SYBR Green vs. Probe-Based Detection for Cancer Genes: A Strategic Guide for Precision Oncology Research
This article provides a comprehensive comparison of SYBR Green and probe-based (e.g., TaqMan) qPCR methods for analyzing cancer genes, tailored for researchers and drug development professionals.
SYBR Green vs. Probe-Based Detection for Cancer Genes: A Strategic Guide for Precision Oncology Research
Abstract
This article provides a comprehensive comparison of SYBR Green and probe-based (e.g., TaqMan) qPCR methods for analyzing cancer genes, tailored for researchers and drug development professionals. It covers foundational principles, including the distinct chemistries of DNA-binding dyes and fluorescent probes, and their application in detecting oncogenes, tumor suppressors, and emerging biomarkers like ctDNA and miRNAs. The content delivers practical methodological protocols, troubleshooting and optimization strategies for sensitive mutation detection, and a critical validation framework for assessing sensitivity, specificity, and cost-effectiveness. By synthesizing current data and real-world applications, this guide supports informed decision-making for robust, reliable genetic analysis in cancer research and diagnostic development.
Core Principles: Understanding SYBR Green and Probe-Based qPCR Chemistries
SYBR Green I represents a fundamental tool in molecular biology, enabling the detection and quantification of nucleic acids through its unique interaction with double-stranded DNA (dsDNA). This technical guide delves into the biophysical mechanism of SYBR Green, detailing how its fluorescence is activated upon binding to the minor groove of dsDNA. We explore the structure-property relationships that govern its binding modes, including intercalation and surface binding, and how these relate to its performance in quantitative real-time PCR (qPCR). Framed within the context of cancer gene research, this review provides a comparative analysis with probe-based detection methods (e.g., TaqMan), evaluating specificity, sensitivity, and cost-effectiveness for applications in gene expression profiling and biomarker validation. The discussion is supported by experimental data, detailed protocols, and analytical workflows relevant to researchers and drug development professionals.
SYBR Green I is an unsymmetrical cyanine dye that serves as a sensitive, fluorescent stain for nucleic acid detection and quantification [1] [2]. Its core structure consists of a benzothiazolium ring system linked by a monomethine bridge to a quinolinium ring system, with a substituent containing a heteroatom [1]. Initially developed as a safer and more quantifiable alternative to ethidium bromide for agarose gel electrophoresis, its applications have expanded to include dsDNA quantification in solution, flow cytometry, and most notably, real-time PCR [2] [1]. In the context of qPCR, SYBR Green I provides a simple and cost-effective means to monitor the amplification of DNA in real-time, which is crucial for gene expression analysis, genotyping, and pathogen detection [3] [4]. When discussing gene expression analysis, particularly for cancer research, the choice of detection chemistry—dye-based versus probe-based—can significantly impact the specificity, reliability, and cost of the experimental outcomes.
The Molecular Mechanism of Fluorescence Activation
The fluorescence of SYBR Green I is critically dependent on its binding to dsDNA. In its free, unbound state in solution, the dye exhibits very low fluorescence [5] [2]. However, upon binding to the minor groove of dsDNA, its fluorescence increases by up to 1,000-fold [5] [2]. This dramatic enhancement makes the fluorescent signal directly proportional to the amount of dsDNA present in the reaction tube [5].
Biophysical studies at defined dye-to-base pair ratios (dbpr) have revealed that SYBR Green I binds to dsDNA through two primary modes:
- Intercalation: This occurs at lower dbprs, where the dye molecules insert themselves between the base pairs of the DNA double helix [1].
- Surface Binding: At dbprs above approximately 0.15, the dye undergoes surface binding, which leads to a significant increase in fluorescence [1].
It is important to note that while SYBR Green I is highly selective for dsDNA, it can also bind to double-stranded RNA and, to a much lesser extent, single-stranded DNA (ssDNA), though the fluorescence from ssDNA complexes is at least 11-fold lower than that from dsDNA complexes [1]. The binding and subsequent fluorescence are also influenced by the DNA sequence, with studies showing sequence-specific binding using poly(dA) · poly(dT) and poly(dG) · poly(dC) homopolymers, and are affected by the salt composition of the buffer [1].
The following diagram illustrates the two binding modes of SYBR Green I and the resultant fluorescence activation.
SYBR Green in qPCR: A Comparative Framework with TaqMan
In quantitative real-time PCR (qPCR), SYBR Green I is used as an intercalating dye that binds to dsDNA PCR products as they are amplified [6] [7]. The accumulation of amplicons in each cycle is measured by the increasing fluorescent signal, allowing for the quantification of the initial target DNA. The cycle at which the fluorescence crosses a predetermined threshold (the Ct value) is used for quantification [7].
Key Advantages and Disadvantages in qPCR
The use of SYBR Green I in qPCR presents a distinct set of benefits and challenges, particularly when compared to probe-based methods like TaqMan.
Table 1: Comparison of SYBR Green and TaqMan qPCR Methods
| Feature | SYBR Green | TaqMan |
|---|---|---|
| Mechanism | Binds non-specifically to any dsDNA [6] [7] | Sequence-specific hydrolysis probe [5] [7] |
| Cost | Relatively cost-effective and inexpensive [5] [4] | More expensive due to labeled probes [5] [8] |
| Experimental Design | Easy setup; requires only primer design [6] | More complex; requires design of primers and probe [6] |
| Specificity | Lower; binds to primer-dimers and non-specific products [2] [6] | High; requires specific binding of both primer and probe [5] [6] |
| Multiplexing Capability | Not possible [3] | Possible with different colored probes [3] [7] |
| Required Post-PCR Analysis | Melting curve analysis essential [2] [7] | Not required [6] |
The Critical Role of Melting Curve Analysis
A mandatory step in any SYBR Green qPCR protocol is the melting curve analysis (also called dissociation curve analysis) [6] [7]. After the amplification cycles are complete, the temperature is gradually increased while fluorescence is continuously monitored. As the DNA melts and transitions from dsDNA to ssDNA, the SYBR Green dye is released, causing a drop in fluorescence. Plotting the negative derivative of this fluorescence change against temperature results in a melting peak specific to the amplicon's melting temperature (Tm) [7]. This analysis is crucial for verifying that a single, specific PCR product was amplified and for identifying the presence of primer-dimers or other non-specific products, which would appear as additional peaks [2] [7].
Application in Cancer Research: SYBR Green vs. Probe-Based Detection
The choice between SYBR Green and TaqMan chemistries is particularly relevant in cancer gene research, where accurately measuring gene expression, gene copy number variations, and mutations is paramount.
Quantitative Performance in Gene Expression Analysis
A 2014 study directly compared SYBR Green and TaqMan methods for quantifying the expression of adenosine receptor subtypes (A1, A2A, A2B, A3) in breast cancer tissues [5]. The researchers found that with the use of high-performance primers and optimized protocols, both methods demonstrated high amplification efficiencies (>95%) and produced positively correlated, significant data (p < 0.05) [5]. This indicates that for well-optimized assays, SYBR Green can yield data quality comparable to TaqMan for gene expression analysis [5].
Table 2: Performance Metrics from a Breast Cancer Gene Expression Study [5]
| Gene | Normalized Expression (SYBR Green) | Normalized Expression (TaqMan) | Correlation (Pearson) |
|---|---|---|---|
| A1 Adenosine Receptor | 1.44 | 1.38 | Positive and Significant (P < 0.05) |
| A2A Adenosine Receptor | 2.38 | 2.43 | Positive and Significant (P < 0.05) |
| A2B Adenosine Receptor | 3.79 | 3.84 | Positive and Significant (P < 0.05) |
| A3 Adenosine Receptor | 3.55 | 3.58 | Positive and Significant (P < 0.05) |
Sensitivity and Detection Limits
However, other studies highlight scenarios where TaqMan may be superior. In the detection of residual host-cell DNA from Chinese Hamster Ovary (CHO) cells—a common production host for biopharmaceuticals including cancer therapeutics—the TaqMan assay demonstrated a lower limit of detection (LOD) of 10 fg, compared to 100 fg for the SYBR Green assay [8]. Similarly, a study on enterotoxigenic Bacteroides fragilis (ETBF), which is associated with colorectal cancer, found that SYBR Green qPCR underperformed in clinical stool samples. It detected only 13 out of 38 positive samples, whereas TaqMan qPCR and digital PCR detected 35 and 36, respectively [9]. The copy numbers reported by TaqMan were 48-fold higher than those from SYBR Green for the same samples, underscoring TaqMan's superior sensitivity and reliability in complex sample matrices [9].
The following workflow outlines the decision process for selecting a detection chemistry in a cancer research qPCR experiment.
Experimental Protocol: SYBR Green qPCR for Gene Expression
The following protocol is adapted from a study analyzing adenosine receptor gene expression in breast cancer tissues, providing a template for cancer-related gene targets [5].
Sample Preparation and RNA Extraction
- Tissue Samples: Breast cancer tissue samples are quickly placed in liquid nitrogen and stored at -80°C [5].
- Homogenization and RNA Extraction: Homogenize 20-40 mg of frozen tissue (e.g., using a bead-milling method in RLT buffer). Extract total RNA using a commercial kit (e.g., RNeasy plus mini kit, Qiagen) [5].
- Quality Control: Determine the concentration and purity of RNA by measuring UV absorption at 260/280 nm (e.g., using a NanoDrop system). Assess RNA integrity by electrophoresis on a denaturing 1% agarose gel [5].
Reverse Transcription and qPCR Setup
- cDNA Synthesis: Reverse transcribe 1 μg of total RNA into complementary DNA (cDNA) using a reverse transcription kit (e.g., Quantitect Rev. transcription kit, Qiagen) [5].
- Primer Design: Design primers to span exon-exon junctions to avoid amplification of genomic DNA. Use software (e.g., Beacon Designer) for design and ensure specificity [5].
- qPCR Reaction:
- Reaction Mixture: 25 μL total volume containing: 2 μL cDNA template, 1.5 μL each of forward and reverse primer, and 12.5 μL of SYBR Green master mix (e.g., Quantitect SYBR Green master mix, Qiagen) [5].
- Thermal Cycling Conditions (Rotor Gene 6000):
- Initial denaturation: 95°C for 10 min.
- 40 cycles of:
- Denaturation: 95°C for 10 s.
- Annealing/Extension: 60°C for 20 s.
- Data Normalization: Include a reference gene (e.g., Beta-Actin, ACTB) in all runs to normalize the data and correct for variations in RNA quality and quantity. Calculate relative expression using the ΔΔCt method [5].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for SYBR Green qPCR
| Item | Function/Description | Example Product/Citation |
|---|---|---|
| SYBR Green Master Mix | Optimized buffer, nucleotides, polymerase, and SYBR Green dye for qPCR. | Quantitect SYBR Green Master Mix (Qiagen) [5] |
| RNA Extraction Kit | Purifies high-quality, DNA-free total RNA from tissues or cells. | RNeasy Plus Mini Kit (Qiagen) [5] |
| Reverse Transcription Kit | Converts RNA template into stable cDNA for PCR amplification. | Quantitect Reverse Transcription Kit (Qiagen) [5] |
| Validated Primers | Sequence-specific oligonucleotides designed to flank the target amplicon. | Custom designed, e.g., by Beacon Designer software [5] |
| Nuclease-Free Water | Solvent free of RNases and DNases to prevent nucleic acid degradation. | Essential component of reaction mix [5] [4] |
SYBR Green I functions through a elegant mechanism of fluorescence enhancement upon binding to dsDNA, involving both intercalation and surface binding. Its utility in qPCR, especially for cancer gene research, is defined by a trade-off between cost-effectiveness and ease of use on one hand, and specificity and sensitivity on the other. While well-optimized SYBR Green assays can produce data comparable to TaqMan methods for standard gene expression analysis, probe-based methods retain a distinct advantage for applications requiring maximal specificity, sensitivity in complex samples, and multiplexing capabilities. The decision to use SYBR Green or a probe-based alternative should be guided by the specific experimental goals, the nature of the target, and the required level of precision.
In the pursuit of reliable biomarkers for cancer research, quantitative polymerase chain reaction (qPCR) stands as a fundamental technology for analyzing gene expression. While DNA-binding dye-based methods like SYBR Green provide a cost-effective solution, probe-based detection systems, with TaqMan chemistry being the most prominent, offer a critical advantage: unparalleled specificity. This specificity is paramount when accurately quantifying cancer-associated genes or identifying single-nucleotide polymorphisms (SNPs) in complex biological samples such as tumor tissues. The TaqMan probe principle, first reported in 1991, relies on the 5´–3´ exonuclease activity of Taq polymerase to cleave a dual-labeled probe during hybridization, enabling fluorophore-based detection [10]. This mechanism, combined with Fluorescence Resonance Energy Transfer (FRET)-based quenching, forms the basis for a highly specific detection system that significantly reduces false-positive signals common in non-specific dye methods [5].
The core components of a TaqMan probe system are an unlabeled primer pair and a dual-labeled oligonucleotide probe. The probe is covalently linked to a fluorophore (reporter dye) at its 5' end and a quencher at its 3' end [10] [11]. Several fluorophores are commonly used, including 6-carboxyfluorescein (FAM) and tetrachlorofluorescein (TET), while the quencher is typically a non-fluorescent molecule such as a Black Hole Quencher (BHQ) or tetramethylrhodamine (TAMRA) [10] [12]. The physical proximity between the fluorophore and quencher when the probe is intact results in quenching of the reporter's fluorescence, primarily through FRET and/or contact quenching mechanisms [13] [12]. FRET is a quantum phenomenon where excitation is transferred from a donor fluorophore to an acceptor quencher through non-radiative dipole-dipole interaction, provided they are within 1-10 nanometers and have sufficient spectral overlap [13]. The efficiency of this energy transfer is inversely proportional to the sixth power of the distance between the donor and acceptor, making it extremely distance-dependent [13] [12].
Mechanism of TaqMan Probes and FRET Quenching
The TaqMan Workflow
The TaqMan detection process is an elegant integration of enzymatic amplification and fluorescent signaling that occurs during the PCR thermal cycling. The mechanism can be broken down into four key stages, illustrated in the diagram below.
- Denaturation: The temperature is raised (typically to 95°C) to separate the double-stranded DNA template into single strands [11].
- Annealing: The temperature is lowered to allow both the forward and reverse primers and the TaqMan probe to anneal to their specific complementary sequences on the template DNA. The probe binds downstream from one of the primers [10] [11].
- Extension and Cleavage: Taq DNA polymerase extends the primer, synthesizing a new DNA strand. When the polymerase reaches the bound TaqMan probe, its 5' to 3' exonuclease activity cleaves the probe [10] [11]. This hydrolysis step is the defining feature of the chemistry.
- Signal Detection: Cleavage of the probe separates the fluorophore from the quencher. Once physically separated, the quencher can no longer suppress the fluorescence of the reporter dye. The fluorophore is now free to emit fluorescence when excited by the real-time PCR instrument's light source [10] [11].
This process repeats in every PCR cycle, leading to an accumulation of fluorescence signal that is directly proportional to the amount of amplified product. The fluorescence is detected in real-time, and the cycle at which the fluorescence crosses a predefined threshold (Ct value) is used for quantitative analysis.
Advanced Quenching Mechanisms
While the core principle involves distance-dependent quenching, the underlying physical mechanisms are nuanced. The quenching in dual-labeled probes operates through two primary pathways, which can occur simultaneously.
- Förster Resonance Energy Transfer (FRET): This is a "through-space" mechanism where energy is transferred from the excited-state fluorophore (donor) to the quencher (acceptor) without photon emission. Efficient FRET requires [13] [12]:
- Proximity: The donor and acceptor must be within approximately 10–100 Å.
- Spectral Overlap: The emission spectrum of the donor must overlap with the absorption spectrum of the acceptor.
- Favorable Orientation: The relative orientation of the donor and acceptor dipoles affects the transfer efficiency.
- Static Quenching (Contact Quenching): This mechanism involves the direct physical contact between the fluorophore and quencher, forming a non-fluorescent ground-state complex or intramolecular dimer. This association is driven by the tendency of hydrophobic, planar dye molecules to stack in an aqueous solution. Static quenching is highly dependent on temperature and solvent conditions [12].
The evolution of quencher technology has been critical to the performance of modern TaqMan assays. Early quenchers like TAMRA were fluorescent themselves, contributing to background noise. The development of dark quenchers like the Black Hole Quencher (BHQ) family, which have broad absorption spectra and no native fluorescence, has significantly improved the signal-to-noise ratio and enabled multiplexing experiments [12]. The following table summarizes the key distinctions between these quenching mechanisms.
Table 1: Comparison of Quenching Mechanisms in Oligonucleotide Probes
| Feature | FRET Quenching | Static Quenching |
|---|---|---|
| Mechanism | Through-space energy transfer | Direct contact and complex formation |
| Physical Contact | Not required | Required |
| Temperature Dependence | Not very temperature sensitive | Decreases with increasing temperature |
| Effect on Absorption Spectrum | Unchanged | Often distorted due to complex formation |
| Dye Pair Example | FAM and TAMRA | FAM and BHQ-1 [12] |
TaqMan vs. SYBR Green: A Quantitative Comparison for Cancer Research
Selecting the appropriate detection chemistry is a critical decision in experimental design. The choice between the probe-based TaqMan method and the dye-based SYBR Green method involves a trade-off between cost, convenience, and specificity.
SYBR Green binds non-specifically to the minor groove of all double-stranded DNA (dsDNA) generated during PCR, leading to a fluorescence increase of over 1,000-fold compared to the unbound state [5]. While this method is relatively cost-effective and easy to use, its major limitation is a lack of inherent specificity; it cannot distinguish between the desired target amplicon and any non-specific PCR products, such as primer-dimers [5]. This can lead to overestimation of the target concentration or false-positive results.
In contrast, TaqMan assays require the specific hybridization of the probe to the target sequence for a fluorescence signal to be generated. This additional layer of specificity ensures that the detected signal originates only from the intended amplicon. This is particularly crucial in cancer research for applications like SNP genotyping, determining viral load, verifying microarray results, and accurately quantifying low-abundance transcripts [10].
Empirical studies directly comparing these methods underscore the performance differences. Research comparing the detection of the enterotoxigenic Bacteroides fragilis (ETBF) bft gene, relevant to colorectal cancer, found that while SYBR Green, TaqMan qPCR, and digital PCR (dPCR) had comparable limits of detection (<1 copy/μl) for purified bacterial DNA, their performance diverged significantly in complex clinical stool samples [9]. SYBR Green qPCR reported only 13 out of 38 samples as positive, whereas TaqMan qPCR and dPCR detected the gene in 35 and 36 samples, respectively. Furthermore, the reported bft copy numbers from TaqMan qPCR and dPCR were 48-fold and 75-fold higher than those from SYBR Green for the same samples, demonstrating SYBR Green's potential for underestimation in the presence of sample-derived inhibitors or background [9].
Another study on adenosine receptor expression in breast cancer tissues found that with high-performance primers and optimized protocols, SYBR Green could produce data comparable to TaqMan, with efficiencies above 95% and significant positive correlations between the methods [5]. This suggests that for well-optimized, single-target assays where non-specific amplification is minimal, SYBR Green can be a valid option.
Table 2: Performance Comparison of SYBR Green vs. TaqMan qPCR
| Parameter | SYBR Green | TaqMan |
|---|---|---|
| Chemistry Basis | Binds to dsDNA minor groove [5] | Sequence-specific hydrolysis probe [10] [5] |
| Specificity | Lower (detects all dsDNA) [5] | Higher (detects only target sequence) [5] [9] |
| Cost | Lower | Higher |
| Ease of Use & Design | Simpler (only primers needed) | More complex (requires probe design) |
| Multiplexing Potential | Not possible | Possible with multiple dye/quencher pairs [11] [12] |
| Signal-to-Noise Ratio | Can be high with optimized primers | Typically very high due to dark quenchers [12] |
| Best Application | Single-target gene expression, initial screening | SNP detection, low-abundance targets, multiplexing [10] |
Experimental Protocols and Applications
Detailed Protocol for TaqMan Gene Expression Assay
The following protocol is adapted from methodologies used in studies of adenosine receptor gene expression in breast cancer tissues and the detection of the bft gene [5] [9].
RNA Extraction:
- Homogenize 20-40 mg of frozen tissue (e.g., breast cancer tissue) in a suitable lysis buffer.
- Extract total RNA using a commercial kit (e.g., RNeasy plus mini kit, Qiagen).
- Determine the RNA concentration and purity by measuring UV absorption at 260/280 nm (optimal ratio: ~2.0). Verify RNA integrity by denaturing agarose gel electrophoresis [5].
Reverse Transcription (cDNA Synthesis):
- Use 1 μg of total RNA for reverse transcription.
- Perform the reaction using a commercial reverse transcription kit (e.g., Quantitect Rev. transcription kit, Qiagen) with a mix of random hexamers and oligo-dT primers to ensure comprehensive cDNA representation [5].
Real-Time Quantitative PCR Setup:
- Reaction Mixture (25 μl total volume):
- For TaqMan Assay: 2 μl of cDNA template, 12.5 μl of TaqMan Universal PCR Master Mix (contains DNA polymerase, dNTPs, and optimized buffer), 1.5 μl of pre-designed primer and probe mix (Assays-on-Demand Gene Expression Products), and nuclease-free water to 25 μl [5] [11].
- For Custom SYBR Green Assay (Comparison): 2 μl of cDNA template, 12.5 μl of SYBR Green Master Mix (e.g., Quantitect SYBR Green master mix, Qiagen), 1.5 μl each of forward and reverse primer (final concentration typically 200-500 nM each), and nuclease-free water to 25 μl [5].
- Include a no-template control (NTC) and a positive control in each run. For absolute quantification, a standard curve of known copy numbers must be included.
- Reaction Mixture (25 μl total volume):
Thermal Cycling Conditions (TaqMan):
- Initial Denaturation: 95°C for 10 minutes (activates the hot-start polymerase).
- Amplification (40-50 cycles):
- Denature: 95°C for 10-15 seconds.
- Anneal/Extend: 60°C for 20-60 seconds (fluorescence data collection occurs at this step).
- The thermal profile for SYBR Green is similar but typically concludes with a melt curve analysis to check amplicon specificity [5] [9].
Data Analysis:
- The qPCR software will generate a Ct (threshold cycle) value for each reaction.
- For relative gene expression analysis, normalize the Ct of the gene of interest (GOI) to the Ct of a reference gene (e.g., Beta-actin, ACTB) using the ΔΔCt method: ΔCt = Ct(GOI) - Ct(Reference Gene). Fold-change can be calculated as 2^(-ΔΔCt) [5].
The Scientist's Toolkit: Essential Reagents for TaqMan Assays
Table 3: Key Research Reagent Solutions for Probe-Based Detection
| Reagent / Tool | Function | Example Products |
|---|---|---|
| TaqMan Master Mix | Provides the essential components for PCR: hot-start Taq DNA polymerase, dNTPs, MgCl₂, and optimized reaction buffer. | TaqMan Universal PCR Master Mix, TaqMan Genotyping Master Mix [11] [9] |
| Assay-on-Demand | Pre-optimized, ready-to-use primer and probe sets for specific gene targets, saving time on design and validation. | Applied Biosystems TaqMan Gene Expression Assays [5] [11] |
| Custom TaqMan Assays | Tailored primer and probe sets designed for unique targets, such as specific mutations or novel transcripts. | Custom TaqMan SNP Genotyping Assays [11] |
| RNA Extraction Kit | Isolates high-quality, intact total RNA from complex starting materials like tumor tissues. | RNeasy Plus Mini Kit (Qiagen) [5] [9] |
| Reverse Transcription Kit | Converts RNA template into stable complementary DNA (cDNA) for subsequent PCR amplification. | Quantitect Reverse Transcription Kit [5] |
| Non-Fluorescent Quencher (NFQ) | A dark quencher that minimizes background fluorescence, leading to a higher signal-to-noise ratio. | Minor Groove Binder (MGB)-NFQ probes [11] [12] |
Advanced Applications and Future Directions in Cancer Research
The specificity of TaqMan chemistry has enabled its adaptation for sophisticated applications beyond standard gene expression quantification, playing a critical role in advancing cancer research and molecular diagnostics.
One of the most significant applications is in SNP genotyping and mutation detection. TaqMan genotyping assays utilize two probes, each labeled with a different fluorophore (e.g., FAM and VIC) and specific for one allele of the SNP. The exonuclease activity leads to fluorescence specific to the allele present in the sample, allowing for clear discrimination [11]. This is vital for identifying somatic mutations in oncogenes and tumor suppressors. Furthermore, technologies like competitive allele-specific TaqMan (castPCR) have been developed to enhance the detection of rare mutations in a background of wild-type DNA, a common challenge in liquid biopsy analysis [11]. This method combines allele-specific qPCR with an allele-specific MGB blocker oligonucleotide to effectively suppress nonspecific amplification of the non-target allele, enabling highly sensitive and specific mutation detection [11].
The principles of nucleic acid probe-based detection continue to evolve. Enzymatic methods using CRISPR-Cas systems or Argonaute (Ago) proteins are being developed for point mutation detection with extremely high sensitivity, capable of detecting mutant alleles at frequencies as low as 0.01% [14]. For instance, the PAND (PfAgo-mediated Nucleic Acid Detection) system uses the Pyrococcus furiosus Argonaute protein to cleave target DNA guided by nucleic acids, generating a fluorescent signal and enabling the detection of SNPs in genes like BRCA1, KRAS, and EGFR [14]. Similarly, primer exchange reaction (PER)-based signal amplification strategies have demonstrated the ability to detect cancer-associated single-nucleotide mutations in circulating tumor DNAs (ctDNAs) with a limit of detection down to 27 fM, even discriminating mutant sequences in the presence of a 1000-fold excess of wild-type DNA [15].
These advanced techniques, building upon the foundational principle of probe-based specificity exemplified by TaqMan chemistry, are pushing the boundaries of cancer diagnostics towards earlier detection, minimal residual disease monitoring, and comprehensive profiling of tumor heterogeneity. The workflow below illustrates how these advanced probe-based methods integrate into a comprehensive cancer research pipeline.
Cancer is fundamentally a genetic disease driven by somatic mutations that alter the normal functioning of critical genes. These alterations are categorized into several classes, with oncogenes, tumor suppressor genes, and fusion genes representing the most critical targets for research and therapeutic development. Oncogenes, such as KRAS and EGFR, are mutated forms of normal proto-oncogenes that promote cell growth and division; their activation is typically a gain-of-function event. In contrast, tumor suppressor genes like TP53 normally function to control cell division and repair DNA damage; their inactivation through loss-of-function mutations removes crucial cellular brakes on tumorigenesis. Fusion genes, created by chromosomal rearrangements, can produce chimeric proteins with potent oncogenic activity. The detection and characterization of mutations in these genes have become central to precision oncology, enabling molecular subtyping of tumors and guiding targeted treatment strategies [16] [17].
The efficacy of any detection methodology is paramount, as accurately identifying these genetic alterations directly impacts diagnostic, prognostic, and therapeutic decisions. This technical guide explores these key genetic targets within the specific context of methodological approaches, particularly comparing SYBR Green versus probe-based detection systems in cancer gene research. Each method offers distinct advantages and limitations in sensitivity, specificity, multiplexing capability, and cost, factors that researchers must carefully balance based on their experimental objectives and resource constraints.
Oncogenes: KRAS and EGFR
KRAS: The Prevalent Oncogene
The Kirsten rat sarcoma viral oncogene homolog (KRAS) encodes a small GTPase that functions as a molecular switch, cycling between an active GTP-bound state and an inactive GDP-bound state to regulate cell proliferation, survival, and differentiation [18]. It is the most frequently mutated oncogene in human cancers, with particularly high prevalence in pancreatic ductal adenocarcinoma (PDAC) (82.1%), colorectal cancer (CRC) (~40%), and non-small cell lung cancer (NSCLC) (21.20%) [19]. Approximately 98% of mutations occur at codons 12, 13, and 61, with G12D (29.19%), G12V (22.17%), and G12C (13.43%) being the most common subtypes [19].
Oncogenic mutations in KRAS impair its GTPase activity or confer resistance to GTPase-activating proteins (GAPs), resulting in constitutive activation of KRAS and sustained downstream signaling through effector pathways like RAF-MEK-ERK and PI3K-AKT-mTOR [19] [18]. This leads to increased cellular survival and proliferation. The G12C mutation, which contains a unique cysteine residue, has been successfully targeted by covalent inhibitors such as sotorasib and adagrasib, approved for treating NSCLC [19] [18]. However, response rates remain around 30-40% with median progression-free survival of approximately 6 months, and resistance mechanisms frequently emerge [19]. Other mutations, like G12D and G12V, currently lack effective targeted therapies, spurring research into novel approaches including pan-KRAS inhibitors, targeted degraders, and RNA-based strategies [18].
EGFR: A Receptor Tyrosine Kinase Oncogene
The Epidermal Growth Factor Receptor (EGFR) is a receptor tyrosine kinase that activates downstream signaling cascades, including the MAPK and PI3K pathways, upon ligand binding. Mutations in EGFR, particularly in NSCLC, lead to constitutive, ligand-independent activation of its kinase domain, driving uncontrolled cell proliferation [20]. Common activating mutations include exon 19 deletions and the L858R point mutation in exon 21. Another critical mutation, T790M, is a major mechanism of resistance to first-generation EGFR tyrosine kinase inhibitors (TKIs) [14]. Detection of EGFR mutations is essential for initiating targeted therapy with EGFR TKIs and for monitoring the emergence of resistance.
Table 1: Prevalence of Key Oncogene Mutations in Solid Tumors
| Cancer Type | KRAS Mutation Prevalence | Common KRAS Subtypes | EGFR Mutation Prevalence |
|---|---|---|---|
| Pancreatic Ductal Adenocarcinoma | 82.1% | G12D (37.0%), G12V | Not a primary driver |
| Colorectal Cancer (CRC) | ~40% | G12D (12.5%), G12V (8.5%) | Less common |
| Non-Small Cell Lung Cancer (NSCLC) | 21.20% | G12C (13.6%) | 23.2% (in Indo-Asian population) [20] |
| Cholangiocarcinoma | 12.7% | Information not specified | Information not specified |
| Uterine Endometrial Carcinoma | 14.1% | Information not specified | Information not specified |
KRAS Signaling Pathway
The following diagram illustrates the KRAS signaling pathway, showing how oncogenic mutations lead to constitutive activation of downstream effectors.
Diagram Title: KRAS Oncogenic Signaling Pathway
Tumor Suppressor Gene: TP53
The TP53 gene encodes the p53 protein, a critical tumor suppressor often dubbed "the guardian of the genome." In response to cellular stress, such as DNA damage, p53 acts as a transcription factor to activate genes that orchestrate cell cycle arrest, DNA repair, senescence, or apoptosis (programmed cell death) [16]. This prevents the propagation of damaged cells and suppresses tumor development.
TP53 is the most frequently mutated gene in human cancer, with alterations occurring in over 50% of all malignancies [20]. Most TP53 mutations are missense mutations that result in a loss of its tumor-suppressive function. Some mutations also confer a "gain-of-function" phenotype, where the mutant p53 protein acquires new oncogenic properties that promote tumor invasion, metastasis, and chemoresistance. In lung cancer, TP53 mutations are among the most common alterations, with a reported frequency of 17.7%, often co-occurring with other driver mutations like EGFR and KRAS [20]. The presence of TP53 mutations is frequently associated with more aggressive disease and poorer prognosis across multiple cancer types.
Fusion Genes in Cancer
Fusion genes are created when chromosomal rearrangements—such as translocations, deletions, or inversions—join parts of two separate genes. This results in a chimeric gene that can produce a fusion protein with oncogenic potential. These aberrant proteins often possess constitutive kinase or transcription factor activity that drives uncontrolled cell growth.
Well-known examples include the BCR-ABL1 fusion in chronic myeloid leukemia (CML), the EML4-ALK fusion in a subset of NSCLC, and TMPRSS2-ERG fusions in prostate cancer. The detection of fusion genes is highly clinically relevant, as it can dictate the use of specific targeted therapies, such as ALK inhibitors for EML4-ALK positive NSCLC. Advanced sequencing technologies, particularly RNA sequencing (RNA-Seq), are highly effective for discovering and detecting these chimeric transcripts [17].
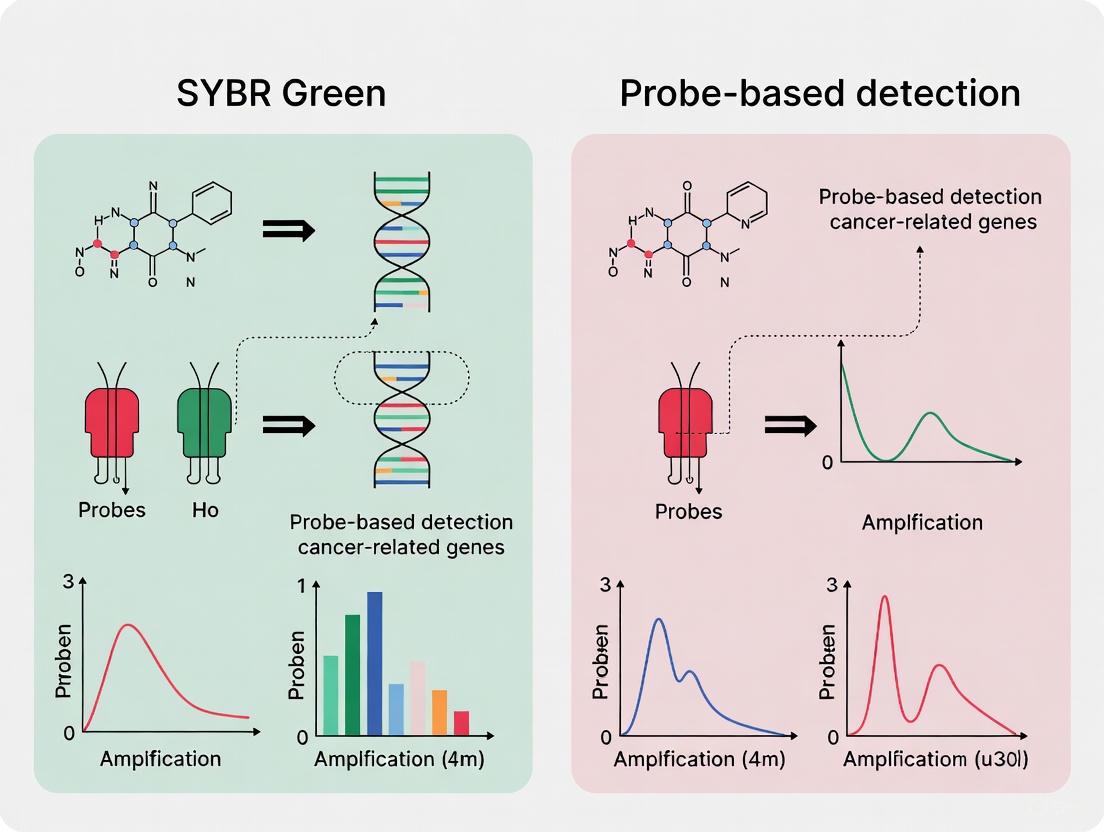
Detection Methodologies: SYBR Green vs. Probe-Based Assays
The accurate detection of mutations in genes like KRAS, EGFR, and TP53, as well as fusion genes, is foundational to cancer research and precision medicine. Quantitative PCR (qPCR) is a cornerstone technique for this purpose, primarily utilizing two detection chemistries: SYBR Green and probe-based systems (e.g., TaqMan).
SYBR Green Chemistry
SYBR Green is a fluorescent dye that intercalates non-specifically into the minor groove of double-stranded DNA (dsDNA). The fluorescence signal increases proportionally with the amount of amplified PCR product.
- Principles and Workflow: The dye binds to any dsDNA generated during PCR amplification. After each cycle, fluorescence is measured. Following amplification, a melt curve analysis is performed by gradually increasing the temperature and measuring the decrease in fluorescence. The temperature at which the dsDNA denatures (Tm) is specific to the amplicon's length, GC content, and sequence, allowing for product identification [16] [21].
- Advantages: It is cost-effective as it does not require a specific probe for each target. The same dye can be used for any assay, making it highly flexible. It is well-suited for gene expression analysis and genotyping when followed by melt curve analysis [16].
- Disadvantages: The lack of sequence specificity is a major drawback. SYBR Green will bind to any non-specific amplification product or primer-dimer, potentially leading to false-positive signals and over-quantification. Melt curve analysis adds time to the protocol and may not always adequately distinguish between amplicons with similar Tm values [16].
Probe-Based Chemistry (TaqMan Assays)
TaqMan assays utilize a sequence-specific oligonucleotide probe labeled with a fluorescent reporter dye at one end and a quencher molecule at the other.
- Principles and Workflow: The probe hybridizes to a specific sequence within the target amplicon. During PCR, the 5' to 3' exonuclease activity of the Taq polymerase cleaves the probe, separating the reporter from the quencher and generating a fluorescent signal. The fluorescence intensity is directly proportional to the amount of probe cleased, and thus, to the amount of target amplified [16].
- Advantages: This method offers superior specificity and reliability because fluorescence is generated only if the specific probe sequence is amplified. It allows for multiplexing—the simultaneous detection of multiple targets in a single reaction by using probes labeled with different reporter dyes. This is crucial for detecting single nucleotide polymorphisms (SNPs) or multiple mutation types from a single sample [14] [16].
- Disadvantages: Probe-based assays are more expensive to design and synthesize. Each target requires a unique, optimized probe, reducing flexibility. The presence of a probe can sometimes slightly reduce amplification efficiency [16].
Comparison for Cancer Gene Detection
For detecting critical cancer mutations, the high specificity of probe-based assays makes them the preferred choice for clinical validation and diagnostic applications. Their ability to distinguish single-nucleotide changes, such as the G12D versus G12V mutation in KRAS, is essential. The multiplexing capability is also vital for efficient profiling of multiple mutation hotspots from limited patient material.
SYBR Green-based assays, while more affordable and flexible, are generally more suitable for initial screening or research applications where absolute specificity is less critical, provided that primer design is highly optimized and melt curve analysis can reliably distinguish between amplicons.
Table 2: Comparison of SYBR Green vs. Probe-Based qPCR Detection Methods
| Feature | SYBR Green | Probe-Based (e.g., TaqMan) |
|---|---|---|
| Principle | Intercalates into any dsDNA | Sequence-specific hybridization and cleavage |
| Specificity | Lower; relies on primers and melt curve | Very High; requires exact probe match [16] |
| Multiplexing | Not possible in a single tube | Yes; with different fluorescent dyes [16] |
| Cost | Lower (universal dye) | Higher (custom probe required) |
| Best For | Gene expression, initial screening, genotyping with clear Tm differences | SNP detection, mutation screening, multiplex assays [14] [16] |
| Sensitivity | Can be compromised by primer-dimers | High and more reliable [21] |
Advanced Detection Technologies and Workflows
While qPCR is a workhorse for targeted detection, advanced technologies are employed for broader genomic profiling. Next-Generation Sequencing (NGS) allows for the parallel sequencing of millions of DNA fragments, providing a comprehensive view of mutations, copy number variations, and gene fusions across hundreds of genes simultaneously [17]. It is becoming the standard of care for patients with advanced solid tumors.
Emerging methods are also showing significant promise. CRISPR-Cas systems can be harnessed for mutation detection. For example, the DASH (Depletion of Abundant Sequences by Hybridization) method uses Cas9 to selectively cleave wild-type sequences, enriching mutant alleles for subsequent detection with sensitivity as low as 0.1% [14]. Similarly, Argonaute (Ago) proteins, such as PfAgo, can be programmed with guide strands to specifically cleave nucleic acids, enabling the development of highly sensitive detection platforms like PAND and NAVIGATER, the latter achieving a detection limit of 0.01% for mutations in liquid biopsies [14].
Furthermore, Surface-Enhanced Raman Scattering (SERS) is being explored as a label-free method to detect epigenetic modifications like DNA methylation, which are also crucial in cancer. A 2025 study identified a SERS band at 790 cm⁻¹ that strongly correlated with 5-methylcytosine levels, demonstrating its potential for cancer diagnosis [22].
Another groundbreaking development is the use of Artificial Intelligence (AI). The SEQUOIA AI tool can predict the activity of thousands of genes, including established cancer gene signatures, directly from standard histopathology biopsy images [23]. This approach could potentially infer genetic alterations from routine images, reducing the need for costly genetic tests.
Experimental Protocol: Detection of a KRAS Point Mutation using SYBR Green qPCR with Melt Curve Analysis
The following workflow outlines a standard protocol for detecting a specific KRAS mutation, such as G12C, using SYBR Green-based qPCR.
Diagram Title: SYBR Green qPCR Workflow for Mutation Detection
Detailed Protocol Steps:
- DNA Extraction and Quantification: Extract high-quality genomic DNA from patient samples (e.g., formalin-fixed paraffin-embedded (FFPE) tissue or circulating tumor DNA from blood). Precisely quantify the DNA using a spectrophotometer or fluorometer [16] [21].
- Primer Design: Design primers that flank the mutation site of interest (e.g., codon 12 of KRAS). Primers should be 18-22 bases long with a Tm of 58-60°C and a GC content of 40-60%. To ensure amplification of cDNA only (and not genomic DNA contamination), design primers to span an exon-exon junction if possible [16].
- qPCR Reaction Setup:
- Prepare a reaction mix containing: 1x SYBR Green Master Mix (containing DNA polymerase, dNTPs, Mg²⁺, and SYBR Green dye), forward and reverse primers (typically 0.2-0.5 µM each), and template DNA (10-100 ng) [21].
- Include no-template controls (NTC) to check for contamination and positive controls (wild-type and known mutant DNA) for calibration.
- Thermal Cycling: Run the qPCR protocol with the following steps:
- Initial Denaturation: 95°C for 5-10 minutes.
- Amplification (40-50 cycles):
- Denaturation: 95°C for 15-30 seconds.
- Annealing: 60°C (or optimized Tm) for 30-60 seconds.
- Extension: 72°C for 30-60 seconds. Fluorescence data is acquired at the end of this step.
- Melt Curve Analysis: After cycling, run the melt curve protocol:
- Gradually increase the temperature from 60°C to 95°C (e.g., 0.3°C increments) while continuously monitoring fluorescence.
- Plot the negative derivative of fluorescence versus temperature (-dF/dT vs. T) to generate distinct melt peaks.
- Data Interpretation: The cycle threshold (Ct) indicates the initial quantity of the target. The melt peak Tm is used for genotyping. A single peak at the wild-type Tm indicates a wild-type sample. A single peak at a lower Tm indicates a homozygous mutant. Two distinct peaks indicate a heterozygous sample containing both wild-type and mutant alleles [16].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Cancer Gene Detection Research
| Reagent / Tool | Function | Example Application |
|---|---|---|
| SYBR Green Master Mix | Provides all components for dsDNA detection in qPCR; enables melt curve analysis. | Gene expression profiling; initial screening for genetic alterations [16]. |
| TaqMan Probe Assays | Provide sequence-specific detection for SNPs and mutations in multiplex qPCR. | Differentiating between KRAS G12D and G12V mutations; EGFR T790M detection [14] [16]. |
| Next-Generation Sequencing (NGS) Panels | High-throughput, parallel sequencing of multiple cancer-related genes from a single sample. | Comprehensive genomic profiling of solid tumors; identifying co-mutations in TP53 and KRAS [17]. |
| CRISPR-Cas9 System (e.g., DASH) | Programmable nuclease for selective depletion of wild-type sequences to enrich mutant alleles. | Enriching low-abundance KRAS mutations in a background of wild-type DNA prior to sequencing [14]. |
| Argonaute Proteins (e.g., PfAgo, TtAgo) | Nucleic acid-guided nucleases for highly specific cleavage and detection of mutant sequences. | Ultra-sensitive detection of KRAS G12D or EGFR T790M in liquid biopsy samples [14]. |
| SERS Substrates (e.g., Silver Nanoparticles) | Plasmonic nanoparticles for label-free, enhanced spectroscopic detection of molecular structures. | Detecting global DNA methylation levels via 5-methylcytosine-associated SERS bands [22]. |
Quantitative Polymerase Chain Reaction (qPCR) stands as a cornerstone technology in molecular biology, providing researchers with a powerful tool for quantifying nucleic acids with exceptional sensitivity and speed. In modern cancer biomarker analysis, qPCR has evolved beyond traditional gene expression studies to become an indispensable technology for liquid biopsy applications, enabling non-invasive cancer detection, prognosis, and treatment monitoring. The method's capacity to detect as little as a single copy of a target sequence makes it particularly valuable for analyzing scarce targets such as circulating tumor DNA (ctDNA) and microRNAs (miRNAs) found in biological fluids [24] [25]. This technical guide explores the fundamental principles, applications, and methodological considerations of qPCR in cancer research, with particular emphasis on the critical comparison between SYBR Green and probe-based detection systems within the context of cancer gene analysis.
The integration of qPCR into liquid biopsy workflows has transformed cancer diagnostics by providing minimally invasive alternatives to traditional tissue biopsies. Liquid biopsy focuses on detecting cancer biomarkers in bodily fluids such as blood, urine, and saliva, offering real-time insights into tumor biology [26]. These biomarkers include circulating tumor cells (CTCs), ctDNA, miRNAs, and exosomes, all of which can be quantified using qPCR technologies [26]. As cancer remains a leading cause of mortality worldwide, with over 10 million deaths annually [25], the development of rapid, sensitive, and specific diagnostic tools is paramount for improving patient outcomes through early detection and personalized treatment strategies.
Fundamental Principles of qPCR Detection Chemistry
Basic qPCR Mechanism and Fluorescence Detection
qPCR operates on the principle of detecting fluorescence signals that increase proportionally with the amount of amplified DNA during PCR cycles. The process involves four distinct phases: linear ground phase, early exponential phase, linear exponential phase (log phase), and plateau phase [24]. The cycle threshold (Ct) value, determined during the early exponential phase when the fluorescence signal rises above background levels, provides the quantitative measurement used to determine initial template concentration [24]. Lower Ct values indicate higher initial target concentrations, enabling precise quantification of nucleic acid targets across a wide dynamic range.
The fundamental workflow of qPCR begins with template preparation, followed by thermal cycling that includes denaturation, annealing, and extension steps. During amplification, the fluorescence signal is monitored in real-time, generating amplification curves that can be analyzed to determine the starting quantity of the target sequence. This process requires essential components including: DNA or cDNA template, thermostable DNA polymerase, primers, dNTPs, MgCl2, buffer, and a fluorescent reporter molecule [24].
Comparison of Detection Chemistries
Table 1: Comparison of SYBR Green vs. Probe-Based qPCR Detection Methods
| Parameter | SYBR Green (Dye-Based) | Probe-Based (TaqMan) |
|---|---|---|
| Detection Mechanism | Binds nonspecifically to double-stranded DNA | Sequence-specific probe hydrolysis or hybridization |
| Specificity | Lower - detects all dsDNA including primer dimers | Higher - only detects specific target sequence |
| Multiplexing Capability | No - single target per reaction | Yes - multiple targets with different fluorophores |
| Cost Considerations | Lower cost - requires only primers | Higher cost - requires labeled probes |
| Workflow Complexity | Simple primer design and optimization | Complex probe and primer design |
| Background Fluorescence | Higher background signal | Lower background fluorescence |
| Post-PCR Verification | Requires melting curve analysis | Not required |
| Best Applications | Gene expression screening, target validation | SNP genotyping, pathogen detection, multiplex assays |
Visualization of qPCR Detection Mechanisms
The following diagram illustrates the fundamental differences in detection mechanisms between SYBR Green and probe-based qPCR approaches:
Probe-Based qPCR Assays: Mechanisms and Applications
TaqMan Probe Chemistry and Variants
Probe-based qPCR relies on the 5'→3' exonuclease activity of Taq DNA polymerase and sequence-specific oligonucleotide probes labeled with reporter and quencher dyes [24]. The most common implementation, TaqMan technology, utilizes dual-labeled probes with a reporter dye at the 5' end and a quencher dye at the 3' end. When the probe is intact, fluorescence resonance energy transfer (FRET) occurs, with the quencher absorbing the light emitted by the reporter [24]. During amplification, the probe binds to the template, and Taq DNA polymerase cleaves the probe via its 5'→3' exonuclease activity, separating the reporter from the quencher and resulting in fluorescence emission [24].
Advanced probe variants include TaqMan minor groove binder (MGB) probes, which incorporate a nonfluorescent quencher and MGB molecule at the 3' end that stabilizes the probe-DNA complex by folding into the minor groove of double-stranded DNA [24]. This enhancement allows for shorter probe designs and improved discrimination of single-base mismatches, particularly valuable for SNP genotyping and mutation detection in cancer research.
Key Applications in Cancer Biomarker Analysis
SNP Genotyping
Probe-based qPCR enables accurate single nucleotide polymorphism (SNP) genotyping, which is crucial for identifying genetic variations associated with cancer susceptibility and treatment response. This approach utilizes allele-specific probes with different reporter dyes (e.g., FAM-labeled for allele 1 and VIC-labeled for allele 2) [24]. The assay capitalizes on the fact that probes with mismatched bases at the SNP position bind unstably and resist cleavage, preventing fluorescence emission. This permits precise allele discrimination in large population studies with high throughput, accuracy, and cost-effectiveness [24].
DNA Methylation Analysis
DNA methylation plays a critical role in gene silencing and carcinogenesis. Probe-based qPCR facilitates methylation-specific analysis after bisulfite treatment of DNA, which converts unmethylated cytosines to uracils while leaving methylated cytosines unchanged [24]. The method employs primers and probes designed to distinguish methylated from unmethylated alleles, enabling quantification of methylation status at specific genomic loci. This application has significant implications for cancer epigenetics, early detection, and monitoring of epigenetic therapies [27] [28].
miRNA Quantification
MicroRNAs (miRNAs) are small non-coding RNA molecules (18-28 nucleotides) that regulate gene expression and serve as promising biomarkers for cancer diagnosis and prognosis [25]. Probe-based qPCR offers superior specificity for miRNA detection compared to dye-based methods, addressing challenges related to their short length, high sequence similarity among family members, and low abundance in biological fluids [25]. Specialized stem-loop primers enhance specificity by trapping and annealing to the 3' end of mature miRNA, followed by probe binding to the stem-loop sequence [24].
Viral Detection in Cancer
Probe-based qPCR serves as a gold standard for detecting oncogenic viruses such as Epstein-Barr virus (EBV), which is associated with lymphomas, nasopharyngeal carcinoma, and gastric cancers [27]. Methylation-specific PCR assays for viral promoters (e.g., MSPCP for EBV C promoter) can distinguish latent from lytic viral states, providing diagnostic and prognostic information in virus-associated malignancies [27].
SYBR Green qPCR: Principles and Implementation
Mechanism and Workflow Considerations
SYBR Green-based qPCR utilizes an intercalating dye that binds nonspecifically to the minor groove of double-stranded DNA, experiencing a 20-100 fold increase in fluorescence upon DNA binding [24]. This method offers simplicity and cost-effectiveness, requiring only the addition of PCR primers without the need for specialized probes [29]. The straightforward design process makes it particularly suitable for initial screening applications and target validation studies in cancer research.
A critical requirement for SYBR Green assays is post-amplification melting curve analysis to distinguish specific products from nonspecific amplification such as primer dimers [29]. This verification step involves gradually increasing temperature while monitoring fluorescence to generate dissociation curves based on the melting temperature (Tm) of amplified products, with specific amplicons exhibiting characteristic Tm values distinct from artifacts.
Advantages and Limitations in Cancer Research
The primary advantage of SYBR Green chemistry is its cost-effectiveness, as it eliminates the expense of fluorescently-labeled probes [30] [29]. This makes it ideal for large-scale screening studies where multiple targets need validation across numerous samples. The simplicity of assay design and optimization also reduces development time, particularly when investigating novel cancer biomarkers with limited sequence information.
However, SYBR Green has significant limitations for complex cancer biomarker applications. Its nonspecific binding to any double-stranded DNA can lead to inaccurate quantification due to off-target amplification and primer dimer formation [29]. This reduced specificity is particularly problematic when analyzing low-abundance targets in complex biological samples like plasma or serum. Additionally, the inability to perform multiplex reactions restricts researchers to single-analyte detection per reaction, limiting throughput for comprehensive biomarker panels [29].
qPCR in Liquid Biopsy Applications
Circulating Tumor DNA (ctDNA) Analysis
Liquid biopsy represents a transformative approach in cancer diagnostics, with qPCR playing a pivotal role in detecting and quantifying ctDNA—short DNA fragments released into circulation by tumor cells [26]. ctDNA typically comprises 0.1-1.0% of total cell-free DNA (cfDNA) in cancer patients and carries tumor-specific genetic alterations including point mutations, copy number variations, and epigenetic modifications [26]. Probe-based qPCR methods like droplet digital PCR (ddPCR) offer exceptional sensitivity for detecting rare mutant alleles in background wild-type DNA, enabling applications in early cancer detection, minimal residual disease (MRD) monitoring, and therapy response assessment [31].
The VICTORI study in colorectal cancer demonstrated the clinical utility of ctDNA analysis, where 87% of recurrences were preceded by ctDNA positivity, while no ctDNA-negative patients relapsed [31]. Similarly, the TOMBOLA trial compared ddPCR and whole-genome sequencing for ctDNA detection in bladder cancer, showing 82.9% concordance between methods with ddPCR exhibiting higher sensitivity in low tumor fraction samples [31].
Advanced Methodologies in Liquid Biopsy
Table 2: qPCR Applications in Liquid Biopsy for Cancer Management
| Application Area | Specific Use Case | qPCR Methodology | Performance Metrics |
|---|---|---|---|
| Multi-Cancer Early Detection | DNA methylation profiling of cfDNA | Methylation-specific probe-based qPCR | 88.2% top prediction accuracy for cancer signal origin [31] |
| Minimal Residual Disease (MRD) | Post-surgical recurrence risk stratification | ddPCR and targeted qPCR panels | 94.3% ctDNA positivity in treatment-naive colorectal cancer patients [31] |
| Treatment Response Monitoring | EGFR mutation detection in NSCLC | Probe-based qPCR (TaqMan) | Baseline EGFR detection in plasma prognostic for shorter PFS and OS [31] |
| Virus-Associated Cancers | EBV-associated lymphoma detection | Methylation-specific PCR for EBV C promoter | High methylation (94-100%) in tumors vs. low methylation in healthy controls [27] |
Workflow for Liquid Biopsy Analysis Using qPCR
The following diagram illustrates a standardized workflow for processing liquid biopsy samples using qPCR technologies:
Experimental Protocols for Key Applications
Methylation-Specific qPCR for Cancer Detection
DNA methylation analysis via qPCR provides crucial information for cancer diagnosis and prognosis. The following protocol outlines methylation-specific PCR for detecting promoter hypermethylation in cancer biomarkers:
Sample Preparation:
- Extract cell-free DNA from 1-4 mL plasma using commercial kits (e.g., QIAamp DNA Blood Mini Kit)
- Treat 500 ng - 1 μg DNA with sodium bisulfite using commercial conversion kits (e.g., EZ DNA Methylation Kit)
- Purify bisulfite-converted DNA and elute in 20-40 μL elution buffer
Primer and Probe Design:
- Design methylation-specific primers to amplify regions of interest with 3-5 CpG sites in binding regions
- Ensure primers specifically recognize bisulfite-converted sequences with converted Cs at non-CpG positions
- Design TaqMan probes with reporter-quencher pairs (FAM-BHQ1 for methylated alleles, VIC-BHQ2 for unmethylated alleles)
- Validate primer specificity using in silico tools and control samples
qPCR Reaction Setup:
- 10 μL TaqMan Universal Master Mix II
- 1 μL bisulfite-converted DNA template (5-10 ng/μL)
- 0.5 μL each forward and reverse primer (10 μM stock)
- 0.5 μL TaqMan probe (5 μM stock)
- Nuclease-free water to 20 μL final volume
Thermal Cycling Conditions:
- Initial denaturation: 95°C for 10 minutes
- 45 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 60 seconds (with fluorescence acquisition)
Data Analysis:
- Calculate ΔCt values between methylated and unmethylated signals
- Determine methylation ratios using standard curves or ΔΔCt methods
- Establish threshold values for positive methylation calls based on control samples
This protocol has been successfully applied to detect EBV C promoter methylation in Hodgkin lymphoma, showing 94-100% methylation in tumor tissues versus minimal methylation in controls [27].
miRNA Quantification Using Stem-Loop qPCR
Mature miRNA quantification requires specialized approaches due to their short length. The stem-loop RT-qPCR method provides enhanced specificity and sensitivity:
RNA Extraction:
- Extract total RNA from 200-500 μL plasma using miRNA-specific kits (e.g., miRNeasy Serum/Plasma Kit)
- Include spike-in synthetic miRNA controls for normalization
- Elute RNA in 15-30 μL nuclease-free water
Stem-Loop Reverse Transcription:
- Design stem-loop RT primers with:
- 3' portion complementary to 6-8 nucleotides at miRNA 3' end
- Stem-loop structure
- Universal sequence at 5' end
- Prepare RT reaction:
- 1-5 μL RNA template
- 1 μL stem-loop RT primer (1 μM)
- 4 μL 5× Reverse Transcription Buffer
- 0.5 μL dNTPs (100 mM)
- 1 μL Reverse Transcriptase
- Nuclease-free water to 20 μL
- Incubate at 16°C for 30 minutes, 42°C for 30 minutes, 85°C for 5 minutes
qPCR Amplification:
- Design forward primer complementary to miRNA 5' region
- Use universal reverse primer complementary to stem-loop sequence
- Utilize TaqMan probe with FAM reporter and BHQ-1 quencher
- Prepare qPCR reaction:
- 10 μL TaqMan Master Mix
- 1 μL cDNA template
- 0.5 μL forward primer (10 μM)
- 0.5 μL reverse primer (10 μM)
- 0.5 μL TaqMan probe (5 μM)
- Nuclease-free water to 20 μL
- Thermal cycling: 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds
This approach enables specific detection of mature miRNA forms, which is crucial given their role as regulatory molecules in cancer pathways [24] [25].
The Scientist's Toolkit: Essential Reagents and Technologies
Table 3: Research Reagent Solutions for qPCR Cancer Biomarker Analysis
| Reagent/Category | Specific Examples | Function in qPCR Workflow |
|---|---|---|
| Fluorescent Reporters | SYBR Green I, FAM, VIC, TET, TAMRA | Detection of amplified products through fluorescence emission |
| Quenchers | BHQ (Black Hole Quencher), TAMRA | Suppress reporter fluorescence until amplification occurs |
| Specialized Probes | TaqMan dual-labeled probes, MGB probes, Molecular Beacons | Provide sequence-specific detection with enhanced specificity |
| DNA Polymerases | Hot-start Taq, Reverse Transcriptase | Catalyze DNA amplification with reduced non-specific amplification |
| Sample Preparation Kits | QIAamp DNA Blood Mini Kit, miRNeasy Serum/Plasma Kit | Isolate high-quality nucleic acids from complex biological samples |
| Bisulfite Conversion Kits | EZ DNA Methylation Kit, MethylEdge Bisulfite Conversion System | Convert unmethylated cytosines to uracils for methylation analysis |
| Reference Assays | RNAse P detection, Reference genes (GAPDH, ACTB) | Normalize sample input and control for extraction efficiency |
Emerging Trends and Future Perspectives
The field of qPCR in cancer biomarker analysis continues to evolve with several emerging trends shaping its future applications. Amplification-free detection technologies are gaining attention for miRNA analysis, addressing limitations of PCR-based methods including labor-intensive workflows, technical variability, and difficulties in quantifying low-abundance targets [25]. These approaches aim to enable direct miRNA interrogation without RNA extraction, reverse transcription, or amplification, potentially improving reliability and scalability for clinical applications [25].
Multiplexing capabilities are expanding through advanced probe systems and digital PCR platforms, allowing simultaneous quantification of multiple biomarkers from limited sample volumes. The development of novel CRISPR-based detection methods like MUTE-Seq (Mutation tagging by CRISPR-based Ultra-precise Targeted Elimination in Sequencing) enables ultrasensitive detection of low-frequency mutations in ctDNA by selectively eliminating wild-type DNA [31]. Such innovations significantly enhance sensitivity for minimal residual disease monitoring.
Integration of multi-analyte approaches represents another frontier, combining ctDNA mutation analysis with methylation profiling, fragmentomics, and protein biomarkers to improve diagnostic accuracy [31]. Studies demonstrate that combining tissue and liquid biopsy significantly increases detection of actionable alterations and improves survival outcomes in patients receiving tailored therapy, despite only 49% concordance between methods [31].
As these technological advances continue to mature, qPCR remains positioned as a fundamental tool in cancer biomarker research, bridging the gap between basic science discovery and clinical application through its unique combination of sensitivity, quantitative accuracy, and practical implementation across diverse laboratory settings.
Application in Oncology: Protocols for Detecting Cancer Biomarkers and Mutations
Designing Primers and Probes for Cancer-Associated SNPs and Point Mutations
Quantitative PCR (qPCR) is a cornerstone technique in molecular diagnostics and cancer research, enabling the sensitive detection and quantification of genetic alterations such as single nucleotide polymorphisms (SNPs) and point mutations. These somatic mutations can act as drivers of oncogenesis, influence therapeutic responses, and serve as biomarkers for minimal residual disease monitoring [32]. The choice between intercalating dye-based methods (e.g., SYBR Green) and probe-based methods (e.g., TaqMan) represents a critical decision point in assay design, with significant implications for specificity, cost, throughput, and robustness [5]. This guide provides an in-depth technical framework for designing primers and probes to detect cancer-associated mutations, contextualized within the comparative performance of SYBR Green and TaqMan chemistries. The principles outlined support applications in research, diagnostic assay development, and therapeutic monitoring for oncology.
SYBR Green vs. TaqMan: Core Principles and Comparative Analysis
Chemistry and Mechanism
SYBR Green is a fluorescent dye that binds non-specifically to the minor groove of double-stranded DNA (dsDNA). During qPCR, the fluorescence increases proportionally to the amount of amplified dsDNA product, allowing for quantification [5]. Its primary advantage is simplicity and cost-effectiveness, as it requires only sequence-specific primers. However, a significant limitation is its inability to distinguish between specific and non-specific amplification products (e.g., primer-dimers), which can lead to false-positive signals [5] [33]. Post-amplification melting curve analysis is, therefore, essential to verify amplicon specificity.
TaqMan Probes (Hydrolysis Probes) rely on a sequence-specific, dual-labeled oligonucleotide probe in addition to PCR primers. The probe is conjugated with a fluorescent reporter dye at the 5' end and a quencher dye at the 3' end. When intact, the quencher suppresses the reporter's fluorescence via Förster Resonance Energy Transfer (FRET). During the amplification cycle, the 5'→3' exonuclease activity of the DNA polymerase cleaves the probe, physically separating the reporter from the quencher and resulting in a permanent fluorescent signal proportional to the target amplification [5]. This mechanism provides inherent specificity, as fluorescence is generated only upon hybridization and cleavage of the correct probe.
Quantitative Performance Comparison
The following table summarizes key performance characteristics of SYBR Green and TaqMan assays as demonstrated in various studies.
Table 1: Comparative Performance of SYBR Green and TaqMan qPCR Methods
| Parameter | SYBR Green | TaqMan | Context and Notes |
|---|---|---|---|
| Assay Efficiency | 94.3% - 99% [8] [34] | 96.6% - 103% [8] [34] | Efficiencies >90% are generally acceptable. Both methods can achieve high, comparable efficiency with proper optimization [5]. |
| Limit of Detection (LOD) | 100 fg - 102 copies/μL [8] [34] | 10 fg - 101 copies/μL [8] [34] | TaqMan often demonstrates a 1-log improved sensitivity due to reduced background signal. |
| Specificity | High, but dependent on primer specificity and requires melting curve analysis [33]. | Very High, conferred by the sequence-specific probe [5]. | SYBR Green can detect 100% of viral sequence variants in one study, whereas TaqMan failed to detect 47% of single-nucleotide variants in its probe-binding site [33]. |
| Cost & Accessibility | Relatively low cost and easy design [5]. | More expensive due to probe synthesis [5]. | SYBR Green is economical for primer screening and assays where target sequence is highly conserved. |
| Multiplexing Potential | None. | High. Enables detection of multiple targets in a single reaction using probes with different fluorophores. | Critical for assays like internal controls or simultaneous detection of a mutation and a reference gene. |
| Impact on HER2 Gene Dose Analysis | Underestimated amplification ratio (5-fold vs. 10-fold) [35]. | Correctly estimated 10-fold amplification [35]. | For gene copy number variation (CNV) analysis, probe-based methods are generally more accurate [35]. |
Primer and Probe Design for SNP and Point Mutation Detection
The accurate detection of single-base changes demands rigorous in silico design and experimental validation.
General Design Parameters
- Amplicon Length: Shorter amplicons (80-200 bp) typically yield higher amplification efficiencies and are more robust when analyzing degraded DNA from formalin-fixed paraffin-embedded (FFPE) samples [36].
- Primer Characteristics: Primers should be 18-25 nucleotides long with a melting temperature (Tm) of 50-65°C. The Tm of both primers in a pair should be within 1-2°C of each other. Avoid long runs of identical nucleotides, secondary structures, and 3'-complementarity to prevent primer-dimer formation.
- Specificity Checking: Use tools like NCBI Primer-BLAST to ensure primers are specific to the intended genomic target and do not amplify homologous sequences or pseudogenes [37].
- Exon-Junction Spanning: When detecting mRNA expression, designing at least one primer to span an exon-exon junction prevents amplification of contaminating genomic DNA [37].
Allele-Specific Primer Design for SNP Genotyping
Allele-Specific PCR (AS-PCR) differentiates alleles based on the preferential amplification by a primer whose 3'-terminal nucleotide is complementary to one allele (e.g., wild-type vs. mutant). To enhance specificity, an additional, deliberate mismatch is often introduced near the 3' end of the allele-specific primer.
Table 2: Strategy for Introducing Artificial Mismatches in Allele-Specific Primers
| SNP Type | Recommended Artificial Mismatch (Base Pair & Position) | Reported Specificity | Rationale |
|---|---|---|---|
| A/T (or T/A) | C-A in the 3rd nucleotide from the 3' end [38]. | 81.9% [38] | This specific transversion creates a strong destabilizing effect that efficiently suppresses amplification of the non-matched template. |
| A/G (or T/C) | T-G or C-A in the 3rd position from the 3' end [38]. | Found to be effective [38]. | These transversions provide sufficient thermodynamic discrimination. |
| A/C (or T/G) | A-G (transition) in the 2nd position from the 3' end [38]. | Effective for improving specificity [38]. | Positioning the mismatch closer to the terminal base increases its discriminatory power. |
| G/C (or C/G) | Mismatch in the 2nd position from the 3' end [38]. | 43% polymorphism rate [38]. | Systematic testing of mismatch position is critical for these SNP types. |
TaqMan Probe Design for Mutation Detection
- Probe Placement: Design the probe to hybridize to the stable, conserved region of the amplicon. For SNP detection, the polymorphic site should be located centrally within the probe sequence to maximize allelic discrimination.
- Probe Characteristics: The probe should have a Tm 5-10°C higher than the primers to ensure it hybridizes before primer extension. Length is typically 15-30 nucleotides.
- Fluorophore-Quencher Selection: Choose reporter dyes (e.g., FAM, VIC) and quenchers (e.g., BHQ, TAMRA) based on the real-time PCR instrument's channels. For multiplexing, ensure the emission spectra of reporters are well-separated.
Diagram Title: Workflow for Designing qPCR Assays for Cancer Mutations
Experimental Protocols for Validation
Protocol: Assay Optimization and Efficiency Calculation
- Standard Curve Preparation: Prepare a serial dilution (e.g., 5- or 10-fold) of the target template (e.g., plasmid DNA or cDNA with known concentration) over a range of at least 5 orders of magnitude.
- qPCR Run: Perform qPCR using both the optimized primer set (for SYBR Green) and the primer-probe set (for TaqMan) with the standard curve dilutions.
- Efficiency Calculation: The qPCR software plots the Cycle Threshold (Ct) values against the logarithm of the template concentration. The slope of the resulting standard curve is used to calculate the amplification efficiency (E) using the formula:
Protocol: Analytical Validation for Diagnostic Sensitivity
- Limit of Detection (LOD) Determination: Perform qPCR on a dilution series of the target mutation in a background of wild-type DNA (e.g., cell line DNA or synthetic fragments). The LOD is the lowest concentration at which the target is detected in ≥95% of replicates [39] [36].
- Specificity Testing: Test the assay against a panel of samples with known genotypes, including wild-type, heterozygous, and homozygous mutant, to confirm that the signal is generated only from the intended allele.
- Analysis of Variant Tolerance: As demonstrated in viral studies, test the assay against known sequence variants to ensure that mismatches do not lead to false negatives, a particular advantage of the SYBR Green method for evolving targets [33].
Table 3: Key Reagents and Resources for Mutation Detection Assays
| Item | Function/Description | Example Use Case |
|---|---|---|
| High-Fidelity DNA Polymerase | Enzyme for accurate amplification of templates for cloning standards. | Generation of plasmid DNA for standard curves [34]. |
| Hot-Start Taq Polymerase | Reduces non-specific amplification and primer-dimer formation by requiring thermal activation. | Essential for both SYBR Green and TaqMan assays to improve specificity and sensitivity [36]. |
| Unique Molecular Identifiers (UMIs) | Molecular barcodes added to each template molecule before amplification. | Allows bioinformatic removal of PCR duplicates and errors, improving quantification accuracy in NGS-based assays [36]. |
| Nicking Enzyme Amplification Reaction (NEAR) | Isothermal amplification method for rapid target enrichment. | Used in novel methods like SPEAR for ultra-sensitive SNP detection without traditional PCR [39]. |
| CRISPR-Cas12b RNP | Ribonucleoprotein complex for sequence-specific recognition and cleavage. | Provides single-base resolution specificity in conjunction with isothermal amplification for detecting cancer SNPs [39]. |
| Panel of Normal (PON) Samples | A collection of sequencing data from normal, non-tumor samples. | Serves as a baseline reference in NGS pipelines to filter out sequencing artifacts and common germline variants [36]. |
Diagram Title: Method Selection Guide for Mutation Detection
The strategic design of primers and probes is fundamental to the success of any qPCR-based assay for cancer-associated SNPs and point mutations. The choice between SYBR Green and TaqMan methodologies is not a matter of superiority but of application-specific suitability. SYBR Green offers a cost-effective and flexible solution for gene expression studies and variant scanning, especially when the target region is prone to sequence variations that could compromise probe binding [33]. In contrast, TaqMan probes provide an unparalleled level of specificity and are the gold standard for high-fidelity diagnostic applications, multiplexing, and accurate copy number analysis [35] [32]. Emerging technologies like CRISPR-Cas-assisted methods (e.g., SPEAR) further push the boundaries of sensitivity and speed for point mutation detection [39]. By adhering to the detailed design rules, validation protocols, and selection framework outlined in this guide, researchers and clinical developers can implement robust, reliable genomic tools to advance cancer research and precision medicine.
Workflow for Absolute Quantification of Viral Loads and Gene Copy Number Variations (e.g., MYC-N amplification)
Absolute quantification is a cornerstone of molecular diagnostics, providing precise measurement of specific nucleic acid sequences within a biological sample. In the context of cancer research and virology, this capability is indispensable for assessing oncogene amplification status, such as MYC-N in neuroblastoma, or determining viral load for disease monitoring. The choice between SYBR Green and probe-based detection methods represents a critical methodological crossroads, each offering distinct advantages and limitations that significantly impact assay design, cost, specificity, and data interpretation [40] [41] [42]. This technical guide provides an in-depth examination of workflows for absolute quantification of viral loads and gene copy number variations (CNVs), with particular emphasis on the comparative analytical considerations between fluorescent DNA-binding dyes and hydrolysis probes within cancer gene research frameworks.
The fundamental principle underlying absolute quantification in real-time PCR revolves around monitoring fluorescence accumulation during amplification, with the point at which fluorescence crosses a predetermined threshold (Cq or Ct value) being inversely proportional to the starting quantity of the target nucleic acid. Probe-based chemistries, such as TaqMan, utilize sequence-specific fluorescent probes that provide enhanced specificity through an additional hybridization step. In contrast, SYBR Green-based methods rely on intercalation with double-stranded DNA, offering a more cost-effective and flexible alternative while requiring rigorous validation to ensure specificity [40] [42]. For clinical applications like MYCN amplification assessment in neuroblastoma—a powerful prognostic marker—accuracy in copy number determination directly impacts therapeutic decisions, making methodological choices particularly consequential [42] [43].
Fundamental Principles of Quantitative PCR
Basic Mechanisms of Fluorescence Detection
Real-time quantitative PCR (Q-PCR) enables target quantification through the continuous monitoring of fluorescence signals throughout the amplification process. Two primary detection chemistries dominate the field: SYBR Green dye and sequence-specific probes. SYBR Green is a cyanine dye that exhibits fluorescence enhancement upon binding to the minor groove of double-stranded DNA. This binding is non-sequence-specific, resulting in fluorescence emission proportional to the total dsDNA present in the reaction [40] [42]. The major advantage of this approach lies in its simplicity and cost-effectiveness, as it requires only sequence-specific primers without the need for specialized fluorescent probes.
Hydrolysis probes (e.g., TaqMan probes) constitute the most common probe-based approach, utilizing oligonucleotides dual-labeled with a fluorescent reporter dye at the 5' end and a quencher molecule at the 3' end. When intact, the proximity of the quencher suppresses reporter fluorescence through Förster resonance energy transfer (FRET). During amplification, the 5'→3' exonuclease activity of DNA polymerase cleaves the probe, separating the reporter from the quencher and resulting in permanent fluorescence emission proportional to target amplification [40]. Molecular beacons represent an alternative probe design that maintains a stem-loop structure in the unbound state, keeping fluorophore and quencher in close proximity. Hybridization to the target sequence separates the pair, generating a fluorescence signal without probe degradation [41].
Mathematical Models for Absolute Quantification
Absolute quantification requires establishing a relationship between fluorescence kinetics and initial template concentration. The exponential amplification phase is characterized by the equation:
[ Nt = N0 \times (1 + E)^t ]
Where (Nt) is the number of amplicon molecules at cycle (t), (N0) is the initial number of target molecules, and (E) is the amplification efficiency. The threshold cycle (Cq) represents the point at which fluorescence exceeds background levels and relates to the initial template concentration by:
[ Cq = -\frac{1}{\log(1+E)} \times \log(N_0) + \text{constant} ]
For viral load quantification using NASBA (Nucleic Acid Sequence-Based Amplification), different kinetic models apply. During the initial phase, amplification proceeds exponentially while primers are not rate-limiting, transitioning to linear growth once primer pools become depleted, with RNA formation described by:
[ \frac{d[\text{aRNA}]}{dt} = c_6[\text{cDNA}] ]
where ([\text{aRNA}]) is antisense RNA concentration and (c_6) is the rate constant for T7 RNA polymerase activity [41]. Quantitation relies on comparing the transcription rates of wild-type viral RNA and a calibrator RNA added at known concentration, with the ratio directly reflecting their relative initial concentrations.
Comparative Analysis: SYBR Green vs. Probe-Based Detection
Table 1: Comparative characteristics of SYBR Green and probe-based detection methods
| Parameter | SYBR Green | TaqMan Probes | Molecular Beacons |
|---|---|---|---|
| Specificity | Moderate (requires melting curve analysis) | High (dual specificity: primers + probe) | High (stem-loop structure provides specificity) |
| Cost | Low (no specialized probes required) | High (fluorescent probes increase cost) | High (specialized beacon design required) |
| Multiplexing Capability | Limited (single target per reaction) | High (multiple probes with different fluorophores) | High (multiple beacons with different fluorophores) |
| Experimental Flexibility | High (easy primer redesign) | Low (probe redesign costly) | Low (beacon redesign costly) |
| Methylation Density Analysis | Possible via melting curves [40] | Not possible without specialized designs | Not possible without specialized designs |
| Best Applications | Mutation scanning, methylation density, single-target studies | Multiplex assays, high-specificity requirements | Live-cell monitoring, multiplex assays |
SYBR Green-Based Approaches
SYBR Green methodologies offer distinct advantages in Scenarios requiring methylation density assessment or when research budgets are constrained. The MethySYBR assay exemplifies an innovative application that leverages SYBR Green's capacity for melting curve analysis to determine CpG methylation density following bisulfite conversion. This technique employs a two-step PCR approach: initial multiplex pre-amplification using methylation-independent primers, followed by nested methylation-specific PCR with SYBR Green detection. The resulting amplicons undergo melting curve analysis, where the melting temperature (Tm) directly correlates with GC content affected by methylation status [40].
A significant advantage of SYBR Green is its cost-effectiveness, particularly when analyzing multiple targets or developing new assays. The absence of expensive target-specific probes substantially reduces per-reaction costs, enabling higher throughput screening applications. Furthermore, the ability to perform melting curve analysis provides an internal specificity control, identifying non-specific amplification or primer-dimer formation that might compromise quantification accuracy. Studies comparing SYBR Green I and TaqMan probes for MYCN copy number determination in neuroblastoma have demonstrated equivalent results, validating SYBR Green as a reliable alternative for gene copy number quantification [42].
Probe-Based Detection Systems
Probe-based detection methods offer enhanced specificity through the requirement of three specific hybridization events (two primers plus probe) for signal generation. This triple specificity mechanism makes probe-based approaches particularly valuable in applications requiring exceptional discrimination between highly similar sequences, such as in viral load monitoring where sequence variations might affect clinical interpretation, or in distinguishing paralogous genes with high homology [41].
The MethyLight assay represents a prototypical probe-based approach for DNA methylation analysis, employing methylation-specific primers and a fluorescent probe to quantitatively assess methylated alleles. This technology demonstrates exceptional sensitivity, capable of detecting one methylated allele amidst a 10,000-fold excess of unmethylated alleles [40]. For viral load quantification, molecular beacons in NASBA assays enable real-time monitoring of amplification through target-specific hybridization, with mathematical modeling separating the kinetics of amplicon formation from beacon binding to accurately determine initial template concentration [41].
A critical consideration for probe-based assays in cancer gene research involves methylation heterogeneity in clinical samples. While probe-based methods excel at detecting completely methylated alleles, they may underestimate heterogeneously methylated sequences unless specifically designed with this limitation in mind. Complex probe designs addressing this issue can substantially increase both cost and implementation complexity [40].
Workflow for Absolute Quantification
Experimental Design Considerations
Robust absolute quantification requires meticulous experimental planning. Assay specificity must be empirically validated through melting curve analysis for SYBR Green-based methods or through no-template controls for probe-based approaches. For gene copy number variation analysis, reference genes with stable copy numbers must be selected and validated for the specific biological system under investigation. The dynamic range of quantification should encompass expected target concentrations, typically requiring 4-5 log units for viral load applications and 2-3 log units for CNV analysis [42].
Primer and probe design represents a critical success factor. For SYBR Green assays, primers should generate amplicons of 75-150 bp with minimal primer-dimer potential. For methylation analysis following bisulfite conversion, primers must account for C→T conversions in unmethylated sequences while incorporating CpG sites at the 3' end to maximize methylation specificity [40]. Probe-based assays require additional validation of probe efficiency and specificity, with particular attention to ensuring fluorophore-quencher compatibility in multiplex applications.
Step-by-Step Protocol for Gene Copy Number Variation Analysis
The following protocol outlines the absolute quantification of MYCN amplification in neuroblastoma samples, adaptable to other gene copy number variations:
DNA Extraction and Qualification
- Extract genomic DNA from tumor samples using standard phenol-chloroform or column-based methods.
- Quantify DNA concentration using fluorometric methods (e.g., Qubit) to ensure accuracy over spectrophotometric approaches.
- Assess DNA quality via agarose gel electrophoresis or genomic quality number (GQN) metrics.
Assay Selection and Validation
- For SYBR Green approach: Design primers flanking the MYCN target region and reference gene (e.g., NAG). Validate primer specificity through melting curve analysis and ensure amplification efficiency between 90-110% relative to standard curves.
- For probe-based approach: Design TaqMan probes with 5' fluorophore (FAM for MYCN, VIC for reference) and 3' non-fluorescent quencher. Validate probe efficiency and ensure no cross-reactivity between channels.
Standard Curve Preparation
- Prepare serial dilutions (typically 1:10) of standard templates with known copy numbers.
- For MYCN quantification, use genomic DNA with known MYCN status or plasmid constructs containing target sequences.
- Include a minimum of five data points across the expected dynamic range.
Q-PCR Amplification
- Prepare reaction mixtures containing:
- 1× SYBR Green Master Mix or TaqMan Universal Master Mix
- Forward and reverse primers (300-900 nM final)
- For probe-based: Target and reference probes (100-200 nM final)
- 10-50 ng genomic DNA
- Perform amplification using the following cycling conditions:
- Initial denaturation: 95°C for 10 min
- 40-45 cycles of: 95°C for 15 sec, 60°C for 1 min
- For SYBR Green only: Melt curve stage: 65°C to 95°C, increment 0.5°C
- Prepare reaction mixtures containing:
Data Analysis and Copy Number Determination
- Calculate Cq values for target (MYCN) and reference (single-copy gene) using platform software.
- Generate standard curves for both target and reference genes from serial dilution data.
- Determine absolute copy numbers for target and reference genes from respective standard curves.
- Calculate copy number variation using the formula: [ \text{Gene Copy Number} = \frac{\text{Target Gene Copy Number}}{\text{Reference Gene Copy Number}} \times \text{Ploidy} ]
- For MYCN amplification, a ratio ≥ 4.0 typically indicates amplification [42] [43].
Table 2: Essential research reagents for absolute quantification workflows
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Detection Chemistries | SYBR Green I, TaqMan Probes, Molecular Beacons | Generate fluorescence signal proportional to amplicon concentration; determine specificity and cost |
| Enzyme Systems | Hot-start DNA polymerases, Reverse transcriptase (viral load) | Catalyze DNA amplification; hot-start enzymes prevent non-specific amplification |
| Standard Materials | Plasmid DNA, Genomic DNA with known copy number, Synthetic oligos | Establish standard curves for absolute quantification; essential for determining copy number |
| Sample Processing Reagents | Bisulfite conversion kits (methylation analysis), DNA extraction kits | Modify and purify nucleic acids for analysis; bisulfite conversion distinguishes methylated cytosines |
| Reference Assays | Single-copy gene primers/probes (ACTB, NAG) [42] | Normalize for DNA input quantity; essential for accurate copy number determination |
| Internal Controls | Calibrator RNA (viral load) [41], Methylated controls | Monitor reaction efficiency; identify inhibition; ensure quantitative accuracy |
Viral Load Quantification Using NASBA with Molecular Beacons
Nucleic Acid Sequence-Based Amplification (NASBA) with molecular beacons provides an isothermal alternative to PCR for viral load quantification, particularly effective for RNA viruses like HIV-1:
RNA Extraction and Calibrator Addition
- Extract viral RNA from patient samples using guanidinium thiocyanate-based methods.
- Add known quantity of calibrator RNA (Q RNA) to each sample prior to extraction to control for variations in extraction efficiency.
NASBA Amplification
- Prepare reaction mixture containing:
- Primers P1 and P2 (1012 molecules per reaction)
- Three enzymes: AMV-RT, RNase H, T7 RNA polymerase
- Two molecular beacons specific for wild-type (WT) and calibrator (Q) RNA
- Incubate at 41°C for 90-120 minutes with continuous fluorescence monitoring.
- Prepare reaction mixture containing:
Data Analysis Principles
- During early amplification phases, RNA formation proceeds exponentially while primers are not rate-limiting: [ \frac{d[\text{aRNA}]}{dt} = c_6[\text{cDNA}] ]
- Once primers become depleted, RNA accumulation becomes linear with time.
- Quantitation is based on comparing transcription rates of WT and Q RNA during the linear phase.
- The ratio of transcription rates directly reflects the initial ratio of WT to Q RNA, enabling absolute quantification [41].
Applications in Cancer Gene Research
MYCN Amplification in Neuroblastoma
MYCN amplification represents one of the most clinically significant applications of gene copy number quantification in oncology. MYCN amplification occurs in approximately 20% of neuroblastoma cases and constitutes a powerful adverse prognostic marker, leading to its incorporation into clinical risk stratification systems [43]. Accurate MYCN copy number determination directly impacts therapeutic decisions, with amplified cases typically receiving more intensive treatment regimens.
Studies have demonstrated that real-time Q-PCR using either SYBR Green I or TaqMan probes provides equivalent results to traditional Southern blot and FISH analyses for MYCN copy number assessment [42]. The Q-PCR approach offers advantages including rapid turnaround time, small DNA requirements, and a large dynamic range of quantification. Furthermore, the ability to simultaneously assess co-amplification of genes frequently co-amplified with MYCN, such as DDX1 and NAG, provides additional prognostic information within the same experimental framework [42].
Recent transcriptional profiling has revealed significant heterogeneity within MYCN non-amplified neuroblastomas, classifying them into three distinct subgroups with markedly different clinical outcomes. Subgroup 2 MYCN non-amplified tumors exhibit a "MYCN" signature despite lacking gene amplification, potentially induced by Aurora Kinase A (AURKA) overexpression, and demonstrate poor prognosis approaching that of MYCN-amplified cases [43]. This complexity underscores the importance of precise quantification methodologies that can complement transcriptional profiling for comprehensive patient stratification.
DNA Methylation Analysis in Cancer
DNA methylation represents another critical application of quantitative PCR in cancer research, where promoter hypermethylation of tumor suppressor genes serves as both a biomarker and potential therapeutic target. The MethySYBR assay exemplifies how SYBR Green-based approaches can advance this field by enabling simultaneous quantification of methylation status and assessment of methylation density through melting curve analysis [40].
This methodology begins with multiplex pre-amplification of bisulfite-converted DNA using methylation-independent primers, enabling analysis of multiple loci from minute DNA quantities (as little as 3 pg). Subsequent nested PCR with methylation-specific primers and SYBR Green detection provides quantitative assessment of methylated alleles, with melting curve analysis of the products revealing methylation density based on the thermal stability differences between methylated and unmethylated sequences [40]. This approach demonstrates exceptional sensitivity, detecting methylated alleles amidst a 100,000-fold excess of unmethylated alleles, making it particularly suitable for analyzing heterogeneous clinical samples or early detection applications.
Troubleshooting and Quality Control
Assay Validation Parameters
Robust absolute quantification requires rigorous validation of multiple assay parameters:
- Amplification Efficiency: Calculated from standard curve slopes using the formula ( E = 10^{-1/slope} - 1 ), should fall between 90-110% for both target and reference assays.
- Linear Dynamic Range: Minimum of 4 orders of magnitude with R² > 0.98 for standard curves.
- Limit of Detection (LOD): Determined by probit analysis, typically 1-10 copies for well-optimized assays.
- Precision: Intra-assay and inter-assay coefficient of variation < 5% for Cq values.
- Specificity: For SYBR Green, single peak in melting curve; for probe-based, no signal in no-template controls.
Common Issues and Solutions
- Poor Amplification Efficiency: Redesign suboptimal primers/probes, optimize annealing temperature, or modify magnesium concentration.
- Inconsistent Replicates: Ensure proper template mixing, check pipette calibration, and include master mixes to minimize preparation variability.
- Non-specific Amplification (SYBR Green): Increase annealing temperature, reduce primer concentration, or incorporate touchdown PCR.
- Inhibition: Dilute samples, implement purification steps, or add amplification facilitators like BSA.
- Discrepant Reference Gene Expression: Validate reference gene stability across sample types or employ multiple reference genes.
Absolute quantification of viral loads and gene copy number variations represents a fundamental capability in modern molecular diagnostics and cancer research. The choice between SYBR Green and probe-based detection methodologies involves thoughtful consideration of research objectives, required specificity, throughput needs, and budgetary constraints. SYBR Green-based approaches offer advantages in cost-effectiveness, flexibility, and ability to assess methylation density through melting curve analysis, while probe-based methods provide enhanced specificity and multiplexing capabilities particularly valuable in complex analytical scenarios.
The workflows described for MYCN amplification analysis and viral load quantification illustrate how these methodologies can be implemented to generate clinically actionable data. As cancer research increasingly recognizes the heterogeneity within molecularly defined subgroups, such as MYCN non-amplified neuroblastoma [43], precise quantification methodologies will play an increasingly important role in refined patient stratification and personalized therapeutic approaches. Similarly, in virology, accurate viral load monitoring continues to guide treatment decisions and disease management across diverse pathogenic contexts.
Quantitative real-time polymerase chain reaction (qRT-PCR or qPCR) remains the gold standard for the detection and quantification of nucleic acids, valued for its exceptional sensitivity, specificity, and broad dynamic range [44]. In molecular biology research, particularly in cancer biomarker validation, a primary application of qPCR is determining changes in gene expression levels between different experimental conditions, such as healthy versus tumor tissue. Relative quantification provides a practical approach to measure these changes without requiring absolute standards for every target gene. Instead, it calculates the expression of a target gene relative to one or more reference genes and compares this ratio between different experimental groups [44].
The core mathematical models for relative quantification are the Livak (2^(-ΔΔCT)) method and the Pfaffl method, which form the foundation for analyzing qPCR data in biomarker studies [44]. The choice between these methods depends heavily on the underlying assumptions about amplification efficiency, which is critically important when working with cancer genes that may exhibit variable expression patterns. Furthermore, the selection between SYBR Green and probe-based detection chemistry directly impacts assay specificity, cost, and workflow complexity—key considerations for researchers validating cancer biomarkers in resource-limited or high-throughput settings [45] [46].
This technical guide provides an in-depth comparison of the ΔΔCT and Pfaffl methods, detailed experimental protocols, and practical considerations for applying these approaches specifically to cancer gene expression studies using both SYBR Green and probe-based detection systems.
Mathematical Foundations of ΔΔCT and Pfaffl Methods
The ΔΔCT (Livak) Method
The Livak method, commonly known as the 2^(-ΔΔCT) method, provides a simplified approach for calculating fold changes in gene expression. This method relies on several key assumptions: (1) the amplification efficiencies of both target and reference genes are approximately 100% (meaning E ≈ 2), and (2) these efficiencies are nearly equal between the target and reference genes [44]. The calculation proceeds through several steps. First, the ΔCT is computed for each sample as the difference between the threshold cycles (CT) of the target gene and the reference gene (ΔCT = CTtarget - CTref). Next, the ΔΔCT is determined by subtracting the ΔCT of the control group from the ΔCT of the treatment group (ΔΔCT = ΔCTTr - ΔCTCo). Finally, the fold change (FC) expression is calculated using the formula FC = 2^(-ΔΔCT) [44].
The widespread adoption of this method stems from its mathematical simplicity and minimal data requirements, needing only CT values without necessitating efficiency calculations for each assay. However, this simplicity comes with significant limitations. The method's accuracy rapidly deteriorates when amplification efficiencies deviate from 100% or when the efficiencies of target and reference genes differ substantially—a common scenario when working with diverse cancer gene targets that may have different secondary structures or amplification characteristics [44] [47].
The Pfaffl Method
The Pfaffl method offers a more flexible approach by explicitly incorporating amplification efficiencies into the fold change calculation, thereby addressing a critical limitation of the ΔΔCT method. This approach is particularly valuable when amplification efficiencies differ between target and reference genes or deviate significantly from 100% [44]. The Pfaffl formula calculates fold change as:
FC = [Etarget^(ΔCTTr - ΔCTCo)] / [Eref^(ΔCTTr - ΔCTCo)]
Where Etarget represents the amplification efficiency of the target gene, Eref denotes the amplification efficiency of the reference gene, ΔCTTr is the ΔCT value for the treatment condition, and ΔCTCo is the ΔCT value for the control condition [44].
The primary advantage of the Pfaffl method lies in its ability to account for efficiency variations, providing more accurate quantification when perfect amplification cannot be assumed. This is particularly relevant for cancer biomarker studies where primer sequences might be constrained by transcript variants or GC-rich regions that affect amplification efficiency. The method requires preliminary experiments to determine actual amplification efficiencies for each primer pair, typically derived from standard curves with serial dilutions of template DNA [44].
Method Comparison and Selection Guidelines
Table 1: Comprehensive Comparison of ΔΔCT and Pfaffl Methods
| Parameter | ΔΔCT Method | Pfaffl Method |
|---|---|---|
| Amplification Efficiency Assumption | Requires 100% efficiency for both target and reference genes | Accommodates different efficiencies for target and reference genes |
| Data Requirements | CT values only | CT values + amplification efficiency values |
| Computational Complexity | Low | Moderate |
| Accuracy Under Ideal Conditions | High | High |
| Accuracy with Efficiency Variations | Low | High |
| Best Applications | High-quality assays with validated near-perfect efficiency | Any scenario, especially with suboptimal primer efficiency |
| Implementation in R | rtpcr package with efficiency=2 |
rtpcr package with actual efficiency values [44] |
Research indicates that the Pfaffl method provides superior accuracy in real-world laboratory conditions where amplification efficiencies frequently deviate from ideal values [44]. A recent evaluation of analysis methods highlighted that analysis of covariance (ANCOVA) approaches, which share the Pfaffl method's consideration of efficiency variations, enhance statistical power compared to the standard 2^(-ΔΔCT) method [47].
Detection Chemistry: SYBR Green vs. Probe-Based Methods
SYBR Green Chemistry
SYBR Green is a fluorescent dye that binds non-specifically to double-stranded DNA generated during PCR amplification. As the PCR product accumulates with each cycle, more dye molecules bind, resulting in increased fluorescence intensity proportional to the amount of DNA present [44]. The key advantage of SYBR Green is its cost-effectiveness, as it requires only sequence-specific primers without the need for expensive fluorescent probes [45] [46]. This makes it particularly suitable for assay development and studies requiring the analysis of multiple targets.
A critical requirement for SYBR Green assays is melting curve analysis following amplification to verify reaction specificity. This analysis distinguishes specific amplification from non-specific products such as primer dimers based on their distinct melting temperatures (Tm) [48]. Recent applications demonstrate the effectiveness of this approach, such as a SYBR Green assay developed for detecting Listeria monocytogenes and Listeria innocua that showed distinct Tm values for different species [45], and another assay for distinguishing toxic Convallaria majalis from edible Allium microdictyon [46].
Probe-Based Chemistry
Probe-based detection systems, such as TaqMan assays, utilize sequence-specific oligonucleotide probes labeled with a fluorescent reporter and quencher dye. When intact, the proximity of the quencher suppresses reporter fluorescence. During amplification, the 5'→3' exonuclease activity of DNA polymerase cleaves the probe, separating reporter from quencher and generating a fluorescent signal proportional to target amplification [44]. These assays offer enhanced specificity through dual recognition (both primers and probe must bind specifically for signal generation), making them ideal for discriminating between highly similar sequences such as single nucleotide polymorphisms or paralogous genes [49].
The primary limitations of probe-based approaches include higher costs and complex probe design, particularly for polymorphic targets [46]. However, for cancer biomarker applications requiring precise discrimination between transcript variants or homologous genes, the additional specificity often justifies these drawbacks.
Chemistry Selection for Cancer Gene Applications
Table 2: Detection Chemistry Comparison for Cancer Biomarker Research
| Consideration | SYBR Green | TaqMan/Probe-Based |
|---|---|---|
| Cost per Reaction | Low | High |
| Specificity | Moderate (requires melt curve verification) | High (dual recognition) |
| Multiplexing Capacity | None (single target per reaction) | High (with different fluorophores) |
| Assay Development Time | Short | Long |
| Optimization Complexity | Moderate | High |
| Ideal for Transcript Variant Discrimination | Possible with careful design | Excellent |
| Sensitivity in Complex Samples | Good | Excellent |
| Best Applications | Primer screening, expression profiling of many targets | Validation studies, low-abundance targets, clinical assays |
For cancer research applications, the choice between SYBR Green and probe-based detection should be guided by experimental objectives and resource constraints. SYBR Green is ideal for initial screening of multiple candidate biomarkers or when working with limited financial resources. Probe-based methods are preferable for clinical validation studies, especially when discriminating between homologous genes or splice variants, or when multiplexing is required to conserve precious patient samples [49] [50].
Experimental Workflow for Biomarker Validation
Sample Preparation and RNA Extraction
The foundation of reliable qPCR data begins with proper sample handling and nucleic acid extraction. For cancer biomarker studies, this typically involves processing tumor tissues, cell lines, or liquid biopsy samples. The extraction method should be selected based on sample type, with commercial kits providing consistent results. For formalin-fixed paraffin-embedded (FFPE) tissues—common in cancer research—specialized extraction protocols are required to address nucleic acid fragmentation and cross-linking [50]. Essential quality control measures include assessing RNA integrity numbers (RIN) and confirming the absence of contaminants using spectrophotometric or microfluidic methods [50] [46].
Reverse Transcription and cDNA Synthesis
Reverse transcription converts RNA to complementary DNA (cDNA) for subsequent qPCR analysis. Consistent input RNA amounts across samples are critical for valid comparisons. For cancer studies with potentially variable RNA quality from clinical specimens, using fixed RNA quantities (e.g., 500ng-1μg) is recommended. The choice between random hexamers and oligo-dT primers depends on experimental goals: random hexamers provide comprehensive representation, while oligo-dT primers enrich for mRNA and may improve detection of long transcripts from partially degraded samples [47].
Reference Gene Selection and Validation
Appropriate reference gene selection is paramount for accurate relative quantification. In cancer studies, reference gene expression should be stable across all experimental conditions, including different tumor types, treatments, or disease stages. At minimum, two validated reference genes should be used to normalize target gene expression [44]. Common reference genes include GAPDH, ACTB, and B2M, but these should be empirically validated for each experimental system, as cancer-associated metabolic changes can alter their expression. The rtpcr package supports analysis with up to two reference genes, providing more robust normalization than single reference genes [44].
qPCR Assay Design and Validation
For SYBR Green assays, primer pairs should be designed to produce amplicons of 75-150 bp with similar melting temperatures (typically 58-62°C). Amplicons should span exon-exon junctions to minimize genomic DNA amplification. Specificity should be verified through melt curve analysis and agarose gel electrophoresis [48] [46]. For probe-based assays, the probe should be located between the forward and reverse primers with a Tm 5-10°C higher than the primers.
Amplification efficiency should be determined for each primer pair using a standard curve with serial dilutions (e.g., 1:10) of cDNA template. Efficiency (E) is calculated from the slope of the standard curve using the formula E = 10^(-1/slope) - 1, with ideal efficiencies ranging from 90-110% (corresponding to slopes of -3.6 to -3.1) [46]. The correlation coefficient (R²) of the standard curve should exceed 0.98.
Data Analysis Workflow
The following workflow diagram illustrates the complete process from experimental design to data interpretation for relative quantification in cancer biomarker studies:
Advanced Applications in Cancer Research
Integration with High-Throughput Technologies
While qPCR remains the standard for targeted gene expression analysis, its utility in cancer research is enhanced when combined with high-throughput technologies. RNA sequencing (RNA-seq) provides unbiased transcriptome-wide discovery of differentially expressed genes, which can subsequently be validated using qPCR on larger sample cohorts [51] [50]. This integrated approach leverages the strengths of both technologies: the discovery power of RNA-seq and the sensitivity, precision, and cost-effectiveness of qPCR for validation studies.
Recent advancements include combined RNA and DNA analysis, exemplified by a validated integrated assay that simultaneously profiles gene expression, mutations, and copy number variations from a single tumor sample [50]. Such approaches enable comprehensive molecular characterization of cancer biomarkers while controlling for sample-to-sample variation.
Machine Learning Approaches
Machine learning algorithms applied to gene expression data can enhance cancer classification and biomarker discovery. Studies have demonstrated that algorithms like Support Vector Machines can achieve high accuracy (up to 99.87%) in classifying cancer types based on RNA-seq data [51]. While these approaches typically utilize genome-wide expression data, validated biomarkers identified through qPCR can be incorporated into focused predictive models with potential clinical utility.
Alternative Quantification Methods
Digital droplet PCR (ddPCR) represents an alternative quantification method that provides absolute quantification without standard curves. Recent studies comparing ddPCR to qPCR for copy number variation analysis found ddPCR demonstrated 95% concordance with gold standard methods, while qPCR showed only 60% concordance, particularly at higher copy numbers [52]. For cancer biomarker applications requiring precise quantification of low-abundance transcripts or subtle expression differences, ddPCR may offer advantages despite higher per-reaction costs.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for qPCR Biomarker Validation
| Category | Specific Items | Function & Importance |
|---|---|---|
| Sample Collection & Preservation | RNAlater, PAXgene Blood RNA Tubes | Stabilizes RNA immediately upon sample collection, prevents degradation |
| Nucleic Acid Extraction | AllPrep DNA/RNA kits (Qiagen), TRIzol, Column-based purification systems | Simultaneous DNA/RNA extraction, maintains nucleic acid integrity |
| Quality Assessment | NanoDrop spectrophotometer, Agilent TapeStation, Qubit fluorometer | Quantifies nucleic acid concentration, assesses purity (A260/280), evaluates integrity (RIN) |
| Reverse Transcription | High-Capacity cDNA Reverse Transcription kits, Random hexamers, Oligo-dT primers | Converts RNA to stable cDNA for repeated qPCR analysis |
| qPCR Reagents | SYBR Green Master Mix, TaqMan Universal Master Mix, ROX reference dye | Provides enzymes, buffers, nucleotides for amplification, contains passive reference dye |
| Assay Design Tools | Primer-BLAST, Beacon Designer, commercial assay libraries | Designs sequence-specific primers/probes, checks specificity |
| qPCR Instruments | Applied Biosystems QuantStudio series, Bio-Rad CFX, Roche LightCycler | Precise thermal cycling and fluorescence detection |
| Analysis Software | rtpcr R package, LinRegPCR, qBASE+, SDS software | Efficiency calculation, fold change determination, statistical analysis |
The rtpcr package for R deserves special emphasis among analysis tools, as it provides a comprehensive framework for calculating amplification efficiency, performing statistical analysis, and generating publication-quality graphical presentations of qPCR data [44]. This package implements both the ΔΔCT and Pfaffl methods and accommodates complex experimental designs with up to three different factors.
The accurate relative quantification of gene expression using qPCR remains an essential methodology for cancer biomarker validation. The choice between ΔΔCT and Pfaffl methods should be guided by careful evaluation of amplification efficiencies, with the Pfaffl method providing more robust results when efficiencies deviate from ideal values. Similarly, the selection between SYBR Green and probe-based detection chemistry involves trade-offs between cost, specificity, and experimental throughput.
For researchers validating cancer biomarkers, rigorous experimental design including proper reference gene validation, replication strategies, and data analysis methods is paramount. The integration of qPCR with emerging technologies like RNA-seq and machine learning approaches strengthens biomarker discovery and validation pipelines. By adhering to these methodologies and leveraging the appropriate tools and reagents, researchers can generate reliable, reproducible gene expression data to advance cancer diagnostics and therapeutic development.
Multiplexing with TaqMan Probes for Simultaneous Analysis of Multiple Gene Targets
In the field of cancer research, the ability to accurately analyze the expression of multiple genes simultaneously provides a significant advantage for understanding complex biological pathways, identifying biomarkers, and evaluating therapeutic responses. This technical guide focuses on multiplexing with TaqMan probes, a powerful qPCR methodology that enables researchers to quantify several genetic targets in a single reaction. When framed within the broader thesis of SYBR Green versus probe-based detection systems, TaqMan multiplexing emerges as a superior approach for applications demanding high specificity, such as profiling cancer gene expression panels, validating oncogenic signatures, and detecting low-abundance transcripts. Unlike dye-based methods that nonspecifically bind to any double-stranded DNA, TaqMan assays utilize target-specific fluorescent probes, ensuring that the measured signal originates exclusively from the gene of interest even in complex genetic backgrounds [53] [7]. This capability is paramount in cancer research where distinguishing between closely related paralogs or detecting minor mutations can have direct diagnostic and prognostic implications.
The transition from single-plex to multiplex qPCR represents a paradigm shift in experimental efficiency and data quality. By enabling the concurrent measurement of multiple targets from the same sample aliquot, multiplex TaqMan assays minimize sample volume requirements, reduce hands-on time, and eliminate inter-assay variability that often plagues studies comparing gene expression across separately run reactions [54]. Furthermore, the inclusion of internal controls within the same reaction tube—such as reference genes and positive controls—provides a robust normalization framework that enhances data reliability for sensitive applications like measuring minimal residual disease or subtle transcriptional changes in response to candidate therapeutics. For research and drug development professionals working with precious clinical samples or conducting high-throughput screening campaigns, these advantages make TaqMan multiplexing an indispensable tool in the molecular diagnostics arsenal.
Technical Foundations: How TaqMan Probes Work
Core Mechanism and Chemistry
The fundamental principle underlying TaqMan probe technology revolves around the 5' nuclease activity of Taq DNA polymerase, which cleaves a target-specific probe during the PCR amplification process [55] [53]. Each TaqMan probe is a short, single-stranded oligonucleotide designed to bind specifically to a complementary DNA sequence located between the forward and reverse primer sites. The probe is dually labeled with two key components: a fluorescent reporter dye at the 5' end and a quencher molecule at the 3' end [55]. When the probe is intact, the proximity of the quencher to the reporter dye suppresses fluorescence emission through a mechanism known as Fluorescence Resonance Energy Transfer (FRET) [53]. This results in minimal background signal at the beginning of the amplification reaction.
During the annealing phase of PCR, both the primers and the TaqMan probe bind to their complementary target sequences. As Taq polymerase extends the primer during the elongation phase, it eventually encounters the bound probe. The polymerase's 5' nuclease activity then cleaves the probe, physically separating the reporter dye from the quencher [55] [7]. This separation eliminates the FRET effect, allowing the reporter dye to fluoresce freely. The fluorescence intensity increases proportionally with each amplification cycle as more probe molecules are cleaved, enabling real-time monitoring of DNA accumulation [53]. This cleavage mechanism is particularly advantageous because it does not inhibit the overall PCR process—after cleavage, the polymerase continues to extend the DNA strand to completion [53].
Comparison with SYBR Green Chemistry
Understanding the distinctions between TaqMan probes and SYBR Green dye-based detection is crucial for selecting the appropriate method for cancer gene research. The table below summarizes the key differences:
Table 1: Key Differences Between TaqMan and SYBR Green qPCR Methods
| Parameter | TaqMan Probes | SYBR Green Dye |
|---|---|---|
| Detection Mechanism | Target-specific fluorogenic probes | Intercalates into any double-stranded DNA |
| Specificity | High - requires binding of both primers and probe [53] | Lower - requires well-designed primers and melt curve analysis [53] [7] |
| Sensitivity | High (detection of 1-10 copies) [53] | Variable, dependent on primer optimization [53] |
| Multiplexing Capability | Yes - multiple targets with different dyes [53] [56] | No - all amplification products detected with same signal [53] |
| Experimental Optimization | Minimal - predesigned assays available [53] | Requires extensive primer validation and optimization [53] |
| Cost Considerations | Higher per reaction due to probe costs | Lower per reaction, but potentially higher optimization costs |
For cancer gene research, the superior specificity of TaqMan probes is particularly valuable when distinguishing between homologous genes, detecting single nucleotide polymorphisms (SNPs) in oncogenes, or quantifying splice variants [57]. The multiplexing capability enables researchers to simultaneously measure multiple genes within a pathway—such as apoptosis regulators or cell cycle controllers—alongside reference genes from the same sample, ensuring more accurate normalization and reliable expression ratios [54].
Implementing TaqMan Multiplexing: Experimental Design and Workflows
Probe Selection and Design Considerations
Successful multiplexing with TaqMan probes begins with careful experimental design and probe selection. Researchers can choose from predesigned assays covering over 2.8 million targets across more than 30 species, or opt for custom assays tailored to specific sequences not available in standard catalogs [55]. When designing custom TaqMan probes for multiplex applications, several critical parameters must be considered:
- Probe Length: Optimal length typically ranges between 20-30 nucleotides to ensure both specificity and efficient hybridization [57].
- Melting Temperature (Tm): The Tm of the TaqMan probe should be approximately 10°C higher than that of the primers (generally 65-70°C) to ensure the probe binds before the primers during annealing [57].
- Sequence Considerations: Avoid repetitive nucleotide sequences, especially stretches of four or more consecutive G bases, which can form secondary structures and impair probe efficiency [57].
- Fluorophore Selection: For multiplex assays, select reporter dyes with non-overlapping emission spectra. Common combinations include FAM, VIC, ABY, JUN, and Cyanine 5 (Cy5) [56].
- Quencher Compatibility: Modern TaqMan probes typically use non-fluorescent quenchers (NFQ) which minimize background signal and increase sensitivity, making them ideal for multiplex applications [56].
For advanced applications requiring discrimination of highly similar sequences—such as mutant versus wild-type alleles in cancer genes—TaqMan MGB (Minor Groove Binder) probes offer enhanced specificity. MGB probes contain a minor groove binder moiety that increases the Tm of the probe, allowing the use of shorter probes while maintaining high specificity for single-base mismatches [53].
Practical Workflow for Multiplex TaqMan Assays
The implementation of a multiplex TaqMan experiment follows a systematic workflow from assay design to data analysis. The diagram below illustrates this process:
Key steps in the workflow include:
Reaction Optimization: When combining multiple primer-probe sets in a single tube, careful optimization of reagent concentrations is essential. Primer concentrations typically range from 50-900 nM, while probe concentrations are generally optimized between 50-250 nM [57]. The ratio between different primer-probe sets should be balanced to ensure similar amplification efficiencies across all targets.
Control Selection: Appropriate controls are critical for validating multiplex assays. These should include:
- No Template Control (NTC): Contains all reagents except cDNA template to detect contamination.
- No Reverse Transcriptase Control (No-RT): Processes RNA without reverse transcriptase to assess genomic DNA contamination.
- Positive Controls: Known expressed genes (e.g., GAPDH, ACTB) to confirm reaction efficiency [55].
Sample Preparation: High-quality RNA extraction is fundamental to successful multiplex qPCR. For gene expression analysis, purified total RNA should be free of contaminants such as genomic DNA, proteins, or enzymatic inhibitors [55]. Alternatively, researchers can use specialized kits like the TaqMan Cells-to-CT Express Kit to perform real-time RT-qPCR directly from cultured cells, eliminating the need for RNA purification [55].
Reagent Solutions and Experimental Setup
Essential Research Reagents
Implementing a robust multiplex TaqMan assay requires several key components. The table below details the essential research reagent solutions and their functions:
Table 2: Essential Research Reagents for Multiplex TaqMan Experiments
| Reagent / Component | Function | Specifications & Considerations |
|---|---|---|
| TaqMan Probes | Target-specific detection | Dual-labeled with reporter dye (FAM, VIC, etc.) and quencher (NFQ, TAMRA); HPLC-purified for optimal performance [56] [57] |
| qPCR Primers | Amplification of target sequences | Sequence-specific; designed to work with corresponding probes; desalted and available in dry or liquid format [56] |
| Reverse Transcriptase | cDNA synthesis from RNA templates | Essential for gene expression studies; high efficiency and fidelity |
| qPCR Master Mix | Provides reaction components | Contains DNA polymerase, dNTPs, buffer, MgCl₂; optimized for probe-based detection [53] |
| RNA Extraction Kit | Isolation of high-quality RNA | Removes contaminants; preserves RNA integrity; can be column-based or direct lysis methods [55] |
| Nuclease-free Water | Reaction preparation | Free of RNases and DNases to prevent sample degradation |
| qPCR Plates/Tubes | Reaction vessels | Compatible with real-time PCR instrument; optical clarity for fluorescence detection |
Multiplexing Configurations and Dye Combinations
The power of TaqMan multiplexing lies in the ability to distinguish multiple targets simultaneously through different fluorescent reporter dyes. Advanced configurations now enable researchers to detect up to six targets in a single reaction using optimized dye-quencher combinations [56]. The most common approaches include:
- MGB Probes: Ideal for multiplexing up to five targets using FAM, VIC, ABY, JUN, and Cyanine 5 (Cy5) dyes with nonfluorescent quenchers [56].
- QSY/QSY2 Probes: Enable multiplexing up to six targets, with QSY2 particularly useful for 5th and 6th targets using Cyanine 5 (Cy5) and Cyanine 5.5 (Cy5.5) dyes [56].
- TAMRA Probes: Traditional option suitable for multiplexing up to two targets, typically with FAM, VIC, and TET dyes [56].
For cancer gene research, this multiplexing capability allows researchers to design comprehensive panels that simultaneously measure oncogenes, tumor suppressor genes, reference genes, and potentially internal controls for sample quality in a single well. This comprehensive approach significantly enhances throughput while conserving precious clinical samples.
Data Analysis and Interpretation in Multiplex Experiments
Quantification Methods and Quality Control
Data analysis from multiplex TaqMan experiments follows the same fundamental principles as single-plex qPCR but requires additional considerations for accurate interpretation of multiple targets. The primary metric in qPCR analysis is the Cycle threshold (Ct) value, which represents the PCR cycle at which the fluorescent signal exceeds background levels [55]. A lower Ct value indicates a higher starting concentration of the target sequence.
Two primary quantification methods are employed:
Absolute Quantification: Determines the exact copy number or concentration of target sequences by comparing Ct values to a standard curve of known concentrations [55] [7]. This method is particularly useful in applications such as viral load quantification or determination of gene copy number variations.
Relative Quantification: Compares the expression levels of target genes between different samples (e.g., treated vs. control, tumor vs. normal tissue) relative to one or more stably expressed reference genes [55]. The 2^(-ΔΔCt) method (Livak method) is widely used for this purpose when amplification efficiencies are near 100% and comparable between targets [55] [7].
For multiplex experiments, quality control is paramount. Researchers should verify that:
- Amplification efficiencies for all targets fall between 90-110% [7]
- No signal is detected in negative controls (NTC and No-RT)
- There is no evidence of signal bleed-through between different fluorescent channels
- Reference genes show stable expression across all sample types
Special Considerations for Cancer Research Applications
In cancer gene research, multiplex TaqMan assays enable sophisticated experimental designs that can provide insights into tumor biology and treatment response. Key applications include:
- Gene Expression Profiling: Simultaneous measurement of multiple genes within a pathway (e.g., cell cycle regulation, apoptosis, metastasis) to understand coordinated regulation in tumor samples [55].
- Biomarker Validation: Confirming potential biomarkers identified through genomic or proteomic screens by quantifying their expression in larger sample cohorts.
- Pharmacodynamic Studies: Monitoring changes in gene expression in response to therapeutic interventions to understand mechanism of action and identify early response indicators.
The high sensitivity and specificity of TaqMan probes make them particularly suitable for detecting low-abundance transcripts often encountered in cancer research, such as transcription factors, regulatory non-coding RNAs, or mutated alleles present in a small subpopulation of cells [55] [57].
Advanced Applications in Cancer Research
TaqMan probe technology has revolutionized molecular diagnostics in oncology by enabling precise detection of genetic alterations driving cancer progression. A prime application is the detection of HER2 gene amplification in breast cancer, where TaqMan probes specific to the HER2 gene and a reference gene are used to determine HER2 status, guiding targeted therapy decisions [57]. The assay design involves two probes labeled with different fluorescent groups—one targeting the HER2 sequence and another targeting a reference sequence—which are reacted with patient DNA samples. The resulting fluorescence signals determine the HER2 amplification status, providing clinicians with critical information for treatment selection [57].
Beyond HER2 testing, multiplex TaqMan assays facilitate:
- SNP Genotyping: Distinguishing single nucleotide polymorphisms in genes involved in drug metabolism (e.g., CYP450 family) to inform personalized treatment strategies [53].
- Fusion Gene Detection: Identifying chromosomal translocations creating oncogenic fusion genes (e.g., BCR-ABL in leukemia) using specially designed probes spanning breakpoint regions.
- MicroRNA Expression Profiling: Quantifying small non-coding RNAs with roles in cancer regulation using modified TaqMan assays specifically designed for short RNA sequences [53].
The following diagram illustrates a representative advanced application—detecting gene amplification in cancer diagnostics:
For drug development professionals, these applications translate into valuable tools for target validation, compound screening, and biomarker identification throughout the drug discovery pipeline. The reproducibility and precision of TaqMan assays make them particularly suitable for regulated environments where data quality and consistency are paramount.
Multiplexing with TaqMan probes represents a sophisticated methodology that offers significant advantages for simultaneous analysis of multiple gene targets in cancer research. When evaluated against the alternative of SYBR Green chemistry, TaqMan probes provide superior specificity, reliable multiplexing capabilities, and enhanced reproducibility—attributes particularly valuable in complex experimental scenarios such as profiling cancer gene expression, detecting somatic mutations, and validating therapeutic targets. The technology's robust performance, combined with its compatibility with high-throughput workflows, makes it an indispensable tool for researchers and drug development professionals seeking to generate high-quality genetic data efficiently.
As molecular diagnostics continues to evolve, TaqMan probe technology is poised to maintain its relevance through ongoing innovations in probe chemistry, instrumentation, and data analysis methods. Emerging advancements such as digital PCR applications, integrated liquid biopsy workflows, and expanded multiplexing panels will further solidify the role of this methodology in precision oncology. For the scientific community focused on unraveling the complexities of cancer genetics and developing targeted therapies, mastering multiplex TaqMan approaches provides a critical foundation for generating translatable research findings that can ultimately inform clinical decision-making and improve patient outcomes.
The management of solid tumors, particularly non-small cell lung cancer (NSCLC) and colorectal cancer (CRC), has been transformed by the identification of driver mutations in genes such as KRAS and EGFR. These mutations serve as critical biomarkers for predicting response to targeted therapies [58] [26]. Traditionally, tumor genotyping has relied on tissue biopsy, an invasive procedure that is not always feasible, may fail to capture tumor heterogeneity, and cannot be easily repeated for disease monitoring [26]. Liquid biopsy has emerged as a powerful, minimally invasive alternative that analyzes tumor-derived components in blood or other body fluids, including circulating tumor DNA (ctDNA), circulating tumor cells (CTCs), and exosomes [58] [26].
The core analytical challenge in liquid biopsy is the detection of very low allele frequency mutations. In early-stage cancer or minimal residual disease, ctDNA can be vanishingly rare, often constituting less than 0.1% of the total cell-free DNA (cfDNA) in plasma [59]. This creates a demanding technical requirement for molecular diagnostics. The choice of detection platform is therefore paramount, balancing sensitivity, specificity, cost, and throughput. This case study focuses on this critical choice, evaluating the performance of the cost-effective SYBR Green dye-based qPCR method against the highly specific TaqMan probe-based qPCR for detecting KRAS and EGFR mutations in liquid biopsy samples, framed within the context of cancer gene research.
Technical Comparison: SYBR Green vs. TaqMan qPCR Chemistries
The fundamental principles of SYBR Green and TaqMan chemistries lead to significant differences in their application, performance, and suitability for detecting low-abundance mutations.
Underlying Mechanisms
SYBR Green Chemistry: This method utilizes a fluorescent dye that binds nonspecifically to the minor groove of all double-stranded DNA (dsDNA). As the target DNA is amplified during PCR, the dye binds to each new copy, resulting in a fluorescence increase proportional to the amount of PCR product. A critical subsequent step is the dissociation (melting) curve analysis, which assesses the specific melting temperature (Tm) of the amplicon to help distinguish specific products from nonspecific amplification or primer-dimers [60] [61] [62].
TaqMan Probe-Based Chemistry: This method employs a target-specific, oligonucleotide probe labeled with a fluorescent reporter dye at the 5' end and a quencher at the 3' end. When the probe is intact, the quencher suppresses the reporter's fluorescence via fluorescence resonance energy transfer (FRET). During PCR, the Taq DNA polymerase's 5' to 3' exonuclease activity cleaves the probe only when it is bound to its complementary target sequence. This cleavage separates the reporter from the quencher, resulting in a permanent fluorescence signal that accumulates with each cycle [61] [62]. TaqMan MGB (Minor Groove Binder) probes are a common refinement that increases the probe's binding affinity (Tm), allowing for the use of shorter probes and better discrimination of single-base mismatches, which is crucial for mutation detection [61].
Performance Comparison for Liquid Biopsy Applications
Table 1: Key Differences Between SYBR Green and TaqMan qPCR Chemistries
| Feature | SYBR Green qPCR | TaqMan qPCR |
|---|---|---|
| Specificity Mechanism | Amplicon melting curve analysis | Sequence-specific hybridization and probe hydrolysis |
| Inherent Specificity | Lower; binds any dsDNA | Higher; requires specific probe binding |
| Multiplexing Capability | No | Yes; with different reporter dyes [62] |
| Assay Development | Simpler; requires only primers | Complex; requires design & validation of probe [62] |
| Cost | Lower; no probe cost | Higher [62] |
| Sensitivity (Theoretical) | Variable; can be compromised by non-specific amplification | High; can detect 1-10 copies, ideal for low-abundance targets [62] |
| Primary Challenge | Risk of false positives from primer-dimers/non-specific amplification [60] [61] | Probe synthesis required for each new target [61] |
Experimental Protocols for Mutation Detection
This section outlines detailed methodologies for detecting KRAS and EGFR mutations from liquid biopsy samples, adaptable to both SYBR Green and TaqMan chemistries.
Pre-Analytical Phase: Plasma and evDNA Preparation
The pre-analytical phase is critical for the success of any liquid biopsy assay, as it directly impacts the quality and yield of the analytes.
Blood Collection and Plasma Separation: Blood (2×10 mL) should be collected into cfDNA BCT (Blood Collection Tubes) from manufacturers like Streck or Roche, which contain cell-stabilizing preservatives. These tubes prevent the release of genomic DNA from white blood cells, allowing for sample stability at room temperature for up to 3-7 days. If using conventional EDTA tubes, plasma must be separated within 2-6 hours of collection. Plasma is obtained by a first centrifugation at 1,600-3,000 × g for 10-20 minutes, followed by a second, higher-speed centrifugation of the supernatant at 16,000 × g for 10 minutes to remove residual cells and debris [59].
Extraction of Cell-Free DNA (cfDNA) and Exosomal DNA (evDNA): For cfDNA, extract from 1-5 mL of plasma using commercial silica-membrane column kits optimized for low-concentration samples. For exosomal DNA (evDNA), which can have a higher mutant allele frequency than cfDNA in early-stage disease [63], exosomes are first isolated from cleared plasma using Size Exclusion Chromatography (SEC). DNA is then extracted from the exosome fractions using the Phenol:Chloroform:Isoamyl Alcohol (PCI) method, followed by ethanol precipitation to recover the DNA [63].
DNA Quantification and Quality Control: Quantify the extracted DNA using a fluorometer (e.g., Qubit) for accuracy at low concentrations. The quality can be assessed by agarose gel electrophoresis or an automated electrophoresis system to confirm the expected fragment size distribution (~160-200 bp for cfDNA).
Analytical Phase: qPCR Setup and Cycling
This phase covers the setup and execution of the qPCR assay itself.
Primer and Probe Design: For both SYBR Green and TaqMan, design primers to generate short amplicons (60-150 bp) suitable for fragmented cfDNA. For TaqMan assays, the probe should be designed to span the mutation site of interest (e.g., KRAS G12D, EGFR L858R). For allelic discrimination, two separate reactions with wild-type and mutation-specific probes are typically used.
Reaction Setup:
- SYBR Green Master Mix: 1X SYBR Green PCR buffer, 3-5 mM MgCl₂, 0.2 mM dNTPs, 0.5-1 U DNA polymerase, 0.2-0.5 µM of each primer, and 2-5 µL of template DNA.
- TaqMan Master Mix: 1X TaqMan buffer, 3-5 mM MgCl₂, 0.2 mM dNTPs, 0.5-1 U DNA polymerase, 0.2-0.5 µM of each primer, 0.1-0.2 µM probe, and 2-5 µL of template DNA.
qPCR Cycling Conditions:
- Initial Denaturation: 95°C for 5-10 min.
- 45-50 Cycles of:
- Denaturation: 95°C for 15 sec.
- Annealing/Extension: 60°C for 1 min (optimize temperature based on primer Tm).
- (For SYBR Green only) Dissociation Curve: 95°C for 15 sec, 60°C for 1 min, then gradual increase to 95°C with continuous fluorescence measurement.
Post-Analytical Phase: Data Analysis
Cycle Threshold (Ct) and Quantification: The Ct value is determined for each reaction. For absolute quantification, a standard curve with known copy numbers of the target is required. For relative quantification (e.g., mutant allele frequency), the results are analyzed based on the differential detection between assays.
Allele-Specific qPCR (AS qPCR) Data Analysis: In methods like Intplex AS qPCR, the mutant allele frequency is calculated based on the differential amplification of mutant and wild-type alleles, potentially with a pre-amplification step to enrich the target [63].
Diagram 1: Liquid Biopsy qPCR Workflow. This diagram outlines the key steps from blood draw to final mutation report, highlighting the pre-analytical, analytical, and post-analytical phases. VAF: Variant Allele Frequency.
Case Study: KRAS Mutation Detection in Colorectal Cancer
A 2025 study investigated the performance of KRAS mutation detection in plasma exosomal DNA (evDNA) from patients with early-stage (I-III) colorectal cancer [63]. The study employed an allele-specific quantitative PCR (AS qPCR) method, a refined approach similar in principle to TaqMan, on evDNA extracted via Size Exclusion Chromatography (SEC).
Key Findings and Performance Data
The study demonstrated that mutation analysis in evDNA is a highly sensitive approach. The assay achieved a sensitivity of 0.01% for mutant allele frequency. Notably, 85% of the tested evDNA samples harbored one or two KRAS mutations, with a median mutant allele frequency of 1.18% (range: 0.01%–63.33%). Furthermore, the study reported a high concordance of mutation detection between evDNA and matched tumor tissue DNA, validating the clinical utility of this liquid biopsy approach [63].
Table 2: Performance Metrics of Detection Technologies for Liquid Biopsy
| Technology | Theoretical Sensitivity | Key Strengths | Key Limitations | Suitability for Low-Abundance Mutation |
|---|---|---|---|---|
| SYBR Green qPCR | Moderate (0.1-1.0%) | Low cost, simple assay development [62] | Low inherent specificity; prone to false positives [61] | Low to Moderate |
| TaqMan qPCR | High (0.1%) | High specificity, suitable for multiplexing [62] | Requires specific probe design, higher cost [61] | High |
| Digital Droplet PCR (ddPCR) | Very High (0.01%-0.001%) [58] | Absolute quantification without standard curve, high sensitivity | Limited multiplexing, higher cost than qPCR | Very High |
| Next-Generation Sequencing (NGS) | Variable (0.1-5%) | Comprehensive profiling, detects novel variants [58] | High cost, complex data analysis, requires high-quality DNA [58] | Moderate to High (depends on depth) |
Diagram 2: qPCR Detection Mechanisms. This diagram contrasts the workflows for SYBR Green and TaqMan qPCR, highlighting the fundamental difference in specificity: SYBR Green binds any dsDNA, while TaqMan requires specific probe hybridization.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Liquid Biopsy qPCR
| Item | Function/Description | Example Brands/Formats |
|---|---|---|
| cfDNA BCT Tubes | Blood collection tubes with preservatives to stabilize nucleated blood cells, preventing contamination of cfDNA with genomic DNA. | Streck cfDNA BCT, PAXgene Blood ccfDNA (Qiagen), Roche cfDNA BCT [59] |
| cfDNA/evDNA Extraction Kits | Silica-membrane columns or chemical methods optimized for maximum yield of short, fragmented DNA from plasma or exosomes. | QIAamp Circulating Nucleic Acid Kit, PCI method for evDNA [63] |
| qPCR Master Mix | Pre-mixed solutions containing buffer, dNTPs, Mg²⁺, hot-start DNA polymerase, and either SYBR Green dye or optimized for probe-based assays. | TaqMan Fast Advanced Master Mix, SYBR Green PCR Master Mix |
| Assay-On-Demand Probes/Primers | Pre-validated, sequence-specific primer and probe sets for known mutations (e.g., EGFR, KRAS), ensuring reproducibility and saving development time. | Thermo Fisher Scientific TaqMan Assays |
| Digital Droplet PCR (ddPCR) Supermix | Master mix formulated for droplet generation and PCR in ddPCR, used for ultra-sensitive absolute quantification of mutant alleles. | Bio-Rad ddPCR Supermix for Probes |
The detection of low-abundance KRAS and EGFR mutations from liquid biopsy samples is a cornerstone of modern precision oncology. This case study highlights that the choice between SYBR Green and TaqMan qPCR is not merely a technical preference but a strategic decision with significant implications for data quality and clinical application. TaqMan probe-based chemistry, with its superior inherent specificity and compatibility with multiplexing, is generally the more reliable and recommended choice for this demanding application, particularly when detecting known, predefined mutations at very low allele frequencies [62]. While SYBR Green offers a cost-effective and flexible alternative, its susceptibility to non-specific amplification makes it less ideal for the critical task of identifying low-level mutations in a high-background of wild-type DNA [61].
The field continues to evolve rapidly. Techniques like ddPCR are being used for their exceptional sensitivity in detecting mutations at frequencies as low as 0.001%–0.01% [58], while NGS provides an unbiased approach for comprehensive mutational profiling and discovery [58] [64]. Furthermore, the analysis of exosomal DNA (evDNA) is emerging as a highly promising source of tumor DNA, with studies suggesting it may offer a sensitivity advantage over cfDNA, especially in early-stage disease [63]. The ongoing development of even more sensitive assays and standardized protocols will be crucial for fully integrating liquid biopsy into routine clinical practice for cancer diagnosis, monitoring, and the early detection of resistance.
Optimization Strategies: Enhancing Sensitivity and Specificity in Cancer Gene Assays
In the context of cancer gene research, the choice between SYBR Green and probe-based detection methods for quantitative PCR (qPCR) presents a significant strategic decision. SYBR Green chemistry offers a cost-effective and flexible approach, binding nonspecifically to all double-stranded DNA (dsDNA), while probe-based methods like TaqMan provide target-specific detection through a fluorogenic probe [65]. However, this nonspecific binding nature of SYBR Green means it cannot distinguish between the desired amplicon and nonspecific products such as primer-dimers—artifactual amplicons formed by self-annealing primers that consume PCR reagents and generate false-positive signals [66] [65]. These primer-dimers pose a particular challenge in cancer research applications where detecting low-abundance transcripts or subtle expression changes in oncogenes and tumor suppressors is common. The formation of primer-dimers can severely compromise data accuracy by reducing amplification efficiency, increasing background noise, and interfering with accurate quantification [66] [67]. This technical guide provides comprehensive strategies for designing and validating high-efficiency primers that minimize dimer formation, with specific consideration for their application in SYBR Green versus probe-based detection systems for cancer gene research.
Understanding Primer-Dimers: Formation Mechanisms and Impact on PCR
Mechanisms of Primer-Dimer Formation
Primer-dimers are short, double-stranded DNA fragments that form when PCR primers anneal to each other rather than to the target DNA template [66] [68]. This process occurs through three sequential mechanisms: First, two primers anneal at their 3' ends due to complementary regions, often involving just a few nucleotides [68] [67]. If this hybridized construct remains stable, DNA polymerase binds and extends both primers according to their complementary sequences [68]. In subsequent PCR cycles, the newly synthesized primer-dimer product serves as a template for further amplification, leading to exponential accumulation of these artifactual products [68]. The stability of the initial primer-primer hybrid is enhanced by high GC-content at the 3' ends and longer complementary overlaps [68].
The risk of primer-dimer formation is highest during reaction setup before thermal cycling begins, when all reaction components are mixed at room temperature—conditions that favor nonspecific primer interactions [67]. Although DNA polymerases exhibit optimal activity around 70°C, they retain some polymerizing capability at lower temperatures, enabling extension of improperly annealed primers before the first denaturation step [68].
Impact on PCR Performance and Data Quality
Primer-dimer formation negatively impacts PCR performance through multiple mechanisms that are particularly problematic in sensitive cancer gene expression applications:
- Resource Competition: Primer-dimers compete with the target amplicon for essential PCR reagents including primers, DNA polymerase, dNTPs, and magnesium ions, reducing amplification efficiency of the actual target [69] [67].
- Signal Interference: In SYBR Green-based detection, primer-dimers bind the fluorescent dye and generate false-positive signals that obscure accurate quantification [65] [67].
- Reduced Sensitivity: The consumption of reaction components by primer-dimers decreases assay sensitivity, potentially preventing detection of low-abundance cancer transcripts [67].
- Inaccurate Quantification: Non-specific amplification can lead to erroneous quantification of gene expression levels, compromising data interpretation in cancer research [66].
The table below summarizes the comparative impact of primer-dimers in SYBR Green versus probe-based detection systems:
Table 1: Impact of Primer-Dimers in Different Detection Systems
| Parameter | SYBR Green Detection | Probe-Based Detection |
|---|---|---|
| Signal Generation | Direct signal from primer-dimer accumulation [65] | No signal unless probe binds to dimer (rare) [67] |
| Resource Depletion | Affects efficiency similarly in both systems [67] | Affects efficiency similarly in both systems [67] |
| Detection Method | Melt curve analysis essential for identification [65] | May not be visible in amplification plots [67] |
| False Positives | Common in non-template controls [67] | Less common [70] |
| Inhibition Pattern | Delayed Ct values, reduced amplification efficiency [67] | Delayed Ct values, reduced amplification efficiency [67] |
Strategic Primer Design for Minimizing Dimer Formation
Fundamental Primer Design Parameters
Careful primer design represents the most effective approach to prevent primer-dimer formation. The following parameters should be optimized during in silico primer design:
Primer Length: Design primers between 18-24 nucleotides to balance specificity and binding efficiency. Longer primers (>30 bp) hybridize more slowly and may reduce amplification yield, while shorter primers increase the risk of nonspecific binding [71] [72].
Melting Temperature (Tm): Maintain primer Tms between 54-65°C, with forward and reverse primers having Tms within 2-5°C of each other. The annealing temperature (Ta) is typically set 2-5°C above the primer Tm for optimal specificity [71] [72]. Tm can be calculated using the formula: Tm = 4(G + C) + 2(A + T) [72].
GC Content: Maintain GC content between 40-60%, with a roughly even distribution of G/C and A/T bases throughout the primer. Avoid stretches of identical nucleotides, particularly multiple Gs or Cs at the 3' end [71] [72].
3' End Specificity: Ensure the last 5 nucleotides at the 3' end contain no more than 3 G/C bases and lack complementarity to other primers in the reaction. The 3' terminus is critical for polymerase extension and particularly prone to dimer initiation [72].
Secondary Structures: Avoid self-complementarity and intra-primer homology that can lead to hairpin formation or self-dimers. Use primer design software to evaluate parameters for "self-complementarity" and "self 3'-complementarity" [71] [72].
Advanced Design Strategies for Challenging Targets
For particularly challenging applications such as high-resolution melting analysis or detection of single-nucleotide polymorphisms (SNPs) in cancer genes, advanced primer engineering strategies may be employed:
Self-Avoiding Molecular Recognition Systems (SAMRS): Incorporating SAMRS nucleobases (modified bases represented as g, a, c, t) into primers enables binding to natural DNA but not to other SAMRS-containing primers. This approach significantly reduces primer-primer interactions while maintaining efficient target binding, particularly valuable in multiplex PCR applications [69] [68].
Homo-Tag Assisted Non-Dimer System (HANDS): This technique adds a nucleotide tail complementary to the 3' end of the primer to its 5' end, creating a stem-loop structure that prevents primer-dimer formation while allowing target binding [68].
RNase H-dependent PCR (rhPCR): Utilizing blocked cleavable primers that only become active after specific cleavage by thermostable RNase HII at high temperatures. This method provides additional selectivity against primer-dimers as the enzyme demonstrates inherent primer:template mismatch discrimination [68].
Table 2: Optimal Primer Design Parameters for Cancer Gene Research
| Parameter | Optimal Range | Rationale | Special Considerations for Cancer Genes |
|---|---|---|---|
| Length | 18-24 nucleotides [71] [72] | Balances specificity and binding efficiency | Longer primers may be needed for GC-rich oncogenes |
| Tm | 54-65°C (within 2°C for pair) [71] [72] | Ensures synchronized annealing | Critical for detecting SNPs in cancer-associated genes |
| GC Content | 40-60% [71] [72] | Prevents overly stable or unstable hybrids | Higher GC content may be unavoidable in some targets |
| 3' End Stability | Max 3 G/C in last 5 bases [72] | Reduces mispriming and dimer initiation | Essential for allele-specific PCR in mutation detection |
| Self-Complementarity | Minimal (low scores in design tools) [72] | Prevents hairpins and self-dimers | Particularly important in multiplexed cancer panels |
Primer Design and Validation Workflow
Experimental Validation and Optimization Techniques
Primer Validation Protocols
Following in silico design, rigorous experimental validation is essential to confirm primer specificity and efficiency, particularly for cancer gene applications where quantification accuracy is critical.
Standard Curve Analysis for Efficiency Determination: Prepare a dilution series of template DNA (e.g., cDNA, gDNA, or cloned target) spanning at least five orders of magnitude (typically from 10 ng/µL to 0.1 pg/µL) [45]. Perform qPCR amplification using the designed primers and plot the Cycle threshold (Ct) values against the logarithm of template concentration. Calculate amplification efficiency using the formula: E = 10^(-1/slope) - 1. Ideal primers demonstrate efficiency between 90-110% (slope of -3.1 to -3.6) with correlation coefficient (R²) > 0.99 [45].
Melt Curve Analysis for SYBR Green Assays: After amplification, gradually increase temperature from 60°C to 95°C while continuously monitoring fluorescence. Analyze the derivative melt curve to identify specific amplicons by their characteristic melting temperatures (Tm). A single sharp peak indicates specific amplification, while multiple peaks, shoulders, or broad peaks suggest primer-dimer formation or nonspecific amplification [65].
Gel Electrophoresis Verification: Separate PCR products on 1.5-2% agarose gels. Specific amplicons should appear as single discrete bands at the expected size, while primer-dimers typically migrate as diffuse bands or smears around 30-50 bp [68] [45].
PCR Optimization Strategies
When primer-dimer formation persists despite careful design, several optimization strategies can be employed:
Hot-Start PCR: Utilize hot-start DNA polymerases that remain inactive until activated by high temperature (typically 95°C). This prevents enzymatic extension of misprimed templates during reaction setup [66] [68]. Various hot-start mechanisms include:
Annealing Temperature Optimization: Perform temperature gradient PCR (e.g., testing 55-70°C) to identify the highest annealing temperature that maintains specific amplification while minimizing dimer formation [66] [71].
Primer Concentration Titration: Systematically vary primer concentrations (typically 0.05-1.0 µM) to determine the lowest concentration that supports robust amplification without dimer formation [71] [67].
Touchdown PCR: Begin with an annealing temperature 5-10°C above the calculated Tm and gradually decrease by 0.5-1°C per cycle until the optimal annealing temperature is reached. This approach favors amplification of specific targets during early cycles when primer-dimer formation is most detrimental [71].
Table 3: Troubleshooting Guide for Primer-Dimer Issues
| Problem | Potential Causes | Solutions | Validation Method |
|---|---|---|---|
| Primer-dimer in NTC | Low annealing temperature, high primer concentration, primer complementarity [67] [70] | Increase Ta, reduce primer concentration, redesign primers [71] [67] | Melt curve analysis, gel electrophoresis [65] [45] |
| Multiple peaks in melt curve | Non-specific amplification, primer-dimer formation [65] | Optimize Mg²⁺ concentration, increase annealing temperature, use hot-start polymerase [66] [71] | Melt curve analysis, sequence verification [65] |
| Reduced amplification efficiency | Resource consumption by primer-dimers, suboptimal primer design [67] | Redesign primers with optimized parameters, use SAMRS modifications [69] [72] | Standard curve analysis [45] |
| False positives in SYBR Green | Nonspecific dye binding to primer-dimers [65] [67] | Switch to probe-based detection, redesign primers, optimize reaction conditions [65] [70] | Melt curve analysis, no-template controls [65] |
Special Considerations for Cancer Gene Research Applications
SYBR Green vs. Probe-Based Detection in Cancer Research
The choice between SYBR Green and probe-based detection methods carries specific implications for cancer gene research:
SYBR Green Applications:
- Ideal for initial screening of multiple cancer biomarkers where probe cost would be prohibitive
- Suitable for expression profiling of well-characterized cancer genes with established specificity
- Enables high-resolution melt analysis for mutation scanning in oncogenes [65]
- Requires extensive validation to ensure specificity for each target transcript
Probe-Based Detection Advantages:
- Superior specificity for distinguishing homologous cancer genes (e.g., RAS family members)
- Enables multiplexing for pathway-focused cancer gene panels
- Reduced false positives in minimal residual disease detection
- More reliable for clinical validation of cancer biomarkers [65] [70]
Advanced Techniques for Mutation Detection
Detection of single-nucleotide polymorphisms (SNPs) and somatic mutations in cancer genes requires exceptional primer specificity:
Allele-Specific PCR: Design primers with the 3' terminal nucleotide complementary to the mutant sequence. Under optimized conditions, extension occurs efficiently only with perfectly matched templates [69].
Enzyme-Assisted Methods: Utilize mismatch-sensitive enzymes such as restriction endonucleases or CRISPR-Cas systems to selectively cleave wild-type sequences, enriching mutant alleles for detection [14].
Digital PCR Applications: Partition reactions into thousands of individual droplets or chambers to detect rare mutations in heterogeneous tumor samples by limiting the impact of primer-dimer formation through endpoint detection [69].
Detection Method Selection for Cancer Research
Successful primer design and validation for cancer gene research requires both foundational reagents and specialized tools. The following table outlines essential resources for establishing robust PCR assays in cancer research settings.
Table 4: Research Reagent Solutions for Primer Design and Validation
| Category | Specific Products/Tools | Function/Application | Considerations for Cancer Research |
|---|---|---|---|
| Polymerase Systems | Hot-start Taq polymerases [66] [68] | Prevents pre-PCR mispriming and dimer formation | Essential for detecting low-abundance cancer transcripts |
| Design Software | PrimerSelect, Eurofins Genomics tools [45] [72] | In silico primer design and validation | Check cross-reactivity with homologous cancer genes |
| Specificity Verification | BLAST algorithms [45] [70] | Validates primer specificity against genomic databases | Critical for avoiding amplification of pseudogenes |
| Quantitation Reagents | SYBR Green master mixes [65] | Nonspecific detection of double-stranded DNA | Cost-effective for screening multiple cancer biomarkers |
| Specific Detection | TaqMan probes, molecular beacons [65] [68] | Target-specific fluorescence detection | Superior for distinguishing mutant vs. wild-type alleles |
| Quality Control | Agarose gels, bioanalyzers [45] | Visual confirmation of amplification specificity | Verify single bands for clean cancer gene amplification |
| Advanced Modifications | SAMRS components [69] [68] | Reduces primer-primer interactions | Particularly valuable in multiplexed cancer panels |
| Contamination Control | UNG treatment, aliquoted reagents [70] | Prevents false positives from amplicon carryover | Critical for sensitive detection of minimal residual disease |
Effective primer design and validation represent foundational elements in generating reliable gene expression data for cancer research. The strategic implementation of careful in silico design following established parameters for length, Tm, GC content, and specificity checks, combined with rigorous experimental validation through standard curves and melt curve analysis, enables researchers to minimize primer-dimer formation and maximize assay robustness. For SYBR Green applications, comprehensive optimization and validation are particularly critical, while probe-based methods offer inherent advantages for specific mutation detection and multiplexing. By adopting these systematic approaches to primer design and validation, cancer researchers can ensure the accuracy and reproducibility of their molecular analyses, ultimately supporting more reliable conclusions in cancer gene expression studies and biomarker development.
Using Melting Curve Analysis to Verify SYBR Green Assay Specificity and Identify Non-Specific Products
In the realm of cancer gene research, the choice between SYBR Green and probe-based detection methods is often dictated by the critical balance between cost and specificity. SYBR Green dye, a cost-effective intercalating dye that binds all double-stranded DNA (dsDNA), offers significant economic advantages for high-throughput screening of cancer biomarkers. However, its non-specific binding nature necessitates a robust mechanism to verify that the fluorescence signal originates from the intended target amplicon and not from spurious products like primer dimers or off-target amplification. Melting curve analysis (MCA) serves as this essential, post-amplification quality control step. This technical guide delves into the principles and applications of MCA, providing researchers with detailed protocols and troubleshooting frameworks to ensure data integrity, thereby positioning optimized SYBR Green assays as a reliable and powerful tool in the molecular oncologist's arsenal.
Quantitative PCR (qPCR) is indispensable in cancer research, enabling everything from the validation of transcriptomic data to the precise quantification of oncogene expression and tumor suppressor genes. The selection of detection chemistry—SYBR Green versus TaqMan probes—is a fundamental consideration. SYBR Green is an economical, intercalating dye that fluoresces upon binding to the minor groove of any dsDNA [5]. This "one-size-fits-most" approach reduces assay costs and simplifies setup but introduces a significant risk: the dye cannot distinguish between the specific PCR amplicon and non-specific products like primer dimers or mis-amplified sequences [65] [3]. These artifacts can lead to overestimation of target concentration and compromise data reliability, a critical concern when assessing gene expression profiles in patient samples.
In contrast, TaqMan assays utilize a sequence-specific probe labeled with a fluorophore and a quencher, generating a fluorescent signal only upon successful hybridization and cleavage of the probe during amplification [5] [3]. This inherent specificity makes them highly robust, but at a significantly higher cost per reaction. For research involving large panels of cancer genes or requiring frequent assay redesign for new targets, this cost can become prohibitive.
Therefore, the integration of melting curve analysis is what makes SYBR Green a viable and trustworthy alternative. MCA transforms SYBR Green from a non-specific dye into a specific assay by verifying the identity and purity of the amplified product based on its characteristic melting temperature (Tm) [65] [73]. This guide will demonstrate how rigorous MCA allows researchers to leverage the cost-effectiveness of SYBR Green while achieving a level of specificity comparable to probe-based methods, a strategy successfully employed in studies measuring gene expression profiles of targets like adenosine receptors in breast cancer [5].
The Principle of Melting Curve Analysis
Fundamental Theory
Melting curve analysis is a powerful technique that leverages the fundamental thermodynamic properties of DNA. After the completion of qPCR amplification, the resulting dsDNA products are subjected to a controlled temperature ramp, typically from around 60°C to 95°C [65]. As the temperature increases, the hydrogen bonds between the two DNA strands break, causing the dsDNA to denature into single strands. The SYBR Green dye, which is intercalated within the dsDNA, is released upon denaturation, resulting in a rapid decrease in fluorescence intensity [65] [73]. The process is monitored in real-time by the qPCR instrument.
The key parameter derived from this analysis is the melting temperature (Tm), which is the temperature at which 50% of the DNA duplexes are denatured [73]. The Tm is a unique property of a DNA sequence, determined by its length, GC content, and base sequence. Longer amplicons and those with higher GC content generally have higher Tm values due to greater stability [73]. Therefore, a specific PCR product with a known and consistent sequence will produce a distinct, predictable Tm peak.
Data Representation and Interpretation
The raw data from MCA, a plot of fluorescence versus temperature, produces a melting curve. However, for easier interpretation, this curve is often converted into a derivative melt curve (also known as a dissociation curve), which plots the negative derivative of fluorescence relative to temperature (-dF/dT) against temperature [65] [73]. This conversion transforms the gradual drop in fluorescence into a sharp, well-defined peak, with the peak maximum representing the Tm of the PCR product.
A single, narrow peak in the derivative plot strongly suggests that a single, specific product has been amplified. Observing multiple peaks, shoulders on the main peak, or unusually wide peaks indicates a mixture of products with different Tm values, signaling potential issues such as non-specific amplification or primer-dimer formation [65] [74]. The typical workflow and decision-making process based on MCA are illustrated below.
Identifying Non-Specific Products and Troubleshooting
A critical function of MCA is to diagnose the source of non-specific amplification. The Tm of the aberrant peak provides a primary clue for troubleshooting.
Common Artifacts and Solutions
Primer-Dimer Formation: This occurs when primers anneal to each other rather than the template, leading to the amplification of a very short product.
- MCA Signature: A peak with a low Tm, typically below 80°C [74]. This peak is often smaller than the specific product peak but can be dominant in reactions with low template concentration or inefficient amplification.
- Solutions: Redesign primers to minimize 3'-end complementarity. Lowering primer concentration or increasing the annealing temperature can also deter primer-dimer formation [65] [75].
Non-Specific Amplification: This involves the amplification of an off-target genomic sequence, often similar in size to the intended product.
- MCA Signature: A peak with a Tm close to or sometimes even higher than the specific product peak (Tm > 80°C) [74]. This can manifest as a distinct second peak or a shoulder on the main peak.
- Solutions: Increase the annealing temperature to enhance stringency. Redesign primers with improved specificity using bioinformatics tools (e.g., Primer-BLAST) to ensure they are unique to the target sequence [65] [75].
Genomic DNA Contamination: If the primer set is not designed to span an exon-exon junction, amplification from contaminating genomic DNA can occur.
- MCA Signature: May produce an additional peak, often with a higher Tm if the genomic amplicon is longer.
- Solutions: Treat RNA samples with DNase I. Design primers that span an intron or, ideally, an exon-exon junction [75].
Troubleshooting Guide for Abnormal Melt Curves
The following table summarizes common melt curve anomalies and their recommended fixes.
Table 1: Troubleshooting Guide for SYBR Green Melt Curve Anomalies
| Melt Curve Profile | Likely Cause | Recommended Solutions |
|---|---|---|
| Single peak, but Tm < 80°C | Only primer-dimer amplified; no specific product. | Redesign primers. Optimize template quality and concentration [74]. |
| Double peaks; minor peak < 80°C | Specific product + primer-dimer. | Lower primer concentration; increase annealing temperature; increase template concentration [74]. |
| Double peaks; minor peak > 80°C | Specific product + non-specific amplification. | Increase annealing temperature; check primer specificity; remove genomic DNA contamination [74]. |
| Multiple, irregular, or noisy peaks | Severe non-specific amplification or template contamination. | Check template quality/purity; redesign primers; run instrument diagnostics [65] [74]. |
| Broad or shallow peak | Multiple products of similar Tm or instrument sensitivity. | Confirm specificity on a high-concentration agarose gel (e.g., 3%). A temperature span ≤ 7°C may be acceptable [74]. |
Experimental Protocol: Implementing MCA for Assay Validation
This section provides a detailed, step-by-step protocol for validating a SYBR Green qPCR assay for a cancer gene target using MCA.
Pre- and Post-Amplification Analysis
A comprehensive validation strategy combines in silico design with empirical post-amplification checks, as outlined in the workflow below.
Step-by-Step Methodology
Primer Design and In Silico Validation:
- Design Parameters: Use software (e.g., Beacon Designer, Primer-BLAST) to design primers that are 19-22 bp long with an annealing temperature (Tm) of 60 ± 1 °C. The amplicon length should be between 70-150 bp for efficient amplification [75] [76].
- Specificity Check: Use BLAST analysis to ensure primers are unique to the target gene and do not bind to related pseudogenes or family members.
- Structural Check: Analyze primers with tools like OligoAnalyzer to avoid stable secondary structures (hairpins) and self-/hetero-dimer formation (ΔG ≤ -9 kcal/mol) [75].
- Exon-Spanning: For cDNA analysis, design primers to span an exon-exon junction to prevent amplification from genomic DNA.
qPCR Setup and Amplification:
- Reaction Mix: Prepare a 10-25 µL reaction containing 1X SYBR Green Master Mix, forward and reverse primers (typically 100-500 nM each, requires optimization), and template cDNA/DNA.
- Cycling Conditions:
- Initial Denaturation: 95°C for 5-10 min.
- 40-50 Cycles of:
- Denaturation: 95°C for 10-15 sec.
- Annealing: 60°C for 20-30 sec (optimize using a temperature gradient).
- Extension: 72°C for 20-30 sec.
Melting Curve Acquisition:
- After the final amplification cycle, immediately run the melt curve protocol on your qPCR instrument. A standard program is:
Data Analysis and Specificity Confirmation:
- View the melting data as a derivative plot (-dF/dT vs. Temperature).
- Identify the Tm of the main peak(s). Compare the observed Tm to the in silico predicted Tm for your amplicon (tools like uMelt can provide predictions) [77].
- A single, sharp peak at the expected Tm suggests a specific product. For final confirmation, analyze the PCR product by agarose gel electrophoresis. A single, discrete band at the expected size provides strong corroborative evidence [65].
SYBR Green vs. TaqMan: A Quantitative Comparison in Cancer Research
The choice between SYBR Green and TaqMan is not merely theoretical. Numerous studies have directly compared their performance in real-world research scenarios, including cancer. The data reveal that with careful optimization, SYBR Green can deliver performance on par with TaqMan, making it a cost-effective choice for many applications.
Table 2: Quantitative Comparison of SYBR Green and TaqMan qPCR Methods from Peer-Reviewed Studies
| Study Context | PCR Efficiency | Limit of Detection (LOD) | Key Finding | Citation |
|---|---|---|---|---|
| Adenosine Receptors in Breast Cancer | SYBR Green: >97%TaqMan: >97% | Not Specified | A significant positive correlation (P<0.05) was found between normalized gene expression data from both methods. | [5] |
| Residual CHO Cell DNA in Biopharmaceuticals | SYBR Green: 94.3%TaqMan: 96.6% | SYBR Green: 100 fgTaqMan: 10 fg | The TaqMan assay showed better (10x) sensitivity, attributed to the additional specificity of the probe. | [8] |
| Enterotoxigenic B. fragilis (bft gene) in Stool | SYBR Green: >98%TaqMan: >98% | <1 copy/µL (both) | SYBR Green under-performed in clinical samples, detecting only 13/38 positives vs. 35/38 with TaqMan. Copy numbers were significantly lower. | [9] |
| SARS-CoV-2 Detection (N and E genes) | Optimized for multiplex | Comparable to commercial TaqMan kit | With high-performance primers and MCA, the SYBR Green assay achieved 97% specificity and 93% sensitivity versus the TaqMan benchmark. | [76] |
The synthesis of these findings indicates that SYBR Green performance is highly dependent on assay optimization and sample type. For well-characterized targets in controlled samples (e.g., cell lines, purified DNA), it can be as efficient and reliable as TaqMan [5]. However, in complex samples with potential inhibitors or high background (e.g., stool), the added specificity of a TaqMan probe may be necessary to avoid false negatives and ensure accurate quantification [9]. The successful deployment of a SYBR Green assay for SARS-CoV-2 diagnosis further underscores that with rigorous validation including MCA, it can meet high diagnostic standards [76].
Successful implementation of a specific SYBR Green assay relies on a suite of carefully selected reagents and bioinformatics tools.
Table 3: Research Reagent Solutions for SYBR Green qPCR and Melt Curve Analysis
| Item | Function/Description | Example Use Case |
|---|---|---|
| SYBR Green Master Mix | A pre-mixed solution containing hot-start DNA polymerase, dNTPs, buffer, and the SYBR Green dye. Often includes ROX as a passive reference dye. | The core reagent for performing the qPCR reaction. Choosing a high-quality, robust master mix is critical for sensitivity and reproducibility. |
| High-Purity Primers | HPLC- or PAGE-purified oligonucleotides designed for high specificity and minimal self-complementarity. | Reduces the risk of primer-dimer formation and non-specific amplification, leading to cleaner melt curves. |
| Nucleic Acid Extraction Kit | Kits for purifying DNA or RNA from various sample types (e.g., tissues, cells, blood). | Ensures high-quality, contaminant-free template, which is vital for accurate Cq values and preventing PCR inhibition. |
| uMelt Software | Free online tool (University of Utah) that predicts the melt curve and derivative plot of a DNA sequence. | Used during assay design to predict the Tm of your amplicon and identify potential issues before ordering primers [77]. |
| Primer-BLAST | NCBI tool that combines Primer3 design with BLAST search to ensure primer specificity. | Verifies that designed primers will amplify only the intended target gene and not other sequences in the genome [75] [76]. |
Melting curve analysis is not merely an optional add-on but a fundamental component of any rigorous SYBR Green qPCR experiment, especially in the precise field of cancer gene research. It provides the necessary quality control to transform a cost-effective but non-specific detection method into a highly reliable and specific assay. By adhering to the detailed protocols for primer design, experimental optimization, and systematic troubleshooting outlined in this guide, researchers can confidently employ SYBR Green assays. This approach delivers data quality comparable to more expensive probe-based methods, thereby maximizing research output and enabling the large-scale screening of cancer biomarkers without compromising scientific integrity.
The accurate detection of single-nucleotide polymorphisms (SNPs) and point mutations is a cornerstone of modern precision oncology, enabling everything from cancer subtyping and prognosis to guiding therapeutic decisions. Genes such as EGFR, TP53, and KRAS frequently harbor mutations that are critical diagnostic and therapeutic targets [14]. In this context, real-time PCR (qPCR) has emerged as a rapid, sensitive, and accessible platform for mutation detection. However, a central challenge in qPCR assay design is achieving sufficient specificity to distinguish between wild-type and mutant sequences that differ by only a single nucleotide. This technical challenge frames a fundamental methodological choice for researchers: the use of intercalating dyes like SYBR Green versus sequence-specific fluorescent probes [14] [8].
While SYBR Green provides a low-cost and flexible detection method, its inability to distinguish between specific and non-specific amplification products can be a significant limitation in complex genotyping applications [8]. Hydrolysis probes, particularly those incorporating Minor Groove Binder (MGB) technology, offer a sophisticated solution to this problem. These probes are engineered to overcome the thermodynamic limitations of traditional TaqMan probes, providing the enhanced specificity required for reliable allelic discrimination in cancer gene research [78] [79] [80].
The Biochemical Basis of MGB Probe Technology
Fundamental Structure and Mechanism
MGB probes are dual-labeled hydrolysis probes that incorporate a unique chemical moiety at their 3' end. The standard configuration consists of:
- A 5' fluorescent reporter dye (e.g., FAM, HEX, VIC)
- A 3' non-fluorescent quencher (e.g., Eclipse Dark Quencher)
- A covalently attached Minor Groove Binder molecule
The MGB moiety is typically the trimer of 1,2-dihydro-(3H)-pyrrolo[3,2-e]indole-7-carboxylate (CDPI3), which binds reversibly to the minor groove of double-stranded DNA [79]. This binding event is a key differentiator from conventional TaqMan probes, fundamentally changing the hybridization dynamics and enabling the design of shorter, more specific oligonucleotides.
Table 1: Comparison of qPCR Detection Chemistries for Genotyping
| Feature | SYBR Green | Traditional TaqMan Probes | MGB Probes |
|---|---|---|---|
| Specificity | Moderate (binds any dsDNA) | High | Very High |
| Probe Length | Not Applicable | Typically 25-40 bases | 8-20 bases |
| Tm Enhancement | Not Applicable | Limited | Significant (+10-20°C) |
| Mismatch Discrimination | Poor | Moderate | Excellent |
| Best Application | Target discovery, initial screening | Known target quantification | SNP detection, allelic discrimination |
| Cost | Low | Moderate | Moderate-High |
Enhanced Allelic Discrimination Through Duplex Stabilization
The exceptional capability of MGB probes to discriminate single-base mismatches stems from two interconnected mechanisms. First, the MGB moiety stabilizes the probe-target duplex through van der Waals forces, hydrogen bonding, and electrostatic interactions with the AT-rich regions of the minor groove. This stabilization effectively increases the melting temperature (Tm) of the probe-target hybrid by approximately 10-20°C, allowing for the use of shorter probes (typically 12-18 nucleotides) while maintaining a practical hybridization temperature [78] [79].
Second, and critically for allelic discrimination, this stabilization effect is highly sequence-dependent. A single base mismatch creates a structural distortion that prevents the MGB moiety from binding optimally to the minor groove. This results in a disproportionately large decrease in Tm (ΔTm) for mismatched hybrids compared to perfectly matched ones. Research demonstrates that this effect is particularly pronounced when the mismatch occurs within the MGB binding region, leading to enhanced discrimination between wild-type and mutant alleles [79] [80]. The shorter probe length further enhances this discrimination, as a single base mismatch represents a larger percentage of the total hybridization sequence.
MGB Probes Versus SYBR Green: A Technical Comparison in Cancer Research
Fundamental Differences in Detection Mechanism
The choice between SYBR Green and probe-based detection represents a fundamental strategic decision in assay design, with significant implications for data quality, specificity, and application suitability.
SYBR Green is an intercalating dye that fluoresces when bound to any double-stranded DNA. While cost-effective and flexible, this mechanism presents notable challenges for genotyping:
- Inability to distinguish specific from non-specific amplification, including primer dimers
- Limited multiplexing capability
- Reduced specificity in detecting single-nucleotide changes [8] [45]
In contrast, MGB probes provide a three-layer specificity check:
- Primer specificity during amplification
- Probe hybridization specificity during the annealing step
- 5'-3' nuclease activity of the DNA polymerase, which cleaves only perfectly hybridized probes
This multi-layered specificity is particularly valuable in cancer research, where samples often contain a mixture of wild-type and mutant DNA (heteroplasmy or heterozygosity) and the mutant allele may be present at low frequencies [14] [80].
Performance Comparison in Sensitive Applications
Direct comparative studies highlight the performance advantages of probe-based methods in demanding applications. In quantifying residual host-cell DNA in biopharmaceuticals, TaqMan assays demonstrated a 10-fold lower limit of detection (10 fg) compared to SYBR Green (100 fg) [8]. This enhanced sensitivity is crucial for detecting low-abundance mutations in liquid biopsies or minimal residual disease monitoring.
Table 2: Quantitative Performance Comparison in Genotyping Applications
| Parameter | SYBR Green qPCR | MGB Probe qPCR |
|---|---|---|
| Limit of Detection (DNA) | 100 fg [8] | 10 fg [8] |
| Detection Sensitivity | 0.1%-1% mutant allele [14] | 0.01%-0.1% mutant allele [14] [80] |
| ΔTm for Single Base Mismatch | Not applicable | 8-12°C [79] |
| Multiplexing Capacity | Low | Moderate (2-4 plex) |
| PCR Efficiency | 94.3% [8] | 96.6% [8] |
For cancer researchers, this translates to tangible benefits in detecting rare mutations. Techniques like NAVIGATER (Nucleic Acid enrichment Via DNA Guided Argonaute from Thermus thermophilus), when combined with downstream detection, enable ultra-sensitive detection of mutations as low as 0.01% in pancreatic cancer blood samples [14].
Experimental Design and Protocol for MGB-Based Allelic Discrimination
Probe and Primer Design Considerations
Successful MGB probe design requires careful attention to several critical parameters:
- Probe Length: Optimal length is typically 13-18 nucleotides, shorter than conventional TaqMan probes (25-30 nucleotides) [79]
- Positioning: For SNP detection, the discriminatory nucleotide should be positioned centrally within the probe sequence, preferably within the MGB binding region (closer to the 3' end) where mismatch discrimination is most effective
- Tm Differential: Design for a ΔTm of ≥8°C between matched and mismatched hybrids to enable clear allelic discrimination
- Fluorophore Selection: Common dyes include FAM, HEX, TET, and Yakima Yellow, with choice dependent on instrument compatibility and multiplexing requirements [78] [79]
Primers should be designed following standard qPCR principles, with amplicons kept relatively short (50-150 bp) for optimal efficiency. The annealing temperature should be optimized to be several degrees below the Tm of the probe for the perfectly matched sequence.
Detailed qPCR Protocol for SNP Genotyping
The following protocol has been successfully applied for genotyping mitochondrial DNA mutations associated with Leber hereditary optic neuropathy [80] and can be adapted for cancer-associated SNPs:
Reaction Setup:
- Total Reaction Volume: 20 µL
- Template DNA: 5 ng (or 10-100 copies for low-abundance mutation detection)
- Master Mix: Commercial TaqMan Genotyping Master Mix
- Primers: 0.1-0.9 µM each (optimized concentration)
- MGB Probes: 0.1-0.2 µM each (wild-type and mutant-specific probes)
Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 10 minutes (1 cycle)
- Amplification: 95°C for 15 seconds → 62°C for 40 seconds (40 cycles)
- Post-read: Fluorescence detection at the end of each annealing/extension step
Data Analysis:
- Endpoint Analysis: Plot fluorescence signals for wild-type vs. mutant probes to generate cluster plots for genotype calling
- Quantitative Analysis: Use standard curves from serial dilutions of control plasmids for absolute quantification of mutation load [80]
Advanced Applications and Emerging Technologies
Multiplex Detection of Cancer Mutations
The high specificity of MGB probes enables the design of multiplex assays for simultaneous detection of multiple mutations. Research has demonstrated successful triplex TaqMan-MGB probe qPCR assays for detecting three primary mitochondrial DNA mutations, with standard curves showing high specificity and sensitivity without cross-reactivity [80]. In cancer research, this capability can be leveraged to screen for multiple hotspot mutations in genes like KRAS, EGFR, and TP53 in a single reaction, conserving precious patient samples and reducing processing time.
Comparison with Emerging Probe Technologies
While MGB probes represent a significant advancement, newer technologies continue to emerge. Recent research describes dark quencher (DQ) probes incorporating dihydropyrroloindole carboxylate (DPI3) analogs, which reportedly offer improved specificity and sensitivity compared to traditional TaqMan MGB probes in multiplex SARS-CoV-2 variant detection [81]. These DQ probes form stable heterozygous complexes with complementary DNA and RNA targets, increasing Tm values without requiring connection to quenched groups like MGB. For cancer researchers, such advancements may eventually translate to even more robust multiplex genotyping panels.
Other advanced detection strategies are also being explored, including:
- Enzyme-assisted methods utilizing ligases and exonucleases for mismatch discrimination
- Nanoparticle-based methods leveraging unique optical and electrical properties
- CRISPR-Cas systems for sequence-specific enrichment and detection [14]
Table 3: Research Reagent Solutions for MGB-Based Genotyping
| Reagent/Resource | Function | Example Products |
|---|---|---|
| MGB Probes | Sequence-specific detection with enhanced discrimination | IDT MGB Eclipse Probes, Biosearch Technologies MGB Probes [78] [79] |
| qPCR Master Mix | Optimized buffer, enzymes, and dNTPs for probe-based detection | TaqMan Genotyping Master Mix, BHQ Probe Master Mix [79] [80] |
| Control Templates | Assay validation and standard curve generation | Plasmid standards, gBlock Gene Fragments [78] [80] |
| Hybridization Capture | Target enrichment for low-frequency mutation detection | xGen Hybridization and Wash Kits [82] |
| DNA Purification Kits | High-quality input DNA preparation | Commercial genomic DNA extraction kits [80] [45] |
Minor Groove Binder technology represents a significant advancement in hydrolysis probe design, directly addressing the critical need for precise allelic discrimination in cancer research. By enabling the use of shorter, more specific probes with enhanced mismatch discrimination, MGB probes provide researchers with a powerful tool for detecting somatic mutations, assessing heteroplasmy, and monitoring minimal residual disease with confidence. While SYBR Green retains utility for initial screening and expression analysis, MGB probe-based assays offer the specificity and sensitivity required for definitive genotyping applications in translational cancer research. As probe technologies continue to evolve alongside complementary enrichment strategies, researchers will be increasingly equipped to meet the analytical challenges of precision oncology.
In the field of cancer gene research, accurate quantification of gene expression is paramount for identifying biomarkers, understanding tumor biology, and developing targeted therapies. Real-time quantitative polymerase chain reaction (qPCR) serves as a cornerstone technology for gene expression analysis, with two predominant detection chemistries: SYBR Green and probe-based methods such as TaqMan. The fundamental principle underlying precise quantification in qPCR is the characterization of reaction efficiency through standard curves, which enables researchers to determine initial template quantities with confidence [5] [53].
Reaction efficiency represents the proportionality between the initial amount of target nucleic acid and the amplified product detected during the exponential phase of PCR. In ideal conditions, where reaction efficiency is 100%, the amount of product doubles with each cycle. However, numerous factors can impact this efficiency, including reagent quality, primer design, template purity, and presence of inhibitors [83]. This technical guide examines the critical role of efficiency calculations and standard curves in ensuring accurate quantification, specifically within the context of SYBR Green versus probe-based detection methods for cancer gene research.
Fundamental Principles of qPCR Efficiency
Mathematical Foundations
The efficiency (E) of a qPCR reaction is defined as the fraction of target molecules that are duplicated in each amplification cycle. This parameter follows a predictable mathematical relationship that forms the basis for quantification:
- Ideal Efficiency: At 100% efficiency (E = 2), the number of amplicons doubles each cycle (2^n)
- Efficiency Calculation: Efficiency can be derived from the slope of a standard curve using the formula: E = 10^(-1/slope) [83]
- Theoretical Slope: A perfect reaction with 100% efficiency yields a slope of -3.32
- Practical Range: Acceptable efficiency typically falls between 90-110% (slope of -3.6 to -3.1)
The relationship between efficiency (E), slope, and starting quantity (Q) is expressed as: Q ∝ E^(-Ct) [83]
Where Ct represents the threshold cycle. This exponential relationship means that even small deviations from ideal efficiency can significantly impact calculated starting quantities. For example, with a Ct of 20, the quantities resulting from 100% versus 80% efficiency differ by approximately 8.2-fold, underscoring the critical importance of accurate efficiency determination [83].
Impact of Chemistry Choice on Efficiency
The selection between SYBR Green and TaqMan chemistries influences the approach to efficiency optimization:
- TaqMan Systems: Commercial TaqMan assays are designed within a universal system that integrates cycling conditions, chemistry, and assay design to consistently deliver near-100% geometric efficiency [83]
- SYBR Green Systems: Efficiency is more variable and dependent on primer design and reaction optimization, though studies demonstrate that with high-performance primers and proper protocols, SYBR Green can achieve efficiencies exceeding 95-97%, comparable to TaqMan [5] [84]
Standard Curves: Construction and Interpretation
Experimental Design for Standard Curve Generation
The standard curve method remains a fundamental approach for assessing qPCR efficiency and enabling absolute quantification. Proper construction requires careful experimental design:
Table 1: Optimal Standard Curve Parameters for Efficiency Assessment
| Parameter | Recommended Specification | Rationale |
|---|---|---|
| Number of Points | 7-point series | Provides sufficient data points for reliable linear regression |
| Dilution Factor | 10-fold serial dilutions | Creates a wide dynamic range for assessment |
| Concentration Range | 6-9 logs | Ensures detection over biologically relevant quantities |
| Replication | Minimum of triplicates | Accounts for technical variability |
| Template Type | Highly concentrated target (e.g., plasmid) | Enables creation of extensive dilution series |
The ideal structure for a standard curve intended to assess efficiency is a 7-point series with a 10-fold dilution factor, though such a series requires highly concentrated target material which may not be naturally available in all biological samples [83].
Practical Implementation
In practice, researchers generate standard curves using serial dilutions of known template quantities. The Ct values obtained from amplifying these standards are plotted against the logarithm of their initial concentrations. The resulting slope and correlation coefficient (R²) provide critical information about reaction performance:
- Slope Interpretation: As detailed in Equation 2, the slope determines theoretical efficiency [83]
- Linearity (R²): Values >0.99 indicate excellent linearity and consistent efficiency across the concentration range
- Y-intercept: Reflects the theoretical Ct value for a single copy of the target
A study comparing SYBR Green and TaqMan methods for analyzing adenosine receptor subtypes in breast cancer demonstrated that both methods could achieve efficiencies above 97% with proper optimization, and showed significant positive correlation between the normalized data from both approaches (p < 0.05) [5].
Efficiency Assessment Methods
Standard Curve-Based Assessment
The traditional approach to efficiency determination relies on standard curves generated from serial dilutions. While this method is widely used, it presents several practical challenges:
- Resource Intensive: Requires additional cost and labor for preparation of dilution series
- Error Prone: Susceptible to inaccuracies from inhibitors, pipetting errors, and dilution point mixing problems [83]
- Concentration Dependency: Requires highly concentrated target material for optimal dilution series
These limitations have prompted the development of alternative assessment methods that can provide reliable efficiency data without extensive dilution series.
Alternative Assessment Strategies
Table 2: Methods for Assessing qPCR Efficiency
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| Standard Curve Slope | Efficiency calculated from dilution series slope (E = 10^(-1/slope)) | Direct calculation of numerical efficiency value | Error-prone; requires significant resources |
| Visual Assessment | Comparison of amplification plot slopes for parallelism | No standard curves needed; not impacted by pipetting errors | Does not produce numerical efficiency value |
| User Bulletin #2 | Subtraction of slopes from two assays using same dilution series | Corrects for potential pipet calibration error | Does not correct for other error types; still error-prone |
The visual assessment method offers particular advantages for routine validation. This approach evaluates the parallelism of geometric amplification slopes in log-linear plots. When multiple assays have 100% geometric efficiency, their geometric slopes should be parallel both within and between assays. This method eliminates the need for standard curves and is not impacted by common technical errors such as pipetting inaccuracies or dilution problems [83].
Comparative Analysis: SYBR Green vs. TaqMan in Cancer Research
Technical Performance Considerations
The choice between SYBR Green and TaqMan chemistries involves trade-offs between specificity, cost, and optimization requirements:
- Specificity: TaqMan provides higher specificity due to sequence-specific probe hybridization, while SYBR Green detects all double-stranded DNA, including non-specific products [5] [53]
- Cost Structure: SYBR Green is significantly more cost-effective as it doesn't require fluorescently-labeled probes [5] [76]
- Multiplexing Capability: TaqMan enables multiplex detection using different reporter dyes, while SYBR Green is limited to single-plex reactions [53]
- Optimization Requirements: SYBR Green demands careful primer design and melt curve analysis to verify specificity, whereas predesigned TaqMan assays require minimal optimization [53] [76]
Experimental Evidence in Cancer Applications
Research directly comparing these methodologies in cancer contexts demonstrates their relative performance. A study on adenosine receptor expression in breast cancer tissues found that with optimized conditions, SYBR Green could generate data comparable to TaqMan, with efficiencies exceeding 95% for both methods and significant correlation in normalized expression data (p < 0.05) [5].
Similarly, a recent study detecting SARS-CoV-2 demonstrated high concordance between SYBR Green and TaqMan methods, with difference in Ct values of 0.72 ± 0.83 (p = 0.392) in naso-oropharyngeal swabs, supporting the reliability of properly optimized SYBR Green assays [84]. These findings reinforce that with appropriate validation, SYBR Green represents a viable, cost-effective alternative for gene expression studies in cancer research.
Implementation Protocols for Cancer Gene Analysis
Standard Curve Method for Absolute Quantification
The following protocol outlines the standard curve approach for absolute quantification of cancer gene targets:
- Standard Preparation: Serially dilute (10-fold) a known quantity of target template (e.g., plasmid, PCR product, or synthetic oligo) spanning 6-7 orders of magnitude
- Amplification: Run qPCR reactions with both standard dilutions and unknown samples in the same plate
- Data Collection: Record Ct values for all reactions
- Curve Generation: Plot log10(initial quantity) against Ct values for standards
- Linear Regression: Calculate slope, y-intercept, and R² value from the standard curve
- Efficiency Calculation: Apply formula E = 10^(-1/slope)
- Quantity Determination: Interpolate unknown sample quantities from the standard curve equation
This method is particularly valuable when absolute copy number determination is required, such as in viral load quantification or gene copy number variation studies relevant to cancer pathogenesis [53] [85].
ΔΔCt Method for Relative Quantification
For most gene expression studies in cancer research, relative quantification suffices. The ΔΔCt method provides a simplified approach with specific efficiency considerations:
- Efficiency Validation: Confirm that both target and reference gene assays display similar, near-100% efficiency
- Ct Determination: Record Ct values for target and reference genes in all samples
- Normalization: Calculate ΔCt = Ct(target) - Ct(reference) for each sample
- Calibration: Calculate ΔΔCt = ΔCt(sample) - ΔCt(calibrator)
- Expression Calculation: Determine relative quantity = 2^(-ΔΔCt)
The traditional ΔΔCt method assumes perfect (100%) efficiency for all assays. When this condition is met, it offers "reduced cost, lower labor, higher throughput and greater accuracy compared to the standard curve method" [83]. Modified equations can accommodate differing efficiencies between assays, but best practice involves using only assays with 100% efficiency [83].
The Scientist's Toolkit: Essential Reagents for qPCR Efficiency
Table 3: Key Research Reagent Solutions for qPCR Efficiency Studies
| Reagent/Kit | Function | Application Context |
|---|---|---|
| RNeasy Plus Mini Kit (Qiagen) | Total RNA extraction with genomic DNA removal | Preparation of high-quality RNA from tissue samples [5] |
| QIAamp DNA FFPE Tissue Kit (Qiagen) | DNA extraction from formalin-fixed paraffin-embedded tissue | Enables analysis of archived clinical cancer specimens [86] [85] |
| QIAamp Circulating Nucleic Acid Kit (Qiagen) | Cell-free DNA extraction from plasma, urine | Liquid biopsy analysis for cancer detection [85] |
| Quantitect SYBR Green Master Mix (Qiagen) | Optimized reaction mix for SYBR Green qPCR | Provides consistent buffer conditions for efficiency monitoring [5] |
| TaqMan Universal PCR Master Mix (ABI) | Optimized reaction mix for TaqMan assays | Ensures consistent 5' nuclease activity for probe hydrolysis [5] |
| DNeasy Blood & Tissue Kit (Qiagen) | Genomic DNA extraction from fresh tissues | Standardized DNA isolation for qPCR template preparation [85] |
Troubleshooting Efficiency Abnormalities
Common Issues and Solutions
Deviations from ideal efficiency (90-110%) require systematic troubleshooting:
- Low Efficiency (<90%): Often results from poor primer design, reaction inhibitors, or suboptimal reaction conditions. Redesign primers or purify template to resolve.
- High Efficiency (>110%): Typically indicates procedural errors in standard curve preparation or presence of contaminants. Verify dilution accuracy and prepare fresh reagents.
- Poor Replicate Consistency: Suggests pipetting inaccuracies or insufficient mixing. Calibrate pipettes and ensure complete homogenization of reaction components.
Efficiency Validation in Published Research
Recent studies demonstrate the critical role of efficiency monitoring in generating reliable data. For example, when developing SYBR Green assays for SARS-CoV-2 detection, researchers achieved detection limits of 25 copies per reaction only after rigorous optimization of reaction efficiency [84]. Similarly, in cancer research, NGS-based studies routinely incorporate quality metrics analogous to qPCR efficiency, such as monitoring variant allele frequencies and read depth to ensure detection sensitivity [86] [85].
Accurate reaction efficiency calculations and proper standard curve implementation are fundamental to reliable gene quantification in cancer research. While TaqMan assays provide consistently high efficiency with minimal optimization, SYBR Green methods offer a cost-effective alternative that delivers comparable performance when properly validated. The selection between these approaches should consider research objectives, resource constraints, and required throughput.
As precision medicine advances in oncology, with growing application of NGS and liquid biopsy technologies [86] [87] [85], the principles of robust quantification remain essential. By implementing rigorous efficiency monitoring and appropriate standard curves, cancer researchers can ensure the accuracy and reproducibility of their gene expression data, ultimately supporting the development of more effective targeted therapies and diagnostic markers.
qPCR Efficiency Assessment Workflow
Addressing Challenges in Detecting Rare Mutations and Low-Abundance Targets in Complex Samples
The detection of rare mutations and low-abundance molecular targets in complex biological samples represents a pivotal challenge in modern cancer research and diagnostic development. The identification of somatic point mutations in key oncogenes and tumor suppressor genes, such as PIK3CA, EGFR, KRAS, and TP53, provides critical information for cancer diagnosis, prognosis, and targeted treatment selection [88] [89] [14]. However, this process confronts significant technical hurdles, primarily due to the low concentration of mutant alleles amidst an overwhelming background of wild-type genetic material, a scenario particularly common in liquid biopsies, early-stage tumors, and minimal residual disease monitoring [90] [91].
Within this context, the choice of detection methodology becomes paramount. This technical guide examines the core challenges and solutions in this field, with a specific focus on the comparative analysis of SYBR Green versus probe-based detection systems for cancer gene research. While SYBR Green-based methods offer a cost-effective and flexible approach, probe-based systems typically provide enhanced specificity and reliability for discriminating single-nucleotide variants, especially in samples with limited tumor content [89]. The evolution of these techniques, alongside emerging technologies including next-generation sequencing (NGS) and CRISPR-Cas systems, provides researchers with a powerful arsenal to advance precision oncology.
Core Challenges in Rare Mutation Detection
The accurate identification of low-abundance targets is complicated by several interconnected factors. Low mutant allele frequency is a primary obstacle; in liquid biopsies or heterogeneous tumor samples, mutant DNA fragments can be present at frequencies as low as 0.01% to 5% amid a vast excess of wild-type sequences, demanding exceptional analytical sensitivity and specificity [89] [14]. This is compounded by the high complexity of sample matrices, such as blood, plasma, or formalin-fixed paraffin-embedded (FFPE) tissues, which contain numerous interfering substances that can inhibit enzymatic reactions and reduce detection efficiency [88]. Furthermore, the limitations of conventional technologies present hurdles. For instance, Sanger sequencing has poor sensitivity (~20%) for low-abundance mutations, while even NGS can be limited by high costs, complex data analysis, and specific issues like PCR amplification biases [92] [88] [89].
Technical Approaches and Methodologies
PCR-Based Detection Strategies
SYBR Green vs. Probe-Based qPCR
Real-time quantitative PCR (qPCR) remains a cornerstone technique for mutation detection due to its speed, simplicity, and cost-effectiveness. The fundamental difference between SYBR Green and probe-based methods lies in their detection mechanism.
SYBR Green is an intercalating dye that fluoresces when bound to double-stranded DNA. While cost-effective and easy to design, its major limitation is non-specific binding; it cannot distinguish between specific amplicons and non-specific products like primer dimers, which is a critical drawback when detecting rare mutations [89]. In contrast, probe-based systems (such as TaqMan) utilize a sequence-specific oligonucleotide probe labeled with a fluorophore and quencher. Fluorescence occurs only upon probe hybridization and cleavage, ensuring that the detected signal originates exclusively from the target sequence.
A study on PIK3CA mutations (H1047R and E545K) highlights this distinction. Researchers developed a probe-based qPCR assay using a mutation-specific primer with the variant base at its 3' end, combined with a phosphate-modified "blocking oligonucleotide" complementary to the wild-type sequence to suppress its amplification. This approach achieved a detection sensitivity of 5% mutant allele fraction for H1047R and 10% for E545K in human genomic DNA, successfully identifying mutations in frozen biopsies, FFPE material, and cancer cell lines [89]. This level of specificity would be challenging to achieve with SYBR Green due to its inherent inability to discriminate sequences post-amplification.
Table 1: Comparison of SYBR Green and Probe-Based qPCR for Mutation Detection
| Feature | SYBR Green qPCR | Probe-Based qPCR |
|---|---|---|
| Principle | Binds double-stranded DNA non-specifically | Sequence-specific hybridization and cleavage |
| Cost | Lower | Higher |
| Assay Design Flexibility | High | Lower (requires specific probe design) |
| Specificity | Lower (cannot distinguish primer dimers) | High (specific probe hybridization required) |
| Multiplexing Capability | Not possible | Possible with different colored fluorophores |
| Sensitivity for Rare Mutations | Limited | Superior (as demonstrated with 5% for PIK3CA H1047R) [89] |
| Best Suited For | Gene expression, presence/absence tests | SNP detection, rare mutation identification, viral load quantification |
Advanced Probe-Based and Enzyme-Assisted Methods
To further enhance specificity and sensitivity, several sophisticated probe-based strategies have been developed:
- Allele-Specific PCR with Blocking Oligos: As used in the PIK3CA assay, this method combines a 3'-mutation-specific primer with a wild-type-blocking oligonucleotide to preferentially amplify mutant sequences [89].
- Nucleic Acid Analog Probes: Using chemically modified probes like Peptide Nucleic Acids (PNA) or Locked Nucleic Acids (LNA) increases the thermal stability of probe-target hybrids. The difference in melting temperature (ΔTm) between a perfectly matched and a single-base-mismatched duplex is magnified, allowing for superior discrimination of point mutations [88] [14].
- Enzyme-Assisted Methods: Enzymes such as ligases and CRISPR-Cas systems can be harnessed for their high specificity.
- Ligase Chain Reaction (LCR): Two adjacent probes hybridize to a target and are ligated only if there is perfect complementarity at the junction, enabling precise discrimination of point mutations [88].
- CRISPR-Cas Based Detection (e.g., DASH): The Cas9 nuclease, guided by a specific RNA (sgRNA), cleaves and depletes wild-type sequences. Mutant sequences, which may lack the required Protospacer Adjacent Motif (PAM) site, remain intact and can be enriched for subsequent PCR detection, achieving sensitivities as low as 0.1% [88] [14]. The NAVIGATER system, which uses the Argonaute (Ago) enzyme, has been reported to detect mutations at an ultra-sensitive level of 0.01% in blood samples [14].
Diagram Title: CRISPR-Cas DASH Method for Mutation Enrichment
Next-Generation Sequencing and Emerging Techniques
Next-Generation Sequencing (NGS) enables the massive parallel sequencing of millions of DNA fragments, providing unparalleled throughput for discovering unknown mutations across multiple genes simultaneously [92] [93]. The basic NGS workflow involves nucleic acid extraction, library preparation (fragmentation and adapter ligation), massive parallel sequencing (often via sequencing-by-synthesis), and bioinformatic data analysis [92] [93]. However, for very low-frequency mutations, error rates during amplification and sequencing can be a limiting factor.
To overcome this, techniques like duplex sequencing are employed. This method tags and sequences both strands of the original DNA molecule independently. Errors occurring during PCR or sequencing are highly unlikely to affect both strands at the same position, allowing for an exponential reduction in false-positive calls [90]. The MethylSaferSeqS protocol is an advanced example that combines duplex sequencing with epigenetic analysis. It copies original DNA strands, separates them from the copies, and then subjects the original strands to bisulfite treatment for methylation analysis while using the copied strands for genetic mutation analysis, all from the same precious sample [90].
Nanopore sequencing, a third-generation technology, offers ultra-long reads and real-time analysis, which can be advantageous for resolving complex genomic regions and detecting structural variations [92].
Table 2: Comparison of Key Genetic Analysis Platforms for Low-Abundance Targets
| Platform | Key Principle | Approx. Sensitivity | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Sanger Sequencing | Dideoxy chain termination | ~20% mutant allele [89] | Low cost, simple data analysis | Low sensitivity, low throughput |
| qPCR (Probe-based) | Tagman probe hydrolysis | 0.1% - 5% [89] [14] | Fast, inexpensive, simple | Limited multiplexing, targeted only |
| Digital Droplet PCR (ddPCR) | Absolute quantification via sample partitioning | 0.001% - 0.1% | Absolute quantification, high sensitivity | Targeted only, higher cost than qPCR |
| Next-Generation Sequencing (NGS) | Massive parallel sequencing | ~1% - 5% (lower with duplex) [90] | Unbiased discovery, high multiplexing | Complex data analysis, higher cost |
| CRISPR-Cas Systems (e.g., DASH) | Guided nuclease cleavage & enrichment | ~0.1% [88] | High specificity, can enrich targets | Complex workflow, PAM sequence dependency |
Detailed Experimental Protocols
Protocol: Allele-Specific qPCR for PIK3CA H1047R Mutation
This protocol is adapted from a study demonstrating reliable detection of the PIK3CA H1047R mutation in breast cancer biopsies and FFPE samples with 5% sensitivity [89].
1. DNA Extraction and Quantification:
- Extract genomic DNA from your sample (e.g., frozen tissue, FFPE sections, or cell lines) using a standard kit. For FFPE samples, ensure deparaffinization and adequate proteinase K digestion.
- Precisely quantify DNA using a fluorometric method (e.g., Qubit) for superior accuracy over spectrophotometry.
- Dilute DNA to a working concentration (e.g., 5-10 ng/μL).
2. Primer and Blocker Design:
- Mutant-Specific Reverse Primer: Design a primer with the mutant nucleotide (A>G for H1047R) at the 3' terminus. This dramatically reduces amplification efficiency from the wild-type template.
- Wild-Type Blocker Oligonucleotide: Design a phosphate-modified oligonucleotide complementary to the wild-type sequence. Position the mutation site near the middle of the blocker sequence to ensure it overlaps with and sterically hinders the mutant-specific primer. This blocker is non-extendable and suppresses amplification of the wild-type allele [89].
- Forward Primer: Design a standard primer upstream of the mutation site.
- Include a separate set of primers for an internal control gene (e.g., a housekeeping gene) to assess DNA quality and loading.
3. qPCR Reaction Setup:
- Prepare two separate reaction mixes for each sample: the Mutant Assay Mix and the Internal Control Mix.
- Mutant Assay Mix (per reaction):
- 1X SYBR Green PCR Master Mix
- Forward Primer (e.g., 200 nM final concentration)
- Mutant-Specific Reverse Primer (e.g., 200 nM)
- Wild-Type Blocker Oligonucleotide (e.g., 400-600 nM, requires optimization)
- DNA template (e.g., 20-50 ng total genomic DNA)
- Nuclease-free water to volume.
- Internal Control Mix (per reaction):
- 1X SYBR Green PCR Master Mix
- Control Forward and Reverse Primers (e.g., 200 nM each)
- Same amount of DNA template as the mutant assay.
- Nuclease-free water to volume.
- Run all reactions in triplicate on a real-time PCR instrument.
4. Thermal Cycling Conditions:
- Initial denaturation: 95°C for 10 minutes.
- 40-50 cycles of:
- Denaturation: 95°C for 15-30 seconds.
- Annealing/Extension: 60-64°C for 1 minute (temperature requires optimization based on primer Tm).
- (Optional) Include a melt curve step at the end to verify amplicon specificity.
5. Data Analysis:
- Use the ΔΔCq method for relative quantification.
- Calculate ΔCq(sample) = Cq(Mutant Assay, sample) - Cq(Internal Control, sample).
- Calculate ΔCq(control) = Cq(Mutant Assay, wild-type control) - Cq(Internal Control, wild-type control).
- Calculate ΔΔCq = ΔCq(sample) - ΔCq(control).
- The fold-enrichment or relative quantity is given by 2^(-ΔΔCq). A sample is considered positive if the relative amplification is statistically significantly higher than that of a known wild-type control [89].
Protocol: Nucleic Acid Detection using CRISPR-Cas9 Enrichment (DASH)
This protocol outlines the use of CRISPR-Cas9 to deplete wild-type sequences and enrich for mutant alleles prior to PCR detection [88] [14].
1. sgRNA Design and Preparation:
- Design a sgRNA that is perfectly complementary to the wild-type target sequence, including the PAM site (NGG). The goal is for the mutation to disrupt the PAM or the seed region, preventing Cas9 cleavage of the mutant DNA.
- Synthesize the sgRNA via in vitro transcription or purchase commercially.
2. Cas9 Cleavage Reaction:
- Incubate the purified genomic DNA sample (100-200 ng) with the following:
- Purified Cas9 nuclease.
- Designed sgRNA (molar excess to Cas9).
- Cas9 reaction buffer.
- Incubate at 37°C for 1-2 hours to allow for complete cleavage of wild-type DNA fragments.
3. Post-Reaction Processing:
- The reaction can be heat-inactivated (e.g., 70°C for 10 minutes).
- The resulting mixture, now enriched for uncleaved mutant DNA, can be used directly as a template for a downstream detection method, such as:
- Allele-Specific qPCR (as described in Protocol 4.1): The enriched template will yield a significantly lower Cq for the mutant assay if the mutation is present.
- Sanger Sequencing: The enrichment increases the relative fraction of mutant DNA, making it more likely to be detected by sequencing.
Diagram Title: Experimental Workflow for CRISPR-Cas9 Enrichment
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Detecting Low-Abundance Targets
| Reagent / Material | Function and Role in Detection |
|---|---|
| High-Fidelity DNA Polymerase | Critical for PCR-based methods to minimize introduction of errors during amplification, which is vital for distinguishing true low-frequency mutations from artifacts [90]. |
| Blocking Oligonucleotides | Phosphate-modified non-extendable DNA strands that bind to and suppress the amplification of wild-type sequences, thereby improving the signal-to-noise ratio for mutant alleles in allele-specific PCR [89]. |
| Nucleic Acid Analogs (PNA, LNA) | Synthetic probes with modified backbones that increase binding affinity and specificity to complementary DNA/RNA. They enhance the discrimination of single-base mismatches in hybridization assays [88] [14]. |
| Combinatorial Peptide Ligand Libraries (CPLLs) | Used primarily in proteomics, these libraries contain millions of hexapeptide ligands that bind and normalize protein concentrations in a sample, enriching low-abundance proteins for easier detection [94]. |
| CRISPR-Cas9 System (Cas9, sgRNA) | Provides programmable nuclease activity to selectively cleave and deplete abundant wild-type DNA sequences, enriching the sample for rare mutant targets prior to detection [88] [14]. |
| Biotin-Streptavidin Beads | Used in pull-down assays and protocols like MethylSaferSeqS for the solid-phase capture and separation of specific nucleic acid strands or biotinylated probes [90]. |
| Methylation-Specific Reagents | Sodium bisulfite is used to convert unmethylated cytosine to uracil, allowing for the parallel detection of epigenetic modifications (methylation) alongside genetic sequences, as in MethylSaferSeqS [90]. |
| Ultrasensitive Chemiluminescent Substrates | Substrates (e.g., SuperSignal West Atto) that provide high signal-to-noise ratios for detecting low-abundance proteins in western blotting, enabling detection down to the attogram level [95]. |
The field of rare mutation and low-abundance target detection is rapidly advancing, driven by the critical needs of cancer research and personalized medicine. The choice between SYBR Green and probe-based detection is not merely a technical preference but a strategic decision guided by the required balance between cost, simplicity, specificity, and sensitivity. For applications demanding high specificity in a complex background of wild-type sequences, such as detecting cancer driver mutations in liquid biopsies, probe-based methods and their advanced derivatives (e.g., those using blocking oligonucleotides, PNAs, or CRISPR-enrichment) are unequivocally superior.
Future progress will likely hinge on the integration of these methodologies, such as combining the enrichment power of CRISPR systems with the sensitivity of probe-based qPCR or the multiplexing capability of NGS. Furthermore, the harmonization of genetic with epigenetic analyses, as exemplified by MethylSaferSeqS, provides a more comprehensive molecular portrait of a tumor from a limited sample. As these technologies continue to evolve, they will undoubtedly enhance our ability to achieve earlier cancer diagnosis, more precise monitoring of treatment response, and a deeper understanding of tumor heterogeneity.
Strategic Selection: A Direct Comparison of Performance and Practical Considerations
The selection of an appropriate detection chemistry is a critical decision in molecular biology, profoundly impacting the reliability, cost, and analytical performance of quantitative PCR (qPCR) assays. This is particularly true in cancer genes research, where the accurate quantification of oncogene expression, tumor suppressor genes, and genetic rearrangements can directly influence diagnostic conclusions and therapeutic strategies. Within this context, two principal chemistries dominate the landscape: the DNA-binding dye SYBR Green and the sequence-specific, hydrolysis-based TaqMan probe system. The former offers a cost-effective and flexible approach, while the latter is often favored for its potential for superior specificity and sensitivity in complex biological samples. Framed within a broader thesis on methodological selection for cancer research, this whitepaper provides an in-depth, technical comparison of these two chemistries. It synthesizes evidence from peer-reviewed studies, focusing squarely on the pivotal metrics of analytical sensitivity and specificity, to guide researchers, scientists, and drug development professionals in making an informed choice for their specific applications.
Fundamental Mechanisms and Key Differences
The core difference between SYBR Green and TaqMan chemistries lies in their mechanism of fluorescence emission and, consequently, their inherent specificity.
SYBR Green Chemistry: This method relies on the intercalation of the SYBR Green I dye into the minor groove of all double-stranded DNA (dsDNA) molecules generated during PCR amplification [5]. The fluorescence of the dye increases over a thousand-fold upon binding to dsDNA, providing a direct measure of total amplified product [5]. A significant technical consideration with this method is the necessity for post-amplification melting curve analysis to verify the specificity of the product by distinguishing it from primer-dimers or other non-specific amplicons based on their melting temperature (Tm) [96].
TaqMan Probe Chemistry: This method utilizes a sequence-specific oligonucleotide probe dual-labeled with a fluorescent reporter dye at the 5' end and a quencher molecule at the 3' end [5]. When intact, the quencher suppresses the reporter's fluorescence via Förster Resonance Energy Transfer (FRET). During the amplification cycle, the 5'→3' exonuclease activity of the DNA polymerase cleaves the probe as it extends the primer, physically separating the reporter from the quencher and resulting in a permanent increase in fluorescence that is proportional to the target amplification [5]. This mechanism ensures that fluorescence is generated only upon the specific amplification of the intended target sequence.
The logical relationship between these mechanisms and their performance outcomes is summarized in the diagram below.
Comparative Performance Data: Sensitivity and Specificity
Direct comparisons in peer-reviewed studies reveal that performance is highly dependent on the specific application, sample type, and protocol optimization.
Table 1: Head-to-Head Comparison of SYBR Green and TaqMan Performance in Selected Studies
| Study Context | Target / Gene | Sensitivity (LOD) | Specificity Notes | Key Findings | Citation |
|---|---|---|---|---|---|
| Residual CHO cell DNA in Biopharmaceuticals | 18s ribosomal RNA | SYBR Green: 100 fgTaqMan: 10 fg | Melting curve showed no unspecific products for SYBR Green. | TaqMan demonstrated a 10-fold higher sensitivity. The LOD of the TaqMan assay showed better sensitivity. | [8] |
| Enterotoxigenic Bacteroides fragilis (ETBF) in Stool | bft gene | All methods: <1 copy/μl (purified DNA).TaqMan & dPCR vastly outperformed SYBR in clinical samples. | SYBR Green under-performed in complex clinical samples. | In clinical stool samples, SYBR reported 13/38 positives vs. 35/38 for TaqMan. TaqMan gave 48-fold higher copy numbers. | [9] |
| her2 Gene Dose in Breast Tumors | her2 oncogene | SYBR Green: 2.5-40 ng DNATaqMan: 1-100 ng DNA | Not explicitly stated, but related to accuracy. | TaqMan detected a 10-fold gene dose increase vs. 5-fold for SYBR. TaqMan is more preferable for correct gene dose evaluation. | [35] |
| Adenosine Receptor Subtypes in Breast Cancer | A1, A2A, A2B, A3 | Efficiencies >95% for both methods. | Significant positive correlation (P<0.05) between data from both methods. | With high-performance primers and proper optimization, SYBR Green performance was comparable to TaqMan. | [5] |
Detailed Experimental Protocols from Key Studies
This protocol is critical in biopharmaceutical development, where sensitive detection of trace DNA impurities is required.
- Primer and Probe Design: Primers were designed against the high-copy-number 18s ribosomal RNA gene of Cricetulus griseus (CHO cells) using PrimerSelect software. For the TaqMan assay, a specific probe was also designed.
- Reaction Setup (SYBR Green):
- Master Mix: SYBR Green Premix.
- Primer Concentration: Optimized for each target.
- Template: Serially diluted purified CHO genomic DNA.
- Reaction Volume: 20 µL.
- Reaction Setup (TaqMan):
- Master Mix: TaqMan Universal PCR Master Mix.
- Primer & Probe Concentration: Optimized for each target.
- Template: Serially diluted purified CHO genomic DNA.
- Reaction Volume: 20 µL.
- Real-time PCR Cycling Conditions (for both):
- Initial Denaturation: 95°C for 10 min.
- 40 Cycles of:
- Denaturation: 95°C for 15 sec.
- Annealing/Extension: 60°C for 60 sec (data acquisition point for both methods).
- Data Analysis: Standard curves were generated from the serial dilutions. The Cycle threshold (Ct) values were plotted against the logarithm of the initial DNA concentration. PCR efficiency (E) was calculated using the formula: E = (10^(-1/slope) - 1) * 100%. The Limit of Detection (LOD) was determined as the lowest concentration at which the target was consistently detected.
This protocol highlights the challenges of working with inhibitor-rich clinical samples like stool.
- Sample Collection and DNA Extraction:
- Samples: Matched pre-operative faecal and luminal stool samples from colorectal cancer patients.
- Extraction Kit: QIAmp DNA Stool Mini Kit (Qiagen).
- DNA Quantification: NanoDrop 2000c spectrophotometer.
- Primer and Probe Sequences:
- SYBR Green Primers (from Odamaki et al.): Forward: 5'-AGT GGA AGA TTT GTG AGA TG-3'; Reverse: 5'-TAC TGC AGG ACA TTA CCA G-3'.
- TaqMan Primers/Probe (from Chen et al.): Forward: 5'-CGC TAC CCT GTC TCA AAG-3'; Reverse: 5'-GCA GTC TTC CAA CAG TTC-3'; Probe: 5'-FAM-CCA GAG CTG CAG TGT GGA-BHQ1-3'.
- Reaction Setup (SYBR Green qPCR):
- Master Mix: 5 µL SYBR Green Master Mix (Roche).
- Primers: 500 nM each.
- Template: 25-35 ng genomic DNA.
- Total Volume: 10 µL.
- Reaction Setup (TaqMan qPCR):
- Master Mix: 5 µL TaqMan Fast Advanced Master Mix (Applied Biosystems).
- Primers/Probe: 900 nM each primer, 250 nM probe.
- Template: 25-35 ng genomic DNA.
- Total Volume: 10 µL.
- Real-time PCR Cycling Conditions:
- SYBR Green:
- Initial Denaturation: 95°C for 5 min.
- 50 Cycles of: 95°C for 10 sec, 65°C for 10 sec, 72°C for 20 sec.
- Melting Curve Analysis: 65°C to 97°C with 5 acquisitions per °C.
- TaqMan:
- Initial Denaturation: 95°C for 10 min.
- 50 Cycles of: 95°C for 10 sec, 60°C for 20 sec (data acquisition).
- SYBR Green:
- Data Analysis: Absolute quantification was performed using a standard curve generated from purified ETBF DNA of known concentration. Genome copy numbers were calculated based on the estimated weight of the ETBF genome.
The workflow for a typical comparative study, from sample preparation to data interpretation, is outlined below.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of either qPCR chemistry requires a suite of optimized reagents.
Table 2: Key Research Reagent Solutions for qPCR Assay Development
| Reagent / Material | Function | Example in Study | Citation |
|---|---|---|---|
| Specialized DNA Polymerase | Enzyme with high processivity and, for TaqMan, robust 5'→3' exonuclease activity. | HotFire Polymerase (Standard PCR); AmpliTaq Gold (SYBR Green); TaqMan Universal PCR Master Mix. | [96] [9] |
| Optimized Primer Pairs | Sequence-specific primers designed for high efficiency and minimal dimer formation. | Primers designed using Beacon Designer or Primer Express software, spanning exon-exon junctions for cDNA. | [5] [96] |
| Fluorescent Detection System | Chemistry for signal generation: intercalating dye or labeled probe. | SYBR Green I dye; Dual-labeled TaqMan probe (FAM/BHQ). | [5] [9] |
| Nucleic Acid Extraction Kit | High-purity, inhibitor-free DNA/RNA isolation from complex samples. | RNeasy plus mini kit (RNA); DNeasy Blood and Tissue Kit; QIAmp DNA Stool Mini Kit. | [5] [9] |
| Standard Curve Template | Serial dilution of known target quantity for absolute quantification. | Purified genomic DNA from target cell line; synthetic oligonucleotide with target sequence. | [9] [8] |
The choice between SYBR Green and TaqMan chemistries is not a simple matter of one being universally superior. The evidence indicates that TaqMan assays generally provide superior analytical sensitivity and inherent specificity, particularly in applications requiring the utmost sensitivity (e.g., detecting trace residual DNA) or when analyzing complex, inhibitor-rich samples (e.g., stool) [9] [8] [35]. This makes TaqMan a strong candidate for high-stakes diagnostic and biopharmaceutical quality control applications.
However, a well-optimized SYBR Green assay can achieve performance comparable to TaqMan for many gene expression and genetic analysis studies, as demonstrated with adenosine receptor subtypes [5]. Its lower cost and greater flexibility make it an excellent choice for assay development, high-throughput screening of limited targets, and for laboratories with budget constraints.
For the cancer gene researcher, the decision should be guided by a balanced consideration of assay requirements, sample type, and available resources. When the target is well-defined, the sample is challenging, and maximum specificity is paramount, TaqMan is the recommended path. For broader discovery screens or when resources are limited, a rigorously optimized SYBR Green assay represents a powerful and validated alternative.
Quantitative PCR (qPCR) is a cornerstone technique in molecular biology, especially in the rapidly advancing field of cancer research. The choice of detection chemistry—SYBR Green or probe-based methods (such as TaqMan)—impacts every aspect of an experiment, from initial budget and setup complexity to data quality and interpretability [5]. For researchers studying cancer genes, this decision is critical, as it influences the feasibility of large-scale screening, the reliability of diagnostic assays, and the overall progress of drug development [97]. This guide provides a detailed cost-benefit analysis framed within the context of cancer gene research, offering a structured comparison of reagent costs, throughput capabilities, and setup complexity to help researchers and drug development professionals make an informed choice.
Quantitative Cost & Performance Comparison
The selection between SYBR Green and probe-based detection is fundamentally a trade-off between cost and specificity. The following table summarizes the core quantitative differences that inform this decision.
Table 1: Direct Comparison of SYBR Green and Probe-Based qPCR Methods
| Parameter | SYBR Green | Probe-Based (e.g., TaqMan) |
|---|---|---|
| Cost Per Reaction | ~$2–$6 [76] | More expensive; specific costs often proprietary, but probes significantly increase reagent price [5] [4] |
| Primer Cost | Low (only primers required) | Moderate to High (requires synthesis of dual-labeled probes) [5] |
| Setup Complexity | Lower (primer design and validation) | Higher (requires design and optimization of both primers and probe) [5] [98] |
| Specificity | High only with rigorous optimization and melt curve analysis [5] [4] | Very High (requires specific hybridization of the probe) [5] [8] |
| Multiplexing Potential | Limited (detects total dsDNA) | Excellent (multiple probes with different dyes can be used in one reaction) [98] [99] |
| Limit of Detection (LOD) | Higher LOD (e.g., 100 fg of DNA in one study) [8] | Lower LOD (e.g., 10 fg of DNA in the same study) [8] |
| Best Applications in Cancer Research | Profiling a large number of genes, initial screening, validating RNA-seq data when a specific sequence variant is not critical | Detecting specific splice variants, single-nucleotide polymorphisms (SNPs), low-abundance transcripts, and clinical diagnostic assays [100] [97] |
Core Methodologies and Experimental Protocols
SYBR Green-Based qPCR Protocol
The SYBR Green method is popular for its cost-effectiveness and straightforward setup. A typical protocol for detecting cancer-associated genes, such as immune markers in colorectal cancer, involves the following steps [101]:
- RNA Extraction: Use a dedicated kit, such as the Stool Total RNA Purification Kit (Norgen), for challenging sample types like stool. For cell cultures, kits like miRNeasy Mini (Qiagen) are standard. Assess RNA concentration and purity via spectrophotometry (A260/A280 ratio of ~1.8-2.0 is ideal) [101] [102].
- Reverse Transcription: Convert purified RNA (e.g., 1 μg) to cDNA using a kit like Quantitect Reverse Transcription (Qiagen). This step includes a genomic DNA wipeout buffer to prevent false positives [5] [101].
- qPCR Reaction Setup:
- Reaction Mix: 2 μL cDNA template, 1.5 μL each of forward and reverse primer (final concentration 0.25 μM each), and 10 μL of a master mix like Quantitect SYBR Green Master Mix (Qiagen) in a total volume of 25 μL [5] [4].
- Primer Design: Design primers to span exon-exon junctions, preventing amplification of genomic DNA. Use software like Beacon Designer and validate specificity with BLAST [5] [76].
- Thermal Cycling: Conditions typically are: 95°C for 10 min (polymerase activation), followed by 40 cycles of 95°C for 10 s (denaturation), 60°C for 20 s (annealing/extension), with fluorescence acquisition at the end of each annealing/extension step [5].
- Post-Amplification Analysis:
- Perform melting curve analysis by gradually increasing the temperature from 60°C to 95°C while monitoring fluorescence. A single sharp peak indicates specific amplification; multiple peaks suggest primer-dimers or non-specific products [4] [76].
- Calculate gene expression using the ΔΔCt method, normalizing to a stable housekeeping gene (e.g., β-actin or GAPDH) [5].
Probe-Based qPCR Protocol
The TaqMan method offers superior specificity for applications like detecting low-frequency cancer mutations. A standard protocol is as follows [5] [98]:
- Sample Preparation and cDNA Synthesis: This initial step is identical to the SYBR Green protocol, emphasizing high-quality RNA extraction and reverse transcription.
- qPCR Reaction Setup:
- Reaction Mix: 2 μL cDNA template, 1.5 μL of a pre-designed primer and probe mix (e.g., Assays-on-Demand from ABI), and 12.5 μL of TaqMan Universal PCR Master Mix in a 25 μL reaction [5].
- Probe and Primer Design: The probe is a dual-labeled oligonucleotide with a 5' reporter dye (e.g., FAM) and a 3' quencher. Design requires specialized software to ensure the probe binds between the PCR primers. For cancer research, probes can be designed to distinguish between wild-type and mutant alleles [98] [97].
- Thermal Cycling: Conditions are similar: 95°C for 10 min, followed by 40 cycles of 95°C for 10 s and 60°C for 20 s. Fluorescence is measured at the annealing/extension step when the probe is cleaved [5].
- Data Analysis: No melt curve is needed. The increase in fluorescence is directly proportional to the probe hydrolysis and target amplification. Data is analyzed using the ΔΔCt method relative to a reference gene [5].
Workflow and Signaling Pathways
The fundamental difference between the two chemistries lies in their mechanism of fluorescence emission. The following diagram illustrates the core signaling pathways for each method.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of qPCR in cancer research relies on a suite of reliable reagents and kits. The following table details essential components for setting up a robust assay.
Table 2: Key Reagent Solutions for qPCR in Cancer Gene Research
| Reagent/Kits | Function/Description | Example Use-Cases |
|---|---|---|
| SYBR Green Master Mix | A pre-mixed solution containing SYBR Green dye, Taq polymerase, dNTPs, and buffer. | High-throughput gene expression profiling of well-characterized cancer genes; initial screening of biomarker panels [4] [76]. |
| TaqMan Master Mix & Assays | Pre-mixed solution optimized for probe-based hydrolysis. Assays include validated primer-probe sets. | Detection of specific mutations (e.g., in EGFR, KRAS), splice variants, and low-abundance transcripts in liquid biopsies [98] [97]. |
| RNA Extraction Kits | Kits designed for specific sample types (e.g., stool, FFPE, plasma) to purify high-quality RNA. | Isulating RNA from challenging clinical samples like stool for colorectal cancer biomarker studies (e.g., Norgen kit) [101]. |
| Reverse Transcription Kits | Kits for converting RNA into cDNA, often including steps for genomic DNA removal. | First-step in two-step RT-qPCR protocols for archivable cDNA; essential for all gene expression studies [5] [101]. |
| One-Step RT-qPCR Kits | Combine reverse transcription and qPCR in a single tube, reducing hands-on time and contamination risk. | Rapid viral detection or high-throughput screening where workflow speed is critical [4] [76]. |
| Inhibitor-Resistant Polymerases | Engineered enzymes that tolerate PCR inhibitors common in clinical samples (e.g., from plasma, FFPE). | Reliable amplification from difficult sample matrices without sacrificing sensitivity or specificity [97]. |
Application in Cancer Research and Future Directions
In oncology, SYBR Green is well-suited for cost-effective, large-scale transcriptional profiling, such as validating gene signatures from RNA-seq data or screening the effects of small molecule libraries on the expression of oncogenes or tumor suppressors [98]. Its lower cost allows for more biological and technical replicates, increasing statistical power.
Conversely, TaqMan's superior specificity makes it the gold standard for clinical diagnostics and validating specific genetic alterations. It is indispensable for detecting single-nucleotide variants (e.g., in BRAF or EGFR), gene fusions, and for liquid biopsy applications where discriminating a mutant allele against a high background of wild-type DNA is required [97]. Its multiplexing capability allows for the simultaneous detection of multiple actionable mutations from a single, precious patient sample, maximizing information from limited material.
Future directions focus on overcoming the limitations of both methods. Innovations like TEQUILA-seq dramatically reduce the cost of probe synthesis, potentially making probe-based enrichment and detection more accessible [100]. Furthermore, the engineering of novel DNA polymerase variants capable of performing highly multiplexed RT-PCR with a single enzyme points toward a future of simpler, cheaper, and more powerful quantitative nucleic acid analyses [99].
The selection of an appropriate detection chemistry is a critical determinant of success in quantitative PCR (qPCR), especially for complex applications such as cancer gene expression profiling. While SYBR Green dye offers a cost-effective and straightforward approach for single-analyte detection, its utility diminishes significantly in multiplexed assays designed to simultaneously quantify multiple transcripts. This technical guide delineates the superior multiplexing capabilities of probe-based methods, specifically TaqMan assays, detailing the mechanistic advantages, experimental validation protocols, and practical implementation strategies that establish them as the preferred choice for robust, reliable multi-analyte panels in oncological research and drug development.
In the realm of cancer biology, the simultaneous quantification of multiple gene targets—such as oncogenes, tumor suppressor genes, and internal controls—from a single, often precious, patient-derived sample is not merely a convenience but a necessity. Multiplex qPCR, defined as the amplification of two or more target genes in the same reaction using the same reagent mix, directly addresses this need [103]. It conserves limited sample material, reduces reagent costs and hands-on time, minimizes pipetting errors, and, crucially, ensures that the gene expression profiles being compared are not subject to well-to-well variation [103] [104]. For cancer researchers and drug development professionals, this technique is indispensable for profiling gene expression signatures, validating biomarkers, and performing companion diagnostics.
The core challenge in multiplex qPCR, however, lies in accurately distinguishing the signals from each specific target. It is here that the fundamental differences between SYBR Green and probe-based chemistries become decisive. While SYBR Green is a viable and economical option for single-plex reactions, its architecture is inherently incompatible with true multiplexing, a limitation that probe-based methods like TaqMan are uniquely designed to overcome.
Fundamental Mechanistic Differences Dictate Multiplexing Capability
The ability to multiplex effectively is dictated by the underlying mechanism of fluorescence detection. The following diagram illustrates the core processes that differentiate these two chemistries.
SYBR Green Chemistry: A Non-Specific Approach
The SYBR Green method relies on a fluorescent dye that binds non-specifically to the minor groove of any double-stranded DNA (dsDNA) molecule [5] [7]. As the PCR progresses, the dye intercalates into all amplified products—including the specific target, non-specific products, and primer-dimers—resulting in a cumulative fluorescent signal [7] [3]. The primary drawback for multiplexing is that all dsDNA products emit an identical fluorescent signal, making it impossible to distinguish between different genetic targets within the same reaction [3]. While melt curve analysis can identify the presence of multiple products post-amplification, it cannot provide real-time, target-specific quantification for each individual analyte [7].
Probe-Based Chemistry: An Inherently Specific Architecture
In contrast, probe-based methods like TaqMan employ target-specific oligonucleotide probes that are dual-labeled with a fluorescent reporter dye at the 5' end and a quencher molecule at the 3' end [5] [7]. When the probe is intact, the quencher suppresses the reporter's fluorescence via proximity. During the annealing/extension phase, the Taq DNA polymerase enzymatically cleaves the probe only when it is bound to its specific complementary sequence. This cleavage physically separates the reporter from the quencher, resulting in a detectable fluorescent signal that is directly proportional to the amplification of that specific target [7] [3]. This core mechanism allows for multiplexing because each unique target can be assigned a probe labeled with a distinct reporter dye (e.g., FAM, VIC, ABY, JUN), whose unique fluorescence emission spectrum can be independently monitored by the qPCR instrument throughout the reaction [103].
Quantitative Performance Comparison
The mechanistic advantages of probe-based chemistries translate into superior quantitative performance in head-to-head comparisons, particularly for complex targets. The table below summarizes key findings from comparative studies.
Table 1: Comparative Performance of SYBR Green and TaqMan qPCR Methods
| Parameter | SYBR Green | TaqMan | Experimental Context |
|---|---|---|---|
| Detection Limit | 100 fg [8] | 10 fg [8] | Quantification of residual CHO host-cell DNA [8] |
| Dynamic Range | 2.5 - 40.0 ng [35] | 1.0 - 100.0 ng [35] | Analysis of her2 gene dose in breast tumors [35] |
| Gene Dose Accuracy | Overestimated at high copy number (10-fold increase appeared as 5-fold) [35] | Correctly reflected actual gene dose (10-fold increase measured as 10-fold) [35] | Analysis of her2 gene dose in breast tumors [35] |
| Amplification Efficiency | 94.3% - >97% [5] [8] | 96.6% - >97% [5] [8] | Various studies on gene expression and DNA quantification [5] [8] |
| Specificity Control | Requires post-amplification melt curve analysis [7] | Inherently specific via probe hybridization; no melt curve needed [7] | Fundamental assay design [5] [7] |
As evidenced by the data, while both methods can achieve high amplification efficiencies, TaqMan consistently demonstrates a lower limit of detection (LOD) and a wider dynamic range [35] [8]. This enhanced sensitivity is critical for detecting low-abundance transcripts often encountered in cancer research, such as those from rare cell populations or liquid biopsies. Furthermore, TaqMan's accuracy in quantifying gene amplification events, as shown in the her2 study, is essential for reliable biomarker validation and clinical assay development [35].
Implementing a Multiplex Probe-Based qPCR Assay
The Scientist's Toolkit: Essential Reagents and Materials
Successful development of a multiplex qPCR panel requires careful selection of core components. The following table outlines the essential research reagent solutions and their functions.
Table 2: Key Research Reagent Solutions for Multiplex qPCR
| Item | Function | Key Considerations |
|---|---|---|
| Sequence-Specific Probes | Provides target-specific detection with a unique fluorescent dye. | Tm should be ~10°C higher than primers (~68-70°C); avoid sequence overlap with other assay components [103]. |
| High-Quality Primer Pairs | Amplifies the specific genomic target region. | Must be specific, non-interacting, and designed to work with the probe; check for dimer formation [103] [104]. |
| Multiplex Master Mix | Provides optimized buffer, enzymes, dNTPs for simultaneous amplification of multiple targets. | Formulated with higher levels of DNA polymerase, dNTPs, and MgCl2 to offset competition; contains a suitable passive reference dye [103] [104]. |
| Optical Plate & Seals | Holds the reaction mix and allows for fluorescence detection. | Must be compatible with the real-time PCR instrument and prevent evaporation. |
| qPCR Instrument | Performs thermal cycling and measures fluorescence in real-time. | Must have optical capabilities to distinguish between the selected dye spectra [103] [3]. |
Critical Experimental Design and Optimization Workflow
Designing a robust multiplex assay is an iterative process that requires meticulous planning and validation. The workflow for this process is outlined below.
Detailed Experimental Protocols:
Assay Design and In Silico Checks:
- Primer/Probe Design: Design primers and probes using specialized software (e.g., Beacon Designer, PrimerQuest). Ensure amplicons are small (e.g., 80-150 bp) and do not overlap. The Tm of TaqMan probes should be approximately 68-70°C, about 10°C higher than the primers [103].
- Specificity Check: Verify primer and probe sequences against the transcriptome (e.g., using UCSC Genome Browser In Silico PCR) to ensure target specificity [103].
- Interaction Check: Use tools like Multiple Primer Analyzer to check for potential primer-dimer formation and other unfavorable interactions across all primer pairs in the multiplex set [103].
Dye Selection and Primer Limitation:
- Fluorophore Selection: Choose reporter dyes (e.g., FAM, VIC, ABY, JUN) with well-separated emission spectra to minimize signal crosstalk [103]. For 3- or 4-plex reactions, a combination of dyes with MGB-NFQ quenchers (e.g., for FAM, VIC) and QSY quenchers (e.g., for ABY, JUN) is recommended [103].
- Primer Limitation: In a duplex reaction where an endogenous control (e.g., a housekeeping gene) is highly abundant, it can consume reagents and impair the amplification of a less abundant target gene. To mitigate this, reduce the primer concentration for the abundant target. A typical reduction is from 900 nM each (for singleplex) to 150 nM each, while keeping the probe concentration at 250 nM [103].
Experimental Validation Protocol:
- Singleplex Confirmation: First, run and optimize each primer/probe set individually in singleplex reactions to confirm efficient amplification [103].
- Multiplex Assembly and Comparison: Combine the optimized assays into a single multiplex reaction. It is critical to validate that the Cycle threshold (Ct) values for each target in the multiplex reaction are identical to those obtained in the singleplex reactions [103]. A significant delay in Ct in the multiplex format indicates competition or inhibition, requiring further optimization of primer/probe concentrations or reaction conditions.
- Efficiency and Sensitivity Testing: Perform a standard curve analysis using serial dilutions of the target template (e.g., cDNA) to confirm that the reaction efficiency for each target in the multiplex remains between 90-110% and that the desired limit of detection is achieved [7] [8].
In the pursuit of understanding complex cancer gene networks and developing robust diagnostic panels, the choice of qPCR detection chemistry is paramount. While SYBR Green has its place in initial, single-target studies, its fundamental inability to distinguish between multiple DNA sequences in a single reaction renders it unsuitable for true multiplexing. Probe-based TaqMan methods, with their inherent specificity, capacity for spectral discrimination, and proven performance in sensitive quantification assays, provide the necessary technological foundation. By adhering to rigorous design principles and optimization protocols, researchers can leverage the full power of multiplex qPCR to accelerate cancer research, biomarker discovery, and the development of precise molecular diagnostics.
Data Reproducibility and Robustness in a Clinical Research Context
In the field of cancer gene research, the choice between SYBR Green and probe-based quantitative PCR (qPCR) detection methods represents a critical decision point that directly impacts data reproducibility and robustness. These methodologies form the analytical backbone for countless studies investigating gene expression profiles, oncogenic biomarkers, and therapeutic targets. Data reproducibility—the ability to replicate experimental findings across independent laboratories and platforms—is fundamental to scientific integrity in clinical research. Similarly, robustness refers to the reliability of results under varying experimental conditions, a non-negotiable requirement when research informs drug development and diagnostic applications.
This technical guide examines the factors influencing reproducibility and robustness when employing SYBR Green versus probe-based detection for cancer gene research. We evaluate performance characteristics through quantitative comparisons, provide detailed experimental protocols to minimize technical variability, and establish frameworks for validation that ensure data quality meets the exacting standards required for clinical translation. As we demonstrate, both methodologies can achieve exceptional performance when properly optimized and validated, though they present different considerations for implementation in research and development pipelines.
Technical Comparison of SYBR Green and Probe-Based Detection
Fundamental Principles and Mechanisms
The core distinction between these detection chemistries lies in their mechanism of target recognition and signal generation:
SYBR Green is an intercalating dye that fluoresces when bound to double-stranded DNA (dsDNA). It is relatively cost-effective and straightforward to implement but binds non-specifically to any dsDNA present in the reaction, including non-specific products and primer-dimers [5]. This fundamental characteristic necessitates rigorous validation steps to ensure specificity.
TaqMan Probes (and similar hydrolysis probes) utilize sequence-specific oligonucleotide probes dual-labeled with a fluorescent reporter dye and a quencher. During amplification, the 5'→3' exonuclease activity of DNA polymerase cleaves the probe, separating the reporter from the quencher and generating a fluorescent signal. This mechanism provides inherent specificity, as fluorescence generates only when the specific target sequence is amplified [5].
Table 1: Fundamental Characteristics of SYBR Green and Probe-Based Detection Methods
| Characteristic | SYBR Green | TaqMan/Probe-Based |
|---|---|---|
| Detection Mechanism | Binds to minor groove of dsDNA | Sequence-specific probe hydrolysis |
| Specificity Level | Lower (requires careful optimization) | Higher (built into the design) |
| Cost Consideration | Relatively cost-benefit | More expensive (requires labeled probe) |
| Experimental Workflow | Simpler primer design | Requires probe design & validation |
| Multiplexing Potential | No (detects all amplicons) | Yes (with multiple reporter dyes) |
| Primary Validation Need | Melting curve analysis | Probe specificity verification |
Quantitative Performance Comparison in Cancer Research
Direct comparative studies provide evidence for the performance parity of these methods when properly optimized. Research on adenosine receptor subtypes (A1, A2A, A2B, A3) in breast cancer tissues demonstrated that both SYBR Green and TaqMan methods achieved amplification efficiencies greater than 95% for all genes studied [5]. The correlation between normalized gene expression data from both methods was positive and statistically significant (P < 0.05), indicating that with high-performance primers and proper protocols, SYBR Green can generate data comparable to TaqMan [5].
Similar performance has been demonstrated in HER2 status determination for breast cancer. Studies have shown that SYBR Green-based qPCR assays for HER2 gene copy number and expression analysis produce results completely identical to immunohistochemistry (IHC) in control samples, underlining the legitimacy of molecular approaches in clinical diagnostics [105].
Table 2: Quantitative Performance Metrics from Comparative Studies
| Performance Metric | SYBR Green Results | TaqMan Results | Experimental Context |
|---|---|---|---|
| Amplification Efficiency | >97% | >97% | Adenosine receptors in breast cancer [5] |
| Correlation Between Methods | Significant (P<0.05) | Significant (P<0.05) | Normalized expression data [5] |
| Limit of Detection (LOD) | 100 fg | 10 fg | Residual CHO host-cell DNA [8] |
| Concordance with IHC | 100% (for positive samples) | 100% (for positive samples) | HER2 status determination [105] |
| Reproducibility (Coefficient of Variation) | Intra-assay <1.1%, Inter-assay <2% | Not specified | SARS-CoV-2 detection [106] |
Establishing Reproducible Experimental Protocols
Optimized Protocol for SYBR Green-Based qPCR
The following detailed protocol ensures robust and reproducible results for SYBR Green assays in cancer gene expression analysis:
Reaction Setup:
- Prepare a 25 µL reaction mixture containing:
- 2 µL of cDNA template
- 1.5 µL each of forward and reverse primer (optimal concentration typically 100-400 nM each, requires empirical determination)
- 12.5 µL of 2X Quantitect SYBR Green master mix
- Nuclease-free water to 25 µL [5]
Thermal Cycling Conditions:
- Initial denaturation: 95°C for 10 minutes
- 40 cycles of:
- Denaturation: 95°C for 10 seconds
- Annealing/Extension: 60°C for 20 seconds (temperature must be optimized for specific primer pairs)
- Melting curve analysis: 60°C to 95°C with continuous fluorescence measurement [5]
Critical Validation Steps:
- Melting Curve Analysis: Post-amplification, perform melting curve analysis to verify amplification of a single, specific product. A single sharp peak indicates specific amplification, while multiple peaks suggest primer-dimer formation or non-specific amplification [5].
- Standard Curve Evaluation: Include a 5-fold serial dilution of cDNA to create a standard curve for calculating amplification efficiency. Efficiency between 90-110% (slope of -3.1 to -3.6) is generally acceptable [5].
- No-Template Controls: Include controls containing all reaction components except template to detect contamination or primer-dimer formation.
Optimized Protocol for Probe-Based qPCR
Reaction Setup:
- Prepare a 25 µL reaction mixture containing:
- 2 µL of cDNA template
- 1.5 µL each of primer and probe mix (Assays-on-Demand Gene Expression Products)
- 12.5 µL of TaqMan Universal PCR master mix
- Nuclease-free water to 25 µL [5]
Thermal Cycling Conditions:
- Initial denaturation: 95°C for 10 minutes
- 40 cycles of:
- Denaturation: 95°C for 10 seconds
- Annealing/Extension: 60°C for 20 seconds [5]
Critical Validation Steps:
- Probe Specificity Verification: Ensure probes span exon-exon junctions where possible to prevent genomic DNA amplification.
- Standard Curve Evaluation: As with SYBR Green, include serial dilutions to calculate amplification efficiency.
- Limit of Detection (LOD) Determination: Establish the minimal detectable quantity using serial dilutions of target nucleic acid. For TaqMan assays, LOD as low as 10 fg has been reported [8].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Their Functions in qPCR Assay Development
| Reagent/Chemical | Function/Purpose | Example in Protocol |
|---|---|---|
| SYBR Green Master Mix | Fluorescent detection of dsDNA; contains buffer, dNTPs, polymerase | Quantitect SYBR Green master mix [5] |
| TaqMan Master Mix | Optimized for probe-based detection; contains buffer, dNTPs, polymerase | TaqMan Universal PCR master mix [5] |
| Sequence-Specific Primers | Target amplification; requires careful design spanning exon junctions | Custom primers for adenosine receptors [5] |
| Hydrolysis Probes | Sequence-specific detection; dual-labeled with reporter/quencher | Assays-on-Demand products [5] |
| Reverse Transcription Kit | cDNA synthesis from RNA templates; critical first step | Quantitect Reverse Transcription kit [5] |
| RNA Extraction Kit | High-quality RNA isolation; fundamental for gene expression | RNeasy plus mini kit [5] |
| Control Genomic DNA | Standard curve generation; enables copy number calculation | Control Genomic Human DNA for HER2 analysis [105] |
Data Analysis and Normalization Strategies
Quantification Methods and Reference Genes
Accurate data analysis is paramount for reproducibility. The most common quantification methods include:
The ΔΔCt Method:
- Normalize target gene Ct values to reference gene(s): ΔCt = Ct(target gene) - Ct(reference gene)
- Compare ΔCt between experimental and control groups: ΔΔCt = ΔCt(experimental) - ΔCt(control)
- Calculate fold-change as 2^(-ΔΔCt) [5]
Standard Curve Method:
- Generate a standard curve using serial dilutions of known template quantities
- Use the curve to determine absolute quantities in experimental samples
- Particularly important for copy number determination, as in HER2 analysis [105]
Reference Gene Selection:
- Reference genes (e.g., ACTB, GAPDH, APP) correct for variations in RNA quality, quantity, and cDNA synthesis efficiency [5] [105].
- Validation of reference gene stability across experimental conditions is essential, as commonly used references can vary under certain conditions.
Troubleshooting for Enhanced Robustness
Common issues affecting reproducibility and their solutions:
SYBR Green-Specific Issues:
- Problem: Multiple peaks in melting curve
- Solution: Optimize primer annealing temperature, redesign primers, or adjust magnesium concentration
- Problem: High background in no-template control
- Solution: Implement hot-start polymerase, redesign primers with minimal complementarity at 3' ends
Probe-Based Method Issues:
- Problem: Reduced fluorescence signal
- Solution: Check probe integrity, optimize probe concentration, verify quencher compatibility with instrument
- Problem: High Ct values with good amplification efficiency
- Solution: Increase template input, check for PCR inhibitors
Visualization of Experimental Workflows and Validation Pathways
SYBR Green qPCR Workflow
Probe-Based qPCR Validation Pathway
Both SYBR Green and probe-based detection methods can generate reproducible, robust data for cancer gene research when implemented with appropriate validation. The choice between methods depends on specific research requirements:
- SYBR Green offers cost-effectiveness and flexibility, making it suitable for initial screening, assay development, and studies with limited target numbers. Its successful implementation requires rigorous optimization and validation through melting curve analysis.
- Probe-based methods provide inherent specificity and are ideal for clinical applications, multiplexing, and when distinguishing highly homologous sequences. They facilitate easier implementation and standardization across laboratories.
For both methodologies, adherence to minimum information for publication of quantitative real-time PCR experiments (MIQE) guidelines enhances reproducibility. Critical elements include detailed reporting of primer/probe sequences, amplification efficiency, normalization strategies, and sample quality metrics. Furthermore, the integration of artificial intelligence and machine learning approaches for primer/probe design and data analysis shows promise for further enhancing reproducibility and analytical precision in cancer gene expression studies [51] [88].
In clinical research and drug development contexts, where decisions impact diagnostic and therapeutic strategies, investing in thorough assay validation and standardization ultimately saves resources and strengthens the scientific foundation for translational applications.
The selection of an appropriate detection method is a critical first step in designing robust and reliable cancer gene research. Quantitative PCR (qPCR) stands as a cornerstone technique for gene expression analysis, mutation detection, and biomarker validation in oncological studies. The decision primarily centers on choosing between two principal chemistries: the non-specific, dye-based approach of SYBR Green and the target-specific, probe-based methods (such as TaqMan). This guide provides a structured framework for researchers to align their choice of qPCR methodology with overarching research goals, with a specific focus on the distinct paradigms of high-throughput screening versus definitive confirmatory analysis. Within the context of a broader thesis on cancer genetics, this selection is paramount for generating data that is not only statistically sound but also biologically relevant and translatable to clinical applications.
Technical Comparison of SYBR Green and Probe-Based Methods
Fundamental Principles and Mechanisms
SYBR Green Chemistry
SYBR Green is a fluorescent dye that intercalates nonspecifically into the minor groove of double-stranded DNA (dsDNA). During the qPCR reaction, the dye binds to each new dsDNA amplicon as it is synthesized, resulting in a cumulative fluorescence signal proportional to the total amount of DNA produced [7]. A critical post-amplification step, the melting curve analysis, is required to verify reaction specificity. This analysis involves gradually denaturing the amplified DNA while monitoring fluorescence. A specific PCR product will yield a single, sharp peak at its characteristic melting temperature (Tm), whereas non-specific products or primer-dimers will generate distinct, separate peaks [7] [107].
Probe-Based Chemistry (TaqMan)
TaqMan assays employ a target-specific oligonucleotide probe labeled with two fluorescent dyes: a reporter at the 5' end and a quencher at the 3' end. When the probe is intact, the quencher suppresses the reporter's fluorescence via Fluorescence Resonance Energy Transfer (FRET). During the amplification cycle, the Taq DNA polymerase's 5' to 3' exonuclease activity cleaves the probe as it extends the primer. This cleavage separates the reporter from the quencher, leading to a permanent increase in the reporter's fluorescent signal, which is detected in real-time [7] [108]. This mechanism ensures that fluorescence is generated only if the specific target sequence is amplified.
Comparative Performance Characteristics
The following table summarizes the core characteristics of both methods to facilitate a direct comparison.
Table 1: Technical and practical comparison between SYBR Green and probe-based qPCR assays.
| Feature | SYBR Green | Probe-Based (TaqMan) |
|---|---|---|
| Specificity | Lower; relies on primers and post-run melt curve [7] | Higher; conferred by both primers and the specific probe [7] |
| Cost | Lower; requires only universal dye and primers [30] | Higher; requires a specific, labeled probe for each target [7] [30] |
| Multiplexing Capability | No; detects total dsDNA [7] | Yes; multiple targets with different colored dyes [7] |
| Experimental Flexibility | High; easy to design and optimize new primer sets [30] | Low; new probe required for each target, more complex design [30] |
| Sensitivity to Sequence Variants | High; can detect and identify variants via melt curve shifts [33] [109] | Low; single nucleotide mismatches in the probe-binding site can prevent detection [33] |
| Best Suited For | Screening: Target discovery, primer validation, mutation scanning [33] [109] | Confirmatory Analysis: Validation of specific targets, clinical diagnostics, multiplex assays [7] [110] |
Method Selection Framework: Screening vs. Confirmatory Analysis
The choice between SYBR Green and probe-based methods is not merely a technical preference but a strategic decision dictated by the research phase.
Screening Phase with SYBR Green
The initial screening phase is characterized by the need to evaluate a large number of genes or samples efficiently and cost-effectively. The primary goal is to identify potential targets, biomarkers, or mutations from a vast pool of candidates.
- Primary Rationale: The low cost and design flexibility of SYBR Green make it ideal for processing hundreds or thousands of reactions where the targets may not be fully defined [30]. For instance, when analyzing the expression profiles of numerous candidate genes across a large cohort of tumor samples, SYBR Green offers an economical solution.
- Variant Discovery: SYBR Green is particularly powerful for detecting genetic variations, such as single nucleotide polymorphisms (SNPs) or emerging viral strains. A seminal study on West Nile Virus demonstrated that a SYBR Green assay successfully detected 100% of possible single-point mutations within the target region, whereas a TaqMan assay failed to detect 47% of them [33]. This inherent ability to reveal sequence variations through melting curve analysis is a key asset in screening for novel cancer gene mutations.
- Workflow: A typical screening workflow involves SYBR Green qPCR followed by melting curve analysis. Amplicons with distinct melting profiles can then be sequenced to identify the exact genetic variant.
Confirmatory Analysis with Probe-Based Methods
Once candidate targets have been identified through screening, the research enters a confirmatory phase. Here, the priorities shift towards high specificity, accuracy, and reproducibility, often in a context that may mimic future clinical applications.
- Primary Rationale: The probe-based system provides an additional layer of specificity that minimizes false-positive signals, ensuring that the quantified signal originates exclusively from the intended target [7]. This is crucial for validating biomarkers before they advance to clinical trial stages.
- Clinical and Diagnostic Applications: Probe-based assays are the standard for clinical diagnostics and companion diagnostics. For example, in colorectal cancer, KRAS mutation status must be accurately determined to guide treatment with EGFR-inhibitor drugs like cetuximab, which are only effective for patients with wild-type KRAS [110]. The high specificity of TaqMan probes is essential for such critical treatment decisions.
- Multiplexing for Normalization and Co-detection: A significant advantage in confirmatory assays is the ability to perform multiplex qPCR. This allows researchers to simultaneously detect multiple targets in a single well, such as co-amplifying a gene of interest with an internal reference gene (e.g., GAPDH) for superior normalization, or detecting several pathogenic mutations concurrently [7] [108].
The following diagram illustrates the decision-making workflow for selecting the appropriate method based on research goals.
Experimental Protocols and Data Analysis
Detailed Protocol for SYBR Green Screening Assay
This protocol is adapted from methods used to investigate genetic polymorphisms, such as the HER2 Ile655Val polymorphism in breast cancer [109] and KRAS mutations in colorectal cancer [110].
- Reaction Setup:
- Master Mix: Prepare a reaction containing 1x SYBR Green PCR buffer, 2.5-5 mM MgCl₂, 0.2 mM dNTPs, 0.3 µM of each forward and reverse primer, 0.5-1 U of hot-start DNA polymerase, and 1x SYBR Green I dye.
- Template: Add 1-5 µL of cDNA or DNA (typically 10-100 ng).
- Total Volume: Adjust to 20-25 µL with nuclease-free water [107] [109].
- Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 5-10 minutes.
- Amplification (40-50 cycles):
- Denaturation: 95°C for 15 seconds.
- Annealing: 60-65°C for 20-30 seconds (temperature must be optimized for each primer set).
- Extension: 72°C for 30 seconds. Fluorescence acquisition occurs at the end of this step.
- Melting Curve Analysis: After the final cycle, heat the product from 60°C to 95°C with continuous fluorescence monitoring [107] [109].
- Data Analysis:
- Quantification Cycle (Cq): Determine Cq values for quantification. For relative gene expression, use the 2^(-ΔΔCt) method (Livak method) if amplification efficiencies are near 100% and similar between target and reference genes [7].
- Melting Curve Analysis: Plot the negative derivative of fluorescence over temperature (-dF/dT) vs. Temperature. A single, sharp peak indicates a specific product. Multiple peaks suggest non-specific amplification or the presence of different sequence variants (e.g., heterozygous vs. homozygous genotypes) [7] [109].
Detailed Protocol for Probe-Based Confirmatory Assay
This protocol is typical for assays requiring high specificity, such as the absolute quantification of a viral load or the validation of a specific gene mutation [110] [108].
- Reaction Setup:
- Master Mix: Prepare a reaction containing 1x PCR buffer, 3-5 mM MgCl₂, 0.2 mM dNTPs, 0.5 µM of each forward and reverse primer, 0.1-0.3 µM of TaqMan probe, and 0.5-1 U of DNA polymerase.
- Template: Add 1-5 µL of DNA/cDNA.
- Total Volume: Adjust to 20-50 µL with nuclease-free water [108].
- Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 2-3 minutes.
- Amplification (40-50 cycles):
- Denaturation: 95°C for 15 seconds.
- Annealing/Extension: 60°C for 60 seconds. Fluorescence acquisition occurs at the end of this step [108].
- Data Analysis:
- Standard Curve for Absolute Quantification: Serially dilute a sample of known concentration (e.g., a plasmid with the target insert) to generate a standard curve. Plot the log of the starting quantity against the Cq value. The equation of the regression line (y = mx + b) allows for the calculation of the unknown quantity: Quantity = 10^((Cq-b)/m) [7].
- Quality Control: Ensure the reaction efficiency, calculated as (10^(-1/slope) - 1) * 100, is between 90-110% for a robust assay [7].
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key research reagents and their functions in qPCR experiments.
| Reagent/Material | Function | Technical Notes |
|---|---|---|
| SYBR Green I Dye | Fluorescent intercalating dye that binds dsDNA. | Core component for dye-based qPCR; requires melt curve analysis for specificity [7]. |
| TaqMan Probe | Target-specific oligonucleotide with reporter and quencher dyes. | Confers high specificity; must be designed for each target, increasing cost [7]. |
| Hot-Start DNA Polymerase | Enzyme engineered to be inactive at room temperature. | Prevents non-specific amplification and primer-dimer formation during reaction setup, improving sensitivity and specificity [107]. |
| dNTPs | Nucleotides (dATP, dCTP, dGTP, dTTP/dUTP) for DNA synthesis. | Essential building blocks for amplification. dUTP can be used with UNG enzyme to prevent carryover contamination [107]. |
| Optical Plates/Tubes | Reaction vessels compatible with real-time cyclers. | Must be optically clear for accurate fluorescence detection without auto-fluorescence. |
| Internal Reference Dye (e.g., ROX) | Passive dye included in the master mix. | Normalizes for non-PCR-related fluorescence fluctuations between wells, crucial for well-to-well correction [107]. |
In cancer gene research, the strategic selection between SYBR Green and probe-based qPCR methods is foundational to success. SYBR Green, with its cost-effectiveness and flexibility, is the superior tool for the screening phase, enabling broad target discovery and variant identification. In contrast, probe-based methods, with their exceptional specificity and multiplexing capabilities, are indispensable for the confirmatory phase, where validating a defined target with high precision is paramount for diagnostic development and clinical translation. By aligning the methodological strengths of each chemistry with the specific research objective, scientists can optimize resources, enhance data reliability, and accelerate the pace of discovery in the fight against cancer.
Conclusion
The choice between SYBR Green and probe-based detection is not a matter of superiority but of strategic alignment with research objectives. SYBR Green offers a cost-effective, flexible solution for initial screening and well-optimized assays, with performance comparable to TaqMan when primers are highly specific. In contrast, TaqMan and similar probe-based methods provide an inherent layer of specificity crucial for multiplexing, discriminating single-nucleotide polymorphisms, and validating biomarkers in complex samples like liquid biopsies. Future directions will see these qPCR methods integrated with advanced technologies like AI-driven data analysis, digital PCR, and third-generation sequencing, further solidifying their role in the era of precision oncology and multi-cancer early detection (MCED) tests. A solid understanding of both chemistries empowers researchers to build reliable, accurate, and efficient pipelines for cancer gene analysis.
References
- 1. Investigations on DNA intercalation and surface binding by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC484200/]
- 2. SYBR Green I - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/sybr-green-i]
- 3. SYBR ® Green dye and PrimeTime™ qPCR Probe Assays [https://www.idtdna.com/pages/technology/qpcr-and-pcr/sybr-green-dye-assay-or-primetime-probe-assays]
- 4. Development and validation of cost-effective SYBR Green ... [https://www.nature.com/articles/s41598-024-52250-w]
- 5. Comparison of SYBR Green and TaqMan methods in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3988599/]
- 6. Intercalating Dyes or Fluorescent Probes For RT-qPCR? [https://bitesizebio.com/36056/dyes-fluorescent-probes-rt-qpcr/]
- 7. How qPCR works: SYBR Green vs TaqMan | INTEGRA [https://www.integra-biosciences.com/united-states/en/blog/article/how-qpcr-works-sybrr-green-vs-taqmanr]
- 8. Comparison of SYBR Green and TaqMan real-time PCR ... [https://www.sciencedirect.com/science/article/abs/pii/S1045105614001171]
- 9. Comparison of standard, quantitative and digital PCR in ... [https://www.nature.com/articles/srep34554]
- 10. TaqMan - Wikipedia [https://en.wikipedia.org/wiki/TaqMan]
- 11. How TaqMan Assays Work | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/how-taqman-assays-work.html]
- 12. Quenching mechanisms in oligonucleotide probes [https://oligos.biosearchtech.com/support/education/quenching-mechanisms]
- 13. DNA Probes Using Fluorescence Resonance Energy Transfer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1941713/]
- 14. Advances in nucleic acid probe-based detection of gene point ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507828/]
- 15. Ultrasensitive detection of cancer-associated nucleic acids ... [https://www.sciencedirect.com/science/article/pii/S0956566324008467]
- 16. Gene Expression Detection Assay for Cancer Clinical Use [https://www.jcancer.org/v09p2249.htm]
- 17. Advancing Precision Medicine: The Role of Genetic Testing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12007469/]
- 18. KRAS-targeted therapies in cancer: novel approaches and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12658495/]
- 19. Targeting KRAS mutations: orchestrating cancer evolution ... [https://www.nature.com/articles/s41392-025-02473-8]
- 20. SOMATIC GENE MUTATIONS AND THEIR ASSOCIATION ... [https://www.sciencedirect.com/science/article/pii/S2468294225001753]
- 21. Development and Comparison of Conventional PCR ... [https://brieflands.com/journals/jjm/articles/18546]
- 22. SERS-based detection of DNA methylation for cancer diagnosis [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0325539]
- 23. AI tool 'sees' cancer gene signatures in biopsy images [https://med.stanford.edu/news/all-news/2024/11/cancer-gene-biopsy.html]
- 24. All About Probe-Based Real-Time qPCR Assays [https://www.goldbio.com/blogs/articles/all-about-probe-based-qpcr?srsltid=AfmBOooA4Qqs_IdRqn8L2RlwNU_Ix_-r2nM7TiQn3xupvGqhXdLH2ozz]
- 25. Amplification-Free Testing of microRNA Biomarkers in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384345/]
- 26. Liquid biopsy in cancer: current status, challenges and ... [https://www.nature.com/articles/s41392-024-02021-w]
- 27. Methylation-specific qPCR for the EBV C promoter to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12574023/]
- 28. Melting curve analyses in the quantitative real-time ... [https://www.sciencedirect.com/science/article/pii/S2405844025006668]
- 29. Should I use probe-based detection or dye ... [https://www.aatbio.com/resources/faq-frequently-asked-questions/should-i-use-probe-based-detection-or-dye-based-detection-for-my-qpcr-analysis]
- 30. A Comprehensive Review of Probe-Based and Non- ... [https://sciencetechindonesia.com/index.php/jsti/article/view/1734]
- 31. Cancer in a Drop: Liquid biopsy insights from AACR 2025 [https://pmc.ncbi.nlm.nih.gov/articles/PMC12223554/]
- 32. Cancer Research to Microbes: Harnessing the Power of ... [https://www.thermofisher.com/blog/behindthebench/harnessing-the-power-of-qpcr/]
- 33. SYBR Green-Based Real-Time Quantitative PCR Assay for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC387603/]
- 34. Development and validation of SYBR Green- and probe- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7488977/]
- 35. Comparison of SYBR Green I and TaqMan real-time PCR ... [https://link.springer.com/article/10.1007/s10517-008-0060-3]
- 36. Clinical application of a cancer genomic profiling assay to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5580078/]
- 37. Primer designing tool - NCBI - NIH [https://www.ncbi.nlm.nih.gov/tools/primer-blast/]
- 38. An improved allele-specific PCR primer design method for ... [https://plantmethods.biomedcentral.com/articles/10.1186/1746-4811-8-34]
- 39. SPEAR: CRISPR-mediated ultrasensitive, specific and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11905141/]
- 40. MethySYBR, a Novel Quantitative PCR Assay for the Dual ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2729837/]
- 41. Principles of quantitation of viral loads using nucleic acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC101370/]
- 42. Quantification of MYCN, DDX1, and NAG Gene Copy ... [https://www.sciencedirect.com/science/article/abs/pii/S0893395222049225]
- 43. Identification of MYCN non-amplified neuroblastoma ... [https://www.nature.com/articles/s41416-024-02666-y]
- 44. rtpcr: a package for statistical analysis and graphical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530193/]
- 45. Novel primers drive accurate SYBR Green PCR detection ... [https://www.nature.com/articles/s41598-024-81508-6]
- 46. Development of a SYBR green-based real-time PCR assay for ... [https://applbiolchem.springeropen.com/articles/10.1186/s13765-025-01032-7]
- 47. Analyzing qPCR data: Better practices to facilitate rigor and ... [https://www.sciencedirect.com/science/article/pii/S2405580825004431]
- 48. Piroplasmida) in hard ticks and cattle blood from Thailand [https://pmc.ncbi.nlm.nih.gov/articles/PMC12380414/]
- 49. Methods Established for EPSPS Gene Mutation Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12348478/]
- 50. Clinical and analytical validation of a combined RNA ... [https://www.nature.com/articles/s43856-025-00934-3]
- 51. Machine learning approach to identify significant genes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12547461/]
- 52. Digital droplet PCR is an accurate and precise method to ... [https://www.nature.com/articles/s41598-025-20944-4]
- 53. TaqMan vs. SYBR Chemistry for Real-Time PCR [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/taqman-vs-sybr-chemistry-real-time-pcr.html]
- 54. Guide to Multiplex Immunoassays: Multiplex ELISA and More [https://www.thermofisher.com/blog/life-in-the-lab/guide-to-multiplex-immunoassays-multiplex-elisa-and-more/]
- 55. What are TaqMan Gene Expression Assays? [https://alitheagenomics.com/blog/what-are-taqman-gene-expression-assays]
- 56. TaqMan Probes and qPCR Primers | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/oligonucleotides-primers-probes-genes/applied-biosystems-custom-primers-probes.html]
- 57. Precision and Accuracy with TaqMan Probes_Synbio Technologies [https://synbio-tech.com/taqman-probe]
- 58. Efficacy of liquid biopsy for genetic mutations determination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11895383/]
- 59. Improvement of the sensitivity of circulating tumor DNA- ... [https://www.explorationpub.com/Journals/etat/Article/1002333]
- 60. Cross-platform comparison of SYBR ® Green real-time PCR with... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-9-328]
- 61. TaqMan® vs. SYBR ® Green Chemistries [https://www.biosyn.com/tew/taqman-vs-sybr-green-chemistries.aspx]
- 62. TaqMan Probe vs SYBR Dye- green qPCR- How to select the... based [https://geneticeducation.co.in/taqman-probe-vs-sybr-green-dye-based-qpcr/]
- 63. Performance of KRAS mutation detection in plasma exosomes ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06710-0]
- 64. Review Liquid biopsy-based multi-cancer early detection [https://www.sciencedirect.com/science/article/pii/S2095927325006565]
- 65. Trust your SYBR Green qPCR Data [https://www.thermofisher.com/blog/behindthebench/trust-your-sybr-green-qpcr-data/]
- 66. PCR Optimization: Strategies To Minimize Primer Dimer [https://genemod.net/blog/pcr-optimization]
- 67. The Pain of Primer Dimer [https://kilobaser.com/post/the-pain-of-primer-dimer]
- 68. Primer dimer [https://en.wikipedia.org/wiki/Primer_dimer]
- 69. Eliminating primer dimers and improving SNP detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7200914/]
- 70. Avoiding false positives with PCR | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-avoid-false-positives-in-pcr-and-what-to-do-if-you-get-them/]
- 71. Proven tips for PCR primer design [https://www.neb.com/en-us/nebinspired-blog/proven-tips-for-pcr-primer-design?srsltid=AfmBOop1XrD4Kzb5YC9L7g8c8yJKMR1spTufkyrdk4S6mdbTecfpYx9j]
- 72. Primer design guide - 5 tips for best PCR results [https://the-dna-universe.com/2022/09/05/primer-design-guide-the-top-5-factors-to-consider-for-optimum-performance/]
- 73. Melting Curve Analysis - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/melting-curve-analysis]
- 74. Why Does Your Melt Curve Look So Strange? - Yeasen [https://www.yeasenbio.com/blogs/molecular-biology/why-does-your-melt-curve-look-so-strange]
- 75. Amplification of nonspecific products in quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5727009/]
- 76. Development and validation of cost-effective one-step ... [https://www.nature.com/articles/s41598-022-10413-7]
- 77. Understanding melt curves for improved SYBR Green ... [https://www.idtdna.com/pages/education/videos/detail/understanding-melt-curves-for-improved-sybr-green-assay-analysis-and-troubleshooting]
- 78. MGB Eclipse Probes | IDT [https://www.idtdna.com/pages/products/gmp-oem-and-integrations/gmp-products/mgb-eclipse-probes]
- 79. Minor Groove Binder (MGB) Probes [https://oligos.biosearchtech.com/products/custom-dna-probes-for-qpcr/minor-groove-binder-mgb-probes]
- 80. TaqMan-MGB probe quantitative PCR assays to genotype ... [https://www.nature.com/articles/s41598-020-69220-7]
- 81. Application of novel dark-quencher labeled probes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12090148/]
- 82. Hybrid Capture Solution to Hasten Cancer Research | IDT [https://www.idtdna.com/pages/about/news/2025/04/21/integrated-dna-technologies-introduces-transformative-hybridization-capture-solution-to-accelerate-cancer-research]
- 83. Efficiency of Real-Time PCR [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/efficiency-real-time-pcr-qpcr.html]
- 84. High Concordance Between SYBR Green and TaqMan ... [https://www.mdpi.com/1999-4915/17/8/1130]
- 85. Evaluating sensitivity of NGS-based mutation detection ... [https://link.springer.com/article/10.1186/s13000-025-01697-0]
- 86. Clinical implementation of next-generation sequencing ... [https://www.nature.com/articles/s41598-024-84909-9]
- 87. Precision cancer medicine 2025: some concerns - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12439214/]
- 88. Advances in nucleic acid probe-based detection of gene ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1672155/full]
- 89. A simple and robust real-time qPCR method for the ... [https://www.nature.com/articles/s41598-018-22473-9]
- 90. Detection of rare mutations, copy number alterations, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10104560/]
- 91. Specific Detection of Cytokeratin 20-Positive Cells in Blood of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1867572/]
- 92. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 93. NGS for Beginners | Learn the basics of NGS [https://www.illumina.com/science/technology/next-generation-sequencing/beginners.html]
- 94. Low-Abundance Protein Enrichment for Medical Applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC10299117/]
- 95. Detecting Low Abundance Proteins in Western Blotting [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/detecting-low-abundance-proteins-in-western-blotting.html]
- 96. Real-time PCR based on SYBR-Green I fluorescence: An alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC270040/]
- 97. qPCR in Early Cancer Detection: Why It Still Leads the Pack [https://www.meridianbioscience.com/lifescience-blog/why-qpcr-still-leads-in-oncology/]
- 98. High-Throughput RT-PCR for small-molecule screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3593604/]
- 99. Engineering of novel DNA polymerase variants for single ... [https://www.nature.com/articles/s41598-025-10211-x]
- 100. Innovative Low-cost Probe Generation Empowers Targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12016554/]
- 101. Selection of optimal extraction and RT-PCR protocols for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11551167/]
- 102. Universal SYBR Green qPCR Protocol [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/qpcr/sybr-green-qpcr?srsltid=AfmBOoq08kXZC2d9qiJnb8VSPktmNa0pxPDcOgkSJMaDC0HAJd_NBYZF]
- 103. What is multiplex qPCR? [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/what-is-multiplex-qpcr.html]
- 104. A Quick Tour Around Probe-based Multiplexing qPCR [https://bitesizebio.com/11203/a-quick-tour-around-probe-based-multiplexing-qpcr/]
- 105. Molecular Biological Determination of HER2 Status Using ... [https://www.mdpi.com/1422-0067/26/5/2148]
- 106. Performance evaluation of a SYBR Green-based real-time ... [https://pubmed.ncbi.nlm.nih.gov/40915542/]
- 107. Universal SYBR Green qPCR Protocol [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/qpcr/sybr-green-qpcr?srsltid=AfmBOoqhoy2VKqFy-GcNIrAKEZ-AQ2jfrKbI6w6VFQJKFS9iFn0YWk-V]
- 108. Probe QPCR for Gene-Level Expression [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/probe-based-quantitative-pcr-to-measure-gene-level-expression?srsltid=AfmBOop0ObvvUcyOutVbigw6BPx-II5eD6TtAmnNnikRB0uYF_U2hhfJ]
- 109. Design and characterization of a SYBR Green I-based ... [https://www.sciencedirect.com/science/article/pii/S1687157X2300776X]
- 110. Analysis of Colorectal Cancer Gene Mutations and Application ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12110167/]