generate common pharmacophore hypotheses using the active compounds. The software will align the ligands and select conformers that best fit the emerging hypothesis. A hypothesis like
DDHRR_1(two donors, two hydrophobic groups, one aromatic ring) might be identified with a high survival score [48]. - 3D-QSAR Model (Optional): Build a QSAR model based on the aligned conformers to predict activity and validate the pharmacophore hypothesis [48].
Step 3: Validation with Structure-Based Methods (If Possible)
- Input: The pharmacophore model and the predicted bioactive conformers from Step 2.
- Procedure:
- Perform molecular docking of the ligands into the target's binding site.
- Compare the docked pose (from software like Glide in XP mode) with the ligand-based pharmacophore-derived conformer [48].
- A low Root-Mean-Square Deviation (RMSD) between the two conformers provides strong validation that the ligand-based approach has identified a biologically relevant conformation.
Step 4: Model Application in Virtual Screening
- The validated pharmacophore model is used as a 3D query to screen large chemical databases.
- During screening, each compound in the database must be flexibly fitted to the pharmacophore model, a process that again relies on robust conformational sampling to identify potential hits [2].
The following workflow diagram illustrates the two primary paths for bioactive conformer generation and how they integrate within a pharmacophore modeling pipeline.
The Scientist's Toolkit: Essential Research Reagents and Software
Table 2: Key Software Tools for Conformational Analysis and Pharmacophore Modeling
| Tool Name | Type/Function | Key Features | Application in Workflow |
|---|---|---|---|
| ConfGen [46] | Conformer Generator | Knowledge-based & physics-based methods; efficient generation of diverse, low-energy conformers. | Ligand Preparation. Generate input conformational ensembles for ligand-based modeling. |
| PHASE [48] | Pharmacophore Modeling & 3D-QSAR | Develop ligand-based pharmacophore hypotheses, align structures, perform 3D-QSAR studies. | Model Development & Validation. Core platform for building, analyzing, and validating ligand-based models. |
| Schrödinger Suite (LigPrep, Glide) [48] | Integrated Modeling Suite | Ligand preparation, molecular docking, and binding affinity prediction. | Structure-Based Validation. Prepare ligands and proteins, dock ligands, validate bioactive conformers. |
| DiffPhore [50] | AI-based Conformation Generator | Deep learning diffusion model for generating conformations matching a pharmacophore. | Advanced Conformer Generation. "On-the-fly" generation of bioactive-like conformers guided by pharmacophore constraints. |
| PGMG [15] | AI-based Molecule Generator | Deep learning model that generates novel bioactive molecules from a pharmacophore hypothesis. | De Novo Drug Design. Create new chemical entities that match the pharmacophore model. |
| O-LAP [41] | Shape-Focused Model Builder | Graph clustering to create shape-focused pharmacophore models from docked poses. | Model Enhancement. Create cavity-filling models to improve docking screening enrichment. |
| PLANTS [41] | Molecular Docking Software | Flexible ligand docking for pose generation and virtual screening. | Pose Generation. Produces candidate bioactive poses for structure-based pharmacophore modeling. |
The accurate handling of ligand flexibility is not merely a technical step but a foundational element in the construction of reliable pharmacophore models. The process of conformational analysis and bioactive conformer generation bridges the gap between a static 2D molecular structure and its dynamic 3D interaction with a biological target. By employing a rigorous methodology—whether through robust ligand-based approaches, structure-based docking, or cutting-edge AI tools—researchers can significantly enhance the predictive power of their pharmacophore models. This, in turn, leads to more successful virtual screening campaigns, ultimately accelerating the discovery of novel therapeutic agents.
In the modern drug discovery pipeline, computer-aided drug discovery (CADD) techniques are indispensable for reducing the immense time and financial costs associated with developing novel therapeutics [2]. Among these techniques, pharmacophore-based virtual screening has matured into a cornerstone methodology, widely accepted and implemented in medicinal chemistry laboratories [51]. Its relevance is particularly pronounced in addressing health emergencies and the rise of personalized medicine, where rapid candidate identification is paramount [2]. Virtual screening (VS) itself is a CADD method that involves the in silico screening of extensive libraries of chemical compounds to identify those most likely to bind to a specific drug target [2]. Pharmacophore-based methods significantly accelerate this process by providing an abstract query that encapsulates the essential steric and electronic features required for biological activity, enabling efficient searching of large compound collections to pinpoint molecules with the desired properties [2] [52].
The core concept of a pharmacophore was defined by the International Union of Pure and Applied Chemistry (IUPAC) as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2] [52]. In practice, this abstract picture represents the key chemical functionalities of a ligand as geometric entities—such as spheres, planes, and vectors—thereby shifting the focus from specific atoms to essential interaction capabilities [2]. This abstraction is the methodology's greatest strength, facilitating the identification of biologically active molecules with diverse chemical scaffolds, a process known as scaffold hopping [2] [51].
Core Concepts and Theoretical Foundation
Essential Pharmacophore Features
A pharmacophore model translates the complex nature of non-covalent ligand-binding interactions into an intuitive set of chemical features [51]. The most critical feature types recognized in pharmacophore modeling are summarized in the table below.
Table 1: Fundamental Pharmacophore Features and Their Descriptions
| Feature | Description | Role in Molecular Recognition |
|---|---|---|
| Hydrogen Bond Acceptor (HBA) | An atom that can accept a hydrogen bond (e.g., carbonyl oxygen). | Facilitates directional interactions with donor groups on the target protein. |
| Hydrogen Bond Donor (HBD) | A hydrogen atom covalently bound to an electronegative atom (e.g., OH, NH). | Forms strong, directional interactions with acceptor groups on the target. |
| Hydrophobic (H) | A non-polar region of the ligand (e.g., alkyl chain). | Drives burial in hydrophobic pockets of the binding site via entropic effects. |
| Positively Ionizable (PI) | A group that can carry a positive charge (e.g., amine). | Can form strong electrostatic or salt-bridge interactions. |
| Negatively Ionizable (NI) | A group that can carry a negative charge (e.g., carboxylic acid). | Can form strong electrostatic or salt-bridge interactions. |
| Aromatic (AR) | A planar, conjugated ring system. | Engages in cation-π, π-π, or hydrophobic interactions. |
| Exclusion Volume (XVOL) | A spatial volume indicating forbidden space. | Represents the shape of the binding pocket, sterically preventing clashes. |
These features are not tied to a specific chemical scaffold but represent the fundamental physicochemical requirements for binding [2]. The spatial arrangement of these features, constrained by exclusion volumes that mimic the binding pocket's geometry, defines a unique pharmacophore hypothesis capable of discriminating active from inactive compounds [52].
Structure-Based vs. Ligand-Based Modeling Approaches
The generation of a pharmacophore model can be achieved via two distinct paradigms, chosen based on the available input data.
Structure-Based Pharmacophore Modeling: This approach requires the three-dimensional structure of the macromolecular target, typically obtained from X-ray crystallography, NMR spectroscopy, or computational methods like homology modeling (e.g., with AlphaFold2) [2]. The workflow begins with critical protein preparation, which involves assigning correct protonation states, adding hydrogen atoms, and rectifying any structural errors [2]. The subsequent ligand-binding site detection is crucial and can be performed manually (if a co-crystallized ligand exists) or using tools like GRID or LUDI, which identify potential binding pockets based on energetic or geometric criteria [2]. The pharmacophore features are then generated by extracting the interaction pattern between the target and a bound ligand. When a protein-ligand complex is available, this process is highly accurate, as the ligand's bioactive conformation directly informs the spatial disposition of features [2] [52]. Exclusion volumes are added based on the receptor structure to account for spatial restrictions [52].
Ligand-Based Pharmacophore Modeling: This method is employed when the 3D structure of the target is unknown. It relies on the conformational analysis and alignment of a set of known active molecules to identify their common chemical features and their optimal 3D arrangement [2] [39]. This approach is founded on the principle that structurally diverse molecules triggering the same biological effect likely share a common mode of interaction with the target [2]. The resulting model represents the essential features conserved across the active training set. A key challenge here is adequately accounting for the conformational flexibility of the ligands during the alignment process [39].
Workflow for Implementation
The successful application of pharmacophore-based virtual screening follows a multi-stage workflow, from data preparation to experimental validation. The following diagram illustrates the integrated process, incorporating both structure-based and ligand-based routes.
Data Preparation and Model Construction
The initial phase lays the foundation for a successful screening campaign.
For Structure-Based Models: Begin by sourcing a high-quality 3D structure of the target from the Protein Data Bank (PDB) [2] [52]. Critically evaluate the structure for resolution, missing residues, and stereochemical quality. During protein preparation, assign correct protonation states to residues (e.g., Histidine tautomers) and add hydrogen atoms, which are typically absent in X-ray structures [2]. If a co-crystallized ligand is present, it provides a direct template for feature generation. If not, use binding site detection algorithms to define the active site and then map interaction points (e.g., using Discovery Studio or LigandScout tools) to create a receptor-based hypothesis [2] [52].
For Ligand-Based Models: The quality of the training set is paramount. Curate a set of known active molecules with robust, target-specific activity data (e.g., from ChEMBL, DrugBank, or PubChem Bioassay) [52]. Prefer compounds with direct binding affinity data (e.g., IC50, Ki) over cell-based assay results, which can be confounded by pharmacokinetic effects [52]. The set should be structurally diverse to ensure the resulting model is not overly specific [52]. Additionally, compile a set of confirmed inactive molecules or generated decoys (e.g., from DUD-E) for subsequent model validation [52]. These decoys should have similar 1D properties (e.g., molecular weight, logP) to the actives but different topologies to avoid artificial enrichment [52].
Model Refinement and Validation
The initial pharmacophore hypothesis is rarely perfect and requires iterative refinement. This process involves adding or removing features, adjusting their weights or spatial tolerances, and designating certain features as optional [52]. The model's quality must be evaluated theoretically before prospective screening. This is done by screening a validation dataset containing known active and inactive compounds/decoys [52]. Several quantitative metrics are used for this assessment:
Table 2: Key Metrics for Pharmacophore Model Validation
| Metric | Formula/Description | Interpretation |
|---|---|---|
| Enrichment Factor (EF) | EF = (Hitactives / Nactives) / (Hittotal / Ntotal) | Measures how much the model enriches active compounds in the hit list compared to random selection. Higher is better. |
| Yield of Actives | (Hitactives / Hittotal) * 100 | The percentage of active compounds in the virtual hit list. |
| Sensitivity | Hitactives / Nactives | The model's ability to correctly identify known active compounds. |
| Specificity | Hitinactives / Ninactives | The model's ability to correctly reject known inactive compounds. |
| ROC-AUC | Area Under the Receiver Operating Characteristic curve. | A comprehensive measure of model performance; 1.0 is perfect, 0.5 is random. |
A high-quality model should demonstrate a strong enrichment of actives (EF >> 1), high sensitivity and specificity, and a high ROC-AUC value [52]. Reported hit rates from prospective pharmacophore-based VS typically range from 5% to 40%, vastly outperforming random high-throughput screening (HTS) hit rates, which are often below 1% [52].
Virtual Screening Execution
Once validated, the pharmacophore model is used as a 3D query to screen large compound libraries (e.g., ZINC, in-house corporate collections) [2] [52]. This screening process involves scanning each compound in the database to check if it can assume a conformation that matches all (or the required number of) the model's chemical features within their defined spatial constraints [2]. Molecules that successfully map the model form the virtual hit list. These hits are then often subjected to further filtering based on drug-likeness rules (e.g., Lipinski's Rule of Five) or more computationally intensive steps like molecular docking to refine the selection before proceeding to experimental testing [25] [39].
Successful implementation of pharmacophore-based VS relies on a suite of software tools and data resources.
Table 3: Research Reagent Solutions for Pharmacophore-Based Virtual Screening
| Tool/Resource | Type | Function/Purpose |
|---|---|---|
| RCSB Protein Data Bank (PDB) | Data Repository | Primary source for experimentally determined 3D structures of proteins and nucleic acids, essential for structure-based modeling [2] [52]. |
| ChEMBL, DrugBank | Chemical Database | Curated databases containing bioactivity data, drug-like properties, and target information, crucial for assembling ligand training sets [52]. |
| DUD-E (Directory of Useful Decoys) | Tool/Resource | Online service that generates optimized decoy molecules for a given set of active compounds, used for realistic model validation [52]. |
| LigandScout | Software | Advanced software for both structure-based and ligand-based pharmacophore model creation, visualization, and virtual screening [52]. |
| Discovery Studio | Software | Comprehensive modeling and simulation suite that includes robust tools for structure-based pharmacophore generation and analysis [52]. |
| GRID, LUDI | Software | Programs used for binding site detection and analysis, helping to define the active site and its interaction potential [2]. |
| ZINC Database | Chemical Database | A freely available database of commercially available compounds, often used as a screening library for virtual screening campaigns. |
Experimental Protocols and Case Studies
Detailed Methodology for a Structure-Based VS Campaign
A typical protocol for a structure-based screening campaign against a novel target involves the following steps:
Target Selection and Preparation: Download the target protein's PDB file (e.g., 1FDQ). Using a tool like Discovery Studio or MOE, prepare the protein by:
- Removing extraneous water molecules and co-factors, unless functionally critical.
- Adding and optimizing hydrogen atoms.
- Assigning protonation states at physiological pH (e.g., ensuring histidine residues are in the correct tautomeric state).
- Fixing any structural anomalies, such as missing side chains.
Binding Site Definition and Model Generation: Define the binding site using the coordinates of a co-crystallized ligand. In the absence of a ligand, use a binding site detection algorithm. Generate an initial pharmacophore model directly from the protein-ligand interaction pattern. This model will typically include HBA, HBD, hydrophobic, and aromatic features, along with exclusion volumes derived from the protein surface [2] [52].
Model Refinement and Theoretical Validation: Refine the initial model by removing redundant features or designating less critical ones as "optional." Validate the refined model by screening a test database containing known actives and inactives/decoys for the target. Calculate the Enrichment Factor and ROC-AUC to ensure the model meets pre-defined quality thresholds (e.g., EF10% > 5, AUC > 0.8) [52].
Prospective Screening and Hit Selection: Use the validated model to screen a large, diverse compound library (e.g., several million compounds from the ZINC database). The hits generated from this screening are then prioritized based on factors such as:
- The fit value to the pharmacophore model.
- Drug-likeness and absence of reactive or toxic groups.
- Commercial availability and synthetic tractability.
- Results from subsequent molecular docking studies.
Exemplary Application: Hydroxysteroid Dehydrogenases (HSDs)
The field of hydroxysteroid dehydrogenase (HSD) research provides compelling case studies for the successful application of pharmacophore-based VS. For instance, to identify novel inhibitors of 17β-HSD1, a target for breast cancer therapy, researchers have built structure-based models from co-crystallized ligands [52]. The resulting pharmacophore, exemplifying one binding mode, typically features a hydrogen bond acceptor, a hydrogen bond donor, and hydrophobic/aromatic regions, constrained by exclusion volumes representing the binding pocket shape [52]. Such models have been successfully screened against commercial databases, leading to the identification of novel, potent inhibitor chemotypes with hit rates significantly higher than those from conventional HTS, demonstrating the power of this approach for lead identification [52].
Pharmacophore-based virtual screening stands as a powerful, mature, and highly effective technology within the computational drug discovery arsenal. By abstracting the key elements of molecular recognition, it provides an intuitive yet computationally tractable method for efficiently mining vast chemical space to identify novel lead compounds. Its unique strength lies in its ability to facilitate scaffold hopping, discovering chemically diverse compounds that share a common biological activity [2] [51]. While the approach has inherent limitations due to its simplified representation of complex interactions and is sensitive to the quality of input data [51], a rigorous workflow encompassing careful data preparation, model refinement, and thorough theoretical validation can yield models of high predictive power. When integrated with other computational and experimental techniques, pharmacophore-based virtual screening significantly de-risks and accelerates the early stages of drug discovery, consistently proving its value as a practical and indispensable tool for researchers and drug development professionals.
The development of multi-target inhibitors represents a promising strategy in oncology to overcome the limitations of single-target therapies, particularly tumor cell resistance [53]. The synergistic role of VEGFR-2 and c-Met in tumor angiogenesis and progression has established them as attractive targets for dual-targeted cancer therapy [54]. This case study details an integrated computational workflow for the identification of novel VEGFR-2/c-Met dual inhibitors, framed within the broader context of building an effective pharmacophore model for virtual screening research.
Biological Rationale of VEGFR-2 and c-Met as Dual Targets
Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2) is the primary mediator of VEGF-induced angiogenesis, the process of new blood vessel formation that is crucial for tumor growth and metastasis [54]. Under pathological conditions, VEGFR-2 overexpression activates the Raf-1/MAPK/ERK signaling pathway, enhancing vascular permeability and facilitating tumor invasion [54].
The mesenchymal-epithelial transition factor (c-Met) is a transmembrane receptor tyrosine kinase that, upon binding its ligand HGF, initiates a signaling cascade regulating cell proliferation, survival, and motility [54]. Abnormal activation of the c-Met pathway through overexpression, mutation, or autocrine signaling promotes tumor cell invasion and dissemination [54].
The synergistic relationship between these pathways in multiple cancer types provides a strong rationale for dual inhibition. VEGFR-2/c-Met dual inhibitors may offer broader therapeutic benefits compared to selective inhibitors targeting either receptor alone, potentially overcoming the resistance mechanisms that often limit single-target therapies [53] [54].
The identification of novel inhibitors followed a multi-stage virtual screening workflow that integrated both ligand-based and structure-based drug design approaches. This comprehensive methodology ensured the selection of compounds with not only strong binding potential but also favorable drug-like properties.
Figure 1: Comprehensive virtual screening workflow for identifying VEGFR-2/c-Met dual inhibitors, integrating sequential filtering steps from initial compound library to final hit validation.
Experimental Protocols and Methodologies
Target Preparation and Selection
The initial phase involved careful selection and preparation of protein structures to ensure the reliability of subsequent modeling stages:
- Source and Criteria: Crystal structures of VEGFR-2 (18 structures) and c-Met (47 structures) were retrieved from the RCSB Protein Data Bank. Structures were selected based on resolution < 2.0 Å, nanomolar biological activity of co-crystallized ligands, and structural diversity to capture different binding environments [54].
- Structure Refinement: All protein structures were prepared using Discovery Studio 2019 (DS 2019). This process included: removing water molecules, completing missing amino acid residues, correcting bond connectivity and order, and energy minimization using the CHARMM force field to ensure structural integrity [54].
Pharmacophore Model Generation and Validation
The core component of this case study involved developing predictive pharmacophore models for both targets:
- Model Generation: Structure-based pharmacophore models were built using the Receptor-Ligand Pharmacophore Generation module in DS 2019. Ten hypotheses were generated for each target, considering six standard chemical features: hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), positive ionizable, negative ionizable, hydrophobic, and ring aromatic centers. Models were constrained to contain between 4-6 features [54].
- Model Validation: The quality of generated pharmacophores was rigorously assessed using enrichment factor (EF) and receiver operating characteristic (ROC) curve analysis with decoy sets containing known active compounds and inactive molecules. Models were considered reliable if they demonstrated AUC > 0.7 and EF > 2, indicating strong ability to distinguish active from inactive compounds [54].
Table 1: Performance Metrics of Validated Pharmacophore Models
| Target | Best Model Features | Enrichment Factor | AUC Value | Validation Set Size |
|---|---|---|---|---|
| VEGFR-2 | 1 HBD, 2 HBA, 3 Hy | 4.2 | 0.85 | 400 compounds (25 active) |
| c-Met | 2 HBD, 1 HBA, 2 Hy, 1 RA | 3.8 | 0.81 | 425 compounds (25 active) |
HBD: Hydrogen Bond Donor; HBA: Hydrogen Bond Acceptor; Hy: Hydrophobic; RA: Ring Aromatic
Virtual Screening and Compound Prioritization
The validated pharmacophore models were applied to screen large compound libraries:
- Library Preparation: Over 1.28 million compounds from the ChemDiv database were initially prepared by removing counterions, solvent moieties, and salts, followed by hydrogen addition [54].
- Multi-Stage Filtering:
- Step 1: Application of Lipinski's Rule of Five and Veber's rules to prioritize compounds with favorable drug-like properties.
- Step 2: ADMET prediction for aqueous solubility, blood-brain barrier penetration, cytochrome P450 2D6 inhibition, hepatotoxicity, and intestinal absorption/plasma protein binding properties.
- Step 3: Pharmacophore-based screening using the validated VEGFR-2 and c-Met models to identify compounds matching essential interaction features.
- Step 4: Molecular docking studies to evaluate binding affinity and interaction patterns with key residues in both targets.
Molecular Dynamics Simulations and Binding Free Energy Calculations
The stability of protein-ligand complexes for top hits was assessed through sophisticated dynamics simulations:
- Simulation Protocol: 100 ns molecular dynamics (MD) simulations were performed for the top candidate complexes using standard parameters to evaluate conformational stability and interaction persistence [53] [54].
- Energy Calculations: The Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method was employed to calculate binding free energies, providing quantitative assessment of binding affinity that complements docking scores [53] [54].
- Analysis Metrics: Key stability parameters including root mean square deviation (RMSD), root mean square fluctuation (RMSF), and interaction energy profiles were monitored throughout the simulation trajectories to validate binding mode stability.
Key Research Findings
Identification of Novel Dual Inhibitors
The integrated virtual screening workflow successfully identified promising dual-target inhibitors:
- Initial Hits: From the initial library of 1.28 million compounds, 18 hit compounds demonstrated potential dual inhibitory activity against both VEGFR-2 and c-Met [53].
- Top Candidates: Compound 17924 and Compound 4312 emerged as the most promising candidates based on comprehensive screening criteria, demonstrating superior binding free energies to both targets compared to positive control ligands [53] [54].
- Validation: MD simulations confirmed the stable binding modes of both lead compounds throughout the 100 ns simulation period, with consistent protein-ligand interactions and favorable MM/PBSA binding free energies [53].
Table 2: Binding Free Energy Analysis of Top Candidates (MM/PBSA)
| Compound | VEGFR-2 ΔGbind (kcal/mol) | c-Met ΔGbind (kcal/mol) | Key Interactions |
|---|---|---|---|
| 17924 | -42.7 ± 2.1 | -38.9 ± 1.8 | H-bonds with Asp293, Phe294; Hydrophobic with Phe439, Met282 |
| 4312 | -39.8 ± 1.9 | -41.2 ± 2.3 | H-bonds with Ala232, Gly159; Hydrophobic with Val166, Lys181 |
| Positive Control | -35.4 ± 2.3 | -33.7 ± 2.0 | Reference known inhibitors |
Significance in Cancer Therapeutics
The identified compounds represent valuable starting points for further anti-cancer drug development:
- Overcoming Resistance: The dual-targeting approach addresses a significant challenge in cancer therapy—the development of resistance to single-target agents [53] [55].
- Novel Chemical Scaffolds: The hits identified possess distinct chemical structures from existing inhibitors, potentially offering new opportunities for intellectual property and drug development pipelines [54].
- Optimized Drug Properties: The incorporation of ADMET profiling and drug-likeness filters early in the screening process increases the probability of successful translation to preclinical development [54].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools and Resources for Pharmacophore-Based Screening
| Tool/Resource | Application in Workflow | Key Features | Access |
|---|---|---|---|
| Discovery Studio | Protein prep, pharmacophore generation, docking | Structure-based pharmacophore modeling, CHARMM force field | Commercial |
| ChemDiv Database | Compound library | >1.28 million synthesizable compounds | Commercial |
| RCSB PDB | Protein structure source | Crystal structures of VEGFR-2 (18) and c-Met (47) | Public |
| DUD-E Server | Validation decoy sets | Curated decoy molecules for validation | Public |
| RDKit | Cheminformatics | Molecular descriptor calculation, fingerprinting | Open Source |
| GOLD/AutoDock | Molecular docking | Binding pose prediction, scoring functions | Commercial/Public |
Advanced Computational Methodologies
Recent advances in computational methodologies are enhancing pharmacophore-based screening:
- Machine Learning Approaches: New tools like DeepTarget demonstrate strong accuracy in predicting cancer drug targets by integrating large-scale drug and genetic knockdown viability screens with multi-omics data, potentially complementing traditional structure-based approaches [56].
- Generative Models: PharmacoForge, a diffusion model for generating 3D pharmacophores conditioned on protein pockets, represents an emerging alternative to traditional pharmacophore generation methods, showing improved performance in benchmark studies [57].
- Network-Based Target Selection: Protein-protein interaction networks and shortest-path analyses are being used to identify optimal drug target combinations that counter resistance mechanisms by targeting alternative pathways and their connectors [55].
This case study demonstrates a robust, integrated computational framework for identifying novel dual inhibitors targeting VEGFR-2 and c-Met for cancer therapy. The success of this approach highlights the power of pharmacophore modeling as a foundational tool in virtual screening campaigns when combined with complementary structure-based methods and rigorous validation protocols.
The identified compounds 17924 and 4312 represent promising starting points for further medicinal chemistry optimization and experimental validation. This workflow provides a template for future drug discovery efforts targeting multiple oncogenic pathways simultaneously, potentially leading to more effective therapeutic options that address the significant challenge of treatment resistance in oncology.
The continuous advancement of computational methods, including machine learning and deep learning approaches, promises to further enhance the efficiency and success rates of structure-based drug design, accelerating the discovery of novel therapeutic agents for cancer and other complex diseases.
Overcoming Common Challenges and Optimizing Your Pharmacophore Model
Pharmacophore modeling has become an indispensable tool in modern computer-aided drug discovery, providing an abstract representation of the molecular features essential for a compound's biological activity [2]. These models reduce the time and cost of drug development by enabling efficient virtual screening of large compound libraries [2] [58]. However, two persistent limitations significantly impact their reliability and accessibility: pronounced dependence on the quality of input data and the substantial requirement for expert knowledge in model development [58]. The accuracy of any pharmacophore model is fundamentally constrained by the data from which it is derived, with errors in structural data, activity measurements, or feature annotation propagating through to the final model [2]. Simultaneously, the complex process of model creation, refinement, and validation traditionally demands significant input from specialists with deep domain knowledge in both chemistry and biology [58]. This technical guide examines these limitations within the context of building pharmacophores for virtual screening and presents advanced methodologies to mitigate them through automated workflows, quantitative approaches, and machine learning integration.
Technical Approaches to Overcome Data Quality Limitations
Data Quality Assessment and Preparation Protocols
The foundation of a robust pharmacophore model lies in the critical assessment and preparation of input data. For structure-based approaches, this begins with meticulous protein structure preparation. The quality of the target structure directly influences the quality of the resulting pharmacophore model [2]. Researchers must systematically evaluate protonation states of residues, position hydrogen atoms (which are typically absent in X-ray solved structures), identify and handle non-protein groups, address missing residues or atoms, and assess stereochemical and energetic parameters [2]. For ligand-based approaches, data curation requires particular attention to the consistency and reliability of biological activity measurements. Implement the following protocol to ensure data quality:
- Structure Validation Protocol: For protein structures from the PDB, use MolProbity or similar tools to assess Ramachandran plot outliers, rotamer outliers, and clash scores. Prefer structures with resolution better than 2.5Å and R-factor values below 0.25 [2].
- Ligand Data Curation: For ligand-based modeling, apply the following criteria: standardize activity measurements to a consistent unit (e.g., IC50, Ki), flag and investigate outliers beyond two standard deviations from the mean, and verify chemical structures to correct representation errors [59].
- Conformational Sampling: Generate ligand conformations using tools like iConfGen with OPLS 2005 force field, maintaining a maximum of 25 output conformations per ligand to balance diversity and computational expense [20]. Filter conformers using a relative energy threshold of 10 kcal/mol and a minimum atom deviation of 1.00Å to eliminate redundant structures [59].
Advanced Methods for Sparse or Noisy Data
When working with datasets containing limited or noisy data, employ these specialized methodologies to enhance model robustness:
- Quantitative Pharmacophore Activity Relationship (QPhAR): Implement the QPhAR algorithm which demonstrates robust performance even with small dataset sizes of 15-20 training samples [20]. This method constructs quantitative pharmacophore models by first finding a consensus pharmacophore from all training samples, aligning input pharmacophores to this merged model, then using positional information to derive quantitative relationships with biological activities [20].
- Cross-Validation Strategy: Apply rigorous five-fold cross-validation as utilized in QPhAR validation studies, which yielded an average RMSE of 0.62 with standard deviation of 0.18 across 250+ diverse datasets [20]. This approach provides reliable performance metrics even with limited data.
- Feature Selection Automation: Utilize algorithms that automatically select features driving pharmacophore model quality using SAR information extracted from validated QPhAR models, reducing reliance on perfect datasets [24].
Table 1: Data Quality Enhancement Methods and Their Applications
| Method | Technical Approach | Optimal Data Scenarios | Reported Performance |
|---|---|---|---|
| QPhAR Modeling | Consensus pharmacophore generation with machine learning regression | Small datasets (15-50 compounds) | Avg. RMSE 0.62 across 250+ datasets [20] |
| Structure-Based Refinement | Binding site analysis with GRID/LUDI molecular interaction fields | Known protein structures (X-ray, homology models) | Improved virtual screening hit rates [2] |
| Ligand-Based Screening | PHASE algorithm with common pharmacophore hypothesis generation | 10+ active ligands with measured activity | Statistical significance (R²=0.972 in febrifugine study) [59] |
Methodologies to Reduce Expert Knowledge Dependency
Automated Workflows and Machine Learning Integration
The development of fully automated pharmacophore modeling workflows represents a significant advancement in reducing the expert knowledge barrier. Implement these approaches to minimize manual intervention:
- End-to-End Automated Workflows: Deploy the workflow exemplified by QPhAR-based automated model generation, which requires only a set of 15-50 ligands with known activity values and proceeds through dataset preparation, model generation, virtual screening, and hit ranking without manual intervention [24]. This workflow transforms the researcher's role from hands-on model builder to decision-maker evaluating automatically generated solutions.
- Diffusion Models for Pharmacophore Generation: Utilize cutting-edge generative approaches like PharmacoForge, a diffusion model that generates 3D pharmacophores conditioned on a protein pocket using E(3)-equivariant neural networks [57]. This method frames pharmacophore creation as a denoising diffusion probabilistic process that progressively refines random feature placements into coherent pharmacophores.
- Reinforcement Learning Applications: Implement PharmRL, a reinforcement learning method for automated pharmacophore generation that identifies potential pharmacophore features through CNN analysis of voxelized pocket representations [57]. Though it requires training with positive and negative examples for each protein system, it significantly speeds up generation compared to manual methods.
Hybrid and Structure-Based Automation Protocols
For scenarios with some structural information, these protocols balance automation with structural insights:
- Apo2ph4 Framework Implementation: Apply the Apo2ph4 fragment-based workflow that docks 1456 lead-like molecular fragments into a target pocket, filters fragments by docking energy (<2 kcal/mol), converts poses to pharmacophores, and generates a final model through clustering and scoring [57]. While requiring some manual checks, this method substantially reduces expert time investment.
- Hypogen Algorithm Application: Utilize the Hypogen algorithm in BioVia's Discovery Studio, which enumerates pharmacophore hypotheses from the most active compounds, removes hypotheses matching inactive compounds, and introduces perturbations to optimize the model [20]. This approach operates directly on pharmacophores without requiring underlying molecules for prediction.
- PHASE with 3D QSAR: Employ the PHASE algorithm which develops 3D-QSAR models using pharmacophore fields and PLS regression, creating predictive models that display favourable and unfavourable regions contributing to activity values [59] [20].
Table 2: Automated Pharmacophore Modeling Solutions Comparison
| Tool/Method | Automation Level | Input Requirements | Knowledge Reduction Mechanism |
|---|---|---|---|
| QPhAR Workflow | Fully automated | 15-50 compounds with activity data | Complete end-to-end automation with hit ranking [24] |
| PharmacoForge | Fully automated | Protein pocket structure | Diffusion model generation without manual feature selection [57] |
| Apo2ph4 | Semi-automated | Protein structure or coordinates | Fragment docking with automated pharmacophore assembly [57] |
| PharmRL | Semi-automated | Voxelized pocket representation + training examples | CNN-based feature identification with reinforcement learning [57] |
Experimental Protocols for Validation and Optimization
Validation Framework for Assessing Model Quality
Implement this comprehensive validation protocol to objectively evaluate pharmacophore model performance while minimizing subjective expert judgment:
- Statistical Validation Protocol: For 3D-QSAR pharmacophore models, calculate the following metrics: correlation coefficient (R²), cross-validated R² (Q²), standard deviation (SD), F-statistics, root mean square error (RMSE), and Pearson-R values [59]. Require R² > 0.9, SD < 0.35, and high F-statistics for model acceptance [59].
- Virtual Screening Assessment: Evaluate pharmacophore models using the Fβ-score and FSpecificity-score rather than traditional accuracy metrics, as these better reflect virtual screening objectives where identifying true positives while reducing false positives is paramount [24]. Calculate the FComposite-score for overall model comparison.
- Retrospective Screening Validation: Conduct retrospective screening using benchmarks like LIT-PCBA and DUD-E datasets to measure enrichment factors and assess the ability to identify active compounds in databases [57]. Compare performance against established methods through docking-based evaluation frameworks.
Implementation Workflow for Robust Pharmacophore Modeling
The following diagram illustrates an integrated workflow addressing data quality and expertise limitations through automation and validation checkpoints:
Integrated Workflow for Robust Pharmacophore Modeling
Table 3: Essential Resources for Advanced Pharmacophore Modeling
| Resource Category | Specific Tools/Software | Application Context | Key Function |
|---|---|---|---|
| Pharmacophore Modeling Suites | PHASE (Schrödinger) [59] [20] | Ligand-based 3D-QSAR | Pharmacophore perception and quantitative activity modeling |
| Catalyst/Hypogen (BioVia) [20] | Structure-based design | Automated pharmacophore hypothesis generation | |
| LigandScout [20] | Structure-based modeling | Automated pharmacophore creation from protein-ligand complexes | |
| Protein Structure Analysis | GRID [2] | Binding site characterization | Molecular interaction field calculation for feature identification |
| LUDI [2] | Structure-based design | Interaction site prediction using geometric rules | |
| ALPHAFOLD2 [2] | Homology modeling | Protein structure prediction when experimental structures unavailable | |
| Validation & Screening | LIT-PCBA benchmark [57] | Method validation | Standardized dataset for pharmacophore evaluation |
| DUD-E dataset [57] | Retrospective screening | Benchmark for virtual screening performance assessment | |
| Conformational Sampling | iConfGen [20] | Ligand preparation | 3D conformation generation for small molecules |
| ConFigureen [59] | Conformer generation | Energy minimization and diverse conformation creation |
The limitations of data quality dependence and expert knowledge requirements in pharmacophore modeling present significant but addressable challenges in virtual screening campaigns. Through the implementation of robust data assessment protocols, automated workflows like QPhAR, and advanced machine learning approaches such as diffusion models, researchers can substantially mitigate these constraints. The integration of quantitative pharmacophore activity relationships with fully automated refinement algorithms represents a paradigm shift from expert-driven to data-driven pharmacophore modeling. Future advancements will likely focus on increasing integration of deep learning architectures, improved handling of multi-conformer representations, and enhanced prediction of binding affinities directly from pharmacophore representations. By adopting these methodologies, researchers can build more reproducible, scalable, and effective pharmacophore models that maintain scientific rigor while reducing barriers to implementation in drug discovery pipelines.
In the context of computer-aided drug discovery, managing molecular flexibility is a cornerstone for building reliable and predictive pharmacophore models. A pharmacophore is defined as the "ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2]. The fundamental challenge lies in accurately representing the bioactive conformation of a ligand—the specific 3D geometry it adopts when bound to its target—when this state is often unknown beforehand. Ligand conformational sampling is the computational process that generates a set of plausible 3D structures for a molecule, aiming to encompass this bioactive conformation. The success of subsequent steps, including pharmacophore model creation, virtual screening, and molecular docking, is critically dependent on the quality and efficiency of this initial sampling [2] [7]. This guide details the core strategies, protocols, and emerging methodologies for managing molecular flexibility to support robust pharmacophore-based research.
Core Strategies for Conformational Sampling
Systematic Search Methods
Systematic search methods exhaustively explore a molecule's conformational space by varying its rotatable bonds. This is typically achieved by rotating bonds at fixed intervals (e.g., every 10, 30, or 60 degrees) and generating combinations of these torsion angles. While guaranteeing comprehensive coverage, this approach suffers from a combinatorial explosion for molecules with many rotatable bonds, making it computationally intensive.
To mitigate this, energy window filters are applied to retain only low-energy, physically realistic conformers. A key tool in this category is OMEGA, which robustly samples conformational space at high speed using a torsion-driving algorithm [60]. It produces diverse ensembles based on RMS deviation and strain energy, is exceptionally fast (approximately 0.08 seconds per molecule), and is highly effective at reproducing known bioactive conformations found in structural databases [60].
Deterministic Flexible Alignment
An alternative strategy is deterministic flexible alignment, which integrates conformational search directly into the pharmacophore elucidation process. The PharmaGist webserver employs this methodology [61]. Its algorithm treats one input ligand as a rigid pivot and flexibly aligns other target ligands onto it. The method explicitly handles ligand flexibility by dividing molecules into rigid groups connected by rotatable bonds. It generates transformations for each rigid group and reassembles them into new, aligned conformations of the target ligand, scoring them based on the overlap of pharmacophoric features like hydrogen bond donors/acceptors and hydrophobic groups [61]. A key advantage of this approach is its deterministic nature and efficiency, typically completing runs for up to 32 molecules in seconds to minutes. Furthermore, it is tolerant of outliers and multiple binding modes, as it can identify pharmacophores common to different subsets of the input ligands [61].
Stochastic and Simulation-Based Methods
Stochastic methods use random or probabilistic sampling to explore the conformational landscape, which can be more efficient for very flexible molecules. Monte Carlo (MC) methods, for example, generate new configurations through random changes to degrees of freedom (e.g., torsion angles, rigid-body rotations), accepting or rejecting them based on the Metropolis criterion at a given temperature [62].
Advanced MC algorithms for proteins incorporate specialized moves, such as Concerted Rotations with Variable Angles (CRA), which perform crankshaft-like motions on a protein backbone by perturbing a chain of five consecutive residues and then analytically closing the chain to maintain connectivity [62]. These methods can efficiently overcome energy barriers and sample disparate local minima, often converging faster than Molecular Dynamics (MD) for specific applications like absolute binding free energy calculations [62]. While MD is a more common sampling tool, MC's discrete moves allow for focused sampling on regions of interest, such as binding-site residues.
Emerging AI-Powered Approaches
Deep learning is revolutionizing conformational sampling and pharmacophore mapping. DiffPhore is a state-of-the-art, knowledge-guided diffusion model designed for "on-the-fly" 3D ligand-pharmacophore mapping [50] [63]. This framework uses a score-based diffusion model, parameterized by an SE(3)-equivariant graph neural network, to generate ligand conformations that maximally align with a given pharmacophore model.
The model is trained on large, high-quality datasets of 3D ligand-pharmacophore pairs (e.g., CpxPhoreSet from protein-ligand complexes and LigPhoreSet from diverse ligand conformations) [63]. It incorporates explicit pharmacophore-ligand matching knowledge, including type and directional alignment rules, to guide the conformation generation process. This allows it to outperform traditional pharmacophore tools and several advanced docking methods in predicting binding conformations and virtual screening [50].
Table 1: Key Parameters for Different Conformational Sampling Strategies.
| Strategy | Key Tunable Parameters | Typical Output Size | Computational Cost |
|---|---|---|---|
| Systematic Search | Torsion angle increment, Energy window threshold, Maximum number of conformers | Hundreds to thousands | Medium to High |
| Deterministic Alignment | Feature matching threshold, Scoring weights for feature types, Minimal number of features | Tens of aligned conformations | Low to Medium |
| Stochastic (MC) | Move step sizes (translation, rotation, torsion), Move frequencies, Simulation temperature | Thousands of snapshots | Medium |
| AI-Powered (DiffPhore) | Sampling steps, Noise schedule, Guidance scale from pharmacophore | A single optimized conformation per pharmacophore | Varies by model size |
Experimental Protocols for Pharmacophore Modeling
Protocol 1: Ligand-Based Pharmacophore Generation with Systematic Sampling
This protocol is used when a set of active ligands is known, but the 3D structure of the target protein is unavailable [64] [2].
- Ligand Preparation: Collect a set of known active compounds with diverse structures but a common mechanism of action. Prepare the 2D structures by adding hydrogen atoms, correcting formal charges, and generating possible tautomers and protonation states at biological pH (e.g., using tools like Epik [65]).
- Conformational Ensemble Generation: For each ligand in the set, generate a representative ensemble of low-energy conformations. Using OMEGA [60]:
- Input: Prepared 2D or 3D molecular structures.
- Parameters: Set the energy window (e.g., 10-15 kcal/mol) and the RMSD threshold for clustering (e.g., 0.5-1.0 Å) to ensure diversity.
- Output: A multi-conformer database for each ligand.
- Common Pharmacophore Identification: Use software like Discovery Studio [64] or PHASE [65] to identify shared pharmacophore features from the multi-conformer database.
- Align the conformers of all training set molecules.
- The algorithm identifies the spatial arrangement of chemical features (HBA, HBD, Hydrophobic, etc.) common to the active molecules.
- A quantitative model (e.g., using HypoGen [64]) can be generated if activity data is available, relating the pharmacophore hypothesis to biological potency.
- Model Validation: Validate the selected pharmacophore model (Hypo1) using a set of test molecules not included in the training set. Assess its ability to predict the activity of these test compounds and its robustness in virtual screening [64].
Protocol 2: Structure-Based Pharmacophore Generation from a Protein-Ligand Complex
This protocol is applicable when a high-resolution 3D structure of the target protein, often with a bound ligand, is available [2].
- Protein Structure Preparation: Obtain the structure from the PDB (e.g., PDB ID: 1T8I for Topoisomerase I [64]). Prepare the protein by adding hydrogen atoms, assigning correct protonation states to residues (especially Histidine), and optimizing hydrogen bonding networks.
- Binding Site Analysis: Define the ligand-binding site. This can be done manually based on the co-crystallized ligand's location or using automated tools like GRID or LUDI to identify energetically favorable interaction sites [2].
- Pharmacophore Feature Generation: Map the key interactions between the protein binding site and a bound ligand to define the pharmacophore features.
- Features are placed based on protein-ligand interactions: Hydrogen bond donors/acceptors from the ligand to protein residues, metal coordination sites, hydrophobic patches, and charged/ionic interactions.
- Exclusion volumes (XVOL) are added to represent the steric constraints of the protein backbone and side chains, preventing ligands from occupying forbidden space [2].
- Feature Selection and Refinement: The initial model may contain many features. Select the most critical ones for bioactivity by analyzing conserved interactions across multiple complex structures or by removing features that do not contribute significantly to binding energy [2].
Protocol 3: Virtual Screening with a Validated Pharmacophore Model
This protocol describes how to use a validated pharmacophore model for virtual screening to identify novel hit compounds [64] [7] [66].
- Database Preparation: Screen a large database of drug-like molecules (e.g., ZINC, containing over a million compounds [64]). Prepare the database by generating a conformational ensemble for each molecule and filtering based on drug-likeness (e.g., Lipinski's Rule of Five [64]).
- Pharmacophore-Based Screening: Use the pharmacophore model as a 3D query to search the prepared database.
- Post-Screening Filtration and Ranking: The top-ranking hits from the pharmacophore screen are subjected to further filtration.
- Molecular Docking and Dynamics: To further validate the hits, perform molecular docking (e.g., with Glide [65]) into the target's binding site to analyze precise binding interactions. Finally, run molecular dynamics (MD) simulations to confirm the stability of the ligand in the binding site over time [64].
Workflow Visualization
The following diagram illustrates the logical relationship and workflow between the different conformational sampling strategies and their application in pharmacophore modeling.
Conformational Sampling Strategies in Pharmacophore Modeling
The Scientist's Toolkit: Essential Reagents and Software
Table 2: Key Software Tools and Resources for Conformational Sampling and Pharmacophore Modeling.
| Tool/Resource Name | Type/Function | Key Features |
|---|---|---|
| OMEGA (OpenEye) [60] | Conformer Generator | High-speed, rule-based sampling; excellent reproduction of bioactive conformations; handles macrocycles. |
| PHASE (Schrödinger) [65] | Pharmacophore Modeling & Screening | Intuitive interface for ligand- and structure-based modeling; integrated with commercial compound libraries. |
| PharmaGist [61] | Pharmacophore Detection Webserver | Ligand-based, deterministic flexible alignment; fast and free; tolerant to multiple binding modes. |
| DiffPhore [50] [63] | AI-based Pharmacophore Mapping | Knowledge-guided diffusion model; state-of-the-art binding pose prediction and virtual screening. |
| MCPRO [62] | Monte Carlo Sampler | Optimized for biomolecules; includes specialized moves (e.g., CRA) for protein backbone/side chains. |
| Discovery Studio [64] | Modeling Suite | Includes HypoGen for 3D QSAR pharmacophore generation and validation from ligand sets. |
| ZINC Database [64] [50] | Compound Library | Publicly available database of commercially available, drug-like molecules for virtual screening. |
Effective management of molecular flexibility through robust conformational sampling is non-negotiable for building predictive pharmacophore models. The field offers a spectrum of strategies, from well-established systematic and stochastic methods to innovative deterministic alignment and cutting-edge AI. The choice of strategy depends on the available data (ligands vs. protein structure), the desired balance between comprehensiveness and speed, and the specific goals of the virtual screening campaign. By following the detailed protocols and leveraging the tools outlined in this guide, researchers can systematically address the challenge of flexibility, thereby increasing the likelihood of successfully identifying novel and potent therapeutic agents. The integration of AI methods like DiffPhore promises to further accelerate and enhance the accuracy of this critical process in drug discovery.
Accounting for Protein Flexibility and Induced-Fit Effects in Models
In structure-based drug discovery, the biomolecular target is often treated as a rigid entity. However, proteins are inherently dynamic systems that undergo a spectrum of conformational changes, from side-chain rotations to large-scale domain movements. Protein flexibility and induced-fit effects—where the binding site adapts to accommodate ligand binding—represent critical challenges in computational modeling, particularly in pharmacophore development [67] [68]. Ignoring these dynamic properties frequently leads to false negatives in virtual screening and poor prediction of ligand binding modes and affinities.
This technical guide provides comprehensive methodologies for incorporating protein flexibility and induced-fit effects into pharmacophore models, framed within the broader context of virtual screening research. We present explicit protocols, quantitative comparisons, and visualization tools to equip researchers with practical strategies for enhancing model accuracy in drug development campaigns.
Theoretical Foundation: The Necessity of Dynamics in Modeling
The Spectrum of Protein Flexibility
Protein flexibility manifests across multiple spatial and temporal scales, each requiring distinct computational approaches:
- Side-chain rotations: Rapid side-chain rearrangements, particularly of residues like tyrosine, phenylalanine, and arginine, can significantly alter binding site characteristics. These are often addressed through rotamer libraries in docking [67].
- Loop movements: Flexible binding site loops can open, close, or reorganize to facilitate ligand entry and binding, a common challenge in kinase and protease targets [67].
- Domain motions: Large-scale hinge movements and domain shifts dramatically reshape binding interfaces, requiring advanced sampling techniques [67].
- Intrinsically disordered regions (IDRs): Some proteins or regions lack stable 3D structure until binding partners are encountered, presenting particular challenges for traditional structure-based methods [67].
Induced-Fit Binding Mechanisms
The induced-fit model describes the reciprocal conformational adaptation between protein and ligand upon binding. This phenomenon violates the rigid "lock-and-key" paradigm and necessitates dynamic modeling approaches [69]. Key aspects include:
- Binding site reshaping: Ligand binding can alter binding site volume and topography by up to 40% in extreme cases [68].
- Interaction network reorganization: Hydrogen bonding patterns, π-π stacking, and hydrophobic contacts can reform during the binding process.
- Allosteric propagation: Conformational changes at the binding site can transmit to distal regions, potentially affecting protein function.
Methodological Approaches
Molecular Dynamics Simulations
Molecular dynamics (MD) simulations provide an atomistic, physics-based approach to sampling protein conformational space by numerically solving Newton's equations of motion for all atoms in the system.
Experimental Protocol: MD Simulation for Pharmacophore Feature Sampling
System Preparation
- Obtain protein structure from PDB or homology modeling
- Add missing hydrogen atoms and assign protonation states using tools like PROPKA
- Embed protein in explicit solvent box (TIP3P water model)
- Add physiological ion concentration (0.15M NaCl)
Equilibration Protocol
- Energy minimization: 5,000 steps steepest descent
- Solvent equilibration: 100ps NVT ensemble (298K)
- System equilibration: 100ps NPT ensemble (1atm, 298K)
Production Simulation
- Run unrestrained MD simulation for 100ns-1μs (timescale dependent on system)
- Apply periodic boundary conditions
- Use 2fs integration time step
- Maintain temperature with Langevin thermostat and pressure with Berendsen barostat
Trajectory Analysis for Pharmacophore Generation
- Cluster frames based on binding site RMSD using algorithms like GROMOS
- Extract representative structures from dominant clusters
- Calculate interaction frequencies for pharmacophore features
- Generate consensus pharmacophore model incorporating high-frequency interactions [17]
Table 1: MD Simulation Parameters for Flexibility Studies
| Parameter | Recommended Setting | Rationale |
|---|---|---|
| Force Field | CHARMM36, AMBERff19SB | Optimized for folded proteins |
| Water Model | TIP3P | Computational efficiency |
| Simulation Length | 100ns minimum | Adequate for side-chain and loop motions |
| Sampling Interval | 10-100ps | Balances storage and resolution |
| Binding Site RMSD Clustering | 1.0-2.0Å cutoff | Identifies significant conformational changes |
Ensemble-Based Docking and Pharmacophore Modeling
The ensemble docking approach utilizes multiple protein structures to account for flexibility, either from experimental structures or computational sampling.
Experimental Protocol: Ensemble Pharmacophore Generation
Ensemble Assembly
- Collect multiple experimental structures (apo/holo forms) from PDB
- Alternatively, generate conformational ensemble from MD trajectory
- Select structurally diverse representatives using RMSD-based clustering
Binding Site Analysis
- Align structures using binding site residues
- Identify conserved and variable regions
- Map interaction potentials for each ensemble member
Pharmacophore Model Generation
- Generate structure-based pharmacophores for each ensemble member
- Identify consensus features present across majority of structures
- Include flexible features with spatial tolerances based on observed variations [68]
Model Validation
- Test ability to retrieve known active compounds from decoys
- Validate against experimental activity data (IC50, Ki)
- Assess screening enrichment factors [70]
Figure 1: Ensemble Pharmacophore Workflow - from structural ensemble to consensus model
Advanced Sampling and Machine Learning Approaches
Recent advances integrate enhanced sampling with machine learning to efficiently capture flexibility.
Kinetic Network Models built from MD data identify metastable states and transition pathways, focusing pharmacophore development on thermodynamically relevant conformations [67].
AI-driven methods like AlphaFold3 and DiffPhore leverage deep learning to predict flexible binding modes. DiffPhore specifically uses a knowledge-guided diffusion framework for 3D ligand-pharmacophore mapping that incorporates flexibility through calibrated sampling [50].
Table 2: Comparison of Methods for Handling Protein Flexibility
| Method | Computational Cost | Timescales Accessible | Best Use Cases | Key Limitations |
|---|---|---|---|---|
| Molecular Dynamics | High (CPU/GPU-intensive) | Nanoseconds to microseconds | Atomistic detail, explicit solvent | Limited by simulation timescale |
| Ensemble Docking | Moderate | Static snapshots | Multiple experimental structures available | Discrete sampling of continuum |
| Machine Learning (DiffPhore) | Low (after training) | Training data-dependent | Large chemical spaces, quick screening | Dependent on training data quality |
| Monte Carlo Simulations | Moderate | Dependent on steps | Side-chain flexibility, local motions | Less accurate dynamics |
Case Studies and Experimental Validation
Liver X Receptor β (LXRβ) Flexibility Modeling
LXRβ exemplifies targets with high binding pocket flexibility, where different ligands assume distinct binding poses and interactions [68].
Experimental Protocol: Multi-Structure Pharmacophore Development
Data Curation
- Collect 8 LXRβ-ligand complex structures from PDB (e.g., 1UPV, 1PQC, 3IPA)
- Extract diverse ligands with measured activation data
Binding Pose Analysis
- Superimpose structures using Cα atoms of binding site
- Identify conserved anchor points (charge interaction with Arg319)
- Map variable interaction regions
Pharmacophore Generation
- Generate individual pharmacophores for each complex
- Identify common features: one cationic interaction, two hydrophobic features
- Define spatial tolerances based on observed variations (1.2-1.8Å)
- Validate model using test set of known LXRβ modulators [68]
Serotonin Transporter (SERT) Induced-Fit Modeling
MDMA ("ecstasy") binding to human serotonin transporter (hSERT) demonstrates pronounced induced-fit effects, with ligand mobility within the central binding site [69].
Experimental Protocol: Induced-Fit Monte Carlo Simulations
System Setup
- Obtain hSERT structures in different states (outward-open, occluded, inward-open)
- Generate conformational pathway through morphing
Ensemble Binding Space Docking
- Dock MDMA and analogs to multiple conformational states
- Identify low-energy binding modes across the ensemble
Pharmacophore Feature Extraction
- Identify conserved ionic interaction with Asp98
- Map edge-to-face π-π interactions with Tyr95
- Define spatial relationships to gating residues (Phe341, Tyr176, Phe335)
- Incorporate flexibility through conformationally-tolerant feature definitions [69]
Implementation Framework
Integrated Workflow for Flexible Pharmacophore Modeling
Figure 2: Method Selection Logic - integrating flexibility in pharmacophore modeling
Table 3: Computational Tools for Flexible Pharmacophore Modeling
| Tool Category | Specific Software/Resource | Key Functionality | Application Context |
|---|---|---|---|
| Molecular Dynamics | GROMACS, AMBER, NAMD | All-atom simulation with explicit solvent | High-accuracy flexibility sampling |
| Ensemble Generation | AlphaFold2/3, MODELLER | Protein structure prediction and modeling | When experimental structures are limited |
| Pharmacophore Modeling | LigandScout, MOE, Phase | Feature mapping and model building | Structure- and ligand-based pharmacophore development |
| Virtual Screening | ZINC, PubChem | Compound libraries for screening | Validation and application of pharmacophore models |
| Machine Learning | DiffPhore, PharmacoNet | AI-powered pharmacophore matching | Large-scale screening with flexibility considerations |
Incorporating protein flexibility and induced-fit effects is no longer optional for accurate pharmacophore modeling—it is essential for successful virtual screening campaigns. The methodologies presented herein, from MD simulations to ensemble-based approaches and emerging AI tools, provide researchers with a comprehensive toolkit for addressing dynamic protein-ligand interactions.
Future developments will likely focus on integrating machine learning more deeply into flexibility prediction, with methods like DiffPhore representing the vanguard of this approach [50]. Additionally, community-wide efforts to create standardized flexibility-annotated datasets will enable more rigorous benchmarking and method development.
As these computational techniques continue to mature, their integration into automated drug discovery pipelines will dramatically improve our ability to identify novel therapeutic compounds targeting highly flexible biological targets, ultimately accelerating the drug development process.
Balancing Model Specificity and Sensitivity to Minimize False Positives and Negatives
In virtual screening for drug discovery, the effectiveness of a pharmacophore model is determined by its ability to correctly identify active compounds (sensitivity) while rejecting inactive ones (specificity). This technical guide provides an in-depth examination of strategies to balance these competing demands, thereby minimizing both false positives and false negatives. Through structured methodologies, quantitative validation metrics, and integrated computational approaches, researchers can optimize pharmacophore models to enhance the efficiency and success rates of lead compound identification.
A pharmacophore is defined by the International Union of Pure and Applied Chemistry (IUPAC) as "the ensemble of steric and electronic features that is necessary to ensure the optimal supra-molecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2]. In practical terms, pharmacophore modeling abstracts molecular structures into essential chemical features—such as hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), hydrophobic areas (H), positively/negatively ionizable groups (PI/NI), and aromatic rings (AR)—required for biological activity [2] [39]. Virtual screening using these models enables rapid in silico assessment of large compound databases, significantly reducing the time and cost associated with experimental high-throughput screening [71] [2]. The core challenge lies in developing a model that is specific enough to avoid false positives (compounds incorrectly identified as active) yet sensitive enough to prevent false negatives (active compounds incorrectly rejected) [71]. This balance is critical for maximizing the identification of true leads while minimizing the experimental validation of non-promising compounds.
Core Concepts: Specificity, Sensitivity, and Their Trade-offs
In the context of pharmacophore modeling, sensitivity refers to the model's ability to correctly identify truly active compounds, measured as the proportion of actual actives successfully retrieved. High sensitivity reduces false negatives. Specificity, conversely, is the model's ability to correctly reject inactive compounds, measured as the proportion of inactives successfully excluded, thereby reducing false positives [71] [72].
The relationship between these metrics is often inverse; increasing a model's strictness to improve specificity (e.g., by adding more constraints or reducing feature tolerances) can inadvertently decrease its sensitivity by excluding some genuinely active compounds that lack non-essential features [71]. This trade-off is quantitatively assessed using enrichment factors and Receiver Operating Characteristic (ROC) curves, which visualize the true positive rate against the false positive rate at various classification thresholds [72] [73]. A model's quality is often summarized by the Area Under the Curve (AUC), where values of 0.71-0.8 indicate excellent performance and 0.51-0.7 indicate good performance [73]. The ultimate goal is to create a model that achieves optimal enrichment of active compounds within the top ranks of virtual screening results.
Methodological Framework for Balanced Pharmacophore Modeling
Model Generation Approaches
The foundation of a robust pharmacophore model lies in the selection of an appropriate generation strategy, which is primarily determined by available structural information.
Structure-Based Pharmacophore Modeling This approach is utilized when a high-resolution 3D structure of the target protein, often in complex with a ligand, is available (e.g., from X-ray crystallography or NMR) [2] [39]. The workflow involves:
- Protein Preparation: Critical evaluation and refinement of the protein structure, including protonation of residues, addition of hydrogen atoms, and correction of any structural errors [2] [72].
- Binding Site Detection and Analysis: Identification of the ligand-binding pocket using tools like GRID or LUDI, which generate molecular interaction fields or predict interaction sites based on geometric rules and statistical data from known structures [2].
- Feature Identification and Selection: Extraction of key pharmacophoric features from the protein-ligand interactions. This includes HBA, HBD, HYD, and ionizable features. The initial feature set is often refined by retaining only those interactions crucial for binding energy and biological activity to prevent over-constraining the model [2] [72].
Ligand-Based Pharmacophore Modeling When the 3D structure of the target is unavailable, this method constructs the model from a set of known active ligands [2] [39]. The process entails:
- Ligand Selection and Conformational Analysis: Curating a diverse set of active ligands with measured biological activity (e.g., IC50) and generating their low-energy conformers to account for flexibility [74] [39].
- Common Feature Identification: Using algorithms to identify the 3D spatial arrangement of chemical features common to all or most active ligands, which are presumed essential for target interaction [39].
- Model Hypothesis Generation: Creating multiple pharmacophore hypotheses, which are then ranked based on their ability to align the training set molecules and correlate with their known activity levels [39].
Advanced and Integrated Modeling Techniques
Dynamic Pharmacophore Modeling Incorporating protein flexibility through Molecular Dynamics (MD) simulations provides a more realistic representation of the binding site. By using snapshots from an MD trajectory, a dynamic pharmacophore model can be generated that accounts for conformational changes in the receptor, capturing transient interaction sites that might be missed in a single static structure [39] [13].
Pharmacophore-Informed Generative Models Emerging deep learning approaches, such as the TransPharmer model, integrate interpretable pharmacophore fingerprints with generative pre-training transformers for de novo molecule generation. This method excels at scaffold hopping—producing structurally distinct compounds that still match the essential pharmacophoric constraints— thereby enhancing the potential for identifying novel chemotypes with the desired bioactivity [23].
Quantitative Assessment of Model Performance
The performance of a pharmacophore model must be rigorously validated using quantitative metrics before application in large-scale virtual screening. The following table summarizes the key performance indicators and their interpretation:
Table 1: Key Quantitative Metrics for Pharmacophore Model Validation
| Metric | Calculation/Description | Optimal Range/Value | Interpretation |
|---|---|---|---|
| ROC-AUC [72] [73] | Area Under the Receiver Operating Characteristic curve. Plots True Positive Rate vs. False Positive Rate. | 0.71-0.8 (Excellent), >0.8 (Outstanding) | Overall ability to discriminate between active and inactive compounds. |
| Enrichment Factor (EF) [72] | (Number of actives found in top % of database) / (Total number of actives in database) | Context-dependent; higher values indicate better early enrichment. | Measures the model's efficiency in concentrating active compounds at the top of the screening list. |
| Sensitivity (Recall) [72] | True Positives / (True Positives + False Negatives) | Ideally close to 1.0 | Model's ability to correctly identify active compounds. |
| Specificity [72] | True Negatives / (True Negatives + False Positives) | Ideally close to 1.0 | Model's ability to correctly reject inactive compounds. |
| Pharmacophore Similarity (Spharma) [23] | Tanimoto coefficient of pharmacophoric fingerprints between generated molecules and target. | Higher values indicate closer adherence to target pharmacophore. | Used in generative models to assess output quality. |
| Feature Count Deviation (Dcount) [23] | Average difference in the number of individual pharmacophoric features between generated molecules and target. | Lower values indicate better control over feature generation. | Used in generative models to assess output quality. |
Validation is typically performed using a decoys set (e.g., from DUD-E database) containing known active compounds and presumed inactive molecules with similar physicochemical properties [73]. The model is used to screen this set, and the results are used to calculate the metrics above. For example, in a study on the sigma-1 receptor (σ1R), a structure-based pharmacophore model (5HK1–Ph.B) achieved a ROC-AUC above 0.8 and enrichment values exceeding 3, indicating its superior performance in discriminating actives from inactives [72].
Experimental Protocols for Key Validation Experiments
Protocol 1: ROC Curve Analysis and Model Validation
This protocol outlines the steps to validate a pharmacophore model's discriminatory power.
- Prepare Test Set: Compile a validation set containing known active compounds (e.g., from ChEMBL or BindingDB) and generate a set of decoy molecules with similar physicochemical properties but dissimilar 2D structures using a tool like the DUD-E server [73].
- Run Virtual Screening: Use the pharmacophore model as a query to screen the combined test set (actives + decoys). Software such as LigandScout, MOE, or Phase can be used for this step [72] [73].
- Rank and Analyze Results: Rank the screened compounds based on their pharmacophore fit score. For each compound, record whether it was classified as a "hit" by the model and its true activity status.
- Calculate Metrics and Plot ROC Curve:
Protocol 2: Integrated Virtual Screening Workflow
This protocol describes a comprehensive screening process that combines pharmacophore modeling with other computational techniques to improve success rates, as demonstrated in studies on SARS-CoV-2 PLpro and neuroblastoma [13] [73].
- Pharmacophore-Based Initial Screening: Apply the validated pharmacophore model to screen a large compound database (e.g., ZINC, CMNPD, or an in-house library). Save all poses that match the model, disregarding the docking program's native scoring [71].
- Pharmacophore Filtering: Filter the docked poses using a receptor-based pharmacophore model. This step rapidly eliminates poses that, despite a good docking score, are not chemically complementary to the binding site (e.g., they leave unpaired buried hydrogen bond donors/acceptors) [71].
- Molecular Docking: Subject the hits from the pharmacophore screen to molecular docking (using programs like GOLD, Glide, or AutoDock) to evaluate binding geometry and complementarity at the atomic level. Using multiple docking engines (e.g., both AutoDock and AutoDock Vina) with consensus scoring can mitigate biases inherent in a single program's scoring function [13].
- ADMET Prediction: Screen the top-ranked compounds for desirable Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profiles using in silico tools to prioritize compounds with a higher likelihood of drug-likeness [73].
- Molecular Dynamics (MD) Simulations: Perform MD simulations (e.g., 100 ns) on the protein-ligand complexes of the final top hits to assess the stability of the binding interaction, calculate binding free energies using MM-GBSA/PBSA methods, and confirm that key pharmacophore interactions are maintained dynamically [74] [13] [73].
Diagram 1: Integrated Virtual Screening Workflow. This flowchart outlines the multi-stage computational protocol for identifying lead compounds, combining pharmacophore screening, docking, and dynamics [71] [13] [73].
Case Studies in Optimizing Specificity and Sensitivity
Case Study: Sigma-1 Receptor (σ1R) Pharmacophore Optimization
A study on the sigma-1 receptor compared a new structure-based pharmacophore model (5HK1–Ph.B) against previous models and direct docking. The new model, generated from the crystal structure 5HK1, was manually refined by fusing two hydrophobic features. When validated against a dataset of over 25,000 experimentally tested compounds, 5HK1–Ph.B achieved a ROC-AUC above 0.8 and enrichment factors above 3. It outperformed direct docking, likely because the pharmacophore model's feature tolerances could better accommodate subtle binding site flexibility and more accurately capture the entropic penalty for desolvating polar atoms, aspects often poorly handled by rigid docking scoring functions. This case highlights how a carefully curated structure-based model can enhance both specificity and sensitivity compared to automated methods [72].
Case Study: Selective hCA IX Inhibitors from Natural Products
Researchers seeking selective inhibitors for carbonic anhydrase IX (hCA IX) developed a ligand-based pharmacophore model from known inhibitors with IC50 values below 50 nM. This model was used for virtual screening, prioritizing sensitivity to capture diverse potential hits. The initial 43 hits were then subjected to molecular docking to assess specificity by examining interactions with key residues (ZN301, HIS94, HIS96, HIS119). This two-step process ensured the model was sensitive enough to retrieve novel scaffolds from a natural product library, while docking provided a stringent check on binding mode specificity. The final four leads showed stable interactions in MD simulations and favorable binding free energies, demonstrating the success of this balanced approach [74].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 2: Key Software and Resources for Pharmacophore Modeling and Validation
| Category | Tool Name | Primary Function | Application Note |
|---|---|---|---|
| Pharmacophore Modeling | LigandScout [71] [73] | Structure- & ligand-based model generation, virtual screening. | Automatically generates models from protein-ligand complexes. |
| MOE [71] [39] | Comprehensive molecular modeling suite with pharmacophore module. | Used for visual inspection-based model generation and screening. | |
| Catalyst (Discovery Studio) [39] [72] | Create, validate, and screen 3D pharmacophore models. | Home of the HypoGen algorithm for ligand-based modeling. | |
| Virtual Screening & Docking | GOLD [71] | Molecular docking with genetic algorithm for pose generation. | Often used for pose generation prior to pharmacophore filtering. |
| Glide [71] | High-throughput molecular docking and scoring. | Provides "Pose-Filter" scripts for post-docking pharmacophore analysis. | |
| AutoDock/AutoDock Vina [13] | Open-source molecular docking suite. | Used in comparative docking for consensus scoring. | |
| Model Validation & Analysis | DUD-E Server [73] | Generates decoy sets for virtual screening validation. | Provides property-matched decoys for known actives. |
| ROC Curve Analysis [72] [73] | Standard method for evaluating classification model performance. | Implemented in various software (e.g., Discovery Studio, R scripts). | |
| Databases | ZINC [71] [73] | Public database of commercially available compounds. | Primary source for virtual screening compounds. |
| ChEMBL [73] | Database of bioactive molecules with drug-like properties. | Source for known active compounds for training/validation. | |
| RCSB PDB [2] | Repository for 3D structural data of proteins and nucleic acids. | Source for obtaining target protein structures. |
Achieving an optimal balance between specificity and sensitivity is a dynamic and critical process in pharmacophore-based virtual screening. This balance is not found through a single universal method but through a strategic, multi-stage workflow that leverages the strengths of various computational techniques. The integration of structure-based and ligand-based insights, rigorous quantitative validation using decoy sets and ROC analysis, and the sequential application of pharmacophore filtering, molecular docking, and dynamics simulations provide a powerful framework for refining models. By systematically applying these principles, researchers can construct pharmacophore models that robustly minimize both false positives and false negatives, thereby significantly accelerating the discovery of novel bioactive leads in drug development.
In modern computer-aided drug design, pharmacophore modeling serves as a crucial methodology for identifying the essential structural features responsible for biological activity. A pharmacophore is defined as an ensemble of steric and electronic features that ensure optimal supramolecular interactions with a specific biological target and to trigger its biological response [75]. While basic pharmacophore models identify favorable interaction points, refinement techniques significantly enhance their predictive power and screening accuracy. Two of the most critical refinement approaches involve the strategic incorporation of exclusion volumes and the precise adjustment of feature tolerances.
The fundamental limitation of basic pharmacophore feature hypotheses is that activity prediction is based purely on the presence and arrangement of pharmacophoric features, leaving steric effects unaccounted for [76]. This oversight can lead to false positives during virtual screening, as molecules that spatially fit the feature arrangement but sterically clash with the receptor cavity are incorrectly identified as potential hits. Recent advances in pharmacophore refinement have addressed this limitation through automated algorithms that enhance model selectivity and enrichment rates in virtual screening campaigns [76] [41].
This technical guide examines the core principles, methodologies, and implementation protocols for incorporating exclusion volumes and adjusting feature tolerances within pharmacophore models. Framed within the context of building effective pharmacophore models for virtual screening research, we provide detailed experimental frameworks and quantitative assessments to equip researchers with practical tools for enhancing their drug discovery pipelines.
Core Concepts and Definitions
Exclusion Volumes: Theoretical Basis
Exclusion volumes (also referred to as excluded volumes) represent regions in space that are sterically forbidden by the receptor structure [76]. These features penalize molecules that occupy spatial regions not occupied by active molecules, thereby incorporating critical steric constraints into the pharmacophore model. The HypoGenRefine algorithm in Catalyst, for instance, automates the addition of excluded volume features to pharmacophores based on the steric constraints observed from ligand information alone [76].
In practical terms, exclusion volumes are generated as spheres or complex shapes that represent the van der Waals surfaces of receptor atoms that line the binding pocket. When a screened molecule's atoms intersect with these excluded volumes, the model assigns penalty points, effectively downgrading that molecule's fit score. This approach has demonstrated significant improvements in virtual screening selectivity by reducing false positives that would otherwise sterically clash with the receptor [76] [41].
Feature Tolerances: Principles and Applications
Feature tolerances define the spatial flexibility permitted for each pharmacophoric feature during the matching process. These tolerances are typically implemented as radii around ideal feature points, allowing for minor deviations in feature positioning while still considering a match successful [75]. Proper adjustment of feature tolerances balances model specificity with necessary flexibility to account for legitimate conformational variations.
The refinement of feature tolerances often employs sophisticated algorithms such as the colored Iterative Closest Point (ICP) method, which extends beyond geometric alignment to incorporate pharmacophore "color" information (feature types) as an extra dimension in the point coordinate data [75]. This approach enables more intelligent matching that considers both spatial arrangement and feature type compatibility, with adjustable parameters for iteration counts, fitness values, and root-mean-square deviation (RMSD) thresholds to optimize pharmacophore alignment for specific screening contexts.
Methodologies and Implementation
Incorporating Exclusion Volumes: Protocol
The integration of exclusion volumes into pharmacophore models follows a structured protocol that can be implemented through various computational platforms:
Structure-Based Approach: When a protein structure is available, exclusion volumes can be derived directly from the binding pocket architecture. The protein structure is prepared through protonation and energy minimization, followed by binding site detection. The van der Waals surfaces of lining residues are converted into exclusion spheres, typically with radii matching corresponding atom types [76] [41].
Ligand-Based Approach: In the absence of structural receptor information, the HypoGenRefine algorithm can generate exclusion volumes automatically from a set of active ligands alone. This method identifies consensus steric constraints by analyzing the spatial occupancy of known active compounds, adding excluded volume features to regions not occupied by these actives [76].
Docking-Based Clustering Approach: Advanced methods like the O-LAP algorithm generate shape-focused pharmacophore models by filling the target protein cavity with flexibly docked active ligands. The overlapping ligand atoms are then clustered using pairwise distance-based graph clustering, effectively forming exclusion volumes that represent sterically constrained regions [41].
The following workflow diagram illustrates the exclusion volume incorporation process:
Adjusting Feature Tolerances: Protocol
The process for optimizing feature tolerances employs sophisticated point cloud alignment algorithms:
Pharmacophore Point Cloud Generation: Each pharmacophore feature is represented as a three-dimensional volume with a point cloud consisting of 1000 uniformly distributed points in a sphere. The radius of each pharmacophore cloud is defined according to the initial tolerance estimate, and different pharmacophore types are color-coded for distinct identification [75].
Global Registration with RANSAC Iteration: Two pharmacophore point clouds are processed to calculate a 33-dimensional Fast Point Feature Histogram (FPFH) vector that describes their geometric characteristics. The Random Sample Consensus (RANSAC) algorithm estimates optimal parameters while handling "noise" from regions with distinct differences between the clouds. This global registration process calculates a preliminary rigid rotation and transformation matrix, providing an initial alignment with an associated fitness score [75].
Colored ICP for Local Alignment: The colored Iterative Closest Point (ICP) algorithm extends standard ICP by incorporating pharmacophore feature type information ("color") as an additional dimension. This algorithm iteratively transforms matrices to find the minimum square distance between clouds while considering both geometric and feature type compatibility. Key parameters including iteration counts, fitness values, and RMSD thresholds can be user-adjusted to optimize alignment for specific applications [75].
Tolerance Refinement and Validation: Following alignment, non-overlapped pharmacophores are removed using a refinement algorithm that calculates Euclidean distances between corresponding points in the aligned clouds. Points without corresponding features within a threshold distance are considered irrelevant and removed. The resulting refined model, with optimized feature tolerances, is validated through fitness score calculation and enrichment testing [75].
The workflow for feature tolerance adjustment is detailed below:
Experimental Validation Protocols
Benchmark Compound Validation
To validate the efficiency of pharmacophore refinement algorithms, researchers should employ a molecular dataset consisting of active inhibitors and inactive decoys targeting specific protein receptors. The Directory of Useful Decoys (DUD-e) dataset serves as an excellent small molecule library for this purpose [75]. Recommended benchmark receptors include:
- Human immunodeficiency virus type 1 protease (HIVPR)
- Acetylcholinesterase (ACES)
- Cyclin-dependent kinase 2 (CDK2)
These proteins represent different families (protease, esterase, and kinase), providing diverse testing scenarios. During validation, all pharmacophore models should be screened on consistent platforms such as Pharmit, with comparative analysis against established refinement tools including LigandScout's "shared pharmacophores" feature and Schrödinger Phase's hypothesis alignment plugin [75].
Enrichment Factor Calculation
The enrichment factor (EF) quantifies a pharmacophore model's ability to identify true positive active inhibitors compared to random selection. Calculate EF using the formula:
Where:
Hitssampled= number of active compounds in the screened subsetNsampled= total number of compounds in the screened subsetHitstotal= number of active compounds in the entire databaseNtotal= total number of compounds in the entire database
Higher EF values indicate better pharmacophore model performance, with refined models typically demonstrating significant improvements over baseline approaches [75].
Statistical Analysis
Comprehensive validation should include comparison of molecular properties between screened active and decoy molecules, including:
- Molecular weight (MW)
- Octanol-water partition coefficient (logP)
- Total polar surface area (TPSA)
These properties can be calculated using tools like Open Babel, with statistical comparisons performed using two-tailed Student's t-test and F-test for equality of variances, setting a significance level of 0.05 (α = 0.05) [75].
Performance Metrics and Comparative Analysis
Quantitative Assessment of Refinement Techniques
Table 1: Performance Metrics of Pharmacophore Refinement Techniques in Virtual Screening
| Refinement Technique | Target Protein | Enrichment Factor (EF) | Screened Actives | Hit Rate | Key Parameters |
|---|---|---|---|---|---|
| HypoGenRefine with Excluded Volumes [76] | CDK2 | 25.4 | 38/55 | 69.1% | Two hydrogen-bond acceptor units, one aromatic hydrophobic unit, one aromatic ring unit, two excluded volumes |
| HypoGenRefine with Excluded Volumes [76] | Human DHFR | 19.8 | 29/55 | 52.7% | Three hydrogen-bond features, one hydrophobic feature, three excluded volumes |
| O-LAP Shape-Focused Models [41] | Neuraminidase (NEU) | 31.2 | 42/50 | 84.0% | Pairwise distance threshold: 1.5Å, atom-type specific radii, 50 docked ligands |
| O-LAP Shape-Focused Models [41] | A2A Adenosine Receptor (AA2AR) | 27.6 | 37/50 | 74.0% | Pairwise distance threshold: 2.0Å, atom-type specific radii, 50 docked ligands |
| ELIXIR-A with Tolerance Adjustment [75] | HIVPR | 23.1 | 31/55 | 56.4% | Colored ICP iterations: 50, fitness score: 0.85, RMSD threshold: 1.2Å |
| ELIXIR-A with Tolerance Adjustment [75] | ACES | 28.9 | 39/55 | 70.9% | Colored ICP iterations: 75, fitness score: 0.92, RMSD threshold: 0.9Å |
Comparative Analysis of Refinement Approaches
Table 2: Comparison of Pharmacophore Refinement Algorithms and Software Tools
| Software Tool | Algorithm Basis | Exclusion Volume Handling | Feature Tolerance Adjustment | Optimal Use Cases | Limitations |
|---|---|---|---|---|---|
| ELIXIR-A [75] | Python-based, Open3D library, point cloud registration | Manual coordinate input or imported models | Colored ICP with adjustable iterations, fitness, RMSD | Multi-target pharmacophore comparison, high-precision alignment | Requires pre-generated pharmacophore models |
| Catalyst/HypoGenRefine [76] | Ligand-based hypothesis generation with steric refinement | Automated addition from ligand information | Fixed tolerances based on training set | Ligand-based design when receptor structure unavailable | Limited to ligand-derived steric constraints |
| O-LAP [41] | C++/Qt5-based graph clustering of docked poses | Generated from clustered overlapping ligand atoms | Implied through centroid generation and clustering | Structure-based design with known active ligands | Requires successful docking of active ligands first |
| LigandScout [75] | Structure and ligand-based with shared pharmacophores | Derived from protein structure | Feature alignment with matching tolerance | Structure-based design with known protein-ligand complex | Limited cross-platform compatibility |
Research Reagent Solutions
Table 3: Essential Computational Tools for Pharmacophore Refinement Research
| Tool/Category | Specific Examples | Function in Research | Implementation Notes |
|---|---|---|---|
| Pharmacophore Modeling Software | ELIXIR-A [75], LigandScout [75], Catalyst [76], O-LAP [41] | Core platform for model development and refinement | ELIXIR-A is open-source; O-LAP released under GNU GPL v3.0 |
| Virtual Screening Platforms | Pharmit [75], ZINCPharmer [75] | Database screening with refined pharmacophore models | Pharmit supports import of refined models from ELIXIR-A |
| Benchmarking Datasets | DUD-E (Directory of Useful Decoys-Enhanced) [75], DUDE-Z [41] | Validation with active compounds and property-matched decoys | Reduces testing bias in method evaluation |
| Molecular Docking Tools | PLANTS [41], AutoDock, GOLD | Generation of input poses for shape-focused models | O-LAP uses top-ranked poses from flexible docking |
| Protein Preparation Tools | REDUCE [41], Maestro Protein Preparation Wizard | Structure protonation and optimization for structure-based approaches | Essential for accurate exclusion volume definition |
| Ligand Preparation Tools | LIGPREP [41], Open Babel | Generation of 3D conformers, tautomers, and partial charges | Open Babel calculates molecular properties for validation |
| Visualization Systems | VMD (Visual Molecular Dynamics) [75], SAMSON [77] | Visualization of refined pharmacophore models and screening results | ELIXIR-A outputs compatible with VMD |
The strategic incorporation of exclusion volumes and precise adjustment of feature tolerances represent fundamental advancements in pharmacophore modeling that significantly enhance virtual screening outcomes. Through the methodologies detailed in this technical guide—including structure- and ligand-based exclusion volume generation, point cloud registration for tolerance refinement, and rigorous validation protocols—researchers can develop highly selective pharmacophore models with improved enrichment factors and reduced false positive rates.
The quantitative performance metrics presented demonstrate that refined pharmacophore models consistently outperform basic feature-based approaches across diverse protein targets. As the field progresses, integration of these refinement techniques with emerging technologies like machine learning and molecular dynamics simulations will further accelerate the drug discovery process, enabling more efficient identification of novel therapeutic candidates through virtual screening workflows.
In the field of computer-aided drug design (CADD), pharmacophore modeling has emerged as a powerful and versatile technique for identifying potential therapeutic compounds. The International Union of Pure and Applied Chemistry (IUPAC) defines a pharmacophore as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2]. This abstract representation of molecular interactions enables researchers to move beyond specific atomic structures and focus on the essential chemical functionalities required for biological activity. Pharmacophore models represent these key functionalities as geometric entities in three-dimensional space—including hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), hydrophobic areas (H), positively and negatively ionizable groups (PI/NI), aromatic rings (AR), and exclusion volumes (XVOL) that define forbidden spaces [2]. The primary strength of this approach lies in its ability to facilitate "scaffold hopping"—identifying chemically distinct compounds that share the same interaction pattern with a biological target—making it invaluable for virtual screening of large compound databases in the search for novel drug candidates [2] [20].
The application of pharmacophore models spans multiple critical phases in drug discovery. They serve as essential queries for virtual screening of chemical databases, aid in lead optimization by highlighting key interactions, enable multi-target drug design, and support de novo drug design efforts [78] [2]. With the continuous growth of chemical databases containing billions of compounds, efficient virtual screening methods have become increasingly important for reducing the time and costs associated with experimental screening [7]. The relevance of these computational approaches has been particularly evident during recent health emergencies, where rapid identification of therapeutic agents is crucial [2]. This technical guide provides an in-depth overview of both commercial and open-source software tools for pharmacophore modeling, with a specific focus on their application in building robust pharmacophore models for virtual screening research.
Commercial Software Solutions
LigandScout
LigandScout, developed by Inte:ligand, represents a sophisticated platform for structure-based pharmacophore modeling and virtual screening. The software excels at automatically extracting detailed pharmacophore features from protein-ligand complex structures available in the Protein Data Bank (PDB) [79]. When a protein-ligand complex structure is imported, LigandScout performs automatic pharmacophore feature assignment, adds exclusion volumes to represent the steric constraints of the binding pocket, and can optionally include an exclusion volume coat (a second shell of exclusion volumes) for more precise shape definition [79]. The software incorporates advanced algorithms such as the Greedy 3-Point Search, which implements a matching-feature-pair maximizing search strategy that is both faster and more accurate than previous methods, particularly beneficial when screening ultra-large compound libraries [79].
A key innovation recently demonstrated with LigandScout is the FragmentScout workflow, which addresses a critical bottleneck in fragment-based drug discovery: evolving primary fragment hits with millimolar potency to lead candidates with micromolar potency [79]. This novel approach aggregates pharmacophore feature information from multiple experimental fragment poses obtained through XChem high-throughput crystallographic fragment screening. By generating a joint pharmacophore query for each binding site that combines features from all fragment poses, FragmentScout effectively mines the growing collection of XChem datasets to identify promising compounds [79]. In a recent application to SARS-CoV-2 NSP13 helicase, this workflow successfully identified 13 novel micromolar potent inhibitors validated in cellular antiviral and biophysical assays, demonstrating its practical utility in drug discovery campaigns against challenging targets [79].
Table 1: Key Features and Applications of LigandScout
| Feature Category | Specific Capabilities | Research Applications |
|---|---|---|
| Pharmacophore Creation | Automatic feature detection from PDB complexes, exclusion volume generation, joint pharmacophore queries from fragment data | Structure-based drug design, fragment-based lead discovery [79] |
| Virtual Screening | Greedy 3-Point Search algorithm, ultra-large library screening, LigandScout XT for high-performance screening | Virtual screening of internal corporate collections, Enamine REAL database screening [79] |
| Specialized Workflows | FragmentScout for fragment-to-lead optimization, protein-ligand interaction analysis | Targeting challenging drug targets like SARS-CoV-2 NSP13 helicase [79] |
BIOVIA Discovery Studio
BIOVIA Discovery Studio provides a comprehensive environment for pharmacophore-based drug design, featuring the established CATALYST Pharmacophore Modeling and Analysis toolset [78]. The software supports both structure-based and ligand-based pharmacophore modeling approaches, offering researchers flexibility depending on available data [78]. For structure-based design, researchers can automatically generate pharmacophores from receptor binding sites or receptor-ligand complexes, while ligand-based approaches enable pharmacophore elucidation from sets of active ligands without requiring structural information about the target protein [78]. A significant advantage of Discovery Studio is its integration with the PharmaDB database, which contains approximately 240,000 receptor-ligand pharmacophore models built from and validated using the scPDB (structural database of protein-ligand complexes), enabling efficient profiling of compounds against known targets [78] [80].
The 2025 release of BIOVIA Discovery Studio introduced several enhancements relevant to pharmacophore modeling and virtual screening. The PharmaDB Profiler protocol now stores function information in CSV files and includes total entries that map for the function as an additional property, improving data analysis capabilities [80]. The Interaction Pharmacophore Generation protocol has been enhanced to support producing a diverse set of pharmacophores in addition to top-scoring pharmacophores, giving researchers more options for virtual screening queries [80]. Furthermore, improvements in the Prepare Protein protocol now allow it to handle inputs with more than 99,999 atoms, including hydrogen atoms added by the protocol, facilitating work with large complex systems [80].
Table 2: Key Features and Applications of BIOVIA Discovery Studio
| Feature Category | Specific Capabilities | Research Applications |
|---|---|---|
| Pharmacophore Modeling | CATALYST hypothesis generation, structure-based and ligand-based approaches, ensemble pharmacophores for diverse compound sets | Multi-target drug design, activity profiling, de novo drug design [78] |
| Virtual Screening | PharmaDB database (~240,000 models), 3D conformation database searching, off-target activity exploration | Drug repurposing, toxicity prediction, virtual screening campaigns [78] [80] |
| Library Design | Combinatorial library enumeration, ionization states and tautomers, physicochemical property calculation | Lead optimization, library design with Lipinski/Véber rule filtering [78] |
Open-Source Tools and Emerging Methodologies
While commercial solutions offer comprehensive features, several open-source and academic methodologies have emerged that provide powerful alternatives for pharmacophore-based drug discovery. Although the search results do not explicitly name specific open-source software, they reference several methodological approaches that are commonly implemented in open-source platforms. The QPhAR (Quantitative Pharmacophore Activity Relationship) methodology represents a significant innovation in the field, enabling the construction of quantitative pharmacophore models that can predict biological activity based purely on pharmacophoric representations [24] [20]. This approach offers distinct advantages over traditional QSAR methods by abstracting molecular interactions and reducing bias toward overrepresented functional groups in small datasets [20].
The integration of machine learning with pharmacophore-based screening represents another emerging trend that accelerates virtual screening procedures. Recent studies have demonstrated that machine learning models can predict docking scores without time-consuming molecular docking procedures, achieving 1000 times faster binding energy predictions than classical docking-based screening [7]. These models learn from docking results, allowing researchers to choose their preferred docking software while dramatically accelerating the screening of ultra-large chemical libraries. Ensemble models that combine multiple types of molecular fingerprints and descriptors further reduce prediction errors and enable highly precise docking score values for target proteins [7].
Another significant advancement is the development of fully automated end-to-end pharmacophore modeling workflows that can derive quality-optimized pharmacophores from input datasets with minimal human intervention [24]. These workflows leverage SAR information extracted from validated QPhAR models to automatically select features that drive pharmacophore model quality, outperforming manually curated models based on shared feature pharmacophore generation from highly active compounds [24]. Such automated systems are particularly valuable for analyzing complex data patterns that may be non-obvious to human researchers, presenting distilled insights to support expert decision-making.
Experimental Protocols and Methodologies
Structure-Based Pharmacophore Modeling Protocol
Structure-based pharmacophore modeling relies on the three-dimensional structure of a macromolecular target to identify key interaction points. The following protocol outlines the standard workflow for generating structure-based pharmacophores using commercial software tools, based on methodologies successfully applied in recent research [79] [40]:
Protein Structure Preparation: Obtain the 3D structure of the target protein from the Protein Data Bank (PDB). Critically evaluate the structure quality, addressing factors such as residue protonation states, positions of hydrogen atoms (typically absent in X-ray structures), presence of non-protein groups, and any missing residues or atoms. Software-specific preparation wizards can automate many of these steps while allowing manual inspection and adjustment [2].
Binding Site Detection and Characterization: Identify the ligand-binding site through analysis of known protein-ligand complexes or using computational binding site detection tools. Programs like GRID and LUDI can predict potential interaction sites by sampling protein regions with functional groups to identify energetically favorable interaction points or by applying geometric rules derived from non-bonded contacts in experimental structures [2].
Pharmacophore Feature Generation: Import the protein-ligand complex or prepared protein structure into the pharmacophore modeling software. For complexes with bound ligands, the software will automatically identify interaction features between the ligand and protein residues. For apo structures, the software will calculate all possible interaction points within the binding site. The initial model typically contains numerous features that require refinement [2].
Feature Selection and Model Refinement: Select only the features essential for ligand bioactivity to create a selective pharmacophore hypothesis. This can be achieved by removing features that don't strongly contribute to binding energy, identifying conserved interactions across multiple protein-ligand structures, incorporating spatial constraints from receptor information, or preserving residues with key functions indicated by sequence alignments or variation analysis [2].
Exclusion Volume Assignment: Add exclusion volumes to represent the shape and steric constraints of the binding pocket. These volumes define forbidden areas where ligand atoms should not be positioned, improving the selectivity of the pharmacophore model during virtual screening [2].
Model Validation: Validate the pharmacophore model using known active compounds and decoy molecules. Calculate enrichment factors and area under the ROC curve (AUC) to quantify the model's ability to distinguish active from inactive compounds. A model with an AUC value of 0.98 and early enrichment factor (EF1%) of 10.0, as demonstrated in a recent XIAP inhibitor study, indicates excellent predictive capability [40].
Diagram 1: Structure-based pharmacophore modeling workflow.
Ligand-Based Pharmacophore Modeling Protocol
When the 3D structure of the target protein is unavailable, ligand-based pharmacophore modeling provides an alternative approach using the structural and chemical features of known active ligands. The following protocol is adapted from successful implementations in recent research [24] [7] [20]:
Ligand Dataset Curation: Compile a set of known active compounds with associated biological activity data (e.g., IC₅₀ or Kᵢ values). Ideally, select 15-50 compounds representing diverse chemical scaffolds and a range of potency values. Include both highly active and moderately active compounds, as weaker actives contain important information for pharmacophore modeling [24].
Conformational Analysis and Generation: Generate representative 3D conformations for each ligand in the dataset. Use conformer generation algorithms that efficiently explore the conformational space while maintaining computational feasibility. Typically, generate 20-50 conformers per compound to ensure coverage of potential bioactive conformations [20].
Pharmacophore Hypothesis Generation: Align the ligand conformations and identify common chemical features and their spatial relationships. Software tools use various algorithms to identify potential pharmacophore hypotheses that explain the observed activity across the compound set. In the Hypogen algorithm, for example, hypotheses are generated from the most active compounds and must fit a minimum subset of remaining active compounds [20].
Hypothesis Validation and Selection: Evaluate generated hypotheses using statistical measures and their ability to distinguish active from inactive compounds. Use methods such as cost function analysis, correlation coefficients between experimental and predicted activities, and receiver operating characteristic (ROC) curves. Select the hypothesis that best explains the structure-activity relationship within the dataset [20].
Database Searching and Virtual Screening: Apply the validated pharmacophore model as a search query against compound databases. The screening process identifies molecules that match the spatial arrangement of chemical features defined in the pharmacophore model. For large database screening, consider pre-filtering by molecular properties or using efficient screening algorithms like the Greedy 3-Point Search implemented in LigandScout [79] [7].
Hit Evaluation and Experimental Validation: Select top-ranking compounds from the virtual screening results for further analysis. Evaluate these hits using molecular docking, assess their drug-like properties, and ultimately proceed with experimental testing to validate the predicted activity [7].
Diagram 2: Ligand-based pharmacophore modeling workflow.
Integrated Virtual Screening Protocol
Combining pharmacophore-based screening with other computational methods enhances the efficiency and success rate of virtual screening campaigns. The following integrated protocol has been successfully applied in multiple recent studies [13] [7] [81]:
Initial Pharmacophore-Based Screening: Use a validated pharmacophore model (structure-based or ligand-based) to screen large compound databases. This step rapidly reduces the chemical space to compounds matching the essential interaction pattern required for target binding.
Molecular Docking of Pharmacophore Hits: Subject the compounds identified through pharmacophore screening to molecular docking studies. Use multiple docking programs or consensus docking approaches to mitigate limitations of individual docking algorithms. For example, in a study searching for SARS-CoV-2 PLpro inhibitors, comparative molecular docking using both AutoDock and AutoDock Vina helped identify true hits through consensus scoring [13].
Binding Mode Analysis and Hit Selection: Analyze the binding poses of top-ranked docking compounds to ensure they form key interactions with the target protein. Select compounds that not only show favorable docking scores but also present logical interaction patterns with functionally important residues in the binding site.
Molecular Dynamics Simulations: Perform molecular dynamics (MD) simulations on the top hits to evaluate the stability of protein-ligand complexes and quantify binding free energies. MD simulations for 100-200 nanoseconds can provide insights into conformational flexibility, interaction persistence, and the dynamic behavior of the complex [13] [80].
ADMET Profiling: Evaluate the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of selected hits using predictive computational models. Filter out compounds with unfavorable pharmacokinetic or toxicity profiles before proceeding to experimental validation.
Experimental Validation: Synthesize or procure the top virtual screening hits for in vitro biological testing. Begin with biochemical assays to confirm target engagement and functional activity, followed by cell-based assays to evaluate efficacy in more physiologically relevant systems.
Research Reagent Solutions
Table 3: Essential Computational Resources for Pharmacophore Modeling and Virtual Screening
| Resource Category | Specific Tools/Databases | Application in Research |
|---|---|---|
| Structural Databases | RCSB Protein Data Bank (PDB), scPDB | Source of protein structures for structure-based modeling; scPDB provides curated binding site annotations [2] [80] |
| Compound Libraries | ZINC database, Enamine REAL, ChEMBL | ZINC contains 230+ million purchasable compounds; REAL offers ultra-large screening collections; ChEMBL provides bioactivity data [7] [40] |
| Validation Resources | DUDe (Database of Useful Decoys), DEKOIS | Provide decoy molecules for pharmacophore model validation and benchmarking [40] |
| Computational Infrastructure | High-performance computing (HPC) clusters, GPU acceleration | Essential for screening large databases and running molecular dynamics simulations [80] [7] |
Pharmacophore modeling represents a powerful approach in the modern drug discovery toolkit, effectively bridging the gap between structural biology and medicinal chemistry. Commercial software solutions like LigandScout and BIOVIA Discovery Studio provide robust, feature-rich environments for both structure-based and ligand-based pharmacophore modeling, offering sophisticated algorithms for feature detection, model validation, and virtual screening at scale [79] [78]. The continuous development of these platforms, evidenced by the recent 2025 release of Discovery Studio with enhanced pharmacophore diversity and screening capabilities, ensures they remain at the forefront of computational drug discovery methodology [80].
Emerging trends in the field point toward increased automation, machine learning integration, and quantitative pharmacophore modeling approaches that enhance the predictive power and efficiency of virtual screening campaigns [24] [7] [20]. The successful application of these methodologies across diverse target classes—from viral proteins like SARS-CoV-2 NSP13 helicase to cancer targets like XIAP and enzyme targets like HPPD—demonstrates their versatility and impact in accelerating drug discovery [79] [13] [40]. As these computational approaches continue to evolve, integrating more sophisticated AI and machine learning capabilities, they promise to further reduce the time and cost associated with identifying novel therapeutic agents, ultimately contributing to more efficient drug development pipelines across a broad spectrum of disease areas.
Robust Validation and Performance Benchmarking of Pharmacophore Models
The development of a pharmacophore model is a critical step in structure-based drug design, providing an abstract representation of the steric and electronic features necessary for molecular recognition by a biological target. However, the predictive power and reliability of any pharmacophore model depend entirely on the rigor of its validation. Internal validation methods serve as the first and most crucial line of assessment, ensuring that the model possesses genuine structure-activity relationship information rather than chance correlation. Without proper validation, pharmacophore models may yield misleading results in virtual screening campaigns, wasting valuable computational and experimental resources.
This technical guide focuses on two fundamental pillars of internal validation for pharmacophore modeling: Leave-One-Out Cross-Validation (LOO CV) and essential statistical metrics. These methods collectively provide researchers with a robust framework for quantifying model quality, assessing predictive performance, and establishing confidence in the model's ability to identify novel bioactive compounds. When implemented within a comprehensive validation strategy that may include external test sets and decoy-based assessments, these internal validation techniques form the foundation for trustworthy pharmacophore models that can effectively prioritize compounds for experimental testing.
Core Statistical Metrics for Model Assessment
A suite of statistical metrics is employed to quantitatively evaluate the quality and predictive power of pharmacophore models. These metrics assess different aspects of model performance, from goodness-of-fit to predictive accuracy and statistical significance.
Table 1: Key Statistical Metrics for Pharmacophore Model Validation
| Metric | Formula/Calculation | Optimal Range | Interpretation |
|---|---|---|---|
| Correlation Coefficient (R²) | R² = 1 - (SS₍res₎/SS₍tot₎) | > 0.8 | Measures how well the model explains variance in training data; higher values indicate better fit [82] [83]. |
| Cross-Validated Correlation Coefficient (Q²) | Q² = 1 - (PRESS/SS₍tot₎) | > 0.5 | Indicates predictive ability via cross-validation; higher values suggest robust predictions [83] [36]. |
| Root Mean Square Error (RMSE) | RMSE = √(Σ(Ŷᵢ - Yᵢ)²/n) | Lower values better | Measures average difference between predicted and experimental activities [82]. |
| Fisher Test (F Value) | F = (SS₍reg₎/p)/(SS₍res₎/(n-p-1)) | Higher values better | Assesses overall statistical significance of the model [83]. |
| Cost Difference | ΔCost = Null Cost - Total Cost | > 60 bits | Large difference suggests a high probability (>90%) of representing a true correlation [82]. |
The configuration cost is another critical parameter that should be monitored during model generation. This cost measures the entropy of the hypothesis space and should remain below 17 bits to indicate that the model was not generated from an excessively flexible set of training compounds [82]. Additionally, the goodness of hit score (GH) integrates multiple assessment parameters to evaluate model performance in database screening, with scores closer to 1.0 (maximum) indicating excellent ability to separate active from inactive compounds [82].
Leave-One-Out Cross-Validation: Protocol and Implementation
Leave-One-Out Cross-Validation (LOO CV) is a robust resampling technique used to assess the predictive capability of a pharmacophore model without requiring an external test set. This method is particularly valuable in early drug discovery stages where the number of known active compounds may be limited.
Theoretical Foundation and Algorithm
LOO CV operates through an iterative process where each compound in the training set is systematically omitted and its activity is predicted using a model built from the remaining compounds. The process begins with a training set of N compounds with known biological activities (e.g., IC₅₀ values). For each iteration i (where i = 1 to N):
- Compound i is temporarily removed from the training set
- A new pharmacophore model is generated using the remaining N-1 compounds
- The generated model predicts the activity of the omitted compound i
- The predicted activity is compared to the experimental value
This cycle repeats until every compound has been omitted exactly once [36]. The complete workflow ensures that each compound serves as both a training and test instance, maximizing the use of limited data while providing a rigorous assessment of predictive performance.
Implementation Workflow
The following diagram illustrates the systematic LOO CV process for pharmacophore model validation:
Interpretation of Results
The primary output of LOO CV is the cross-validated correlation coefficient Q², which is calculated as:
Q² = 1 - (PRESS / SS₍total₎)
Where PRESS is the Predictive Residual Sum of Squares (Σ(yᵢ - ŷᵢ)²) and SS₍total₎ is the total sum of squares (Σ(yᵢ - ȳ)²) [83]. A Q² value > 0.5 generally indicates a model with good predictive ability, while Q² > 0.7 suggests excellent predictive power [83]. The difference between R² and Q² is also informative; a small difference (Δ < 0.3) suggests the model is not overfit to the training data.
Complementary Internal Validation Methods
While LOO CV provides essential information about predictive performance, comprehensive internal validation requires additional methods to address different aspects of model robustness and statistical significance.
Fischer Randomization Test
The Fischer randomization test, also known as the randomization test or Y-scrambling, assesses the probability that the model emerged by chance rather than representing a true structure-activity relationship [82] [36]. The test involves:
- Randomly shuffling the activity values among the training set compounds
- Generating new pharmacophore models using the scrambled data
- Repeating this process numerous times (typically 19-99 iterations)
- Comparing the original model's cost and correlation to those from randomized datasets
A successful test demonstrates that the original model has significantly better statistical metrics (higher R², lower costs) than models built from randomized data. For a 95% confidence level, none of the 19 randomized datasets should produce a model with comparable or better cost values [82].
Test Set Validation
Although sometimes categorized as external validation, test set validation using compounds withheld from model generation provides crucial evidence of predictive ability [82] [36]. The protocol involves:
- Dividing available compounds into training (typically 70-80%) and test (20-30%) sets prior to model generation
- Ensuring both sets span similar activity ranges and structural diversity
- Generating the model exclusively from the training set
- Predicting activities of test set compounds and calculating predictive R² (R²₍pred₎)
A high R²₍pred₎ value (>0.6) indicates the model can accurately predict activities of compounds not used in its construction [83]. The test set method is particularly valuable for estimating how the model will perform when screening truly novel compounds.
Table 2: Comparison of Internal Validation Methods
| Method | Primary Function | Key Outputs | Advantages | Limitations |
|---|---|---|---|---|
| LOO CV | Predictive ability assessment | Q², RMSE of prediction | Maximizes training data usage, no separate test set needed | Can overestimate performance for structurally similar compounds |
| Fischer Randomization | Chance correlation assessment | Statistical significance (p-value) | Directly tests hypothesis of random correlation | Requires multiple model generations, computationally intensive |
| Test Set Validation | External predictive ability | R²₍pred₎, RMSE of test set | Most realistic assessment of predictive performance | Requires withholding compounds, challenging with small datasets |
| Cost Analysis | Model significance evaluation | Total cost, cost difference | Provides probabilistic interpretation of model quality | HypoGen-specific, requires understanding of cost calculation |
Experimental Protocol: Implementing a Comprehensive Validation Strategy
This section provides a step-by-step protocol for implementing internal validation methods based on established practices in pharmacophore modeling research [82] [83] [36].
Preliminary Data Preparation
- Compound Collection: Gather a structurally diverse set of compounds with consistent biological activity data (e.g., IC₅₀ values from the same assay protocol). The activity range should ideally span at least 4 orders of magnitude [82].
- Chemical Feature Identification: Perform feature mapping to identify relevant pharmacophore features (hydrogen bond donors/acceptors, hydrophobic regions, aromatic rings, etc.) present in active compounds [36].
- Conformational Generation: Generate representative conformational ensembles for each compound using poling algorithms to ensure coverage of accessible conformational space [36].
Model Generation and Validation Workflow
- Training-Test Set Division: Randomly divide compounds into training (80%) and test (20%) sets, ensuring both sets contain representative active and inactive compounds and span similar activity ranges [83].
- Hypothesis Generation: Use the HypoGen algorithm or equivalent to generate multiple pharmacophore hypotheses from the training set [82].
- Initial Model Selection: Rank hypotheses based on total cost, correlation coefficient (R²), and RMSD values [82].
- LOO CV Implementation: Perform Leave-One-Out Cross-Validation on the top-ranked model(s) using the protocol described in Section 3.
- Fischer Randomization: Execute Fischer randomization at 95% confidence level with 19 random spreadsheets to verify model significance [82].
- Test Set Prediction: Use the final model to predict activities of the test set compounds and calculate predictive R² [83].
Table 3: Essential Tools and Resources for Pharmacophore Modeling and Validation
| Tool/Resource | Type | Primary Function | Application in Validation |
|---|---|---|---|
| Discovery Studio (DS) | Commercial Software | Comprehensive drug discovery platform | HypoGen algorithm for model generation; cost calculation [82] [36] |
| Schrödinger Suite | Commercial Software | Molecular modeling and drug design | Phase module for 3D-QSAR pharmacophore modeling [83] |
| R Statistical Computing | Open-source Environment | Statistical analysis and modeling | Calculation of validation metrics; custom analysis scripts [84] |
| Galaxy Workflow System | Open-source Platform | Reproducible computational analysis | GCAC pipeline for predictive model building and validation [84] |
| Decoy Sets (DUD-E, MUBD) | Benchmarking Databases | Validation with known actives and decoys | Goodness of Hit (GH) score calculation [85] [86] |
| Python/RDKit | Open-source Cheminformatics | Molecular descriptor calculation | Preprocessing and feature calculation for model building [87] |
Robust internal validation is not merely an optional step in pharmacophore modeling but an essential component that determines the real-world utility of the resulting models. Leave-One-Out Cross-Validation provides a rigorous assessment of predictive performance while making efficient use of typically limited training data. When complemented by statistical metrics such as R², Q², and cost analysis, along with specialized tests like Fischer randomization, researchers can develop pharmacophore models with verified predictive power and statistical significance.
The implementation of these validation methods within a structured workflow ensures that pharmacophore models generated for virtual screening will have the highest probability of identifying novel bioactive compounds. As the field advances with new technologies such as deep geometric reinforcement learning for pharmacophore elucidation [87] and maximal unbiased benchmarking sets [86], the fundamental principles of internal validation remain cornerstone to building trustworthy computational models that can effectively guide experimental efforts in drug discovery.
In the discipline of computer-aided drug discovery, a pharmacophore model serves as an abstract representation of the steric and electronic features essential for a molecule to interact with a biological target and trigger a pharmacological response. While the construction of a robust model is a critical first step, the true assessment of its utility for virtual screening lies in a rigorous validation process. External validation, which involves testing a model's predictive power on a completely separate, independent set of compounds not used during model generation, represents the gold standard for evaluating its real-world applicability and capacity for scaffold hopping. This process moves beyond internal validation metrics, providing a realistic estimate of a model's performance in prospective screening campaigns and ensuring that the model captures the fundamental principles of molecular recognition rather than merely memorizing the training data.
The critical importance of external validation is underscored by its role in mitigating overfitting. A model may appear excellent when tested on its training data but fail catastrophically when confronted with novel chemical structures. By using an independent test set, researchers can obtain an unbiased estimate of the model's generalization ability. Furthermore, for pharmacophore models intended to identify new lead compounds, a successful external validation on a diverse compound set demonstrates a potential for "scaffold hopping"—the ability to identify structurally distinct compounds that share the same essential pharmacophoric features. This guide provides a technical deep-dive into the methodologies for designing independent test sets, executing external validation, and interpreting the results within the context of building a reliable pharmacophore model for virtual screening.
Designing a Rigorous Independent Test Set
The foundation of any meaningful external validation is the careful construction of the independent test set. The composition of this set directly influences the scope and reliability of the validation conclusions.
Sourcing and Curation of Active and Inactive Compounds
A robust independent test set should comprise both active and inactive compounds. Actives are molecules with confirmed biological activity against the target of interest, typically with half-maximal inhibitory concentration (IC~50~) or inhibition constant (K~i~) values below a defined threshold (e.g., < 1 µM). These active compounds should be sourced from scientific literature or public databases such as ChEMBL [20] and must be entirely distinct from those used in the pharmacophore generation and training phases. To challenge the model's specificity, the test set must also include inactive compounds or "decoys"—molecules that are drug-like but presumed or confirmed to be inactive against the target. Databases like the Directory of Useful Decoys, Enhanced (DUD-E) are specifically designed for this purpose, providing pharmaceutically relevant decoys that are structurally similar to actives but topologically different to avoid true activity [30] [54]. Before validation, all compounds, both active and inactive, must undergo standard preparation steps including removal of salts, neutralization of charges, and generation of plausible 3D conformations.
Ensuring Chemical Diversity and Applicability Domain
To avoid bias and ensure the model is tested across a broad chemical space, the test set must encompass significant structural diversity. This can be achieved by clustering compounds based on molecular fingerprints (e.g., ECFP4) and selecting representatives from different clusters. The concept of the Applicability Domain (AD) is crucial; it defines the chemical space area where the model's predictions are considered reliable. A model should not be expected to perform well on compounds outside its AD. In quantitative studies, the AD can be defined using methods like Euclidean distance or PCA-based boundaries in descriptor space to identify whether new compounds fall within the domain of the model's training data [88] [89].
Table 1: Key Components of an Independent Test Set for External Validation
| Component | Description | Source Examples | Purpose in Validation |
|---|---|---|---|
| Active Compounds | Molecules with confirmed potency against the target. | ChEMBL, Published literature [20] | Tests model's sensitivity (ability to identify true actives). |
| Inactive/Decoy Compounds | Drug-like molecules with no expected activity. | DUD-E database [30] [54] | Tests model's specificity (ability to reject inactives). |
| Diverse Scaffolds | Actives with varied core structures/Bemis-Murcko scaffolds. | Chemical database clustering [88] | Assesses potential for scaffold hopping. |
| Applicability Domain | Defined chemical space for reliable prediction. | Euclidean distance, PCA [88] | Establishes boundaries for model's reliable use. |
Quantitative Metrics for Assessing Predictive Power
Once the independent test set is screened against the pharmacophore model, a set of quantitative metrics is used to evaluate its performance. These metrics are derived from the classification of test compounds into true positives (TP), false positives (FP), true negatives (TN), and false negatives (FN).
Primary Classification Metrics
The most straightforward metrics include Sensitivity (True Positive Rate) and Specificity (True Negative Rate). Sensitivity, calculated as TP/(TP+FN), measures the model's ability to correctly identify active compounds. Specificity, calculated as TN/(TN+FP), measures its ability to correctly reject inactive compounds [30]. A good model should simultaneously exhibit high sensitivity and high specificity. Another critical metric is the Enrichment Factor (EF), which quantifies how much more likely you are to find active compounds at a given top fraction of the screened database compared to a random selection. For example, EF~1%~ is calculated as (Number of actives in top 1% / Total number of actives) / 0.01. A model is generally considered reliable if it has an EF value exceeding 2 [54].
Comprehensive Performance Indicators
The Receiver Operating Characteristic (ROC) curve, which plots the True Positive Rate against the False Positive Rate at various classification thresholds, provides a visual representation of model performance. The Area Under the ROC Curve (AUC) is a single, widely-used metric to summarize this performance; an AUC of 1.0 represents a perfect model, while 0.5 represents a random model. A model with an AUC greater than 0.7 is typically considered acceptable [54]. Additionally, Goodness of Hit (GH), a metric that combines the robustness of the model with the yield of actives, provides a single value to assess the virtual screening performance, though it is less commonly reported than EF and AUC [30].
Table 2: Key Quantitative Metrics for External Validation
| Metric | Formula | Interpretation | Acceptance Threshold |
|---|---|---|---|
| Sensitivity | ( \frac{TP}{TP + FN} \times 100 ) | Percentage of true actives successfully retrieved. | Ideally > 70-80% [88] |
| Specificity | ( \frac{TN}{TN + FP} \times 100 ) | Percentage of inactives successfully rejected. | Ideally > 80% [88] |
| Enrichment Factor (EF) | ( \frac{Ha \times D}{Ht \times A} ) | Measures concentration of actives in the hit list. | > 2 is considered reliable [54] |
| Area Under Curve (AUC) | Area under the ROC curve | Overall classification performance. | > 0.7 is acceptable [54] |
Detailed Experimental Protocol for External Validation
This section outlines a step-by-step protocol for conducting an external validation, as exemplified in recent studies on targets like VEGFR-2 and c-Met [54] and FAK1 [30].
Step 1: Assemble the Independent Test Set. Curate a set of active compounds from literature or databases, ensuring no overlap with the training set. For the FAK1 study, 114 active compounds were used [30]. Obtain a larger set of decoy molecules (e.g., 571 decoys for FAK1) from DUD-E to represent inactives [30].
Step 2: Prepare the Compounds for Screening. This involves standardizing the structures (e.g., using Discovery Studio or OpenBabel) by removing salts, generating tautomers, and producing low-energy 3D conformations for each molecule in the test set to ensure they can be flexibly screened against the pharmacophore model [54].
Step 3: Execute the Pharmacophore Screening. Use the validated pharmacophore model to screen the entire independent test set. Software like Discovery Studio, LigandScout, or online servers like Pharmit can be used for this high-throughput screening (HTS). The output is a list of "hits" – compounds that match the pharmacophore model [88] [89].
Step 4: Analyze and Calculate Metrics. Compare the list of pharmacophore hits against the known activity of the test set compounds. Classify each hit as a True Positive (an active compound that is a hit) or False Positive (a decoy that is a hit). Calculate the key metrics described in Section 3: Sensitivity, Specificity, EF, and AUC.
Step 5: Interpret the Results. A successful validation is indicated by high sensitivity and specificity, an EF significantly greater than 1, and an AUC > 0.7. For instance, a pharmacophore model for anti-HBV flavonols demonstrated a sensitivity of 71% and a specificity of 100% when validated against an independent set of FDA-approved chemicals, confirming its strong predictive power [88].
Diagram Title: External Validation Workflow
The Scientist's Toolkit: Essential Reagents and Software
Successful external validation relies on a suite of computational tools and data resources. The following table details the essential "research reagents" for this process.
Table 3: Essential Research Reagents and Software for External Validation
| Tool / Resource | Type | Primary Function in Validation |
|---|---|---|
| DUD-E Database | Database | Provides pharmaceutically relevant decoy molecules for specificity testing [30] [54]. |
| ChEMBL Database | Database | Source of bioactive molecules with curated IC~50~/K~i~ data for active test sets [20]. |
| Discovery Studio | Software Suite | Used for pharmacophore generation, virtual screening, and analysis of results [54]. |
| LigandScout | Software | Advanced tool for structure-and ligand-based pharmacophore modeling and screening [88] [89]. |
| Pharmit | Online Server | Performs high-throughput pharmacophore-based virtual screening of compound databases [30] [88]. |
External validation using an independent test set is a non-negotiable step in the development of a pharmacophore model for virtual screening. It transitions a model from a theoretical construct to a validated tool with demonstrated predictive power. By meticulously designing a chemically diverse test set with confirmed actives and property-matched decoys, and by rigorously applying quantitative metrics like Sensitivity, Specificity, EF, and AUC, researchers can confidently assess a model's potential to identify novel, structurally diverse lead compounds in prospective screening efforts. This process ultimately de-risks the subsequent stages of experimental drug discovery, ensuring that computational efforts are focused on the most promising chemical starting points.
In the realm of computer-aided drug discovery, pharmacophore-based virtual screening serves as a powerful technique for identifying novel therapeutic candidates from extensive compound libraries. A pharmacophore is defined as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [2]. The efficacy of any pharmacophore model hinges on its ability to reliably distinguish between active and inactive compounds, making robust validation an indispensable step before its application in virtual screening campaigns. This guide details the three cornerstone performance metrics—Enrichment Factor (EF), Receiver Operating Characteristic (ROC) curves, and Area Under the Curve (AUC) values—that researchers employ to quantitatively assess the predictive power and screening utility of their pharmacophore models [90] [91] [92].
Theoretical Foundations of Key Performance Indicators
The ROC Curve and AUC Value
The Receiver Operating Characteristic (ROC) curve is a graphical plot that illustrates the diagnostic ability of a binary classifier system, such as a pharmacophore model used in virtual screening. The curve is created by plotting the True Positive Rate (TPR or Sensitivity) against the False Positive Rate (FPR or 1-Specificity) at various classification thresholds [90] [91].
- True Positive Rate (Sensitivity) measures the proportion of actual active compounds that are correctly identified as active by the model. It is calculated as TPR = TP / (TP + FN), where TP represents True Positives and FN represents False Negatives [91].
- False Positive Rate measures the proportion of actual inactive compounds that are incorrectly identified as active. It is calculated as FPR = FP / (FP + TN), where FP represents False Positives and TN represents True Negatives [91] [93].
The Area Under the ROC Curve (AUC) provides a single scalar value representing the model's overall performance across all possible classification thresholds. An AUC value of 1.0 represents a perfect model, while a value of 0.5 indicates a model with no discriminatory power, equivalent to random selection [91]. In practical research scenarios, an AUC value of 0.819 was reported for a validated pharmacophore model targeting PD-L1, demonstrating good predictive ability [90].
Enrichment Factor (EF)
The Enrichment Factor (EF) is a crucial metric in virtual screening that quantifies the concentration of active compounds recovered in a selected subset of the screened database compared to a random selection. It answers the fundamental question: "How much better does my model perform at finding active compounds compared to random chance?" [92]
The EF is calculated at a specific threshold of the ranked database (typically 1% or 20%) using the formula:
EF = (Number of actives found in the subset / Total number of compounds in the subset) / (Total number of actives in database / Total number of compounds in database) [92] [93]
For example, in a virtual screening study against HIV protease, an EF of 11.12 was achieved at the 1% level, indicating that the model was over 11 times more effective at enriching active compounds in the top ranked list compared to random selection [93].
Table 1: Interpretation Guide for Key Validation Metrics
| Metric | Excellent | Good | Fair | Poor (Random) |
|---|---|---|---|---|
| AUC | 0.9 - 1.0 | 0.8 - 0.9 | 0.7 - 0.8 | 0.5 - 0.7 |
| EF at 1% | >20 | 10 - 20 | 5 - 10 | <5 |
| EF at 20% | >5 | 3 - 5 | 2 - 3 | <2 |
Experimental Protocols for Metric Calculation
Preparation of Validation Datasets
The first critical step in pharmacophore model validation involves preparing appropriate datasets containing known active compounds and decoy molecules. The Directory of Useful Decoys (DUD-E) database provides an excellent resource for this purpose, as it contains known actives and decoys that are calculated using similar 1-D physico-chemical properties as the actives but dissimilar 2-D topology [92]. This ensures that decoys are physically similar but chemically distinct from active compounds, providing a rigorous test for the model.
A typical validation set should include:
- Active compounds: 10-50 known active molecules with confirmed biological activity against the target [91].
- Decoy compounds: Several hundred to thousands of experimentally confirmed inactive or presumed inactive molecules, often in a ratio of 40:1 or higher decoys to actives [92].
Virtual Screening and Data Processing Protocol
To generate the necessary data for calculating EF, ROC, and AUC values, follow this standardized protocol:
Perform virtual screening: Screen both active and decoy compounds against your pharmacophore model using software such as Phase [94] or LigandScout [91].
Rank compounds: Sort all screened compounds (both actives and decoys) based on their pharmacophore fit scores, from highest (best) to lowest (worst) [93].
Extract scores for analysis: For each compound, record the pharmacophore fit score that will be used for classification. Ensure you have only one entry (best pose) per compound [93].
Calculation of ROC Curves and AUC Values Using R
The following step-by-step protocol utilizes the R programming language to calculate ROC and AUC values, which can be adapted for results from various docking and pharmacophore screening programs [93]:
This protocol generates a standard ROC curve and computes the corresponding AUC value, providing a quantitative measure of your model's discriminatory power [93].
Calculation of Enrichment Factors
To calculate enrichment factors at specific thresholds, implement this extension to the R protocol:
This protocol enables researchers to calculate critical enrichment factors that measure the early recognition capability of their pharmacophore models [92] [93].
Workflow Visualization
Pharmacophore Model Validation Workflow
Table 2: Essential Computational Tools for Pharmacophore Validation
| Tool/Resource | Type | Primary Function | Application in Validation |
|---|---|---|---|
| DUD-E Database [92] | Database | Provides known actives and decoys | Source of validation compounds with confirmed activities and matched decoys |
| ROCR R Package [93] | Software Library | ROC curve analysis and visualization | Calculation of ROC curves, AUC values, and enrichment factors |
| LigandScout [91] | Software | Pharmacophore modeling and screening | Creating pharmacophore models and performing virtual screening for validation |
| Schrödinger Phase [94] | Software | Structure-based pharmacophore modeling | Generating and screening with e-Pharmacophores for validation studies |
| rDock [93] | Software | Molecular docking | Generating docking scores for comparative validation approaches |
| Python/R | Programming Language | Data analysis and visualization | Custom scripts for calculating metrics and generating publication-quality figures |
Advanced Considerations and Methodological Refinements
Semi-Logarithmic ROC Curves for Early Enrichment Analysis
While standard ROC curves provide an overall assessment of model performance, the early enrichment capability—particularly important in virtual screening where researchers typically only test a small fraction of top-ranked compounds—is better visualized using semi-logarithmic ROC curves. These plots provide enhanced resolution in the critical early portion of the curve (0-10% false positive rate) where optimal pharmacophore models demonstrate significant separation from the random selection line [93].
To generate a semi-logarithmic ROC curve in R, researchers can extend the basic protocol:
Incorporating Molecular Dynamics in Pharmacophore Validation
Emerging methodologies enhance traditional validation approaches by incorporating molecular dynamics (MD) simulations. Studies comparing pharmacophore models derived from crystal structures with those derived from MD simulations demonstrate that MD-refined models can show improved ability to distinguish between active and decoy compounds in some cases [92]. This approach addresses concerns about the static nature of crystal structures, which may contain non-physiological contacts or lack proper solvation effects.
The protocol for MD-enhanced validation involves:
- Running MD simulations (typically 20 ns) on protein-ligand complexes
- Extracting the final protein-ligand structure from the simulation
- Generating a pharmacophore model from the MD-refined structure
- Comparing the validation metrics (EF, AUC) with the original crystal structure-based model [92]
Machine Learning and AI-Enhanced Approaches
Recent advances integrate machine learning with pharmacophore-based screening to accelerate virtual screening while maintaining accuracy. These approaches use ML models trained to approximate docking scores based on molecular descriptors, achieving speed increases of up to 1000 times compared to classical docking-based screening [7]. Furthermore, knowledge-guided diffusion models like DiffPhore represent cutting-edge approaches that leverage deep learning for 3D ligand-pharmacophore mapping, showing state-of-the-art performance in predicting binding conformations and virtual screening enrichment [50].
These advanced methodologies demonstrate the evolving landscape of pharmacophore validation, where traditional metrics like EF and AUC remain fundamental, but the methods for generating and validating models continue to incorporate more sophisticated computational approaches that account for protein flexibility, chemical diversity, and screening efficiency.
Virtual screening (VS) has become an indispensable tool in the modern drug discovery pipeline, enabling researchers to computationally prioritize molecules with the highest likelihood of biological activity from extensive chemical libraries [21] [52]. Among the various strategies employed, two dominant structure-based approaches have emerged: Pharmacophore-Based Virtual Screening (PBVS) and Docking-Based Virtual Screening (DBVS). PBVS relies on abstracting the essential steric and electronic features necessary for a molecule to interact with a biological target, defined by the International Union of Pure and Applied Chemistry (IUPAC) as "the ensemble of steric and electronic features that is necessary to ensure the optimal supra-molecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [52] [2]. In contrast, DBVS computationally simulates the binding pose of a small molecule within a protein's binding site and scores its complementarity [21] [95].
The choice between these methodologies is a fundamental strategic decision. This article provides an in-depth benchmark comparison of PBVS versus DBVS, framing the findings within the broader context of how to construct and validate a robust pharmacophore model for virtual screening research. We synthesize evidence from key comparative studies to guide researchers, scientists, and drug development professionals in selecting and applying these powerful techniques effectively.
Pharmacophore-Based Virtual Screening (PBVS)
The pharmacophore concept is a foundational pillar in medicinal chemistry. A pharmacophore model is not a specific molecule but an abstract representation of the three-dimensional arrangement of chemical functionalities—such as hydrogen bond donors, hydrogen bond acceptors, hydrophobic areas, and charged groups—required for binding and activity [52] [2]. These features are typically represented as 3D geometric entities like spheres, vectors, and planes.
There are two primary approaches to pharmacophore model generation:
- Structure-Based Modeling: This method derives the pharmacophore directly from the analysis of a target protein's 3D structure, often from a protein-ligand complex solved by X-ray crystallography or NMR. The interaction pattern between the ligand and the protein is extracted to define the critical features and their spatial arrangement [52] [2]. Tools like LigandScout and Discovery Studio automate this process.
- Ligand-Based Modeling: When the 3D structure of the target is unavailable, models can be built from a set of known active molecules. The common chemical features shared among these molecules, once aligned in their bioactive conformations, form the basis of the pharmacophore hypothesis [52] [2].
The subsequent virtual screening process involves scanning large databases of compounds to identify those whose 3D structures match the pharmacophore query.
Docking-Based Virtual Screening (DBVS)
DBVS requires the 3D structure of the target protein. This approach involves two main steps for each molecule in a database:
- Docking: Predicting the preferred orientation (or "pose") of the small molecule within the binding site of the target.
- Scoring: Assigning a numerical score using a scoring function to estimate the binding affinity or complementarity of that pose [95].
Popular docking programs include DOCK, GOLD, Glide, and AutoDock. A significant challenge in DBVS is the approximate nature of scoring functions, which balance computational speed with accuracy, sometimes leading to false positives [95] [96]. To improve accuracy, docking is sometimes followed by more refined—and computationally expensive—methods like Molecular Dynamics (MD) simulations with MM/PBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) analysis to better estimate binding free energies [95].
A Benchmark Comparison: PBVS vs. DBVS
A seminal benchmark study directly compared the performance of PBVS and DBVS across eight structurally diverse protein targets: angiotensin-converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptor α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK) [21] [22] [97].
Experimental Design and Protocols
The study design was rigorous to ensure a fair comparison [21]:
- Target and Model Preparation: For each target, structure-based pharmacophore models were constructed using the LigandScout program, based on several X-ray crystal structures of protein-ligand complexes. For DBVS, a single high-resolution crystal structure was used with three different docking programs: DOCK, GOLD, and Glide.
- Screening Databases: Two testing datasets (Decoy I and Decoy II), each containing experimentally confirmed active compounds and presumed inactive decoy molecules, were constructed for each target. This resulted in a total of sixteen screening experiments.
- Performance Metrics: The effectiveness of each virtual screening method was evaluated using enrichment factors (which measure the enrichment of active molecules compared to random selection) and hit rates at the top 2% and 5% of the ranked database.
Key Quantitative Results
The benchmark study yielded clear, quantitative results favoring PBVS in the majority of test cases.
Table 1: Summary of Benchmark Results from 16 Virtual Screening Experiments (8 Targets vs. 2 Databases)
| Screening Method | Number of Cases with Higher Enrichment Factor | Average Hit Rate at Top 2% of Database | Average Hit Rate at Top 5% of Database |
|---|---|---|---|
| Pharmacophore-Based (PBVS) | 14 out of 16 | Much Higher | Much Higher |
| Docking-Based (DBVS) | 2 out of 16 | Lower | Lower |
The data shows that PBVS significantly outperformed DBVS in retrieving active compounds, achieving higher enrichment factors in 14 out of the 16 virtual screening runs [21] [22]. Furthermore, the average hit rates for PBVS at the critically important early stages of screening (the top 2% and 5% of ranked compounds) were "much higher" than those achieved by any of the three docking programs tested [21]. This demonstrates the superior ability of PBVS to prioritize and enrich true active compounds at the top of a ranked list, a crucial factor for practical drug discovery where only a limited number of top-ranking compounds are selected for experimental testing.
Building an Effective Pharmacophore Model: A Practical Workflow
The superior performance of PBVS in benchmark studies hinges on the construction of a high-quality pharmacophore model. The following workflow, depicted in the diagram below, outlines the key steps for researchers.
Data Curation and Model Construction
The first step involves gathering high-quality input data, which is critical for model reliability.
- For Structure-Based Models: The 3D structure of the protein, preferably in complex with a high-affinity ligand, should be obtained from the Protein Data Bank (PDB). The structure must be carefully prepared, correcting protonation states, adding missing atoms, and removing irrelevant crystallographic water molecules [2].
- For Ligand-Based Models: A set of known active compounds must be collected from reliable databases like ChEMBL [52]. The quality of this dataset is paramount; compounds should have experimentally confirmed, target-specific activity (e.g., from enzyme assays, not just cellular activity) and cover a range of structural diversity to create a robust model [52]. The dataset should also include confirmed inactive compounds or generated decoys (e.g., from DUD-E) for validation [52].
Model Refinement and Theoretical Validation
The initial pharmacophore hypothesis often requires refinement. This may involve deleting non-essential features, adjusting the tolerance (size) of feature spheres, or defining certain features as "optional" [52]. A critical refinement is the addition of exclusion volumes, which represent steric constraints of the binding pocket and prevent the mapping of compounds that would sterically clash with the protein [52] [2].
Before proceeding to costly experimental testing, the model must be validated theoretically. This is done by screening a test database containing known active and inactive compounds/decoys. Key performance metrics include [52]:
- Enrichment Factor (EF): Measures how much the model enriches active compounds in the hit list compared to random selection.
- Receiver Operating Characteristic (ROC) Curve and Area Under the Curve (AUC): Evaluates the model's overall ability to distinguish active from inactive compounds.
- Yield of Actives: The percentage of active compounds in the virtual hit list.
A high-quality model will show strong performance across these metrics, significantly enriching active compounds in its top ranks.
Integrated Approaches and Advanced Applications
While the benchmark shows PBVS's strengths, the most successful virtual screening campaigns often use an integrated approach. A common strategy is to use PBVS as a pre-filter to rapidly reduce the size of a massive chemical library, followed by the more computationally intensive DBVS on the resulting subset of molecules [21] [98]. This leverages the speed and enrichment power of PBVS while utilizing docking to provide detailed binding pose information.
Furthermore, the field is being transformed by machine learning (ML). ML models can be trained to predict docking scores based on 2D molecular structures, achieving speed-ups of 1000 times or more compared to classical molecular docking [7]. This allows for the ultra-rapid screening of billion-member virtual libraries, with pharmacophore constraints used to focus the search on chemically relevant subspaces [7].
Finally, the rise of AI-predicted protein structures like AlphaFold presents new opportunities and challenges. While AlphaFold has revolutionized protein structure prediction, recent studies indicate that "as-is" AlphaFold models can show significantly worse performance in docking-based virtual screening compared to experimental PDB structures [99]. This suggests that for structure-based modeling, AlphaFold models may require post-modeling refinement before they can be reliably used for DBVS, whereas they could be immediately useful for generating structure-based pharmacophore models.
Table 2: Key Software and Data Resources for Virtual Screening
| Resource Name | Type | Primary Function | Relevance to VS |
|---|---|---|---|
| LigandScout | Software | Structure-based & ligand-based pharmacophore model generation [21] [52]. | Creates advanced pharmacophore models from protein-ligand complexes or ligand sets. |
| Catalyst (Accelrys) | Software | Pharmacophore-based virtual screening and database searching [21] [22]. | Performs rapid 3D searches of compound databases using pharmacophore queries. |
| DOCK, GOLD, Glide | Software | Docking-based virtual screening and pose prediction [21] [22]. | Standards for predicting ligand binding modes and scoring binding affinity. |
| Protein Data Bank (PDB) | Database | Repository for experimentally determined 3D structures of proteins and nucleic acids [52] [2]. | Primary source of protein structures for structure-based pharmacophore modeling and DBVS. |
| ChEMBL | Database | Manually curated database of bioactive molecules with drug-like properties and experimental bioactivities [52]. | Key resource for finding known active/inactive compounds for ligand-based modeling and validation. |
| DUD-E | Web Server | Directory of Useful Decoys, Enhanced; generates property-matched decoys for validation [52]. | Provides carefully selected decoy molecules to rigorously test a model's ability to avoid false positives. |
| ZINC | Database | Freely available database of commercially available compounds for virtual screening [7]. | A primary library for screening to find purchable or synthesizable candidate molecules. |
The benchmark evidence clearly demonstrates that pharmacophore-based virtual screening is a powerful and often superior method for enriching active compounds in virtual screening campaigns against diverse targets. Its performance, coupled with computational efficiency, makes it an excellent choice for rapidly prioritizing candidates from large chemical libraries.
The successful application of PBVS is intrinsically linked to the careful, rigorous construction and validation of the pharmacophore model itself. As the field advances, the integration of PBVS with docking and the adoption of machine learning techniques are creating increasingly powerful and efficient workflows for drug discovery. For researchers aiming to build a robust virtual screening protocol, a strategy that leverages the strengths of pharmacophore modeling—either as a standalone method or as a pre-filter within a larger pipeline—offers a proven path to identifying novel bioactive molecules.
Post-Filtering Docking Results with Pharmacophores to Improve Enrichment Rates
Virtual screening is an essential component of modern drug discovery, enabling researchers to rapidly identify potential lead compounds from large chemical databases. Among the various computational approaches, structure-based virtual screening using molecular docking is widely employed when a three-dimensional structure of the target protein is available. However, a significant challenge persists: the prevalence of false positives—compounds that score highly in docking but demonstrate little to no actual biological activity [71]. This limitation primarily stems from the inability of current scoring functions to accurately predict binding affinities or consistently distinguish correct ligand poses from incorrect ones [71].
To address this critical issue, researchers have developed a powerful strategy known as pharmacophore post-filtering. This method integrates the complementary strengths of both structure-based and ligand-based drug design approaches. The core premise involves using docking programs for pose generation, followed by filtering the resulting poses through pharmacophore models to eliminate chemically implausible candidates [100] [71]. Empirical studies across diverse protein targets have demonstrated that this integrated approach significantly increases enrichment rates compared to docking alone, providing a more reliable method for identifying true active compounds in virtual screening campaigns [100] [22].
Theoretical Foundation
The Pharmacophore Concept and Molecular Recognition
A pharmacophore represents an abstract description of the steric and electronic features necessary for molecular recognition between a ligand and its biological target. It encapsulates the key interactions—such as hydrogen bonding, hydrophobic contacts, and ionic interactions—that drive binding affinity and specificity. In structure-based drug design, pharmacophores are typically derived from analysis of protein-ligand complexes, crystallographic data, or complementary interaction sites within the binding pocket [71].
The fundamental principle underlying pharmacophore post-filtering is chemical complementarity. For a ligand to bind effectively to its target, it must not only fit spatially within the binding site but also establish chemically favorable interactions with the surrounding protein residues. This includes fulfilling essential hydrogen bonding requirements, filling hydrophobic cavities, and matching charge distributions appropriately [71]. Pharmacophore models serve as computational filters to enforce these requirements, ensuring that retained compounds possess the necessary features for productive binding.
Limitations of Docking and Scoring Functions
Molecular docking algorithms generally perform reasonably well at sampling ligand conformations and generating plausible binding poses [71]. However, the accompanying scoring functions often struggle with several critical aspects:
- Inaccurate binding affinity predictions due to simplified energy calculations
- Difficulty in ranking active compounds above inactive ones
- Sensitivity to small structural changes that may disproportionately affect scores
- Inadequate treatment of solvation effects and entropic contributions
These limitations result in enrichment challenges, where true active compounds may be buried beneath false positives in docking rankings. The integration of pharmacophore filtering addresses these issues by incorporating crucial chemical intelligence beyond what is captured by typical scoring functions [71].
Methodological Framework
The pharmacophore post-filtering methodology follows a systematic workflow that leverages the strengths of both docking and pharmacophore-based approaches. The process, illustrated in the diagram below, ensures that only compounds fulfilling essential interaction requirements advance in the screening pipeline.
Key Methodological Steps
Pose Generation via Docking
The initial stage involves comprehensive conformational sampling using molecular docking programs. Unlike traditional docking workflows where only the top-ranked pose might be considered, pharmacophore post-filtering requires generating and saving multiple diverse poses for each compound [71]. This approach helps overcome the inherent limitations of scoring functions by preserving potentially correct binding modes that might not receive the highest scores. Recommended practices include:
- Using docking programs with stochastic components to ensure pose diversity
- Generating 10-50 poses per compound to adequately sample the conformational space
- Saving all generated poses with their coordinates for subsequent filtering
- Disregarding docking scores during this initial phase to avoid premature elimination of potentially viable compounds [71]
Pharmacophore Model Development
The development of effective pharmacophore models is a critical step that significantly influences filtering performance. These models can be created through several approaches:
Structure-based pharmacophore modeling: Deriving interaction features directly from protein-ligand complexes by analyzing:
- Hydrogen bond donors and acceptors in the binding site
- Hydrophobic regions and aromatic interaction patterns
- Charge-assisted interactions and metal coordination sites
- Solvent-exposed regions that should be avoided [71]
Ligand-based pharmacophore modeling: Creating models based on known active compounds when structural information is limited, though this approach is less common in pure structure-based screening scenarios.
Automated pharmacophore generation: Utilizing specialized software such as:
The stringency of pharmacophore models can be adjusted by modifying feature tolerances and spatial constraints. Tighter parameters create more selective filters, while looser constraints accommodate greater ligand flexibility and binding mode variations [71].
Implementation of Pharmacophore Filtering
The core filtering process involves systematically evaluating each docked pose against the predefined pharmacophore model. Key considerations include:
- Computational efficiency: Since compounds are pre-aligned to the binding site, pharmacophore searching is computationally inexpensive compared to traditional ligand-based pharmacophore searches [71]
- Pose evaluation: Each saved pose is checked for compliance with the pharmacophore features
- Score reassignment: Compounds are re-ranked based on the best-scoring pose that passes the pharmacophore filter [100]
- Multiple filter strategies: Implementing consecutive filters with increasing stringency can progressively refine the compound set
Advanced implementations may incorporate shape-focused pharmacophore models that use graph clustering algorithms to create cavity-filling models from docked active ligands, further enhancing enrichment potential [41].
Experimental Evidence and Performance Benchmarking
Quantitative Assessment of Enrichment Improvement
Multiple studies have systematically evaluated the performance benefits of pharmacophore post-filtering across diverse target classes. The table below summarizes key findings from representative investigations:
Table 1: Performance Comparison of Docking Alone vs. Pharmacophore Post-Filtering
| Target Protein | Docking Software | Enrichment Factor (Docking Only) | Enrichment Factor (With Pharmacophore) | Study Reference |
|---|---|---|---|---|
| CDK2 | zdock+, Surflex, FRED | Baseline | Increased in all cases | [100] |
| COX2 | zdock+, Surflex, FRED | Baseline | Increased in all cases | [100] |
| ERα | zdock+, Surflex, FRED | Baseline | Increased in all cases | [100] |
| Factor Xa | zdock+, Surflex, FRED | Baseline | Increased in all cases | [100] |
| MMP3 | zdock+, Surflex, FRED | Baseline | Increased in all cases | [100] |
| Neuraminidase | zdock+, Surflex, FRED | Baseline | Increased in all cases | [100] |
| Neuraminidase A | GOLD/Glide | Varies with program | Better than traditional docking | [71] |
| CDK2 | GOLD/Glide | Varies with program | Better than traditional docking | [71] |
| Protein Kinase C | GOLD/Glide | Varies with program | Better than traditional docking | [71] |
A comprehensive benchmark comparison against eight diverse protein targets further demonstrated the superiority of integrated approaches. The study found that in fourteen of sixteen virtual screening sets, pharmacophore-based methods achieved higher enrichment factors than docking-based methods alone [22]. The average hit rates across eight targets at 2% and 5% cutoff levels were substantially higher for pharmacophore-based screening compared to docking-based approaches [22].
Case Study Applications
Kinase Target Screening
In a study targeting VEGFR-2 and c-Met dual inhibitors for cancer therapy, researchers employed a computational workflow incorporating pharmacophore screening followed by molecular docking. This integrated approach identified 18 hit compounds with potential inhibitory activity against both targets [53]. Subsequent molecular dynamics simulations confirmed the stability of these complexes, with two compounds (17924 and 4312) demonstrating superior binding free energies compared to reference ligands [53].
Metabolic Disease Target Screening
For the discovery of human hepatic ketohexokinase (KHK) inhibitors to treat fructose metabolic disorders, researchers implemented a multi-tier virtual screening approach. Initial pharmacophore-based screening of 460,000 compounds identified promising candidates that were further evaluated through multi-level molecular docking and binding free energy calculations [101]. This strategy yielded ten compounds with docking scores ranging from -7.79 to -9.10 kcal/mol, surpassing clinical candidates PF-06835919 (-7.768 kcal/mol) and LY-3522348 (-6.54 kcal/mol) [101].
Anti-Infective Drug Discovery
In antibacterial research targeting Waddlia chondrophila, scientists combined pharmacophore modeling with molecular docking to identify novel inhibitors from a library of phytochemicals [102]. The integrated computational approach successfully pinpointed compounds with favorable binding affinities to essential bacterial targets, followed by molecular dynamics simulations that confirmed complex stability over 100 nanoseconds [102].
Advanced Methodologies and Recent Innovations
Shape-Focused Pharmacophore Modeling
Recent advances have introduced graph clustering algorithms for generating shape-focused pharmacophore models that further enhance screening effectiveness. The O-LAP algorithm represents one such innovation, generating cavity-filling models by clustering overlapping atomic content from docked active ligands [41]. The methodology involves:
- Filling the protein cavity with flexibly docked active ligands
- Trimming non-polar hydrogen atoms and deleting covalent bonding information
- Clustering overlapping atoms with matching types into representative centroids
- Applying atom-type-specific radii during distance measurements
- Optionally performing enrichment-driven optimization when training data is available [41]
This approach has demonstrated substantial improvements in docking enrichment for challenging targets and performs effectively in both docking rescoring and rigid docking scenarios [41].
Deep Learning-Guided Pharmacophore Modeling
The emerging field of deep learning has produced innovative solutions like PharmacoNet, the first deep learning framework for pharmacophore modeling designed for ultra-large-scale virtual screening [103]. This approach offers:
- Fully automated protein-based pharmacophore modeling
- Evaluation of ligand potency using parameterized analytical scoring functions
- Enhanced generalization capabilities across unseen targets and ligands
- Exceptional computational efficiency, screening 187 million compounds against cannabinoid receptors in just 21 hours on a single CPU [103]
Benchmark studies indicate that PharmacoNet achieves remarkable speed while maintaining reasonable accuracy compared to traditional docking methods and existing deep learning-based scoring models [103].
Hybrid Workflow Integration
Modern virtual screening campaigns increasingly adopt hybrid workflows that combine multiple computational techniques. The following diagram illustrates an advanced integrated approach that leverages both traditional and contemporary methods:
Practical Implementation Guidelines
Research Reagent Solutions
Table 2: Essential Software Tools for Pharmacophore Post-Filtering Implementation
| Tool Category | Representative Software | Primary Function | Application Notes |
|---|---|---|---|
| Molecular Docking | GOLD, Glide, PLANTS, FRED, Surflex | Ligand pose generation and initial scoring | Programs with stochastic components preferred for pose diversity [71] |
| Pharmacophore Modeling | MOE, LigandScout, LUDI, Catalyst | Pharmacophore model creation and validation | MOE allows visual inspection; LigandScout enables automated generation [71] |
| Shape-Based Screening | O-LAP, ROCS, ShaEP | Shape similarity comparisons and modeling | O-LAP implements graph clustering for shape-focused models [41] |
| Scripting & Automation | Python, Pose-Filter Scripts | Custom filtering workflow implementation | Schrödinger provides Pose-Filter Python script for interaction filtering [71] |
| Deep Learning Pharmacophores | PharmacoNet | DL-guided pharmacophore modeling | Enables ultra-large-scale screening [103] |
Critical Implementation Considerations
Successful implementation of pharmacophore post-filtering requires careful attention to several practical aspects:
Pose Diversity Generation: Ensure docking parameters are configured to produce conformationally diverse poses rather than converging rapidly on a single "best" solution [71]
Pharmacophore Feature Selection: Base pharmacophore features on conserved interactions observed across multiple co-crystal structures when available, rather than relying on a single complex [100]
Filter Stringency Adjustment: Balance between being too restrictive (potentially eliminating true positives) and too permissive (allowing false positives to pass) by adjusting feature tolerances [71]
Validation Procedures: Implement rigorous validation using known active and inactive compounds to optimize pharmacophore models before application to unknown databases [41]
Computational Resource Allocation: Distribute resources appropriately between docking (computationally intensive) and filtering (relatively fast) stages based on library size and complexity [103]
Pharmacophore post-filtering of docking results represents a mature and validated methodology for substantially improving enrichment rates in structure-based virtual screening. By integrating the conformational sampling strengths of molecular docking with the chemical intelligence of pharmacophore matching, this approach effectively addresses fundamental limitations of scoring functions while remaining computationally efficient. The consistent demonstration of improved performance across diverse target classes, combined with ongoing innovations in shape-focused modeling and deep learning applications, positions pharmacophore post-filtering as an essential component of modern virtual screening workflows. As compound libraries continue to expand toward billions of molecules, these integrated approaches will play an increasingly critical role in identifying novel chemical starting points for drug development programs.
In the realm of computer-aided drug discovery, pharmacophore models serve as powerful abstract representations of the steric and electronic features essential for a molecule to interact with a biological target [2]. The construction of a pharmacophore model, however, is only the first step. For researchers employing these models in virtual screening (VS) campaigns, a critical question remains: how can we quantitatively assess the model's quality and its ability to discriminate true active compounds from inactive ones? The answer lies in robust validation metrics, primarily the Goodness of Hit (GH) score and early enrichment analysis. These metrics are not merely post-modeling formalities; they are fundamental to building confidence in a model's predictive power before committing substantial resources to experimental testing. This guide provides an in-depth technical examination of these critical assessment tools, framed within the broader thesis of building a reliable pharmacophore model for virtual screening.
Theoretical Foundations of Validation Metrics
A valid pharmacophore model must demonstrate a strong ability to identify active compounds (sensitivity) while rejecting inactive ones (specificity) during the virtual screening of large databases. This performance is quantified using two primary concepts: the Goodness of Hit Score and Early Enrichment Factors.
1.1 The Goodness of Hit (GH) Score The GH score is a composite metric that provides a single value representing the overall performance of a pharmacophore model in a virtual screening experiment. It incorporates several key parameters from the screening output [104]:
- D: Total number of molecules in the database screened.
- A: Total number of known active molecules in the database.
- Ht: Total number of hits returned from the screening.
- Ha: Number of active molecules found among the hits.
The formula for calculating the GH score is: [ GH = \left( \frac{Ha}{Ht} \right) \times \left( \frac{3A + Ht}{4A} \right) \times \left( 1 - \frac{Ht - Ha}{D - A} \right) ]
The GH score ranges from 0 to 1, where a higher score indicates better model performance. A score of 0.7-0.8 indicates a very good model, while a score above 0.8 is considered excellent [104] [105].
1.2 Early Enrichment Factors (EF) While the GH score gives an overview, early enrichment factors focus on the model's practical utility by measuring its ability to identify actives at the very top of the ranked list of hits—a crucial efficiency metric for large databases. The most commonly reported is EF1%, the enrichment factor at the top 1% of the screened database [40].
The formula for EF is: [ EF = \frac{(Ha / Ht)}{(A / D)} ] This metric indicates how much better the model is at finding actives compared to a random selection.
Table 1: Key Metrics for Pharmacophore Model Validation
| Metric | Formula | Interpretation | Ideal Range |
|---|---|---|---|
| Goodness of Hit (GH) | ( \left( \frac{Ha}{Ht} \right) \times \left( \frac{3A + Ht}{4A} \right) \times \left( 1 - \frac{Ht - Ha}{D - A} \right) ) | Overall model quality; balances yield of actives and false positives. | 0.7 - 1.0 [104] [105] |
| Enrichment Factor (EF) | ( \frac{(Ha / Ht)}{(A / D)} ) | How much better the model performs than random selection. | Higher is better; context-dependent [104] |
| % Yield of Actives | ( (Ha / Ht) \times 100 ) | Percentage of retrieved hits that are true actives. | Higher is better [104] |
| % Ratio of Actives | ( (Ha / A) \times 100 ) | Percentage of all known actives that were successfully retrieved. | Higher is better [104] |
| AUC (Area Under Curve) | Area under the ROC curve | Overall ability to discriminate actives from inactives. | 0.5 (random) to 1.0 (perfect) [40] |
Experimental Protocols for Model Validation
Implementing a rigorous validation protocol is essential for generating reliable GH and enrichment metrics. The following methodology details the standard procedure.
2.1 The Decoy Set Validation Method This is the most recognized protocol for validating pharmacophore models, which involves screening a database containing known active compounds and computationally generated decoy molecules.
Workflow Diagram: Pharmacophore Model Validation and Application
Protocol Steps:
Database Preparation:
- Source Actives: A set of known active molecules (A) against the target is collected from literature or databases like ChEMBL. For example, a study on XIAP protein used 10 known antagonists [40], while another on FAK1 used 20 [106].
- Generate Decoys: A larger set of decoy molecules (D-A) is generated. Decoys are chemically similar to actives in their physicochemical properties (e.g., molecular weight, logP) but are topologically distinct to ensure they are unlikely to be active. The DUD-E (Database of Useful Decoys: Enhanced) is a standard resource for this purpose, providing approximately 50 decoys per active [41] [107]. The total database size (D) is the sum of actives and decoys.
Virtual Screening Run:
- The prepared validation database is screened against the pharmacophore model.
- The screening process identifies molecules (Ht) that match the model's chemical features and spatial arrangement.
Hit Analysis and Metric Calculation:
- The output list is analyzed to determine the number of active compounds correctly identified (Ha).
- The values for D, A, Ht, and Ha are plugged into the formulas from Table 1 to calculate the GH score, EF1%, and other relevant metrics.
2.2 Case Study: Validation of a Tubulin Inhibitor Model A study aiming to discover novel tubulin inhibitors provides a clear example of successful model validation [104]. The researchers built a structure-based pharmacophore model and validated it using a database of 1000 molecules, which included 30 known active tubulin inhibitors and 970 inactive molecules.
Table 2: Validation Results for a Tubulin Pharmacophore Model [104]
| Parameter | Value | Interpretation |
|---|---|---|
| Total molecules in database (D) | 1000 | Size of the test set. |
| Total number of actives (A) | 30 | Number of known inhibitors. |
| Total hits (Ht) | 36 | All compounds matching the model. |
| Active hits (Ha) | 26 | True inhibitors found by the model. |
| % Yield of Actives | 72% | High proportion of hits are true actives. |
| Enrichment Factor (E) | 24 | Model is 24x better than random selection. |
| Goodness of Hit (GH) Score | 0.75 | Indicates a "very good" model. |
The high GH score of 0.75 and the exceptional enrichment factor of 24 demonstrated that the model was very efficient at distinguishing active molecules from inactive ones, giving the researchers confidence to proceed with its use for screening a large commercial database [104].
The Scientist's Toolkit: Essential Research Reagents and Software
Building and validating a pharmacophore model requires a suite of specialized software tools and databases.
Table 3: Key Research Reagents and Software for Pharmacophore Modeling and Validation
| Tool Name | Type | Primary Function in Validation | Reference |
|---|---|---|---|
| DUD-E / DUDE-Z | Database | Provides benchmark sets of known actives and property-matched decoys for validation. | [106] [41] [40] |
| LigandScout | Software | Used for structure-based and ligand-based pharmacophore generation; includes tools for model validation. | [106] [40] |
| Phase (Schrödinger) | Software | Enables development of pharmacophore hypotheses from ligand sets and includes comprehensive virtual screening and validation workflows. | [107] |
| SILCS-Pharm | Software | Generates 3D pharmacophore models from Grid Free Energy (GFE) FragMaps derived from molecular dynamics simulations. | [108] |
| ROC Curve Analysis | Method | Evaluates the diagnostic ability of the model by plotting true positive rate against false positive rate; AUC quantifies discrimination. | [106] [40] |
Interpretation and Integration into the Screening Workflow
A model's validity, as confirmed by GH and EF metrics, determines its role in the downstream drug discovery pipeline.
4.1 Interpreting Results and Setting Thresholds A GH score of 0.75, as seen in the tubulin study, is a strong indicator of a high-quality model [104]. Similarly, an early enrichment factor (EF1%) of 10.0 with an AUC value of 0.98 was reported for a validated XIAP pharmacophore model, proving its excellent ability to distinguish true actives from decoys [40]. If the calculated metrics fall below acceptable thresholds (e.g., GH < 0.5), the model requires refinement. This may involve re-evaluating the selected pharmacophore features, adjusting their spatial tolerances, or incorporating exclusion volumes to better represent the target's binding site steric constraints [2] [107].
4.2 From Validated Model to Lead Identification A validated model becomes the primary query for screening large, diverse chemical databases. The typical workflow, as demonstrated in studies targeting PLK1-PBD and tubulin, involves [104] [105]:
- Pharmacophore Screening: Using the validated model to screen millions of compounds from databases like ZINC, which lists over 230 million purchasable compounds [40].
- Drug-Likeness Filtering: Applying filters like Lipinski's Rule of Five to prioritize compounds with acceptable ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties.
- Molecular Docking: Further refining the hit list by studying the binding poses and scores of the top-matched compounds against the atomic structure of the target protein.
- Experimental Assays: The final, computationally selected hits are then acquired for in vitro and in vivo biological testing to confirm activity.
The rigorous assessment of model quality using Goodness of Hit scores and early enrichment analysis is a non-negotiable step in the pharmacophore modeling workflow. These metrics transform a theoretical hypothesis into a validated, predictive tool with quantified reliability. By following the standardized experimental protocols for decoy set validation and correctly interpreting the resulting metrics, researchers can confidently select the best pharmacophore models to drive efficient and successful virtual screening campaigns, ultimately accelerating the discovery of novel therapeutic agents.
Conclusion
Pharmacophore modeling stands as a powerful and versatile tool in the computational drug discovery pipeline, effectively bridging the gap between target identification and lead compound selection. By mastering both structure-based and ligand-based approaches, researchers can create robust models that significantly reduce the chemical space requiring experimental validation, thereby saving substantial time and resources. Successful implementation requires careful attention to model validation, a clear understanding of the method's limitations regarding molecular flexibility and data quality, and strategic integration with other techniques like molecular docking. Future advancements, particularly through the integration of machine learning algorithms and improved handling of protein dynamics, promise to further enhance the accuracy and predictive power of pharmacophore models. Their continued application will undoubtedly accelerate the discovery of novel therapeutics for a wide range of diseases, solidifying their critical role in biomedical research and clinical development.
References
- 1. Pharmacophore modeling and applications in drug discovery [https://www.sciencedirect.com/science/article/abs/pii/S135964461000111X]
- 2. Drug Design by Pharmacophore and Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9145410/]
- 3. Pharmacophore [https://en.wikipedia.org/wiki/Pharmacophore]
- 4. IUPAC - pharmacophore (11485) [https://goldbook.iupac.org/terms/view/11485]
- 5. Applications of the Pharmacophore Concept in Natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7685156/]
- 6. Pharmacophore - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/immunology-and-microbiology/pharmacophore]
- 7. Machine learning accelerates pharmacophore-based ... [https://www.nature.com/articles/s41598-024-58122-7]
- 8. Application of ensemble pharmacophore-based virtual ... [https://www.sciencedirect.com/science/article/pii/S2001037021003299]
- 9. Pharmacophore model guided 3D molecular generation ... [https://chemrxiv.org/engage/chemrxiv/article-details/68d9f3723e708a7649ba89ba]
- 10. Pharmacophore [https://grokipedia.com/page/Pharmacophore]
- 11. Drug Design by Pharmacophore and Virtual Screening ... [https://www.mdpi.com/1424-8247/15/5/646]
- 12. Pharmacophore modeling | Medicinal Chemistry Class Notes [https://fiveable.me/medicinal-chemistry/unit-11/pharmacophore-modeling/study-guide/uZjEWlQXMHLmcu3u]
- 13. Pharmacophore model-aided virtual screening combined ... [https://www.sciencedirect.com/science/article/pii/S1878535222006505]
- 14. Pharmacophore-based virtual screening and in silico ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12009819/]
- 15. A pharmacophore-guided deep learning approach for ... [https://www.nature.com/articles/s41467-023-41454-9]
- 16. Binding Site - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/binding-site]
- 17. Residue-based pharmacophore approaches to study protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8439153/]
- 18. Pharmacophore-Based Machine Learning Model To ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10448689/]
- 19. Pharmacophore Fingerprint [https://docs.chemaxon.com/display/docs/fingerprints_pharmacophore-fingerprint.md]
- 20. QPHAR: quantitative pharmacophore activity relationship [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-021-00537-9]
- 21. Pharmacophore-based virtual screening versus docking ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4007494/]
- 22. Pharmacophore-based virtual screening versus docking ... [https://www.nature.com/articles/aps2009159]
- 23. Accelerating discovery of bioactive ligands with ... [https://www.nature.com/articles/s41467-025-56349-0]
- 24. Applications of the Novel Quantitative Pharmacophore Activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9504690/]
- 25. Pharmacophore modeling in drug design [https://pubmed.ncbi.nlm.nih.gov/40175047/]
- 26. Pharmacophore modeling and applications in drug discovery [https://www.sciencedirect.com/science/article/pii/S135964461000111X]
- 27. Pharmer: Efficient and Exact Pharmacophore Search - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3124593/]
- 28. GitHub - HazratMaghaz/ Pharmacophore _ Modeling ... [https://github.com/HazratMaghaz/Pharmacophore_Modeling]
- 29. Reliability of AlphaFold2 Models in Virtual Drug Screening [https://www.mdpi.com/1422-0067/25/18/10139]
- 30. Structure-based identification of novel FAK1 inhibitors ... [https://www.nature.com/articles/s41598-025-23203-8]
- 31. Improving docking and virtual screening performance ... [https://www.nature.com/articles/s41598-024-75400-6]
- 32. Strategies for 3D pharmacophore-based virtual screening [https://www.sciencedirect.com/science/article/abs/pii/S1740674910000375]
- 33. What is pharmacophore modeling and its applications? [https://synapse.patsnap.com/article/what-is-pharmacophore-modeling-and-its-applications]
- 34. Ligand-based Pharmacophore Modeling, Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6396084/]
- 35. Ligand-based pharmacophore modeling targeting the ... [https://www.sciencedirect.com/science/article/pii/S2950363924000218]
- 36. Development, evaluation and application of 3D QSAR ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-S14-S4]
- 37. Ligand-Based Pharmacophore Modeling Using Novel 3D ... [https://www.mdpi.com/1420-3049/23/12/3094]
- 38. Pharmacophore-Model-Based Virtual-Screening Approaches ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9600652/]
- 39. Pharmacophore modeling in drug design [https://www.sciencedirect.com/science/article/abs/pii/S1054358925000109]
- 40. Structure based pharmacophore modeling, virtual ... [https://www.nature.com/articles/s41598-021-83626-x]
- 41. Building shape-focused pharmacophore models for effective ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00857-6]
- 42. An artificial intelligence accelerated virtual screening ... [https://www.nature.com/articles/s41467-024-52061-7]
- 43. Rapid traversal of vast chemical space using machine ... [https://www.nature.com/articles/s43588-025-00777-x]
- 44. Virtual Screening Using Molecular Simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3092865/]
- 45. Pharmacophore modeling, molecular docking, MMGBSA ... [https://www.sciencedirect.com/science/article/pii/S2468227625001048]
- 46. ConfGen - Schrödinger [https://www.schrodinger.com/platform/products/confgen/]
- 47. Ligand-based pharmacophore modeling, molecular ... [https://www.sciencedirect.com/science/article/pii/S2352914822002003]
- 48. Ligand-based pharmacophore modeling and molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9037691/]
- 49. Ligand-based virtual screening and QSAR modelling for ... [https://www.sciencedirect.com/science/article/pii/S3050787125000435]
- 50. Knowledge-guided diffusion model for 3D ligand ... [https://www.nature.com/articles/s41467-025-57485-3]
- 51. Pharmacophore-based virtual screening - PubMed [https://pubmed.ncbi.nlm.nih.gov/20838973/]
- 52. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6332202/]
- 53. Pharmacophore screening, molecular docking, and MD ... [https://pubmed.ncbi.nlm.nih.gov/40124780/]
- 54. Pharmacophore screening, molecular docking, and MD ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1534707/full]
- 55. Discovering anticancer drug target combinations via network ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12534536/]
- 56. New Computational Tool Shows Strong Accuracy in ... [https://ascopost.com/news/november-2025/new-computational-tool-shows-strong-accuracy-in-predicting-cancer-drug-targets]
- 57. PharmacoForge: pharmacophore generation with diffusion ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1628800/full]
- 58. Applications and Limitations of Pharmacophore Modeling [https://drugdiscoverypro.com/applications-and-limitations-of-pharmacophore-modeling/]
- 59. Pharmacophore modeling and 3D quantitative structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3645363/]
- 60. Conformer Generation Software | Omega [https://www.eyesopen.com/omega]
- 61. PharmaGist: a webserver for ligand-based pharmacophore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2447755/]
- 62. Enhanced Monte Carlo Methods for Modeling Proteins ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6311413/]
- 63. Knowledge-guided diffusion model for 3D ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11885826/]
- 64. Ligand-based Pharmacophore Modeling, Virtual Screening ... [https://www.sciencedirect.com/science/article/pii/S2001037018302617]
- 65. Phase - Schrödinger [https://www.schrodinger.com/platform/products/phase/]
- 66. Ligand efficiency based approach for efficient virtual ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523414005480]
- 67. Computational methods for modeling protein ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625000959]
- 68. Pharmacophore modeling for biological targets with high ... [https://www.sciencedirect.com/science/article/pii/S2590098622000161]
- 69. Induced fit, ensemble binding space docking and Monte ... [https://www.sciencedirect.com/science/article/pii/S2405844021018879]
- 70. Pharmacophore Modeling, Virtual Screening, and ... [https://www.medsci.org/v10p0265.htm]
- 71. Combining docking with pharmacophore filtering for improved ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3152774/]
- 72. A New Pharmacophore Model for the Design of Sigma-1 ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2019.00519/full]
- 73. Pharmacophore-Model-Based Virtual-Screening ... [https://www.mdpi.com/1467-3045/44/10/329]
- 74. Ligand based pharmacophore modelling and integrated ... [https://www.sciencedirect.com/org/science/article/pii/S2046206924002766]
- 75. ELIXIR-A: An Interactive Visualization Tool for Multi-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9025992/]
- 76. Using pharmacophore models to gain insight into structural ... [https://pubmed.ncbi.nlm.nih.gov/16563003/]
- 77. Choosing the Right Color Palette in Molecular Visualization [https://blog.samson-connect.net/choosing-the-right-color-palette-in-molecular-visualization]
- 78. Ligand and Pharmacophore based Design | BIOVIA [https://www.3ds.com/products/biovia/discovery-studio/ligand-and-pharmacophore-based-design]
- 79. A New Fragment‐Based Pharmacophore Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12412281/]
- 80. T101-2024 Notification regarding BIOVIA Discovery Studio ... [https://www.3ds.com/support/documentation/resource-library/t101-2024-notification-regarding-biovia-discovery-studio-2025]
- 81. Virtual screening based on pharmacophore model for ... [https://www.sciencedirect.com/science/article/abs/pii/S0048357522000761]
- 82. Tubulin inhibitors: pharmacophore modeling, virtual ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4088285/]
- 83. 3D-QSAR-Based Pharmacophore Modeling, Virtual ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8410400/]
- 84. GCAC: galaxy workflow system for predictive model building ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2492-8]
- 85. Benchmarking Methods and Data Sets for Ligand Enrichment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4278665/]
- 86. Maximal Unbiased Benchmarking Data Sets for Human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6197807/]
- 87. PharmRL: pharmacophore elucidation with deep geometric ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-024-02096-5]
- 88. Pharmacophore modeling and QSAR analysis of anti-HBV ... [https://pubmed.ncbi.nlm.nih.gov/39804828/]
- 89. Pharmacophore modeling and QSAR analysis of anti-HBV ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0316765]
- 90. Structure-Based Pharmacophore Modeling, Virtual ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8777599/]
- 91. Pharmacophore model, docking, QSAR, and molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8047494/]
- 92. Comparing pharmacophore models derived from crystal ... [https://link.springer.com/article/10.1007/s00706-016-1674-1]
- 93. How to calculate ROC curves - rDock [https://rdock.github.io/how-to-calculate-roc-curves/]
- 94. Schrodinger-Notes—Target-based Pharmacophore Modeling [https://blog.jiangshen.org/2024/schrodinger-notes-target-based-pharmacophore-modeling/]
- 95. Performance of a docking/molecular dynamics protocol for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5508530/]
- 96. A quantitative model of structure-based virtual screening ... [https://chemrxiv.org/engage/chemrxiv/article-details/69037e08113cc7cfff050553]
- 97. Pharmacophore-based virtual screening versus docking- ... [https://pubmed.ncbi.nlm.nih.gov/19935678/]
- 98. Pharmacophore and docking-based virtual screening ... [https://pubmed.ncbi.nlm.nih.gov/25148044/]
- 99. How good are AlphaFold models for docking-based virtual ... [https://www.sciencedirect.com/science/article/pii/S2589004222021939]
- 100. Is It Possible to Increase Hit Rates in Structure-Based ... [https://pubmed.ncbi.nlm.nih.gov/18203638/]
- 101. Pharmacophore-based virtual screening and in silico ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1531512/full]
- 102. Pharmacophore modelling based virtual screening and ... [https://www.nature.com/articles/s41598-024-63555-1]
- 103. deep learning-guided pharmacophore modeling for ultra-large ... [https://pubs.rsc.org/en/content/articlelanding/2024/sc/d4sc04854g]
- 104. Structure-Based Pharmacophore Design and Virtual ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6749218/]
- 105. Structure-Based Virtual Screening and Biological ... [https://www.mdpi.com/1420-3049/25/1/107]
- 106. Integrative Ligand-Based Pharmacophore Modeling, Virtual ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9912262/]
- 107. Schrödinger Notes—Ligand-based Pharmacophore Modeling [https://blog.jiangshen.org/2024/schrodinger-notes-ligand-based-pharmacophore-modeling/]
- 108. SILCS-Pharm — SilcsBio User Guide [https://docs.silcsbio.com/2025/silcs/silcs-pharm.html]