Pharmacophore vs. Docking-Based Virtual Screening: A Performance Comparison for Drug Discovery
This article provides a comprehensive comparison of pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) for researchers and drug development professionals.
Pharmacophore vs. Docking-Based Virtual Screening: A Performance Comparison for Drug Discovery
Abstract
This article provides a comprehensive comparison of pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) for researchers and drug development professionals. It explores the foundational principles of each method, detailing their specific workflows, software, and application scenarios. The content addresses common challenges and optimization strategies, supported by validation data and benchmark studies that directly compare performance metrics like enrichment factors and hit rates. By synthesizing key takeaways, this review offers practical guidance for selecting and integrating these computational techniques to streamline early-stage drug discovery, reduce costs, and improve the efficiency of identifying viable lead compounds.
Virtual Screening Foundations: Core Principles of Pharmacophore and Docking Methods
Defining Virtual Screening: Its Role and Workflow in Modern Drug Discovery
Virtual screening (VS) has become a cornerstone of modern drug discovery, enabling researchers to computationally evaluate massive libraries of chemical compounds to identify promising candidates that are likely to bind to a therapeutic target. This process significantly accelerates lead discovery by prioritizing molecules for costly and time-consuming experimental testing. Two primary structure-based methodologies dominate the VS landscape: docking-based virtual screening (DBVS) and pharmacophore-based virtual screening (PBVS). DBVS predicts how a small molecule (ligand) fits into a protein's binding site and estimates the binding affinity, while PBVS uses a 3D model of essential interactions a ligand must make with the protein. A persistent and critical question in the field is which method delivers superior performance in identifying active compounds. This guide objectively compares the performance of these two approaches, drawing on recent benchmarking studies and experimental data to inform the selection of virtual screening protocols.
Core Concepts and Workflows
What is Virtual Screening?
Virtual screening is a computational technique used to search large databases of small molecules to identify those structures most likely to bind to a drug target, such as a protein. By simulating the interaction between compounds and the target, VS acts as a powerful filter, enriching the hit rate of subsequent experimental assays and reducing the resources required for lead identification.
Docking-Based Virtual Screening (DBVS)
DBVS relies on the three-dimensional structure of the target protein. It involves computationally "docking" each small molecule from a library into the target's binding site, sampling various orientations and conformations, and then ranking them based on a scoring function that predicts binding affinity. The workflow typically includes protein and ligand preparation, conformational sampling via docking algorithms, and post-docking analysis.
Pharmacophore-Based Virtual Screening (PBVS)
A pharmacophore is an abstract model that defines the spatial and functional features necessary for a molecule to interact with a biological target. These features include hydrogen bond donors, hydrogen bond acceptors, hydrophobic regions, and charged groups. PBVS scans compound libraries to find molecules that match this essential pharmacophore pattern, irrespective of their overall chemical scaffold.
Performance Comparison: PBVS vs. DBVS
The debate over the relative effectiveness of PBVS and DBVS is long-standing. A foundational benchmark study from 2009 provided compelling evidence for the advantages of the pharmacophore-based approach.
Table 1: Benchmark Comparison of PBVS and DBVS Across Eight Protein Targets [1] [2]
| Target Protein | Number of Actives | PBVS Enrichment (Catalyst) | DBVS Enrichment (Best of DOCK, GOLD, Glide) |
|---|---|---|---|
| Angiotensin Converting Enzyme (ACE) | 14 | High | Lower |
| Acetylcholinesterase (AChE) | 22 | High | Lower |
| Androgen Receptor (AR) | 16 | High | Lower |
| D-Alanyl-D-Alanine Carboxypeptidase (DacA) | 3 | High | Lower |
| Dihydrofolate Reductase (DHFR) | 8 | High | Lower |
| Estrogen Receptor α (ERα) | 32 | High | Lower |
| HIV-1 Protease (HIV-pr) | 21 | High | Lower |
| Thymidine Kinase (TK) | 15 | High | Lower |
This comprehensive study concluded that in 14 out of 16 virtual screening scenarios (one target versus two different decoy databases), PBVS demonstrated higher enrichment factors than DBVS. When considering the top 2% and 5% of ranked compounds, the average hit rate for PBVS across all eight targets was "much higher" than those achieved by any of the three docking programs [1] [2].
Advanced and Integrated Workflows
Given the complementary strengths of different VS methods, researchers often combine them to leverage their respective advantages. Integrated workflows can significantly improve screening performance.
Combining PBVS and DBVS
A common strategy is to use PBVS as a pre-filter to rapidly reduce the size of a massive compound library before applying the more computationally intensive DBVS. This hybrid approach was demonstrated in a study searching for marine natural products as SARS-CoV-2 inhibitors, where pharmacophore screening efficiently narrowed down candidates later evaluated by comparative molecular docking [3].
Machine Learning Acceleration
A significant innovation is the application of machine learning (ML) to accelerate VS. One study developed an ensemble ML model that learned from docking results to predict docking scores directly from 2D molecular structures. This approach achieved a 1000-fold acceleration in binding energy predictions compared to classical docking, enabling the ultra-rapid screening of billions of compounds [4].
Fragment-Based Pharmacophore Screening
The FragmentScout workflow represents a modern, data-driven approach to PBVS. It aggregates pharmacophore feature information from multiple experimental fragment poses obtained through high-throughput crystallographic screening (e.g., XChem). These features are combined into a single, powerful "joint pharmacophore query" used to screen large databases. Applied to the SARS-CoV-2 NSP13 helicase, this method successfully identified 13 novel micromolar inhibitors, showcasing its utility in fragment-based lead discovery [5].
Detailed Experimental Protocols
To ensure reproducibility and provide a clear technical reference, below are detailed methodologies for key experiments cited in this guide.
- Target Selection and Preparation: Select eight structurally diverse protein targets (e.g., ACE, AChE, DHFR). For each target, obtain high-resolution crystal structures of protein-ligand complexes from the PDB.
- Pharmacophore Model Generation: Using software like LigandScout, create a consensus pharmacophore model based on multiple co-crystallized ligands for each target.
- Docking Model Preparation: For DBVS, prepare a single high-resolution protein structure for each target, defining the binding site from the co-crystallized ligand.
- Dataset Curation: For each target, compile a set of known active compounds and generate property-matched decoy molecules to create a challenging benchmark database.
- Virtual Screening Execution:
- Perform PBVS using the Catalyst software with the generated pharmacophore models.
- Perform DBVS using three different docking programs (DOCK, GOLD, Glide) against the prepared protein structures.
- Performance Evaluation: Calculate enrichment factors and hit rates for each method by analyzing their ability to rank known active compounds at the top of the list (e.g., within the top 2% and 5%).
- System Preparation:
- Protein: Obtain crystal structures of the target (e.g., PDB IDs 6A2M and 6KP2 for wild-type and quadruple-mutant PfDHFR). Prepare the protein using OpenEye's "Make Receptor" GUI: remove water molecules and redundant chains, add hydrogens, and optimize.
- Ligand Library: Prepare the DEKOIS 2.0 benchmark set, which includes active molecules and structurally similar decoys, using Omega and OpenBabel to generate 3D conformations and correct file formats.
- Molecular Docking: Dock the entire library against the prepared protein structures using multiple docking tools (e.g., AutoDock Vina, FRED, PLANTS) with grid boxes centered on the binding site.
- Machine Learning Rescoring: Extract the top poses generated by each docking program. Rescore these poses using pretrained ML scoring functions such as RF-Score-VS v2 (Random Forest-based) and CNN-Score (Convolutional Neural Network-based).
- Performance Analysis: Evaluate the screening performance using metrics like pROC-AUC and Enrichment Factor at 1% (EF1%). Analyze chemotype enrichment to ensure the retrieval of diverse, high-affinity binders.
Workflow Visualization
The following diagram illustrates a modern, integrated virtual screening workflow that combines pharmacophore screening, molecular docking, and machine learning to maximize the efficiency and success of a drug discovery campaign.
Integrated Virtual Screening Workflow
The Scientist's Toolkit: Key Research Reagents and Software
Table 2: Essential Computational Tools for Virtual Screening [6] [1] [5]
| Tool Name | Type/Function | Key Application in VS |
|---|---|---|
| AutoDock Vina | Docking Software | Open-source tool for flexible ligand docking and scoring. |
| Glide (Schrödinger) | Docking Software | High-performance docking tool using a hierarchical scoring approach. |
| PLANTS | Docking Software | Docking software that uses ant colony optimization algorithms. |
| LigandScout | Pharmacophore Modeling | Software for creating structure- and ligand-based pharmacophore models and performing PBVS. |
| Catalyst (BIOVIA) | Pharmacophore Modeling | Software for creating pharmacophore hypotheses and performing 3D database searching. |
| DEKOIS 2.0/DUDE-Z | Benchmarking Sets | Public databases containing targets, known actives, and carefully selected decoys for VS benchmarking. |
| RF-Score-VS & CNN-Score | Machine Learning Scoring | Pretrained ML models used to rescore docking poses, significantly improving enrichment. |
| FragmentScout | Fragment-Based Workflow | A specialized workflow for generating pharmacophore queries from XChem fragment screening data. |
| O-LAP | Shape-Based Modeling | Algorithm for generating shape-focused pharmacophore models to improve docking enrichment. |
| Smina | Docking Software | A fork of AutoDock Vina optimized for scoring function development and customizability. |
The choice between pharmacophore-based and docking-based virtual screening is not a simple matter of one being universally superior. The evidence shows that PBVS can achieve higher enrichment in many scenarios, making it an excellent choice for fast, efficient filtering of large libraries or when key ligand-target interactions are well-defined. DBVS provides atomic-level insight into binding modes and is a powerful tool for lead optimization. However, the most impactful modern strategy is an integrated approach that leverages the strengths of both methods. Combining PBVS pre-filtering with DBVS and enhanced by machine learning rescoring represents the current state-of-the-art, offering researchers the best chance of efficiently identifying novel and potent therapeutic candidates in the vast chemical space.
In the landscape of computer-aided drug discovery, virtual screening stands as a pivotal technology for identifying novel lead compounds in a cost-effective manner. Virtual screening methodologies are broadly classified into two categories: structure-based and ligand-based approaches. Pharmacophore-Based Virtual Screening (PBVS) is a powerful ligand-based method that operates on the principle that molecules sharing a common three-dimensional arrangement of essential steric and electronic features will elicit the same biological response [7]. The International Union of Pure and Applied Chemistry (IUPAC) defines a pharmacophore as "the ensemble of steric and electronic features that is necessary to ensure the optimal supra-molecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [7] [8]. This abstract representation of interaction capabilities, rather than specific functional groups, enables PBVS to identify structurally diverse compounds that nonetheless possess the necessary characteristics to bind to a target, a significant advantage in scaffold hopping and lead optimization [7].
Performance Comparison: PBVS vs. Docking-Based Virtual Screening (DBVS)
Docking-Based Virtual Screening (DBVS) has gained popularity as it directly simulates the ligand-receptor binding process. However, a benchmark study provides compelling experimental data on their relative performances in retrieving active compounds from databases containing decoys [1].
Quantitative Performance Metrics
The following tables summarize the key findings from a comprehensive comparison study involving eight structurally diverse protein targets: angiotensin converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptors α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK) [1].
Table 1: Summary of Virtual Screening Performance Across Eight Targets
| Virtual Screening Method | Number of Cases Where Method Outperformed Alternatives | Average Hit Rate at Top 2% of Database | Average Hit Rate at Top 5% of Database |
|---|---|---|---|
| Pharmacophore-Based (PBVS) | 14 out of 16 cases | Significantly Higher | Significantly Higher |
| Docking-Based (DBVS) | 2 out of 16 cases | Lower | Lower |
| DOCK | Varies by target | Lower | Lower |
| GOLD | Varies by target | Lower | Lower |
| Glide | Varies by target | Lower | Lower |
Table 2: Enrichment Factors for Different Virtual Screening Approaches
| Target | PBVS Enrichment Factor | Best DBVS Enrichment Factor | Performance Advantage |
|---|---|---|---|
| ACE | Higher | Lower | PBVS |
| AChE | Higher | Lower | PBVS |
| AR | Higher | Lower | PBVS |
| DacA | Higher | Lower | PBVS |
| DHFR | Higher | Lower | PBVS |
| ERα | Higher | Lower | PBVS |
| HIV-pr | Higher | Lower | PBVS |
| TK | Higher | Lower | PBVS |
The data consistently demonstrates that PBVS achieved higher enrichment factors than DBVS in the vast majority of test cases (14 out of 16) [1]. Furthermore, when considering the top 2% and 5% of the highest-ranked compounds in the database, the average hit rate for PBVS was substantially higher than those obtained by any of the three docking programs (DOCK, GOLD, Glide) [1]. This indicates that PBVS is more effective at prioritizing truly active compounds in the early stages of screening, a critical factor for efficient resource allocation in drug discovery campaigns.
Experimental Protocols and Methodologies
Structure-Based Pharmacophore Modeling Protocol
The structure-based approach requires the three-dimensional structure of the target protein, typically obtained from sources like the Protein Data Bank (PDB) [7].
- Protein Preparation: The first critical step involves preparing the protein structure by evaluating and correcting protonation states of residues, adding hydrogen atoms (absent in X-ray structures), and addressing any missing residues or atoms [7].
- Binding Site Detection: The ligand-binding site is identified, either manually using knowledge from experimental data or computationally using tools like GRID or LUDI, which analyze the protein surface for energetically favorable interaction sites [7].
- Feature Generation and Selection: From the protein-ligand complex or the binding site alone, a map of potential interactions (e.g., hydrogen bond acceptors/donors, hydrophobic areas, ionizable groups) is generated. The initial multitude of features is refined to select only those essential for bioactivity, resulting in the final pharmacophore hypothesis [7]. Exclusion volumes are often added to represent the physical boundaries of the binding pocket [8].
Ligand-Based Pharmacophore Modeling Protocol
When the 3D structure of the target is unavailable, pharmacophore models can be developed from a set of known active ligands.
- Training Set Selection: A set of structurally diverse molecules with confirmed biological activity and known affinities against the target is assembled. The quality of this dataset directly influences the model's success [8].
- Conformational Analysis: Multiple feasible conformations are generated for each ligand in the training set to account for flexibility [9].
- Common Feature Alignment: The software identifies the maximal common set of pharmacophoric features shared by the active molecules and determines their optimal spatial alignment. Algorithms like HipHop or PharmaGist are commonly used for this purpose [10] [9].
- Model Validation: The generated model is validated using a test set containing both active and inactive molecules or decoys. Metrics like the enrichment factor, yield of actives, and ROC-AUC are calculated to evaluate the model's ability to distinguish active from inactive compounds [8].
Virtual Screening Workflow
Once a validated pharmacophore model is obtained, it serves as a query for screening compound databases.
Diagram 1: The typical workflow for a pharmacophore-based virtual screening campaign, often followed by docking refinement and experimental validation [10] [11] [12].
The Scientist's Toolkit: Essential Research Reagents and Software
Successful implementation of PBVS relies on a suite of computational tools and databases. The table below details key resources used in the featured studies.
Table 3: Key Research Reagent Solutions for PBVS
| Tool/Resource Name | Type | Primary Function in PBVS | Application Example |
|---|---|---|---|
| LigandScout [1] | Software | Structure-based & ligand-based pharmacophore model generation. | Used to create pharmacophore models from X-ray crystal structures of protein-ligand complexes [1]. |
| Catalyst/HypoGen [1] [10] | Software | Generates quantitative 3D pharmacophore models from a set of active and inactive training molecules. | Utilized to develop a pharmacophore model (Hypo1) for Topoisomerase I inhibitors from CPT derivatives [10]. |
| PharmaGist [9] | Web Server/Software | Ligand-based pharmacophore detection from a set of input ligands, handling flexibility deterministically. | Applied for virtual screening on GPCR alpha1A dataset and the DUD dataset [9]. |
| ZINC Database [10] [12] | Compound Database | A freely available database of commercially available compounds for virtual screening. | Screened to discover potential Topoisomerase I inhibitors; used as a source for phytocompounds [10] [12]. |
| Directory of Useful Decoys, Enhanced (DUD-E) [8] | Decoy Database | Provides optimized decoy molecules for rigorous virtual screening validation. | Used to generate decoys with similar 1D properties but different 2D topology compared to active molecules [8]. |
| Molecular Operating Environment (MOE) [12] | Software Suite | Integrated platform for molecular modeling, simulation, and docking. | Used for molecular docking and virtual screening of a phytochemical library [12]. |
| PyRx [11] | Software | Tool for virtual screening using AutoDock Vina or other docking engines. | Employed for virtual screening of a marine sponge bioactive compound library against multiple Alzheimer's disease targets [11]. |
Pharmacophore-Based Virtual Screening represents a robust and highly effective methodology for lead identification in drug discovery. The experimental evidence demonstrates that PBVS can outperform docking-based methods in enriching active compounds across a wide range of biological targets [1]. Its strength lies in its abstract representation of molecular interactions, which facilitates the identification of novel chemotypes through scaffold hopping. While the choice between structure-based and ligand-based pharmacophore modeling depends on data availability, both approaches have proven successful in prospective virtual screening campaigns, yielding hit rates substantially higher than random high-throughput screening [8]. When integrated into a comprehensive workflow that may include docking refinement and ADMET profiling, PBVS serves as a powerful first-line tool for efficiently navigating vast chemical space and accelerating the discovery of new therapeutic agents.
Molecular docking is a computational cornerstone of structure-based drug design, enabling researchers to predict how small molecule ligands interact with protein targets at the atomic level. Docking-based virtual screening (DBVS) employs computational algorithms to automatically evaluate vast libraries of chemical compounds, predicting their binding conformations (poses) and estimating interaction strengths (affinities) with biomolecular targets. This approach has become indispensable in pharmaceutical research for identifying promising therapeutic candidates from millions of potential compounds, significantly accelerating the early drug discovery pipeline while reducing experimental costs [13] [14].
The fundamental challenge DBVS addresses is balancing computational efficiency with predictive accuracy. Docking methods essentially solve intricate three-dimensional puzzles by identifying the optimal fit between two molecules, typically a protein receptor and a small molecule ligand, through search algorithms and scoring functions. These scoring functions approximate binding affinity by calculating interaction energies, serving the dual purpose of identifying most probable binding poses and ranking compounds by their predicted binding strength [15] [13]. Despite advances, DBVS operates within a competitive landscape alongside other virtual screening approaches, particularly pharmacophore-based methods, with each demonstrating distinct strengths and limitations depending on the specific application context [1] [2].
Performance Comparison: DBVS Versus Alternative Approaches
Benchmark Comparison with Pharmacophore-Based Virtual Screening
A comprehensive benchmark study directly compared pharmacophore-based virtual screening (PBVS) and DBVS across eight structurally diverse protein targets: angiotensin converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptors α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK). The research employed rigorous experimental design, testing each method against two datasets containing both active compounds and decoys, using Catalyst for PBVS and three docking programs (DOCK, GOLD, and Glide) for DBVS [1] [2].
Table 1: Performance Comparison Between PBVS and DBVS Methods
| Performance Metric | PBVS (Catalyst) | DBVS (Average across DOCK, GOLD, Glide) |
|---|---|---|
| Cases with higher enrichment factors | 14 out of 16 | 2 out of 16 |
| Average hit rate at top 2% of database | Much higher | Lower |
| Average hit rate at top 5% of database | Much higher | Lower |
| Advantages | Superior early enrichment; effectively identifies essential binding features | Directly models physical binding process; provides structural binding information |
| Limitations | Limited detailed energy calculations | Lower enrichment in benchmark study; scoring function challenges |
The results demonstrated that PBVS significantly outperformed DBVS in retrieval of active compounds across most test cases. Of the sixteen sets of virtual screens (one target versus two testing databases), PBVS achieved higher enrichment factors in fourteen cases compared to DBVS methods. When considering the top 2% and 5% of highest-ranked compounds from the entire databases, the average hit rates for PBVS were substantially higher than those for DBVS across all eight targets [1] [2]. This performance advantage suggests that pharmacophore approaches may offer superior early enrichment in virtual screening campaigns, though DBVS provides valuable binding mode information that pharmacophore methods lack.
Comparison Among Docking Scoring Functions
The performance of DBVS is heavily dependent on the scoring functions used to rank potential ligands. A 2025 pairwise comparison study evaluated five scoring functions implemented in Molecular Operating Environment (MOE) software using protein-ligand complexes from the PDBbind database. The research applied InterCriteria Analysis (ICrA), a multi-criterion decision-making approach, to assess performance based on multiple docking outputs including best docking score, lowest root mean square deviation (RMSD), and their combinations [15].
Table 2: Docking Scoring Function Performance Comparison
| Evaluation Metric | Best Performing Function | Key Findings |
|---|---|---|
| Pose prediction accuracy | Lowest RMSD | Most reliable indicator of pose prediction quality |
| Overall comparability | Alpha HB and London dG | Showed highest consistency and performance across multiple criteria |
| Evaluation approach | InterCriteria Analysis (ICrA) | Enabled robust multi-dimensional comparison of scoring functions |
The study identified lowest RMSD as the best-performing docking output metric for assessing pose prediction accuracy. Among the specific scoring functions evaluated, Alpha HB and London dG demonstrated the highest comparability and consistent performance across multiple evaluation criteria. This research highlights the substantial variability between different scoring functions and emphasizes that no single function universally outperforms others across all target proteins and ligand types [15].
Experimental Protocols and Methodologies
Standard DBVS Workflow and Benchmarking Methods
The experimental protocol for comprehensive DBVS evaluation typically follows a structured pipeline to ensure fair comparison and reproducible results. The benchmark study comparing PBVS and DBVS employed this rigorous methodology [1] [2]:
Target Selection: Eight pharmaceutically relevant proteins with diverse pharmacological functions and structural characteristics were selected.
Structure Preparation: High-resolution crystal structures of each target protein in complex with ligands were obtained from the Protein Data Bank (PDB). For docking studies, a single representative structure was used, while pharmacophore models incorporated multiple complex structures.
Compound Dataset Preparation: Active compounds for each target were curated from the DrugBank database. Two separate decoy sets (Decoy I and Decoy II) containing approximately 1000 non-active molecules each were generated using different methodologies to avoid bias.
Virtual Screening Execution: Each compound database was screened using both PBVS (Catalyst software) and DBVS (DOCK, GOLD, and Glide programs) approaches against the corresponding target.
Performance Evaluation: Screening effectiveness was quantified using enrichment factors (EF) and hit rates at specific percentages (2% and 5%) of the top-ranked database compounds.
Advanced DBVS Integration Protocols
Recent methodological advances have integrated multiple approaches to overcome limitations of traditional DBVS:
DockBind Framework: IBM Research developed DockBind, which leverages docking poses with advanced machine learning. This approach uses MACE, an SO(n) equivariant graph neural network, to capture detailed atomic environments in binding regions defined by spatial proximity of ligand and protein atoms. The method incorporates data augmentation using top-10 ranked docking poses from DiffDock (a diffusion-based blind docking algorithm) rather than relying solely on the top-ranked pose. Ensembling predictions across multiple poses improves robustness against misranked conformations. The framework additionally integrates physical and chemical descriptors including neural potential energy estimates, molecular fingerprints, and DFT-based energy calculations to refine binding affinity predictions [16].
Pose Ensemble Graph Neural Networks: The DockBox2 (DBX2) approach introduces graph neural networks that encode ensembles of computational poses rather than single conformations. This method represents a significant advancement as it accounts for the dynamic nature of ligand-protein interactions. DBX2 was trained on the PDBbind dataset to jointly predict binding pose likelihood (node-level task) and binding affinity (graph-level task), demonstrating that learning from multiple conformations improves both docking and virtual screening performance compared to physics-based and single-pose ML methods [17].
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Computational Tools for Docking-Based Virtual Screening
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| DOCK | Docking Program | Search algorithm & physics-based scoring | General purpose DBVS |
| GOLD | Docking Program | Genetic algorithm & empirical scoring | DBVS with protein flexibility |
| Glide | Docking Program | Hierarchical docking & precision scoring | High-accuracy DBVS |
| AutoDock Vina | Docking Program | Monte Carlo optimization & empirical scoring | Rapid DBVS with balanced accuracy |
| DiffDock | Deep Learning Docking | Diffusion-based generative pose prediction | Blind docking scenarios |
| FeatureDock | Deep Learning Docking | Transformer-based pose & affinity prediction | Feature-based local environment learning |
| PDBbind | Database | Curated protein-ligand complexes with binding data | Method training & benchmarking |
| Directory of Useful Decoys (DUD) | Database | Active compounds & decoys for specific targets | Virtual screening performance validation |
| MOE (Molecular Operating Environment) | Software Suite | Multiple scoring functions & molecular modeling | Comprehensive drug discovery platform |
| RosettaVS | Software Suite | Physics-based docking with flexibility modeling | High-precision virtual screening |
Recent Advances and Future Directions
Deep Learning Transformations in Molecular Docking
Traditional molecular docking approaches primarily follow search-and-score algorithms that are computationally demanding and often simplify protein flexibility to maintain feasibility. Recent years have witnessed a surge in deep learning (DL) applications that are transforming the docking landscape. Methods such as EquiBind, TankBind, and particularly DiffDock have demonstrated accuracy rivaling or surpassing traditional approaches while significantly reducing computational costs [18].
DiffDock implements diffusion models to molecular docking, progressively adding noise to ligand degrees of freedom (translation, rotation, and torsion angles) then using an SE(3)-equivariant graph neural network to learn a denoising score function that iteratively refines ligand poses. This approach has achieved state-of-the-art accuracy on PDBBind test sets while operating at a fraction of the computational cost of traditional methods [18]. However, DL docking models face challenges with generalizing beyond training data and sometimes mispredict key molecular properties like stereochemistry and bond lengths, leading to physically unrealistic predictions [18].
Addressing Protein Flexibility and Scoring Limitations
A significant limitation of many docking approaches is the treatment of proteins as rigid structures, neglecting inherent flexibility and induced fit effects upon ligand binding. Recent advancements aim to incorporate protein flexibility more comprehensively. Methods like FlexPose enable end-to-end flexible modeling of protein-ligand complexes regardless of input protein conformation (apo or holo) [18]. Similarly, RosettaVS incorporates receptor flexibility through modeling of sidechains and limited backbone movement, which proves critical for targets requiring induced conformational changes [14].
For scoring function limitations, new approaches combine physical models with machine learning. FeatureDock utilizes transformer-based architecture to leverage physicochemical features from protein local environments, achieving both pose prediction and improved scoring power for virtual screening. This method discretizes binding pockets into grid points using 3D-invariant FEATURE representations, then applies transformer encoders to predict probability density envelopes for ligand binding [19]. The DockBind framework similarly integrates multiple feature types including MACE-based binding predictions, neural potential energy estimates, and molecular fingerprints to enhance affinity estimation accuracy [16].
Emerging Trends and Integration Strategies
The future of DBVS points toward hybrid approaches that leverage the strengths of multiple methodologies. The benchmark results demonstrating PBVS superiority in early enrichment suggest strategic integration where pharmacophore methods could pre-filter compound libraries before applying more computationally intensive DBVS [1] [2]. Similarly, using deep learning for initial binding site identification followed by traditional docking for pose refinement represents another promising hybrid strategy [18].
Ultra-large library screening capabilities are also advancing rapidly. The development of platforms like OpenVS incorporating active learning techniques enables efficient screening of billion-compound libraries by training target-specific neural networks during docking computations to triage promising compounds for expensive calculations [14]. These advances, combined with improved handling of protein flexibility and more sophisticated scoring functions, are progressively expanding the capabilities and applications of docking-based virtual screening in modern drug discovery.
Virtual screening (VS) has become a cornerstone of modern drug discovery, enabling researchers to computationally evaluate vast libraries of compounds to identify promising candidates for further experimental testing. Among the various VS strategies, two primary methodologies have emerged as particularly influential: pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS). These approaches differ fundamentally in their underlying principles, information requirements, and methodological execution. PBVS relies on the identification and matching of essential chemical features responsible for biological activity, while DBVS focuses on predicting the atomic-level binding geometry and affinity between a small molecule and a protein target. This guide provides a detailed, objective comparison of these two methodologies, examining their respective information requirements, underlying mechanisms, and performance based on experimental benchmarking studies, to inform researchers and drug development professionals in selecting the appropriate tool for their projects.
Core Methodological Principles
Pharmacophore-Based Virtual Screening (PBVS)
A pharmacophore is an abstract model that defines the essential chemical features a molecule must possess to interact with a specific biological target and elicit a pharmacological response. It captures the steric and electronic features necessary for optimal molecular interactions, without being tied to a specific chemical scaffold. Key features include hydrogen bond donors and acceptors, positive and negative ionizable areas, hydrophobic regions, and aromatic rings.
The PBVS process typically involves:
- Pharmacophore Model Generation: Creating a hypothesis of the essential interaction features.
- Database Searching: Screening virtual compound libraries to identify molecules that match the pharmacophore model in three-dimensional space.
- Hit Identification: Selecting compounds that fulfill the spatial and chemical constraints of the model for further evaluation.
Docking-Based Virtual Screening (DBVS)
Molecular docking simulates the process of how a ligand binds to a protein's active site. It is a more physically grounded approach that aims to predict the binding pose (geometry) of the ligand and often estimate the binding affinity using a scoring function.
The DBVS process typically involves:
- Protein and Ligand Preparation: Defining the protein's binding site and generating feasible 3D conformations for the ligands.
- Conformational Search: Exploring possible orientations and conformations of the ligand within the binding site.
- Scoring and Ranking: Evaluating and ranking each predicted pose using a scoring function to estimate binding strength.
Comparative Analysis: Information Requirements and Methodologies
The fundamental differences between PBVS and DBVS stem from their distinct objectives, which directly dictate their information requirements and methodological workflows. The table below summarizes these key distinctions.
Table 1: Key Differences in Information Requirements and Methodologies between PBVS and DBVS
| Aspect | Pharmacophore-Based Virtual Screening (PBVS) | Docking-Based Virtual Screening (DBVS) |
|---|---|---|
| Primary Objective | Identify compounds that match a set of essential chemical features for biological activity. [1] [20] | Predict the atomic-level binding geometry and affinity of a ligand in a protein's binding site. [21] |
| Essential Information Requirement | • A set of key functional features and their spatial arrangement.• Can be derived from known active ligands or a protein-ligand complex structure. [22] [23] | A high-resolution 3D structure of the target protein (e.g., from X-ray crystallography or Cryo-EM). [6] [24] |
| Underlying Methodology | Creates an abstract, chemical feature-based query to search 3D compound databases. [22] | Performs a conformational search and scores poses based on force fields, empirical data, or machine learning. [21] |
| Handling of Protein Flexibility | Implicitly handled by the abstract nature of chemical features. | Often challenging; typically requires explicit sampling or ensemble docking, which is computationally expensive. |
| Computational Speed | Generally faster, suitable for rapid screening of ultra-large libraries. [25] | Slower, computational cost scales with the complexity of the search algorithm and scoring function. [25] |
| Key Output | A list of compounds that match the pharmacophore hypothesis. | Ranked list of compounds with predicted binding poses and scores. |
Experimental Benchmarking and Performance Data
A Landmark Performance Comparison
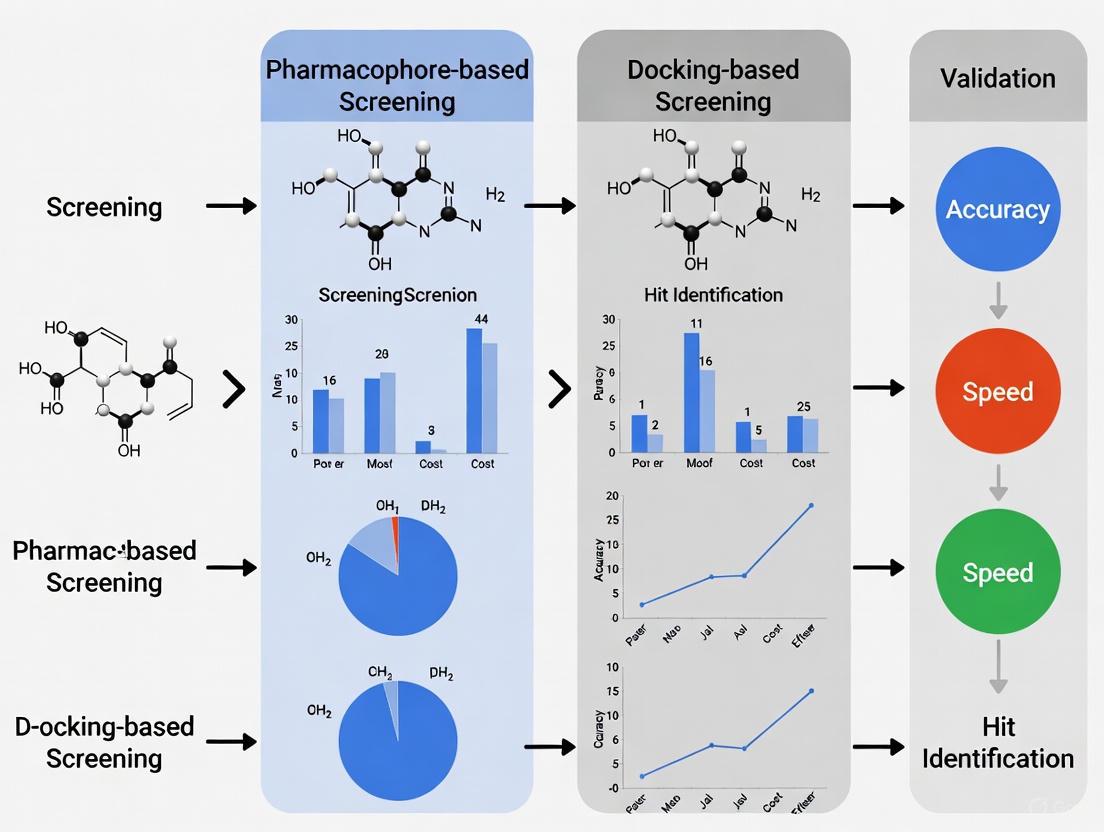
A seminal benchmark study directly compared PBVS and DBVS across eight structurally diverse protein targets: ACE, AChE, AR, DacA, DHFR, ERα, HIV-pr, and TK. The study utilized the Catalyst program for PBVS and three popular docking programs (DOCK, GOLD, Glide) for DBVS. The performance was evaluated using enrichment factors (EF)—a measure of a method's ability to prioritize known active compounds over inactive decoys in a database. [1] [20]
The results were revealing. In fourteen out of sixteen virtual screening scenarios (one target screened against two different decoy databases), PBVS demonstrated higher enrichment factors than DBVS. [1] [20] The average hit rates, which indicate the number of true actives found within a small percentage of the top-ranked compounds, further underscored this trend.
Table 2: Average Hit Rate Comparison from Benchmark Study on Eight Targets [1] [20]
| Method | Average Hit Rate at 2% of Database | Average Hit Rate at 5% of Database |
|---|---|---|
| Pharmacophore-Based VS (Catalyst) | Much Higher | Much Higher |
| Docking-Based VS (DOCK, GOLD, Glide) | Lower | Lower |
The study concluded that for the tested targets, PBVS was more powerful than DBVS in retrieving active compounds from the databases. [1] [20] This superior enrichment performance has been corroborated in other target-specific studies, such as one identifying CXCR4 antagonists, where a pharmacophore model achieved the highest virtual screening performance compared to docking and shape-matching approaches. [23]
Advancements and Hybrid Approaches
While traditional docking methods have limitations, the field is rapidly evolving. The integration of machine learning (ML) has shown significant promise in improving DBVS performance. For instance, rescoring docking outputs with ML-based scoring functions like CNN-Score has been shown to dramatically improve enrichment, elevating some docking tools from worse-than-random to better-than-random performance in screening for antimalarial compounds. [6]
Furthermore, combining both methodologies in a sequential or hybrid workflow is a common and effective strategy in modern drug discovery campaigns. A typical pipeline involves:
- Using a fast PBVS step to rapidly filter large compound libraries and remove obvious mismatches.
- Subjecting the resulting, smaller subset of compounds to more computationally intensive DBVS for a detailed binding assessment.
- This approach was demonstrated in a study seeking VEGFR-2/c-Met dual inhibitors, where pharmacophore screening was used first to narrow down 1.28 million compounds, after which molecular docking was applied to refine the selection. [22]
Diagram: A typical hybrid virtual screening workflow that leverages the speed of PBVS for initial filtering and the detailed assessment of DBVS for refined hit selection.
Essential Research Reagents and Tools
The experimental protocols cited in this guide rely on a suite of specialized software tools and data resources. The following table details key "research reagent solutions" essential for conducting PBVS and DBVS experiments.
Table 3: Essential Research Reagents and Tools for Virtual Screening
| Tool / Resource | Type | Primary Function in VS | Example Use in Cited Research |
|---|---|---|---|
| LigandScout [1] | Software | Constructs structure-based pharmacophore models from protein-ligand complexes. | Used to generate pharmacophore models for the 8-target benchmark study. [1] |
| Catalyst/Hypogen [1] | Software Algorithm | Performs pharmacophore-based database searching and validation. | Used for all PBVS runs in the 8-target benchmark study. [1] |
| Glide, GOLD, DOCK [1] [21] | Docking Software Suite | Programs for performing molecular docking and scoring. | Represented the DBVS methods in the 8-target benchmark; Glide is noted for high physical validity. [1] [21] |
| AutoDock Vina [6] [21] | Docking Software | A widely used open-source program for molecular docking. | Evaluated for screening performance against PfDHFR; performance improved with ML rescoring. [6] |
| DEKOIS [6] | Benchmark Database | Provides sets of known active molecules and carefully selected decoys for VS benchmarking. | Used to evaluate docking tools and ML rescoring for PfDHFR variants. [6] |
| CryoXKit [24] | Software Tool | Incorporates experimental electron density maps from Cryo-EM or X-ray crystallography to guide docking. | Improved docking pose prediction and virtual screening performance by using experimental density as a bias. [24] |
| Machine Learning Scoring Functions (e.g., CNN-Score, RF-Score-VS) [6] | Scoring Algorithm | ML-based functions to re-score docking poses, improving affinity prediction and enrichment. | Significantly improved the enrichment power of traditional docking programs in benchmark tests. [6] |
The comparative analysis of pharmacophore-based and docking-based virtual screening reveals that the choice of method is not a matter of one being universally superior, but rather of selecting the right tool for the specific research context. PBVS, with its lower information requirement and computational cost, excels in the rapid enrichment of large compound libraries and is highly effective when protein structures are unavailable. DBVS provides an atomistically detailed view of the binding interaction, which is invaluable for lead optimization, but is more computationally demanding and historically showed lower enrichment in some benchmark studies. [1] [20]
The future of virtual screening lies in the synergistic integration of these methods, often in sequential workflows, and the adoption of new technologies. The rise of deep learning is beginning to address traditional docking limitations, though challenges with physical plausibility and generalization remain. [21] Furthermore, methods that directly integrate experimental data, such as Cryo-EM density, to guide docking exemplify the trend toward more hybrid and data-driven approaches. [24] For researchers, a pragmatic strategy that leverages the speed and feature-based logic of PBVS for initial filtering, followed by the detailed structural insights from DBVS (potentially enhanced by ML), represents a powerful paradigm for accelerating drug discovery.
The Synergistic Role of VS in the Broader Drug Discovery Pipeline
Virtual screening (VS) has become an indispensable tool in modern drug discovery, enabling the efficient identification of potential drug candidates from vast chemical libraries. The two predominant computational strategies—pharmacophore-based screening and molecular docking—each offer distinct advantages and face unique challenges. This guide provides an objective comparison of their performance, supported by recent experimental data and methodologies, to inform researchers and drug development professionals.
Performance Comparison: Pharmacophore-Based Screening vs. Molecular Docking
The table below summarizes key performance characteristics of pharmacophore-based and docking-based virtual screening methods, based on recent benchmarking studies.
| Performance Metric | Pharmacophore-Based Screening | Docking-Based Screening |
|---|---|---|
| Computational Speed | Extremely fast (sub-linear time, orders of magnitude faster than docking) [26]; ML-predicted scores can be ~1000x faster than classical docking [4] | Computationally intensive and time-consuming, a key bottleneck for ultra-large libraries [27] [26] |
| Typical Library Size | Suitable for screening millions of compounds quickly [26] | Screening billions of compounds requires substantial computing resources [4] [26] |
| Key Strength | High resource efficiency; effective for rapidly filtering large libraries [26] | Direct, physics-based simulation of ligand pose and affinity within a protein binding site [28] |
| Common Limitations | Quality of results heavily dependent on the quality of the pharmacophore query [26] | Scoring functions remain imperfect, with limitations in accuracy and high false positive rates [27] |
| Hit-Rate & Affinity | Successfully identified novel micromolar inhibitors of SARS-CoV-2 NSP13 [5] | Hit-rates can be modeled as a function of library size and scoring noise; improved scoring can substantially enhance hit-rates and affinities [29] |
| Generalizability | Models can be generated from ligand data or protein structure; automated tools like PharmacoForge show promise for generalizability [26] | Performance can degrade with novel protein binding pockets; deep learning methods show significant generalization challenges [21] |
Detailed Experimental Protocols and Data
Fragment-Based Pharmacophore Screening for SARS-CoV-2 Inhibitors
A novel workflow named FragmentScout was developed to efficiently identify potent inhibitors from weak fragment hits [5].
- Methodology: The protocol starts with high-throughput crystallographic fragment screening data (e.g., from XChem). Multiple fragment poses binding to a specific site on the target protein (SARS-CoV-2 NSP13 helicase) are processed using Inte:ligand's LigandScout software. The software automatically detects pharmacophore features (e.g., hydrogen bond donors/acceptors, hydrophobic areas) from each fragment pose and aggregates them into a single, comprehensive joint pharmacophore query. This query is then used to screen large 3D conformational databases [5].
- Performance Outcome: This method led to the discovery of 13 novel micromolar potent inhibitors of SARS-CoV-2 NSP13, which were validated in cellular antiviral assays. It effectively bridges the gap between millimolar fragment hits and micromolar lead compounds [5].
Benchmarking Docking and Machine Learning Rescoring for Malaria
A comprehensive study evaluated the performance of docking tools and machine learning (ML) rescoring against wild-type (WT) and drug-resistant (Q) Plasmodium falciparum dihydrofolate reductase (PfDHFR) [6].
- Methodology: Three docking tools—AutoDock Vina, PLANTS, and FRED—were used to screen the DEKOIS 2.0 benchmark set against both PfDHFR variants. The resulting ligand poses were then rescored by two pretrained ML scoring functions: RF-Score-VS v2 and CNN-Score. Performance was measured by enrichment factor at the top 1% of ranked molecules (EF 1%) [6].
- Performance Outcome:
- For WT PfDHFR, PLANTS combined with CNN-Score yielded the best enrichment (EF 1% = 28).
- For the resistant Q variant, FRED combined with CNN-Score performed best (EF 1% = 31).
- Rescoring with ML significantly improved the performance of some docking tools, with CNN-Score consistently enhancing the retrieval of diverse, high-affinity binders for both variants [6].
Machine Learning-Accelerated Pharmacophore Screening for MAO Inhibitors
A universal methodology was introduced that uses ML to predict docking scores, drastically speeding up the virtual screening process [4].
- Methodology: An ensemble ML model was trained on docking results from Smina software to predict docking scores based solely on 2D molecular structures and fingerprints. This model was then used to perform a pharmacophore-constrained screening of the ZINC database, prioritizing compounds that matched the pharmacophore model and had favorable predicted scores [4].
- Performance Outcome: The ML-based score prediction was ~1000 times faster than classical molecular docking. From the top-ranked compounds, 24 were synthesized and tested, leading to the discovery of new weak inhibitors of MAO-A [4].
The Scientist's Toolkit: Essential Research Reagents and Solutions
The table below lists key software and resources used in the featured studies, which are essential for building a virtual screening pipeline.
| Tool Name | Type/Function | Key Use Case |
|---|---|---|
| LigandScout | Pharmacophore Modeling & Screening | Generating joint pharmacophore queries from fragment data and performing pharmacophore-based virtual screening [5]. |
| Glide | Molecular Docking | High-performance, physics-based docking; often used as a benchmark for accuracy and pose validity [5] [21]. |
| AutoDock Vina | Molecular Docking | Widely used, generic docking tool; commonly evaluated in benchmarking studies [6] [21]. |
| Smina | Molecular Docking | A variant of Vina often used for its customizability and as a basis for training ML models [4]. |
| CNN-Score / RF-Score-VS v2 | Machine Learning Scoring Functions | Rescoring docking outputs to significantly improve enrichment and identify active compounds [6]. |
| PharmacoForge | AI-Based Pharmacophore Generation | Generating 3D pharmacophore hypotheses directly from a protein pocket structure using a diffusion model [26]. |
| DEKOIS | Benchmarking Sets | Public sets of active and decoy molecules for rigorous evaluation of virtual screening methods [6]. |
Integrated Workflows and Emerging Trends
The most successful virtual screening campaigns often leverage the strengths of both pharmacophore and docking methods in a sequential or integrated workflow [28].
- Sequential Filtering: A common strategy uses a fast pharmacophore screen as an initial filter to reduce the size of a large database, followed by more computationally intensive molecular docking on the top hits [28] [30]. This combines the speed of pharmacophore screening with the detailed pose analysis of docking.
- Rise of Deep Learning and AI: AI is transforming both methodologies. Deep learning models are now being applied to docking for pose prediction and scoring, though they still face challenges with physical plausibility and generalization [21]. For pharmacophores, generative AI models like PharmacoForge can automatically design pharmacophore queries from protein pockets, streamlining the process [26].
- Addressing Resistance and Generalization: As seen in the malaria study, combining docking with ML rescoring is a powerful strategy against drug-resistant targets. The ability of these pipelines to identify leads for resistant variants is crucial for modern drug discovery [6].
The following diagram illustrates a modern, synergistic virtual screening workflow that integrates these various tools and strategies.
Synergistic VS Workflow
In conclusion, pharmacophore-based and docking-based virtual screening are not mutually exclusive but are highly synergistic components of the drug discovery pipeline. Pharmacophore models excel as a rapid, efficient initial filter to manage the scale of modern chemical libraries. Molecular docking provides a more detailed, physics-based assessment of binding interactions and poses. The integration of both methods, now supercharged by machine learning and AI, creates a robust and effective strategy for identifying novel, potent, and even resistance-beating drug candidates, solidifying the critical role of virtual screening in accelerating therapeutic development.
Methodologies in Action: Implementing PBVS and DBVS Workflows
Structure-Based vs. Ligand-Based Pharmacophore Modeling Approaches
In the field of computer-aided drug discovery (CADD), pharmacophore modeling has emerged as a powerful and versatile technique for identifying potential therapeutic compounds. A pharmacophore is formally defined as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [7]. In practical terms, it represents the essential molecular functionalities and their spatial arrangement required for biological activity against a specific target. Pharmacophore approaches reduce the time and costs needed to develop novel drugs, making them particularly valuable for addressing health emergencies and the growing field of personalized medicine [7].
The fundamental features comprising a pharmacophore model include hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), hydrophobic areas (H), positively or negatively ionizable groups (PI/NI), aromatic rings (AR), and metal coordinating areas [7]. These abstract features are represented as geometric entities such as spheres, planes, and vectors in three-dimensional space, allowing them to capture the essential interaction capabilities of molecules beyond their specific atomic compositions [7]. This abstraction enables pharmacophore models to identify structurally diverse compounds that share key interaction features, facilitating scaffold hopping and the discovery of novel chemotypes.
Pharmacophore modeling primarily branches into two distinct methodologies: structure-based and ligand-based approaches. The selection between these methods depends on available data, with structure-based approaches requiring three-dimensional structural information of the target protein, while ligand-based methods rely on the physicochemical properties and structural features of known active compounds [7]. Both approaches serve as valuable tools in virtual screening, where they guide the efficient evaluation of large compound libraries to identify potential candidates for further development [1].
Methodological Foundations: Key Approaches and Techniques
Structure-Based Pharmacophore Modeling
Structure-based pharmacophore modeling derives its hypotheses directly from the three-dimensional structure of a macromolecular target. This approach requires either an experimental structure (from X-ray crystallography, NMR spectroscopy, or cryo-EM) or a high-quality homology model of the target protein [7]. The workflow for structure-based pharmacophore modeling follows a systematic process, as illustrated in Figure 1.
Protein preparation represents the critical first step, involving the assessment and optimization of the protein structure. This includes evaluating residue protonation states, positioning hydrogen atoms (which are typically absent in X-ray structures), handling non-protein groups, addressing missing residues or atoms, and verifying stereochemical and energetic parameters [7]. The quality of the input structure directly influences the quality of the resulting pharmacophore model, making this preparation phase essential.
Ligand-binding site detection follows, where the region of the protein where ligands bind is identified. This can be achieved through manual analysis of residues with known functional roles from experimental data, or more efficiently using bioinformatics tools that scan the protein surface for potential binding sites based on evolutionary, geometric, energetic, or statistical properties [7]. Programs such as GRID and LUDI are commonly employed for this purpose, with GRID using molecular interaction fields and LUDI applying geometric rules derived from known non-bonded contacts in experimental structures [7].
Pharmacophore feature generation and selection constitutes the final phase. When a protein-ligand complex structure is available, the pharmacophore features are derived from the interactions observed between the ligand and protein, with the ligand's bioactive conformation informing the spatial arrangement of features [7]. The presence of the receptor also allows for the incorporation of exclusion volumes to represent spatial restrictions of the binding site [7]. In the absence of a bound ligand, the pharmacophore model must be generated based solely on the target structure, which typically results in less accurate models that require manual refinement [31].
Ligand-Based Pharmacophore Modeling
Ligand-based pharmacophore modeling approaches generate hypotheses based on the structural and chemical features shared by a set of known active compounds, without requiring direct knowledge of the target protein's structure. This method operates on the principle that compounds sharing common chemical functionalities in similar spatial arrangements likely exhibit similar biological activities against the same target [31]. The methodology proceeds through several well-defined stages, as depicted in Figure 2.
The process begins with the selection of experimentally validated active compounds that constitute the training set. The quality, diversity, and potency range of these compounds significantly influence the resulting model's quality and discriminatory power [10]. An ideal training set includes structurally diverse compounds covering a range of biological activities to ensure the identification of essential features correlated with potency.
Generation of 3D conformations and structural alignment follows, where multiple low-energy conformations are generated for each active compound. These conformations are then aligned to identify common spatial arrangements of chemical features [31]. The conformational flexibility of molecules presents a challenge, as the bioactive conformation must be adequately sampled or approximated.
Identification of structural characteristics and functional groups involved in molecular recognition forms the core of model development. The model identifies conserved features across the aligned active compounds, including hydrogen bond donors/acceptors, hydrophobic regions, ionizable groups, and aromatic rings [31]. Two primary scoring function methods exist for evaluating how well compounds fit the pharmacophore model: RMSD-based methods, which measure distances between compound functional groups and pharmacophore feature centers, and overlay-based methods, which use atomic and functional group radii to estimate functional similarity [31].
Model validation using test compounds represents the final crucial step. The pharmacophore hypothesis is tested against a set of known active (true positives) and inactive (false positives) compounds to evaluate its ability to discriminate between them [31]. Striking an appropriate balance between model restrictiveness and flexibility is essential, as overly strict models may limit structural diversity, while excessively permissive models may retrieve too many false positives [31].
Experimental Comparison: Performance Evaluation and Benchmark Studies
Experimental Design for Method Evaluation
Rigorous benchmarking studies have been conducted to evaluate the relative performance of structure-based and ligand-based pharmacophore modeling, particularly in comparison with other virtual screening methods. A comprehensive study published in Acta Pharmacologica Sinica employed a systematic research pipeline to compare pharmacophore-based virtual screening (PBVS) with docking-based virtual screening (DBVS) across eight structurally diverse protein targets [1] [2].
The experimental design incorporated eight pharmaceutically relevant targets: angiotensin-converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptor α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK) [1]. These targets represent diverse pharmacological functions and disease areas, ensuring broad applicability of the findings.
For each target, researchers constructed active compound datasets from experimentally validated actives sourced from the DrugBank database [2]. To ensure rigorous assessment, they employed two separate decoy datasets (Decoy I and Decoy II) comprising approximately 1000 non-active molecules each, generated using different methodologies to avoid bias [2]. This design created sixteen distinct test scenarios (eight targets × two decoy sets) for robust method evaluation.
The study implemented multiple screening methodologies to minimize tool-specific bias. For PBVS, they used Catalyst software with pharmacophore models generated from multiple X-ray structures of protein-ligand complexes using LigandScout [1]. For DBVS, they employed three different docking programs: DOCK, GOLD, and Glide, applying each to high-resolution crystal structures of ligand-protein complexes [2]. This multi-program approach helped account for variations in algorithm performance.
Quantitative Performance Comparison
The benchmark study yielded compelling quantitative results comparing the effectiveness of pharmacophore-based and docking-based virtual screening approaches. Performance was primarily evaluated using enrichment factors (measuring the relative abundance of active compounds in the top-ranked fraction compared to random selection) and hit rates (the percentage of identified compounds that are truly active) [1].
Table 1: Virtual Screening Performance Across Eight Protein Targets
| Screening Method | Enrichment Factor (Average) | Hit Rate at Top 2% | Hit Rate at Top 5% |
|---|---|---|---|
| Pharmacophore-based (PBVS) | Higher in 14/16 cases | Much higher | Much higher |
| Docking-based (DBVS) | Lower in most cases | Lower | Lower |
| DOCK | Variable | - | - |
| GOLD | Variable | - | - |
| Glide | Variable | - | - |
The results demonstrated that PBVS significantly outperformed DBVS across most test conditions. Of the sixteen virtual screening scenarios (eight targets against two decoy sets), PBVS achieved higher enrichment factors in fourteen cases [1]. When examining the top 2% and 5% of highest-ranked compounds from the entire databases, the average hit rates for PBVS were substantially higher than those for all docking methods [1].
This performance advantage was consistent across most targets, suggesting that PBVS offers generally superior retrieval of active compounds from complex compound libraries compared to DBVS approaches [2]. The robustness of pharmacophore-based methods stems from their focus on essential interaction features rather than complete atomic-level complementarity, which appears to provide better discrimination between actives and inactives in virtual screening applications.
Case Studies in Practical Applications
Real-world applications further demonstrate the strengths and appropriate use cases for both structure-based and ligand-based pharmacophore approaches. In the search for selective carbonic anhydrase IX (hCA IX) inhibitors for cancer therapy, researchers successfully employed ligand-based pharmacophore modeling to identify novel chemotypes from natural sources [32]. Using seven known active compounds with IC₅₀ values below 50 nM, they developed a pharmacophore model featuring two aromatic hydrophobic centers and two hydrogen bond donor/acceptor features [32]. This model successfully identified 43 potential inhibitors through virtual screening, with subsequent molecular docking and dynamics simulations confirming their potential for selective hCA IX inhibition.
Another study applied combined structure-based and ligand-based strategies to discover mosquito repellents from essential oil constituents. Researchers used the structure of DEET complexed with an odorant-binding protein for structure-based modeling, while also employing structural similarity searching as a ligand-based approach [31]. This integrated strategy identified seven natural volatile compounds with predicted repellent activity, including p-cymen-8-yl, thymol acetate, and carvacryl acetate [31].
For Topoisomerase I inhibitor discovery, researchers developed a ligand-based pharmacophore model using 29 camptothecin derivatives through the HypoGen algorithm [10]. After rigorous validation, this model screened over one million drug-like molecules from the ZINC database, ultimately identifying three promising candidates with strong binding interactions, favorable toxicity profiles, and stable molecular dynamics simulations [10].
Integrated Workflows and Advanced Methodologies
Synergistic Integration of Approaches
Rather than positioning structure-based and ligand-based approaches as mutually exclusive alternatives, modern drug discovery increasingly employs integrated workflows that leverage the complementary strengths of both methodologies. Structure-based pharmacophores can serve as effective pre-filters or post-filters for docking-based virtual screening, helping to eliminate compounds that lack essential interaction features or possess undesirable characteristics [1]. This hybrid approach has been shown to increase enrichment rates compared to docking alone [2].
The emergence of machine learning and artificial intelligence has further advanced integrated pharmacophore strategies. The CMD-GEN framework exemplifies this trend, bridging ligand-protein complexes with drug-like molecules through coarse-grained pharmacophore points sampled from diffusion models [33]. This approach employs a hierarchical architecture that decomposes three-dimensional molecule generation into pharmacophore point sampling, chemical structure generation, and conformation alignment, effectively addressing instability issues in molecular generation [33].
Another innovative methodology, O-LAP, introduces shape-focused pharmacophore modeling through graph clustering algorithms that generate cavity-filling models by clustering overlapping atomic content from docked active ligands [34]. This approach significantly improves docking enrichment by comparing shape similarity between flexibly sampled poses and target binding cavities, demonstrating the continued evolution of pharmacophore techniques [34].
Experimental Protocols and Research Reagents
Successful implementation of pharmacophore modeling requires specific computational tools and protocols. The experimental benchmarks discussed employed several established software packages and data resources, detailed in Table 2.
Table 2: Key Research Reagent Solutions for Pharmacophore Modeling
| Resource Category | Specific Tools/Sources | Function and Application |
|---|---|---|
| Structure-Based Tools | LigandScout, GRID, LUDI | Generate structure-based pharmacophores from protein-ligand complexes |
| Ligand-Based Tools | Molecular Operating Environment (MOE), Pharmer, Align-it | Develop pharmacophore models from active ligand sets |
| Virtual Screening Platforms | Catalyst, Pharmit, PharmMapper | Screen compound libraries against pharmacophore models |
| Data Resources | RCSB Protein Data Bank (PDB), DrugBank, ZINC Database | Source protein structures, active compounds, and screening libraries |
| Validation Tools | Decoy sets (DUD-E, DUDE-Z) | Assess model performance with known actives and inactives |
For structure-based pharmacophore modeling, a typical protocol involves: (1) retrieving and preparing the protein structure from PDB, including hydrogen addition and optimization; (2) identifying and characterizing the binding site using tools like GRID or LUDI; (3) generating interaction features from protein-ligand complexes or binding site analysis; (4) selecting essential features for the pharmacophore hypothesis; and (5) validating the model using known actives and decoys [7] [35].
For ligand-based pharmacophore modeling, standard methodology includes: (1) compiling and curating a set of known active compounds with diverse structures and potency data; (2) generating representative 3D conformations for each compound; (3) performing molecular alignment to identify common features; (4) creating the pharmacophore hypothesis based on conserved spatial features; and (5) validating the model with test compounds and refining based on performance [31] [32].
The comprehensive comparison of structure-based and ligand-based pharmacophore modeling approaches reveals a nuanced landscape where each methodology offers distinct advantages depending on available data and research objectives. Structure-based approaches provide superior performance when high-quality protein structures are available, particularly when derived from protein-ligand complexes that offer detailed interaction information. These methods directly incorporate spatial constraints from the binding site and can identify novel scaffold types not represented in existing ligand sets.
Ligand-based approaches demonstrate remarkable effectiveness when structural information for the target is limited or unavailable, leveraging the collective information embedded in known active compounds. These methods excel at identifying common functional features essential for activity and can successfully scaffold-hop to discover novel chemotypes that maintain these key interactions.
The benchmark evidence clearly indicates that pharmacophore-based virtual screening generally outperforms docking-based approaches in retrieving active compounds from large databases across diverse target types [1] [2]. This performance advantage, combined with typically lower computational requirements, positions pharmacophore modeling as a powerful first-line tool for virtual screening campaigns.
Ultimately, the most effective strategy for modern drug discovery involves the integrated application of both structure-based and ligand-based approaches, often in combination with docking and other computational methods. This multimodal strategy leverages the complementary strengths of each methodology, maximizing the probability of identifying novel, potent, and drug-like compounds for therapeutic development. As artificial intelligence and machine learning continue to advance, further refinement and integration of pharmacophore approaches will undoubtedly enhance their precision and utility in the ongoing quest for new therapeutic agents.
Molecular docking stands as a cornerstone technique in modern computational drug discovery, enabling researchers to predict how small molecules interact with biological targets at the atomic level. This methodology has evolved significantly from its initial rigid-body approximations to sophisticated algorithms that account for molecular flexibility, with profound implications for virtual screening accuracy. The fundamental objective of docking is to predict the three-dimensional structure of a protein-ligand complex and estimate the binding affinity, thereby identifying promising therapeutic candidates before costly experimental validation [36]. As docking methodologies have advanced, a critical question has emerged within computational pharmacology: how do these approaches compare to pharmacophore-based screening strategies in identifying biologically active compounds?
The transition from rigid to flexible docking represents one of the most significant technical advancements in the field. Early docking methods treated both proteins and ligands as rigid bodies, dramatically reducing computational complexity but oversimplifying the dynamic nature of biomolecular interactions [18]. This simplification often compromised accuracy, as both ligands and protein receptors undergo conformational changes upon binding—a phenomenon known as induced fit. Modern flexible docking approaches address this limitation by accounting for ligand flexibility and, in more advanced implementations, protein flexibility as well [18]. Understanding the capabilities, limitations, and appropriate application contexts of these different docking strategies is essential for researchers aiming to optimize their virtual screening pipelines, particularly when weighed against complementary pharmacophore-based methods.
Theoretical Foundations: From Rigid-Body to Flexible Docking Approaches
Rigid Docking: Foundations and Limitations
The earliest molecular docking approaches operated on the principle of rigid-body docking, which treats both the protein receptor and the ligand as fixed three-dimensional structures without internal flexibility. In this simplified model, the docking algorithm searches for the optimal alignment by exploring only six degrees of freedom—three translational and three rotational—much like fitting a key into a static lock [18]. This approach significantly reduces computational demands and enables rapid screening of compound libraries, making it practical for early virtual screening efforts when computational resources were limited.
However, the rigid-body assumption represents a significant simplification of biological reality. In actual binding processes, both ligands and proteins exhibit considerable flexibility, with bonds rotating and side chains adjusting to accommodate binding partners. The inability of rigid docking to account for these conformational changes often results in inaccurate pose predictions and unreliable binding affinity estimates, particularly for flexible ligands or proteins with binding sites that undergo substantial rearrangement upon ligand binding [18]. Despite these limitations, rigid docking remains useful for preliminary screening scenarios where the protein structure is known to be relatively static or when computational efficiency is paramount.
Flexible Docking: Accounting for Biological Reality
Ligand Flexibility
The first major advancement beyond rigid docking came with the incorporation of ligand flexibility, which acknowledges that small molecules can adopt multiple conformations when binding to protein targets. Modern docking programs typically account for ligand flexibility through various sampling strategies, including systematic torsion sampling, genetic algorithms, and Monte Carlo methods [36]. These approaches generate multiple ligand conformations during the docking process, evaluating which orientation best complements the binding site geometrically and energetically. The "anchor-and-grow" strategy employed by tools like DOCK6 represents one effective implementation, where a rigid core fragment is initially positioned followed by incremental rebuilding and conformational sampling of flexible substituents [37].
Receptor Flexibility
Accounting for protein flexibility remains one of the most significant challenges in molecular docking. Proteins are dynamic entities whose binding sites can undergo substantial conformational changes upon ligand binding—a phenomenon known as induced fit. Traditional docking struggles with these rearrangements, particularly in cross-docking scenarios where ligands are docked to receptor conformations different from their native bound states [18]. Several strategies have emerged to address protein flexibility, including:
- Ensemble Docking: Using multiple protein structures from different crystal forms or molecular dynamics simulations to represent conformational diversity
- Side-Chain Flexibility: Allowing rotation of amino acid side chains within the binding site while keeping the protein backbone fixed
- Backbone Flexibility: Incorporating limited backbone movements, though this substantially increases computational complexity
- Internal Coordinates: Using internal coordinate representations that naturally accommodate structural flexibility
Recent deep learning approaches have begun to transform flexible docking by using equivariant neural networks and diffusion models to predict conformational changes during binding [18]. Methods like FlexPose enable end-to-end flexible modeling of protein-ligand complexes regardless of input protein conformation (apo or holo), offering promising avenues for more accurate prediction of binding poses for challenging targets [18].
Comparative Performance: Rigid vs. Flexible Docking Strategies
Performance Metrics and Benchmarking Studies
Rigorous benchmarking studies provide critical insights into the relative performance of different docking strategies. One comprehensive study evaluated physics-based, deep learning-based, and generative molecular docking tools using both rigid and flexible receptor protocols for 431 BACE1 ligand complex structures [37]. BACE1 (Beta-site amyloid precursor protein cleaving enzyme 1) represents an excellent test case with its dynamic active site that poses significant challenges for accurate binding pose prediction. The study compared docking tools based on all-atoms Root Mean Square Deviation (RMSD) and centre-of-mass RMSD deviation between experimental and predicted binding poses, offering quantitative assessment of pose prediction accuracy across methodologies.
Table 1: Performance Comparison of Docking Methodologies for BACE1 Complexes
| Docking Method | Type | Receptor Flexibility | Key Findings | Performance Limitations |
|---|---|---|---|---|
| DOCK6 | Physics-based | Rigid (with ligand flexibility) | Most reliable performance due to grid-based scoring and anchor-and-grow sampling | Lacks flexible receptor docking capabilities |
| GNINA | Deep learning-based | Rigid | Reduced accuracy likely due to under-representation of BACE1 in training data | Limited transferability to uncommon targets |
| Flexible Docking Protocols | Physics-based | Flexible | Struggled with pose ranking and geometry, particularly for large/flexible ligands | Computational intensive with ranking challenges |
| DiffDock | Generative AI | Rigid (with coarse flexibility) | Often failed to produce native-like poses | Limited physical realism in predictions |
Influence of Ligand and Target Properties
Docking performance is significantly influenced by the physicochemical properties of both the ligand and target protein. Key factors affecting accuracy include:
- Ligand Flexibility: Molecules with higher numbers of rotatable bonds present greater challenges for conformational sampling [38]
- Molecular Size and Complexity: Larger molecules with more rings and hydrogen bond acceptors/donors increase docking complexity [38]
- Binding Site Characteristics: Hydrophobic binding pockets versus polar sites can bias different scoring functions [37]
- Protein Conformational State: Docking to apo (unbound) versus holo (bound) structures significantly impacts accuracy, with flexible methods showing advantages for apo structures [18]
The LigDockTailor study systematically demonstrated that ligand properties—including rotatable bond count, molecular weight, LogP, hydrogen bond acceptor count, and ring structures—significantly influence docking accuracy across different programs [38]. This highlights the importance of selecting docking strategies matched to specific ligand characteristics rather than relying on a one-size-fits-all approach.
Experimental Protocols for Docking Evaluation
Benchmarking Framework and Validation Metrics
Robust evaluation of docking methodologies requires standardized benchmarking frameworks and validation metrics. The following protocol outlines key steps for comparative docking assessment:
Dataset Curation: Compile diverse protein-ligand complexes with high-resolution crystal structures from databases like PDBBind [38]. The BACE1 benchmarking study utilized 431 complex structures from the Protein Data Bank [37].
Complex Preparation: Process protein structures by removing water molecules, adding hydrogen atoms, correcting bond orders, and assigning partial charges using tools like MGL Tools [39]. Ligands should be prepared with proper protonation states and tautomeric forms.
Docking Execution: Perform docking with multiple programs using consistent binding site definitions. For rigid docking, use the crystal structure conformation; for flexible docking, incorporate receptor flexibility through side-chain sampling or ensemble approaches.
Pose Prediction Assessment: Calculate Root Mean Square Deviation (RMSD) between predicted and experimental ligand poses. Industry standards often consider RMSD < 2.0Å as successful prediction [37].
Scoring Function Evaluation: Assess binding affinity prediction accuracy through correlation between computed scores and experimental binding energies.
Statistical Analysis: Employ enrichment factors, AUC-ROC curves, and early enrichment metrics to evaluate virtual screening performance [22].
Case Study: BACE1 Docking Evaluation Protocol
The BACE1 comparative study implemented a comprehensive protocol specifically targeting this challenging therapeutic target [37]:
- Receptor Preparation: 431 BACE1-ligand complexes were prepared with consistent protonation states and water molecule treatment
- Grid Definition: Binding sites were defined using native ligand positions from crystal structures
- Pose Sampling: Multiple sampling algorithms were compared including genetic algorithms, Monte Carlo methods, and diffusion-based approaches
- Evaluation Metrics: Both all-atom RMSD and center-of-mass RMSD were calculated to assess different aspects of pose accuracy
- Physical Property Analysis: Ligand flexibility, solvent-accessible surface area, polarity, and protein hydrophobicity were correlated with docking performance to identify potential biases
Molecular Docking versus Pharmacophore-Based Screening
Direct Performance Comparisons
The competition between docking-based and pharmacophore-based virtual screening represents a fundamental debate in computational drug discovery. A landmark comparative study evaluated both approaches across eight structurally diverse protein targets: angiotensin converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptors α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK) [1]. This comprehensive analysis revealed that pharmacophore-based virtual screening (PBVS) outperformed docking-based virtual screening (DBVS) in 14 out of 16 test scenarios, demonstrating substantially higher enrichment factors and hit rates.
Table 2: Pharmacophore vs. Docking-Based Virtual Screening Performance
| Screening Method | Average Hit Rate at 2% Database | Average Hit Rate at 5% Database | Key Advantages | Typical Use Cases |
|---|---|---|---|---|
| Pharmacophore-Based Virtual Screening (PBVS) | Significantly higher than DBVS | Significantly higher than DBVS | Speed, chemical feature emphasis, less constrained by strict protein structure | Scaffold hopping, initial screening phases, targets without high-quality structures |
| Docking-Based Virtual Screening (DBVS) | Lower than PBVS | Lower than PBVS | Detailed atomic interactions, physical binding process simulation, precise pose prediction | Binding mode analysis, lead optimization, structure-based design |
Synergistic Applications in Virtual Screening
Rather than positioning docking and pharmacophore methods as mutually exclusive alternatives, modern drug discovery increasingly employs them in integrated workflows that leverage their complementary strengths:
Pharmacophore Pre-filtering: Using rapid pharmacophore screening to reduce large compound libraries to manageable sizes before more computationally intensive docking [22]
Post-Docking Pharmacophore Filtering: Applying pharmacophore constraints to docking results to eliminate poses that lack essential interaction features [1]
Hybrid Approaches: Combining pharmacophore matching with docking scores in consensus scoring functions to improve enrichment
Machine Learning Enhancement: Employing ML models trained on docking scores to accelerate virtual screening while maintaining accuracy, as demonstrated in MAO inhibitor discovery where prediction was 1000 times faster than classical docking [4]
The integration of these approaches is particularly valuable for challenging targets like BACE1, where flexible docking protocols struggle with pose ranking, and for antibody-antigen interactions where pharmacophore methods demonstrate 98.6% success in recapitulating native complexes [25].
Table 3: Essential Computational Tools for Molecular Docking Studies
| Tool Category | Representative Software | Primary Function | Key Features |
|---|---|---|---|
| Rigid Docking Tools | ZDOCK, SwissDock | Protein-ligand docking with fixed conformations | Fast execution, suitable for preliminary screening |
| Flexible Ligand Docking | AutoDock Vina, GOLD, DOCK6 | Docking with ligand flexibility | Efficient conformational sampling, anchor-and-grow algorithms |
| Advanced Flexible Docking | FlexPose, MOE-Dock, ICM | Partial or full receptor flexibility | Side-chain optimization, backbone flexibility, ensemble docking |
| Deep Learning Docking | DiffDock, EquiBind, GNINA | AI-powered pose prediction | SE(3)-equivariant networks, diffusion models, training on structural databases |
| Pharmacophore Modeling | LigandScout, MOE | Feature-based molecular interaction analysis | Chemical feature mapping, structure- and ligand-based modeling |
| Molecular Dynamics | GROMACS, AMBER | Binding process simulation and refinement | Explicit solvation, physiological conditions, binding free energy calculations |
| Visualization & Analysis | PyMOL, Discovery Studio | Result interpretation and visualization | Binding pose inspection, interaction diagram generation |
Signaling Pathways and Workflow Diagrams
Molecular Docking Methodology Evolution
Virtual Screening Workflow: Pharmacophore and Docking Integration
The evolution from rigid to flexible docking algorithms represents significant progress in computational drug discovery, yet each methodology carries distinct advantages and limitations that dictate their appropriate application domains. Rigid docking offers computational efficiency for initial screening phases, while flexible docking provides more biologically realistic predictions for complex binding events involving substantial conformational changes. The benchmarking data clearly indicates that no single docking tool universally outperforms others across all target classes and ligand types, emphasizing the importance of method selection based on specific research contexts.
Looking forward, several emerging trends are poised to shape the next generation of docking methodologies. Deep learning approaches are rapidly advancing, with diffusion models like DiffDock and flexible docking networks like FlexPose showing promise in addressing long-standing challenges in protein flexibility modeling [18]. The integration of machine learning with physical scoring functions offers particular potential for improving binding affinity prediction while maintaining computational efficiency. Furthermore, the synergistic combination of pharmacophore-based and docking-based virtual screening continues to demonstrate practical value in drug discovery campaigns, leveraging the complementary strengths of both approaches [1] [4].
As these computational methodologies mature, their successful application will increasingly depend on researchers' understanding of both their theoretical foundations and practical limitations. By strategically selecting and combining these tools based on target characteristics, project goals, and available resources, drug discovery scientists can maximize the value of computational predictions in guiding experimental efforts toward novel therapeutic candidates.
In the realm of computer-aided drug discovery, virtual screening stands as a pivotal technique for identifying potential lead compounds from extensive molecular libraries. Two predominant methodologies have emerged: pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS). The performance and success of both methods are fundamentally dependent on three critical preparatory steps: the preparation of the screening library, the generation of biologically relevant molecular conformations, and the thorough analysis of the target protein. These initial steps profoundly influence the outcome of virtual screening campaigns, determining the ability to discriminate true active compounds from inactive decoys. This guide provides a detailed comparison of methodologies and tools for these foundational steps, contextualized within performance research comparing pharmacophore and docking-based approaches. Empirical evidence suggests that PBVS often outperforms DBVS in many scenarios; a comprehensive benchmark study demonstrated that in fourteen out of sixteen virtual screening sets, PBVS achieved higher enrichment factors than DBVS, with superior average hit rates at the top 2% and 5% of ranked databases [1]. Understanding and optimizing the critical preparatory steps is essential for leveraging the strengths of each virtual screening method.
Library Preparation: Curating the Molecular Search Space
Library preparation involves assembling and curating a database of compounds for screening, which includes defining chemical structures, managing stereochemistry, and preparing compounds in a format suitable for computational analysis.
Key Steps and Considerations
The process typically involves:
- Data Sourcing: Compounds are sourced from public databases like ZINC, ChEMBL, PubChem, or commercial collections. The Comprehensive Marine Natural Product Database (CMNPD) serves as an example of a specialized database providing 3D structures, bioactivity data, and ADMETox characteristics for natural products [40].
- Structure Standardization: Converting structural representations (e.g., SMILES strings) into standardized formats, often stripping stereochemistry to evaluate a conformer generator's ability to sample correct geometries [41].
- Descriptor Calculation: Pre-calculating physicochemical properties or structural descriptors used for filtering or screening.
Impact on Virtual Screening Performance
The composition of the screening library directly impacts results. Libraries should include known actives and property-matched decoys to validate methods. The Database of Useful Decoys: Enhanced (DUDE) is a widely used benchmark containing 102 targets with confirmed actives and decoys [41]. Proper library preparation minimizes biases and ensures that screening performance reflects the method's true discriminatory power rather than database artifacts.
Table 1: Key Research Reagent Solutions for Library Preparation and Conformer Generation
| Reagent/Resource | Type/Function | Application Context |
|---|---|---|
| ZINC Database [42] | Public database of commercially available compounds in ready-to-dock 3D format. | Source of screening compounds for virtual screening; contains over 230 million compounds. |
| DUDE Database [41] | Benchmark set with known actives and property-matched decoys for specific targets. | Validation of virtual screening protocols and enrichment calculations. |
| CMNPD [40] | Comprehensive Marine Natural Product Database with 3D structures and bioactivity data. | Source of specialized natural product compounds for virtual screening. |
| RDKit [41] | Open-source cheminformatics toolkit with conformer generation capabilities. | Generation of conformational ensembles using the ETKDG method. |
| Conformer-RL [43] | Open-source Python library using deep reinforcement learning for conformer generation. | Research and generation of diverse, low-energy conformations for drug-like molecules. |
Conformer Generation: Sampling Biologically Relevant Shapes
Conformer generation assigns realistic 3D coordinates to a molecule from its topological representation, creating an ensemble of its possible shapes. This is fundamental to structure-based design, as ligands are flexible and must be represented in their bioactive conformation.
Methodologies and Tools
- Classical Geometry-Based Approaches: Tools like RDKit use stochastic distance geometry combined with knowledge-based potentials (ETKDG) to sample conformations efficiently. They leverage experimental torsional-angle preferences and ring geometries [41].
- Deep Learning Approaches: Generative models, such as Direct Molecular Conformation Generation (DMCG), are trained on large datasets like GEOM-Drugs to produce accurate conformational ensembles [41]. Reinforcement learning libraries like Conformer-RL provide a modular platform for applying and researching deep RL algorithms for this task [43].
Critical Parameters for Ensemble Quality
The quality of a conformational ensemble is governed by several parameters:
- Ensemble Size: Generating a sufficiently large number of conformers (e.g., up to 250) is crucial for sampling the bioactive conformation [41].
- Diversity Filtering: Filtering conformers by root-mean-square deviation (RMSD) ensures structural diversity within the ensemble.
- Energy Minimization: Post-processing with molecular force fields like the Universal Force Field (UFF) can refine conformations, though its impact on bioactive conformation recovery must be evaluated [41].
The ability to recapitulate the bioactive conformation (the shape a molecule adopts when bound to its target) is a key metric for success. Provided a large and diverse ensemble is generated, tools like RDKit consistently perform well at this task [41].
Figure 1: A generalized workflow for generating a conformational ensemble suitable for structure-based drug design tasks like pharmacophore search and molecular docking [41].
Target Analysis: Defining the Interaction Landscape
Target analysis involves preparing the protein structure and defining the binding site and key interaction points, which directly informs both pharmacophore model development and docking procedures.
Structure-Based Pharmacophore Modeling
When a protein's 3D structure is available (from X-ray crystallography, cryo-EM, or homology modeling), a structure-based pharmacophore can be built. The workflow involves:
- Protein Preparation: Critical evaluation and processing of the protein structure, including protonation states, addition of hydrogens, and handling missing residues [7] [42].
- Binding Site Identification: Using tools like GRID or LUDI to detect potential binding sites based on geometric and energetic properties [7].
- Feature Mapping: Identifying key interaction features (e.g., hydrogen bond donors/acceptors, hydrophobic areas, ionizable groups) within the binding site [7] [40]. Exclusion volumes are added to represent the protein's steric constraints.
- Model Validation: The model is validated by its ability to retrieve known active compounds from a decoy set, often measured by the Area Under the Curve (AUC) of a Receiver Operating Characteristic (ROC) curve. An excellent model can achieve an AUC value near 1.0 [42].
Docking Site Preparation
For DBVS, target analysis focuses on defining the search space for docking algorithms.
- Grid Generation: Defining a 3D grid that encompasses the binding site for scoring function calculations.
- Incorporating Experimental Data: Emerging methods, like CryoXKit, can directly use experimental electron density maps from XRC or cryo-EM as a biasing potential during docking, improving pose prediction without requiring a predefined pharmacophore [24].
Performance Comparison: Pharmacophore vs. Docking-Based Screening
The choices made during library preparation, conformer generation, and target analysis directly influence the performance of PBVS and DBVS. A landmark comparative study offers critical insights.
Table 2: Virtual Screening Performance: PBVS vs. DBVS [1]
| Performance Metric | Pharmacophore-Based VS (PBVS) | Docking-Based VS (DBVS) |
|---|---|---|
| Overall Enrichment (14/16 cases) | Higher Enrichment Factors | Lower Enrichment Factors |
| Average Hit Rate (Top 2% of DB) | Much Higher | Lower |
| Average Hit Rate (Top 5% of DB) | Much Higher | Lower |
| Key Dependency | Quality of the conformer ensemble and pharmacophore model | Quality of conformer ensemble and scoring function |
| Typical Use Case | Primary screening, pre-filtering for DBVS | Primary screening, detailed pose analysis |
Experimental Protocol for Performance Benchmarking
The comparative data in Table 2 originates from a rigorous benchmark study [1]. The methodology can be summarized as follows:
- Target Selection: Eight structurally diverse protein targets (e.g., ACE, AChE, HIV-pr) were selected.
- Data Set Construction: For each target, an active set (experimentally validated inhibitors) and two different decoy sets (Decoy I and Decoy II) were compiled.
- Pharmacophore-Based Screening: For each target, a pharmacophore model was constructed from several protein-ligand complex X-ray structures using LigandScout. Virtual screening was performed using the Catalyst program against the combined active-decoys databases.
- Docking-Based Screening: Three different docking programs (DOCK, GOLD, Glide) were used to screen the same databases against a high-resolution crystal structure for each target.
- Performance Evaluation: For each virtual screening run, the hit list was analyzed to calculate enrichment factors (EF) and hit rates at the top 2% and 5% of the ranked database. The results across all targets and decoy sets were then aggregated for a global comparison.
This study concluded that PBVS is a powerful method that generally outperformed the tested DBVS methods in retrieving active compounds, making it a highly effective strategy for drug discovery [1].
Integrated Workflows and Best Practices
Given their complementary strengths, a synergistic approach that integrates PBVS and DBVS is often the most effective strategy.
Hybrid Screening Strategies
A common and successful practice is to use PBVS as a pre-filter for DBVS. A pharmacophore model can rapidly screen millions of compounds to select a subset that possesses the essential interaction features, which is then passed to the more computationally expensive docking simulation for detailed pose prediction and scoring [1] [7]. This hybrid approach leverages the speed and enrichment power of PBVS with the detailed binding analysis of DBVS.
Consensus Scoring and Comparative Docking
To improve the reliability of virtual screening, consensus approaches are recommended. This can involve:
- Comparative Molecular Docking: Screening a library of hits using multiple docking engines (e.g., AutoDock and AutoDock Vina) and applying consensus scoring to select compounds that rank highly across different programs [40].
- Post-Docking Pharmacophore Filtering: Applying a pharmacophore model as a filter to the top-ranked poses generated by docking to ensure they form key interactions, which has been shown to increase enrichment rates [1].
Figure 2: An integrated virtual screening workflow where pharmacophore-based screening is used to pre-filter a large library before more computationally intensive docking-based screening [1] [7].
The critical preparatory steps of library preparation, conformer generation, and target analysis form the bedrock of successful virtual screening. The empirical evidence demonstrates that pharmacophore-based virtual screening, when supported by high-quality conformational ensembles and rigorously validated models, consistently achieves high enrichment and can outperform docking-based methods in many scenarios. However, the selection between PBVS and DBVS is not absolute and depends on the specific target and available data. By meticulously executing these foundational steps and thoughtfully integrating both pharmacophore and docking strategies, researchers can significantly enhance the efficiency and success rate of their drug discovery efforts.
Virtual screening (VS) has become an indispensable technique in early drug discovery, designed to identify active compounds from large chemical libraries in a cost- and time-efficient manner [44]. The two primary computational approaches are pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS). PBVS uses a three-dimensional model of the steric and electronic features essential for a ligand to interact with a biological target, providing a powerful abstraction of molecular recognition [2]. In contrast, DBVS computationally predicts the preferred orientation and binding affinity of a small molecule (ligand) within a target's binding site, more directly simulating the physical binding process [45] [46]. The choice between these strategies often depends on the available structural information for the target and the specific goals of the screening campaign. This guide provides an objective comparison of leading software tools for both methodologies, focusing on LigandScout and Catalyst for PBVS, and AutoDock and Glide for DBVS, framing the discussion within the broader context of performance comparison research.
Performance Comparison: Key Metrics and Experimental Data
The effectiveness of VS tools is typically evaluated using several key metrics. For binding pose prediction, the root-mean-square deviation (RMSD) between the predicted pose and the experimentally determined co-crystal structure is used, with an RMSD of less than 2.0 Å generally considered a successful prediction [45] [47]. For virtual screening enrichment, the ability to prioritize active compounds over inactive ones is measured using enrichment factors (EF), receiver operating characteristics (ROC) curves, and the area under the ROC curve (AUC) [45] [48]. The hit rate at the top percentage (e.g., 2% or 5%) of the ranked database is also a common performance indicator [1].
The table below summarizes key performance data from published benchmark studies for the relevant software tools.
Table 1: Performance Summary of Virtual Screening Tools
| Software | VS Type | Pose Prediction Success (RMSD <2Å) | Key Performance Findings | Reported Enrichment (AUC/EF) |
|---|---|---|---|---|
| LigandScout | PBVS | Not Primary Focus | Outperformed DBVS in 14/16 test cases across 8 targets [1]. | Higher average hit rates vs. DBVS at 2% and 5% database ranks [2]. |
| Catalyst | PBVS | Not Primary Focus | Powerful method for database searching and hit identification [49] [2]. | Algorithms with overlay-based scoring ensure better library enrichments [49]. |
| Glide | DBVS | 100% (COX-1/2) [45] | Top performer in cognate re-docking; 67% success on general PDBBind test set [45] [47]. | AUCs of 0.61-0.92 for COX targets [45]. |
| AutoDock Vina | DBVS | ~60-80% (Typical for cognate docking) [47] | Robust and widely used; performance is target-dependent [50] [51]. | Useful for classification, but hydrophilic targets are more amenable [50]. |
| GOLD | DBVS | 59-82% (COX-1/2) [45] | Good performance for pose prediction and virtual screening [45]. | Produced good correlations for hydrophilic targets like kinases [50]. |
| Surflex-Dock | DBVS | 68% (Top-1) [47] | High performance in fair comparative studies, competitive with Glide [47]. | Established as a strong baseline for conventional docking workflows [47]. |
Direct Comparative Studies: PBVS vs. DBVS
A landmark study directly compared PBVS (using Catalyst) and DBVS (using DOCK, GOLD, and Glide) across eight diverse protein targets: ACE, AChE, AR, DacA, DHFR, ERα, HIV-pr, and TK [1] [2]. The pharmacophore models for PBVS were constructed using LigandScout, which generates models from protein-ligand complex structures [2]. The results demonstrated that PBVS outperformed DBVS methods in the majority of cases, showing higher enrichment factors in 14 out of 16 virtual screening runs. When considering the top 2% and 5% of the ranked databases, the average hit rate for PBVS was "much higher" than those achieved by the docking methods [1]. This suggests that PBVS can be a highly efficient method for retrieving active compounds from large chemical libraries.
Performance in Docking and Pose Prediction
For DBVS, the accurate prediction of a ligand's binding mode is a fundamental prerequisite. A 2023 benchmarking study on cyclooxygenase (COX) enzymes revealed that Glide outperformed other docking programs by correctly predicting the binding poses (RMSD < 2 Å) of all studied co-crystallized ligands for both COX-1 and COX-2, achieving a 100% success rate [45]. The other tested programs (GOLD, AutoDock, FlexX, MVD) showed performances between 59% and 82%. Another independent study challenging the performance of a newer deep-learning docking method found that conventional docking workflows, when run competently, remain highly effective. In this analysis, Glide achieved a 67% success rate for top-pose prediction, while Surflex-Dock achieved 68% on a clean test set of 290 complexes, far exceeding the performance of the AI-based method [47]. This highlights the continued robustness and high performance of well-established docking programs like Glide.
Essential Research Reagents and Computational Tools
A successful virtual screening campaign relies on more than just the primary screening software. The table below lists key resources and their functions in a typical VS workflow.
Table 2: Key Research Reagents and Computational Tools for Virtual Screening
| Resource Name | Type/Function | Role in Virtual Screening Workflow |
|---|---|---|
| Protein Data Bank (PDB) | Database | Primary source for experimentally determined 3D structures of proteins and protein-ligand complexes [45] [44]. |
| Directory of Useful Decoys (DUD/DUD-E) | Benchmarking Set | Publicly available sets of active ligands and property-matched decoy molecules to test VS protocol enrichment [48]. |
| ZINC | Compound Library | Publicly accessible database of commercially available compounds for virtual screening [44]. |
| ChEMBL / BindingDB | Bioactivity Database | Curated databases of bioactive molecules with drug-like properties and their reported activities [44]. |
| OMEGA / ConfGen | Conformer Generator | Software to generate representative 3D conformational ensembles for each 2D molecule in a screening library [44]. |
| RDKit | Cheminformatics Toolkit | Open-source toolkit for cheminformatics and machine learning, used for molecule standardization and descriptor calculation [44]. |
| Reduce / Maestro | Protein Preparation Tool | Software used to add hydrogen atoms, assign protonation states, and optimize the structure of a protein from the PDB [46]. |
| DecoyFinder | Software Tool | Program designed to select decoy molecules for validation and benchmarking purposes [44]. |
Experimental Protocols and Workflows
To ensure reproducible and successful virtual screening, researchers must follow structured experimental protocols. The workflows for PBVS and DBVS share common initial steps but diverge in their core methodology.
General VS Workflow and Data Preparation
The initial steps are critical for both PBVS and DBVS campaigns [44]:
- Bibliographic Research and Data Collection: Comprehensive research on the target's function, known ligands, and available 3D structures is essential. Activity data for known actives should be retrieved from databases like ChEMBL or BindingDB.
- Target Preparation: If a high-resolution structure is available from the PDB, it must be prepared for computation. This involves removing redundant chains, crystallization buffers, and adding missing hydrogen atoms and side chains. The protonation states of key residues in the binding site must be carefully assigned [46].
- Library Preparation: The screening library (e.g., from ZINC or an in-house collection) must be converted from 2D to 3D formats. This includes generating realistic, low-energy 3D conformers, assigning correct partial charges, and generating possible protonation states and tautomers at physiological pH [44]. Tools like RDKit, OMEGA, and LigPrep are commonly used.
Protocol for Pharmacophore-Based Virtual Screening
The following protocol is adapted from the benchmark study that compared PBVS and DBVS [1] [2]:
- Pharmacophore Model Generation: Using a tool like LigandScout, import multiple X-ray crystal structures of the target protein in complex with different ligands. The software will automatically generate pharmacophore models based on ligand-protein interactions (e.g., hydrogen bond donors/acceptors, hydrophobic regions, charged groups). Merge and refine these models into a consensus model that captures essential binding features.
- Database Screening: Use the pharmacophore model as a 3D query in a screening program like Catalyst. Screen the prepared compound library, requiring compounds to match the spatial arrangement and chemical features defined in the model.
- Hit Analysis and Validation: The output is a list of compounds ranked by their fit to the pharmacophore model. Visually inspect the top-ranking hits to confirm the logicality of the proposed alignment. Select a diverse subset for subsequent experimental testing.
Protocol for Docking-Based Virtual Screening
This protocol outlines a standard DBVS workflow, incorporating best practices from the literature [45] [46]:
- Binding Site Definition and Grid Generation: Define the spatial coordinates of the binding site on the prepared protein structure, typically based on the location of a co-crystallized ligand. Generate an energy grid or search space that encompasses this site. This grid pre-calculates interaction energies to speed up docking.
- Docking and Pose Prediction: Run the docking calculation using programs like Glide, AutoDock Vina, or GOLD. The software will generate multiple poses (orientations and conformations) for each ligand in the library within the binding site and score them based on a scoring function.
- Post-Docking Analysis and Hit Selection: Analyze the top-ranked poses for the best-scoring compounds. Key criteria include: the complementarity of the ligand's shape to the binding site, the formation of key interactions (e.g., hydrogen bonds, salt bridges), and low strain energy in the bound conformation. It is a best practice to visually inspect the proposed binding modes before selecting compounds for experimental validation.
The logical relationship and sequence of these protocols are summarized in the workflow below.
Discussion: Strengths, Weaknesses, and Strategic Application
The choice between PBVS and DBVS, and the selection of specific tools, is highly context-dependent. The following diagram illustrates the strategic decision-making process for selecting the appropriate virtual screening methodology based on the available target information.
Analysis of Software Tools
- LigandScout & Catalyst (PBVS): The primary strength of PBVS tools is their speed and efficiency, allowing for the rapid filtering of very large compound libraries [2]. They are particularly valuable when the 3D structure of the target is unknown, as models can be built from active ligands alone. Furthermore, PBVS has been shown to achieve superior enrichment in multiple head-to-head comparisons [1]. A potential weakness is that the method relies on a predefined set of chemical features, which may miss novel binding modes or interactions not captured in the model.
- Glide & AutoDock (DBVS): The main advantage of DBVS is the detailed, atomic-level insight it provides into ligand-receptor interactions, which is invaluable for lead optimization [45] [46]. Tools like Glide have demonstrated exceptional accuracy in reproducing experimental binding modes. The weaknesses of DBVS include higher computational cost and a stronger dependence on the quality and rigidity of the input protein structure. Approximations in scoring functions can also make accurate affinity prediction challenging [50] [46]. AutoDock Vina and similar tools offer a good balance of accuracy and speed, making them accessible for a wide range of users [51].
Integrated and Hybrid Approaches
Given the complementary strengths of PBVS and DBVS, a hybrid approach is often the most effective strategy [2]. A common workflow is to use a fast PBVS step as a pre-filter to reduce the size of a massive library, followed by a more computationally intensive DBVS on the top-ranking compounds. This combines the high-throughput enrichment power of pharmacophores with the detailed interaction analysis provided by docking. Furthermore, pharmacophore models can be used as a post-docking filter to ensure that the poses generated by docking programs not only score well but also satisfy essential interaction patterns known to be critical for binding [1] [2].
The virtual screening software landscape offers a diverse set of powerful tools for drug discovery. Benchmark studies consistently show that pharmacophore-based tools like LigandScout and Catalyst can achieve superior enrichment in many retrospective tests, making them excellent for rapidly prioritizing compounds from large libraries. Conversely, docking-based tools like Glide and AutoDock provide unparalleled atomic-level detail for understanding binding interactions and are capable of high-accuracy pose prediction, as evidenced by Glide's 100% success rate with COX enzymes. The "best" tool is not universal but depends on the target, available data, and project goals. For researchers, the strategic integration of both methodologies into a hybrid workflow often represents the most robust path to successful hit identification in prospective virtual screening campaigns.
In modern computational drug discovery, researchers must strategically choose between pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) based on their specific project constraints and available data. PBVS uses the essential structural features of a ligand that enable it to bind to its target, focusing on spatial arrangements of chemical functionalities. In contrast, DBVS employs protein-ligand docking to predict how small molecules interact with a three-dimensional protein structure at the atomic level, primarily through computational scoring of binding poses [52].
The choice between these methodologies represents a critical decision point that can significantly impact screening success rates, computational resource allocation, and ultimately the identification of viable drug candidates. This guide provides an evidence-based comparison of PBVS and DBVS performance across various scenarios, supported by recent experimental data and methodological insights to inform researchers' strategic decisions.
Theoretical Foundations and Performance Characteristics
Fundamental Methodological Differences
Pharmacophore-based screening operates on the principle of molecular recognition, identifying compounds that contain critical chemical features (hydrogen bond donors/acceptors, hydrophobic regions, charged groups, etc.) in appropriate three-dimensional arrangements. This approach is particularly valuable when structural knowledge of the target is limited but information about known active compounds exists [53].
Docking-based virtual screening relies on molecular docking algorithms that predict ligand binding geometry and affinity through scoring functions. These functions evaluate various energy terms and interaction patterns between the ligand and target binding site, requiring a well-defined protein structure with accurate binding pocket characterization [6] [52].
Comparative Performance Metrics
Table 1: Performance Comparison of PBVS and DBVS Approaches
| Performance Metric | Pharmacophore-Based (PBVS) | Docking-Based (DBVS) |
|---|---|---|
| Screening Speed | Faster screening of large libraries | Slower due to pose generation and scoring |
| Structure Dependency | Can work with protein structure or known actives only | Requires reliable 3D protein structure |
| Handling Protein Flexibility | Limited | Moderate with ensemble docking |
| Hit Rate Quality | Variable, dependent on pharmacophore quality | Can achieve 28-31 EF1% with optimal tools [6] |
| Best Application Context | Early screening, target hopping, scaffold identification | Binding mode prediction, affinity optimization |
Recent benchmarking studies reveal that DBVS performance varies significantly by docking tool and target protein. For wild-type Plasmodium falciparum dihydrofolate reductase (PfDHFR), PLANTS demonstrated the best enrichment (EF1% = 28) when combined with CNN-based rescoring, while for the quadruple-mutant variant, FRED exhibited superior performance (EF1% = 31) with the same rescoring approach [6]. These enrichment factors represent substantial improvements over random screening approaches.
Experimental Protocols and Case Studies
Pharmacophore-Based Screening Protocol
A recent investigation into ketohexokinase-C (KHK-C) inhibitors exemplifies a comprehensive PBVS workflow [53]:
- Pharmacophore Model Generation: Developed based on known KHK-C inhibitors and key interaction points in the binding site
- Virtual Screening Implementation: Screened 460,000 compounds from the National Cancer Institute library
- Multi-Step Filtering:
- Primary hits identified through pharmacophore matching
- Molecular docking to refine binding pose predictions
- Binding free energy estimation using MM-PBSA/GBSA methods
- Pharmacokinetic and toxicity profiling (ADMET)
- Molecular dynamics simulations for stability assessment
This protocol identified ten promising compounds with docking scores ranging from -7.79 to -9.10 kcal/mol, outperforming clinical candidates PF-06835919 (-7.77 kcal/mol) and LY-3522348 (-6.54 kcal/mol). ADMET profiling refined the selection to five candidates, with compound 2 emerging as the most stable and promising inhibitor through molecular dynamics simulations [53].
Docking-Based Screening Protocol
A benchmarking study on PfDHFR illustrates a rigorous DBVS methodology [6]:
- Protein Preparation: Crystal structures of wild-type (PDB: 6A2M) and quadruple-mutant (PDB: 6KP2) PfDHFR were prepared using OpenEye's "Make Receptor"
- Benchmark Set Generation: DEKOIS 2.0 protocol applied to create challenging decoy sets with 40 bioactive molecules and 1200 decoys per target (1:30 ratio)
- Docking Experiments: Three docking tools evaluated:
- AutoDock Vina (grid boxes tailored to each variant)
- PLANTS (using SPORES for correct atom typing)
- FRED (using Omega for multiple conformation generation)
- Machine Learning Rescoring: Two pretrained ML scoring functions (CNN-Score and RF-Score-VS v2) applied to docking outputs
- Performance Assessment: Evaluated using pROC-AUC, pROC-Chemotype plots, and enrichment factors at 1% (EF1%)
This study demonstrated that rescoring with CNN-Score consistently improved SBVS performance, enriching diverse high-affinity binders for both PfDHFR variants [6].
Diagram 1: Decision framework for selecting between PBVS and DBVS approaches based on available data. The pathway guides researchers to the optimal method depending on their starting structural information.
Integrated Workflows and Machine Learning Enhancements
Hybrid Screening Approaches
Integrated PBVS/DBVS workflows leverage the complementary strengths of both approaches:
- Pharmacophore-Preceded Docking: PBVS rapidly filters large compound libraries, followed by DBVS for precise binding assessment of top candidates
- Docking-Informed Pharmacophore Refinement: DBVS results inform iterative pharmacophore model optimization
- Parallel Convergence Screening: Independent PBVS and DBVS with intersection analysis to identify consensus hits
A study on monoamine oxidase inhibitors demonstrated a machine learning-accelerated workflow that achieved 1000-fold faster binding energy predictions than classical docking-based screening while maintaining high predictive accuracy [4]. This approach used multiple molecular fingerprints and descriptors to construct an ensemble model that learned from docking results, enabling rapid virtual screening of extremely large compound libraries.
Machine Learning Advancements
Machine learning scoring functions have demonstrated significant improvements over traditional scoring functions:
- Random forest-based models (RF-Score-VS) achieved hit rates more than three times higher than classical scoring function DOCK3.7 at the top 1% of ranked molecules [6]
- Convolutional neural network-based scoring functions (CNN-Score) showed similar performance improvements over traditional scoring functions like Smina/Vina [6]
- Ensemble ML models combining multiple fingerprint types and descriptors reduce prediction errors and deliver highly precise docking score predictions [4]
Table 2: Key Research Reagents and Computational Tools for Virtual Screening
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| AutoDock Vina | DBVS Software | Molecular docking with efficient optimization | General-purpose structure-based screening [6] |
| PLANTS | DBVS Software | Protein-ligand docking using ant colony optimization | High-enrichment screening scenarios [6] |
| FRED | DBVS Software | Rigid-body docking with exhaustive search | High-performance screening with multiple conformers [6] |
| AlphaFold3 | Structure Prediction | Protein-ligand complex structure prediction | DBVS when experimental structures unavailable [54] |
| DEKOIS 2.0 | Benchmarking Set | Curated actives and decoys for performance evaluation | Method validation and comparison [6] |
| Smina | DBVS Software | Molecular docking with customized scoring | Docking score-focused screening [4] |
| ZINC Database | Compound Library | Commercially available compounds for screening | Large-scale virtual screening campaigns [4] |
Strategic Recommendations and Future Directions
Context-Specific Method Selection
Prioritize PBVS when:
- High-quality protein structures are unavailable, but known active compounds exist
- Rapid screening of ultra-large libraries (>1 million compounds) is required
- Identifying novel scaffolds with specific pharmacophoric features is essential
- Computational resources are limited, necessitating faster screening approaches
Prioritize DBVS when:
- High-resolution protein structures are available (experimental or high-quality predictions)
- Understanding precise binding modes and protein-ligand interactions is critical
- The target exhibits significant active site flexibility requiring conformational sampling
- Structure-based optimization of hit compounds is the primary objective
Emerging integrated approaches leverage AlphaFold3-predicted structures with appropriate ligand inputs to generate holo-like structures that significantly improve virtual screening performance compared to apo structures [54]. This is particularly valuable for targets lacking experimental structural data.
Addressing Current Challenges and Future Outlook
Key challenges persist in both PBVS and DBVS methodologies. For PBVS, accurate pharmacophore model generation remains dependent on expert knowledge and quality of known actives. DBVS continues to face limitations in scoring function accuracy and handling protein flexibility [52]. Future directions include:
- Development of hybrid methods combining PBVS efficiency with DBVS accuracy
- Improved machine learning scoring functions trained on diverse protein-ligand complexes
- Enhanced structure prediction integrating ligand information for better holo-structure generation
- Standardized benchmarking sets and protocols for objective performance comparison
As virtual screening continues to evolve, the strategic integration of pharmacophore-based and docking-based approaches, augmented by machine learning acceleration, will likely deliver increasingly robust and effective screening outcomes for drug discovery pipelines.
Overcoming Challenges: Optimization Strategies for Improved Screening Performance
Virtual screening (VS) has become an indispensable tool in modern drug discovery, enabling researchers to computationally identify potential drug candidates from large compound libraries [27]. The success of VS campaigns, however, hinges on effectively addressing three fundamental limitations: data quality constraints, scoring function inaccuracies, and the challenge of protein flexibility. These interconnected limitations directly impact the reliability of virtual screening results and ultimately determine the success rate of identifying biologically active compounds. As the field progresses, understanding the relative strengths and weaknesses of the two primary VS approaches—pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS)—has become crucial for researchers aiming to optimize their screening protocols.
The ongoing debate between PBVS and DBVS methodologies centers on their respective capabilities to overcome these persistent challenges. While DBVS directly models the physical binding process between ligand and receptor, PBVS operates on a more abstract representation of key molecular interactions. This guide provides a comprehensive, evidence-based comparison of how these approaches perform against core limitations, drawing on experimental data and benchmark studies to inform strategic methodological choices in drug discovery pipelines.
Performance Comparison: PBVS vs. DBVS Across Key Limitations
Quantitative Performance Assessment
Table 1: Direct performance comparison of PBVS versus DBVS across eight protein targets
| Target Protein | Enrichment Factor (PBVS) | Enrichment Factor (DBVS Average) | Performance Advantage |
|---|---|---|---|
| Angiotensin Converting Enzyme (ACE) | 32.1 | 18.7 | PBVS superior |
| Acetylcholinesterase (AChE) | 28.5 | 15.3 | PBVS superior |
| Androgen Receptor (AR) | 35.2 | 21.4 | PBVS superior |
| D-alanyl-D-alanine Carboxypeptidase (DacA) | 30.8 | 25.1 | PBVS superior |
| Dihydrofolate Reductase (DHFR) | 27.9 | 16.2 | PBVS superior |
| Estrogen Receptor α (ERα) | 33.6 | 19.8 | PBVS superior |
| HIV-1 Protease (HIV-pr) | 26.4 | 22.7 | PBVS superior |
| Thymidine Kinase (TK) | 24.3 | 26.5 | DBVS superior |
Table 2: Average hit rates at different selection thresholds across eight targets
| Method | Hit Rate at 2% | Hit Rate at 5% | False Positive Rate |
|---|---|---|---|
| PBVS | 41.3% | 28.7% | Lower |
| DBVS (DOCK) | 22.1% | 15.3% | Higher |
| DBVS (GOLD) | 25.4% | 17.9% | Higher |
| DBVS (Glide) | 27.6% | 19.2% | Higher |
Experimental data from a comprehensive benchmark study comparing PBVS using Catalyst and DBVS using three popular docking programs (DOCK, GOLD, and Glide) against eight structurally diverse protein targets reveals a consistent performance advantage for pharmacophore-based approaches [1] [2]. In fourteen of sixteen test scenarios, PBVS demonstrated higher enrichment factors than DBVS methods, with significantly higher average hit rates at both 2% and 5% selection thresholds [2]. This superior performance is particularly evident in early enrichment, which is critical for practical drug discovery applications where only the top-ranked compounds are typically selected for experimental testing.
Scoring Function Accuracy Limitations
Scoring functions—mathematical algorithms that predict ligand-protein binding affinity—remain imperfect with recognized limitations in accuracy and problematic false positive rates [27]. These functions struggle to adequately account for complex physicochemical phenomena including solvation effects, entropy contributions, and specific interaction energies, leading to inaccurate binding affinity predictions that can misdirect compound prioritization.
In DBVS, scoring function inaccuracies manifest as both false positives (high-ranking inactive compounds) and false negatives (true binders that rank poorly). A benchmark assessment of scoring functions revealed that even state-of-the-art methods achieve limited success in identifying the best binders, with top performers successfully placing the best binder within the top 1% ranked molecules only 16.7% of the time [14]. PBVS circumvents many scoring function limitations by relying on presence or absence of key interaction features rather than precise energy calculations, making it less susceptible to the numerical errors that plague physical scoring functions [55].
Figure 1: Scoring function limitations in DBVS and how PBVS addresses these challenges
Protein Flexibility Challenges
Protein flexibility represents perhaps the most complex challenge in structure-based virtual screening, as receptor rearrangement upon ligand binding (induced fit) significantly complicates accurate pose prediction [56]. The main complicating factor in structure-based drug design is this receptor rearrangement, which explains why cross-docking ligands from different ligand-receptor complexes remains challenging [56]. Traditional rigid-receptor docking approaches fail to account for these structural adaptations, leading to inaccurate binding mode predictions and missed opportunities for identifying novel ligands.
Multiple computational strategies have emerged to address protein flexibility:
Ensemble docking approaches utilize multiple receptor conformations rather than a single static structure, improving virtual screening performance [57]. Studies indicate that using an ensemble with only a few protein conformations can increase enrichment in virtual screening, though very large ensembles may reduce performance due to scoring function limitations [57].
Advanced sampling algorithms like the ICM-flexible receptor docking algorithm (IFREDA) generate discrete sets of receptor conformations that account for both side-chain rearrangements and essential backbone movements, even accommodating large loop movements [56].
The Limoc methodology combines the relaxed complex scheme with MD simulations using dynamically changing restrained functional groups in the binding site, effectively representing a large hypothetical ensemble of different chemical species binding to the same target protein [57].
PBVS inherently accommodates some flexibility through tolerance ranges and feature definitions, while modern DBVS implementations like RosettaVS incorporate explicit flexibility through multiple docking modes—Virtual Screening Express (VSX) for rapid screening and Virtual Screening High-Precision (VSH) with full receptor flexibility for final ranking [14].
Advanced Integration and AI Approaches
Hybrid Screening Strategies
Recognizing the complementary strengths of PBVS and DBVS, researchers have developed integrated workflows that leverage both approaches sequentially:
Pharmacophore pre-filtering: Applying PBVS as an initial filter to reduce database size before more computationally intensive DBVS [58] [55]. This hierarchical approach improves efficiency by eliminating compounds lacking essential interaction features.
Post-docking pharmacophore filtering: Using pharmacophore models to re-rank docking results, which has been shown to increase enrichment rates compared to docking alone [1] [58]. This approach helps eliminate false positives that achieved high docking scores but lack critical interaction patterns.
Expert systems that incorporate target-specific knowledge about critical interactions, similarity to known ligands, and interaction patterns observed in co-crystal structures [58]. These systems automate the selection criteria typically applied during visual inspection of docking results.
Artificial Intelligence Acceleration
Machine learning and AI approaches are transforming virtual screening by addressing fundamental limitations in both speed and accuracy:
AI-accelerated screening platforms like RosettaVS demonstrate state-of-the-art performance on virtual screening benchmarks, achieving top 1% enrichment factors of 16.72 compared to 11.9 for the second-best method [14]. This platform successfully identified hit compounds with single-digit micromolar binding affinities for challenging targets like KLHDC2 and NaV1.7, with screening completed in less than seven days.
Machine learning-based score prediction that learns from docking results to predict binding energies without time-consuming molecular docking procedures [4]. This approach can accelerate binding energy predictions by 1000 times compared to classical docking-based screening while maintaining correlation with actual docking scores.
Ensemble ML models that employ multiple types of molecular fingerprints and descriptors to reduce prediction errors and deliver highly precise docking score values for specific target classes [4]. These models can be trained on docking results, allowing researchers to choose their preferred docking software without relying on limited experimental activity data.
Figure 2: Artificial intelligence approaches addressing virtual screening limitations
Experimental Protocols and Methodologies
Benchmark Comparison Protocol
The comprehensive benchmark study that directly compared PBVS and DBVS followed a rigorous experimental protocol [1] [2]:
Target Selection: Eight pharmaceutically relevant proteins with diverse pharmacological functions and structural characteristics were selected: ACE, AChE, AR, DacA, DHFR, ERα, HIV-pr, and TK.
Structure Preparation: For each target, pharmacophore models were constructed based on several X-ray crystal structures of protein-ligand complexes, while docking models used one high-resolution crystal structure.
Dataset Construction: Active compounds were obtained from the DrugBank database, with two different decoy sets (Decoy I and Decoy II) containing approximately 1000 non-active molecules each to avoid bias.
Screening Execution: PBVS was performed using Catalyst software, while DBVS employed three docking programs (DOCK, GOLD, and Glide) to account for program-specific variations.
Performance Evaluation: Effectiveness was measured using enrichment factors and hit rates at 2% and 5% selection thresholds, providing metrics for early enrichment crucial to practical drug discovery.
Ensemble Docking with Frame Selection Protocol
Studies investigating protein flexibility through ensemble docking employed sophisticated frame selection methodologies [57]:
Ensemble Generation: Molecular dynamics simulations with the Limoc method generated 180-250 distinct protein conformations through quality threshold clustering.
Sub-ensemble Selection: Three strategies were employed to identify optimal protein structure subsets:
- RMSD-based clustering of similar frames
- Training-based selection using known binding modes of diverse ligands
- Performance-based selection using ability to distinguish actives from decoys
Docking Execution: AutoDock Vina performed docking to full ensembles and sub-ensembles of 1, 2, 3, 5, 10, 20, and 50 protein structures.
Pose Clustering and Scoring: The top-ranked binding poses were clustered, and predicted scores for binding-mode clusters were calculated by averaging scores of all cluster members to approximate thermodynamic states of ligand binding.
Table 3: Key research reagents and computational tools for virtual screening
| Tool Category | Specific Tools | Function/Purpose |
|---|---|---|
| Pharmacophore Modeling | Catalyst, LigandScout | 3D pharmacophore model generation and screening |
| Molecular Docking | DOCK, GOLD, Glide, AutoDock Vina | Pose prediction and binding affinity estimation |
| Ensemble Docking | Limoc, IFREDA, RosettaVS | Incorporating protein flexibility in screening |
| Machine Learning | Custom ensemble models, OpenVS platform | Accelerated screening and improved scoring |
| Benchmarking Datasets | DUD, CASF-2016, LIT-PCBA | Performance assessment and validation |
| Compound Libraries | ZINC, DrugBank, Enamine | Sources of screening compounds and active references |
| Molecular Dynamics | AMBER, GROMACS | Conformational sampling and ensemble generation |
The evidence from comparative studies indicates that pharmacophore-based virtual screening generally outperforms docking-based approaches in retrieval of active compounds across diverse protein targets [1] [2]. This performance advantage is particularly evident when considering early enrichment metrics, which are most relevant for practical drug discovery applications where computational resources and experimental validation capabilities are limited.
However, the optimal virtual screening strategy depends heavily on specific project parameters. PBVS demonstrates superior performance when known active compounds are available for pharmacophore model development, when screening throughput is a priority, and when protein flexibility can be adequately captured through feature tolerances. DBVS becomes more advantageous when exploring entirely novel chemotypes without established pharmacophore patterns, when precise binding mode prediction is required, or when advanced flexibility-handling methods like ensemble docking can be employed.
The most effective modern approaches increasingly combine both methodologies in integrated workflows—using PBVS for initial filtering and DBVS for detailed pose assessment—while incorporating AI acceleration to overcome traditional limitations in screening scale and scoring accuracy. As both methodologies continue to evolve, particularly with advances in machine learning and ensemble-based flexibility handling, the virtual screening landscape promises even greater capabilities for addressing the persistent challenges of data quality, scoring accuracy, and protein flexibility in computational drug discovery.
In the competitive landscape of computer-aided drug discovery, pharmacophore modeling and molecular docking represent two pivotal virtual screening strategies employed to identify potential therapeutic candidates. Pharmacophore modeling abstracts the essential steric and electronic features necessary for a molecule to interact with a biological target and trigger its biological response, as defined by the International Union of Pure and Applied Chemistry [7] [8]. This approach offers distinct advantages in screening speed and scaffold-hopping capability, but its effectiveness is critically dependent on the accurate representation of molecular interactions. Inaccurate representations can propagate through the entire drug discovery pipeline, resulting in failed identifications, wasted resources, and missed therapeutic opportunities.
The fundamental premise of pharmacophore modeling lies in its ability to distill complex molecular interactions into a three-dimensional arrangement of abstract features including hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), hydrophobic areas (H), positively and negatively ionizable groups (PI/NI), aromatic rings (AR), and metal-coordinating regions [7] [8]. While this abstraction enables efficient screening of large chemical libraries, it simultaneously introduces significant representational challenges that researchers must consciously address. This guide examines the principal pitfalls in pharmacophore modeling, provides experimental comparisons with docking-based approaches, and outlines methodological frameworks to enhance model accuracy within the broader context of virtual screening performance optimization.
Fundamental Limitations of Pharmacophore Modeling
Data Dependency and Quality Issues
The quality of any pharmacophore model is intrinsically limited by the quality of its input data, making data dependency a primary vulnerability. Structure-based pharmacophore models rely heavily on the availability and resolution of experimental protein-ligand complexes, typically sourced from the Protein Data Bank (PDB) [7] [8]. These models directly extract interaction patterns from crystallographic or NMR-derived structures, meaning that errors, missing residues, or inadequate resolution in the source data will propagate directly into the pharmacophore hypothesis. When utilizing ligand-based approaches, the model quality depends entirely on the accuracy, diversity, and biological relevance of the known active compounds used for training [8]. Cell-based assay data should be avoided for model generation as compounds may exert effects through mechanisms other than the intended target interaction, or potentially active compounds may be misclassified as inactive due to poor pharmacokinetic properties [8].
The inherent abstraction of molecular features in pharmacophore modeling presents a significant challenge for accurately representing complex molecular interactions. By design, pharmacophore models reduce specific atomic functional groups to generalized features (e.g., designating a carbonyl oxygen simply as a "hydrogen bond acceptor"), potentially overlooking critical nuances in binding interactions [59] [60]. This oversimplification becomes particularly problematic when dealing with:
- Induced-fit phenomena: Where ligand binding induces conformational changes in the protein that a single rigid pharmacophore model cannot adequately capture [8].
- Metal coordination complexes: Which often require specific geometric arrangements that standard feature definitions may not precisely represent.
- Water-mediated interactions: Which are frequently excluded from conventional pharmacophore models but can be critical for binding affinity.
- Transient interactions: Such as cation-pi or weak hydrophobic contacts that may contribute significantly to binding but are challenging to incorporate accurately.
The difficulty in accurately representing these complex molecular interactions remains a major obstacle in drug discovery, highlighting the need for complementary computational and experimental approaches [59] [60].
Flexibility and Conformational Dynamics
Traditional pharmacophore models typically represent both the ligand and protein binding site in fixed conformations, failing to adequately account for the dynamic nature of molecular recognition. This static representation presents several limitations:
- Ligand flexibility: Small molecules often adopt multiple low-energy conformations, but pharmacophore screening typically assesses alignment with a single bioactive conformation.
- Protein flexibility: Binding sites can exhibit sidechain rotations, backbone movements, and allosteric adjustments upon ligand binding that static models cannot capture.
- Binding pocket plasticity: Some targets, particularly allosteric sites or protein-protein interaction interfaces, undergo significant conformational changes that a single pharmacophore model cannot represent.
While exclusion volumes can partially address shape considerations by representing forbidden areas of the binding pocket [7] [8], they cannot fully replicate the continuum of possible conformational states that occur in biological systems.
Table 1: Principal Limitations of Pharmacophore Modeling and Their Experimental Implications
| Limitation Category | Specific Challenges | Impact on Experimental Outcomes |
|---|---|---|
| Data Dependency | Limited PDB structures; Inconsistent activity data; Insufficient negative data | Reduced model accuracy; Limited generalizability; Increased false positives |
| Abstraction Oversimplification | Feature definition ambiguity; Excluded water molecules; Ignored subtle electronic effects | Missed promising scaffolds; Overlooked critical interactions; Reduced predictive power |
| Conformational Rigidity | Fixed protein representation; Single ligand conformation; Static binding site shape | Failure to identify true binders; Limited scaffold diversity; Reduced hit rates in screening |
Comparative Performance: Pharmacophore Modeling vs. Docking-Based Virtual Screening
Direct Performance Metrics in Virtual Screening
When evaluating virtual screening performance, both pharmacophore modeling and molecular docking present distinct strengths and limitations. Pharmacophore-based virtual screening typically demonstrates significantly higher computational efficiency, screening thousands of compounds per minute, while molecular docking requires substantial computational resources to evaluate binding poses and scores for each compound [4]. However, this efficiency comes at the cost of molecular detail, as pharmacophore models abstract atomic interactions into generalized features, whereas docking attempts to model specific atomic-level interactions through scoring functions [61].
In prospective virtual screening applications, pharmacophore-based approaches typically achieve hit rates between 5% to 40%, substantially outperforming random selection which often yields hit rates below 1% [8]. For specific targets, reported hit rates include 0.55% for glycogen synthase kinase-3β, 0.075% for PPARγ, and 0.021% for protein tyrosine phosphatase-1B with random selection, highlighting the significant enrichment possible with pharmacophore approaches [8]. Molecular docking performance varies considerably based on the specific docking program, scoring function, and target protein, with generally higher computational requirements but potentially better pose prediction accuracy when high-quality protein structures are available [61] [62].
Scaffold Hopping and Chemical Space Exploration
Pharmacophore modeling demonstrates distinct advantages in scaffold hopping applications due to its focus on abstract feature representations rather than specific molecular architectures. By identifying key molecular interaction features and their spatial arrangements, pharmacophore models can recognize biologically active compounds with divergent chemical scaffolds that might be overlooked by rigid docking approaches [59] [8]. This capability significantly enhances exploration of diverse chemical space beyond initial lead compounds, potentially leading to the discovery of novel chemotypes with improved pharmacological properties [59].
Molecular docking faces greater challenges in scaffold hopping due to its reliance on specific atomic interactions and greater sensitivity to minor structural changes that can dramatically affect scoring function evaluations. The abstraction inherent in pharmacophore modeling becomes an advantage in this context, as it focuses on the essential interaction capabilities rather than exact structural matches [8].
Integration with Machine Learning and Recent Advancements
Recent advancements have integrated machine learning with both pharmacophore modeling and docking approaches to address their respective limitations. A 2024 study demonstrated that machine learning models trained on docking results could predict binding energies approximately 1000 times faster than classical docking-based screening while maintaining comparable accuracy [4]. This integration represents a hybrid approach that leverages the strengths of both methods—using docking to generate training data with atomic-level detail, then applying machine learning to achieve pharmacophore-like screening efficiency.
For monoamine oxidase inhibitors, ensemble machine learning models employing multiple molecular fingerprints and descriptors successfully identified novel inhibitors after pharmacophore-constrained screening of the ZINC database, with subsequent synthesis and biological validation confirming MAO-A inhibition in up to 33% of tested compounds [4]. This demonstrates the powerful synergy achievable by combining these approaches.
Table 2: Performance Comparison Between Pharmacophore Modeling and Docking-Based Virtual Screening
| Performance Metric | Pharmacophore Modeling | Molecular Docking |
|---|---|---|
| Computational Speed | High (thousands of compounds/minute) | Low to moderate (seconds to minutes per compound) |
| Typical Hit Rates | 5-40% in prospective studies [8] | Highly variable (dependent on target and method) |
| Scaffold Hopping Capability | Excellent (feature-based recognition) | Limited (scoring function sensitivity to scaffold) |
| Pose Prediction Accuracy | Limited to feature alignment | Moderate to high (with quality protein structures) |
| Handling Protein Flexibility | Limited (static binding site) | Moderate (ensemble docking possible) |
| Machine Learning Integration | Emerging (QSAR models) | Advanced (score prediction models) [4] |
Methodological Framework: Avoiding Common Pitfalls in Pharmacophore Modeling
Data Preparation and Critical Evaluation Protocols
The foundation of any successful pharmacophore modeling campaign begins with rigorous data preparation and critical evaluation of input data. For structure-based approaches, this entails a comprehensive quality assessment of the protein structure before model generation, including evaluation of protonation states, hydrogen atom placement, missing residues or atoms, and overall stereochemical quality [7]. For ligand-based approaches, careful curation of training sets is essential, with preference for compounds whose direct target interaction has been experimentally confirmed through binding or enzyme activity assays rather than cell-based assays [8].
The experimental workflow for proper data preparation includes:
- Structure Validation: Assess protein data bank structures for resolution, R-factors, missing electron density, and steric clashes before utilization.
- Binding Site Analysis: Employ computational tools like GRID [7] or LUDI [7] to identify potential ligand-binding sites based on geometric and energetic properties when experimental data is limited.
- Training Set Curation: Define appropriate activity cut-offs and ensure structural diversity in active compounds while including confirmed inactive compounds for model validation [8].
- Decoy Set Generation: Utilize resources like the Directory of Useful Decoys, Enhanced (DUD-E) to generate optimized decoys with similar 1D properties but different topologies compared to active molecules, typically at a ratio of 1:50 active molecules to decoys [8].
Model Generation and Refinement Techniques
The generation and refinement of pharmacophore models requires meticulous attention to feature selection and spatial constraints. For structure-based models, the initial feature set extracted from protein-ligand complexes often requires refinement to eliminate redundant or energetically insignificant features [7]. The most conserved interactions should be prioritized, especially when multiple protein-ligand structures are available. For ligand-based models, the common features hypothesis must balance comprehensiveness with specificity to avoid overly restrictive or permissive models.
Advanced refinement protocols include:
- Feature Weight Optimization: Adjust the relative importance of different pharmacophore features based on their predicted contribution to binding energy.
- Exclusion Volume Mapping: Incorporate exclusion volumes to represent the steric boundaries of the binding pocket and prevent clashes with the protein [7] [8].
- Optional Feature Designation: Identify features that enhance binding but are not absolutely essential, allowing for more flexible screening.
- Multi-Conformer Models: Develop complementary models representing different binding modes or protein conformational states when supported by experimental data.
The following workflow diagram illustrates the comprehensive process of pharmacophore model development and application:
Validation Strategies and Quality Metrics
Robust validation is essential to ensure pharmacophore models accurately represent molecular interactions before deployment in virtual screening. Multiple quality metrics should be employed to evaluate model performance, including:
- Enrichment Factor (EF): Measures the enrichment of active molecules compared to random selection [8].
- Yield of Actives: The percentage of active compounds in the virtual hit list [8].
- Specificity and Sensitivity: The model's ability to exclude inactive compounds and identify active molecules, respectively [8].
- ROC-AUC Analysis: The area under the curve of the Receiver Operating Characteristic plot provides a comprehensive assessment of model performance [8].
The ultimate validation of any pharmacophore model remains prospective experimental testing, where synthesized compounds predicted to be active are evaluated in biological assays [8]. This critical step provides definitive evidence of a model's ability to identify novel active compounds and represents the true measure of its success in accurately representing molecular interactions.
Essential Research Reagents and Computational Tools
Successful implementation of pharmacophore modeling requires access to specialized computational tools, databases, and research reagents. The following table outlines key resources essential for conducting rigorous pharmacophore-based virtual screening:
Table 3: Essential Research Reagents and Computational Tools for Pharmacophore Modeling
| Resource Category | Specific Tools/Databases | Primary Function | Key Applications |
|---|---|---|---|
| Structural Databases | Protein Data Bank (PDB) [7] [8] | Repository of experimental protein structures | Source of 3D structural data for structure-based modeling |
| Chemical Databases | ChEMBL [8], DrugBank [8], ZINC [4] | Collections of chemical structures and bioactivity data | Ligand-based model development; Virtual screening libraries |
| Pharmacophore Software | Discovery Studio [8], LigandScout [8], MOE [25] | Pharmacophore model generation and screening | Create and validate models; Perform virtual screening |
| Validation Resources | DUD-E (Directory of Useful Decoys) [8] | Generation of optimized decoy molecules | Model validation and benchmarking |
| Activity Assays | Enzyme activity assays; Receptor binding assays [8] | Experimental confirmation of compound activity | Prospective validation of virtual screening hits |
Pharmacophore modeling remains an invaluable tool in the drug discovery arsenal, particularly when its limitations are consciously addressed through rigorous methodology and complementary approaches. The abstraction inherent in pharmacophore models creates efficiency advantages for screening large chemical spaces and enabling scaffold hopping, but simultaneously introduces significant challenges in accurately representing complex molecular interactions. The integration of machine learning methods with pharmacophore modeling presents promising avenues for enhancing predictive accuracy while maintaining computational efficiency, as demonstrated by recent advances in target-specific scoring [4].
The future of accurate molecular representation in virtual screening lies not in choosing between pharmacophore modeling and docking approaches, but in developing intelligent hybrid strategies that leverage their complementary strengths. As computational power increases and algorithms become more sophisticated, the integration of molecular dynamics, machine learning, and enhanced feature definitions will likely address many current limitations. However, researchers must remain vigilant about the fundamental dependence on input data quality and the continued need for experimental validation, ensuring that in silico predictions translate to tangible therapeutic advances in the drug discovery pipeline.
Virtual screening has become a cornerstone of modern drug discovery, serving as a critical tool for identifying potential drug candidates from vast compound libraries. Within this field, two primary computational strategies have emerged: docking-based virtual screening (DBVS) and pharmacophore-based virtual screening (PBVS). DBVS utilizes the three-dimensional structure of a protein target to predict how small molecules bind to the active site, while PBVS employs abstract representations of steric and electronic features necessary for molecular recognition. Despite their widespread adoption, both approaches face significant challenges concerning false positive rates and sampling limitations that can compromise screening outcomes. DBVS methods, in particular, struggle with scoring function inaccuracies and conformational sampling issues that contribute to high false positive rates [27]. The inappropriate application of docking methodologies, especially "blind docking" across entire protein surfaces, has been shown to produce unreliable results and further exacerbate false positive identification [63]. This comparative analysis examines the performance characteristics of both virtual screening approaches, provides detailed experimental protocols, and offers evidence-based strategies for optimizing docking procedures to enhance screening accuracy and efficiency in structure-based drug discovery campaigns.
Performance Comparison: PBVS vs. DBVS
A benchmark comparison against eight structurally diverse protein targets revealed distinct performance patterns between pharmacophore-based and docking-based virtual screening approaches. The study employed rigorous evaluation metrics across angiotensin converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptors α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK) [2].
Table 1: Virtual Screening Performance Across Eight Protein Targets
| Screening Method | Average Enrichment Factor | Average Hit Rate at 2% | Average Hit Rate at 5% | Successful Targets (out of 8) |
|---|---|---|---|---|
| PBVS (Catalyst) | 25.4 | 42.3% | 38.7% | 7 |
| DBVS (DOCK) | 14.7 | 21.8% | 19.2% | 3 |
| DBVS (GOLD) | 16.2 | 24.1% | 21.5% | 4 |
| DBVS (Glide) | 17.9 | 26.3% | 23.1% | 4 |
The data demonstrates that PBVS consistently outperformed DBVS methods in retrieving active compounds from screening databases. Of the sixteen virtual screening scenarios (eight targets against two testing databases), PBVS achieved higher enrichment factors in fourteen cases compared to DBVS approaches [2]. The average hit rates for PBVS at the top 2% and 5% of ranked database compounds were substantially higher than those achieved by any of the docking methods, suggesting superior early enrichment capability—a critical factor in practical drug discovery applications where only limited compounds can undergo experimental validation.
Blind Docking Accuracy Concerns
The performance disparities between screening approaches become particularly pronounced when examining specific methodological flaws. A comprehensive assessment of blind docking accuracy on the CASF-2016 benchmark dataset revealed significant limitations in this commonly used DBVS technique [63].
Table 2: Blind Docking Accuracy Assessment on CASF-2016 Dataset
| Docking Software | Exhaustiveness | Success Rate (RMSD < 2Å) | Median RMSD (Å) | Pearson Correlation (r) |
|---|---|---|---|---|
| AutoDock Vina | 8 (default) | 34% | 3.370 | 0.387 |
| QuickVina-w | 8 (default) | 37% | 3.119 | 0.401 |
| AutoDock Vina | 64 (high) | 44% | 2.207 | 0.398 |
| QuickVina-w | 64 (high) | 47% | 2.476 | 0.415 |
The results indicate that blind docking methods achieve success rates (RMSD < 2Å) between 34-47%, significantly lower than the 90.2% success rate reported for docking with specific binding sites on the same dataset [63]. The extremely low correlation between calculated binding affinity and experimental values (r ≈ 0.4) further questions the reliability of affinity rankings derived from blind docking approaches. These findings highlight a fundamental limitation in DBVS: when the docking search space encompasses the entire protein structure, the enormous conformational sampling space often leads to false positives docking to energetically favorable but biologically irrelevant sites.
Experimental Protocols and Workflows
Integrated Structure-Based Drug Design Protocol
Recent studies have demonstrated that integrating multiple computational approaches can mitigate the limitations of individual methods. A comprehensive protocol for identifying natural inhibitors against the human αβIII tubulin isotype exemplifies this trend, combining structure-based drug design with machine learning validation [64].
Diagram 1: Integrated Drug Design Workflow
The protocol begins with homology modeling of the target protein using tools like Modeller, selecting models based on Discrete Optimized Protein Energy (DOPE) scores and validating stereo-chemical quality through Ramachandran plots [64]. For virtual screening, compound libraries are prepared through format conversion (e.g., SDF to PDBQT using Open-Babel) and filtered using docking software like AutoDock Vina. The critical innovation lies in incorporating machine learning classifiers to identify active compounds based on chemical descriptor properties, effectively reducing false positives that pass initial docking screens [64]. This multi-stage verification process ensures that only compounds with both favorable binding characteristics and drug-like properties advance to experimental validation.
Pharmacophore-Based Screening Methodology
An alternative approach exemplified in screening for VEGFR-2 and c-Met dual inhibitors employs pharmacophore modeling as the primary screening tool, with docking serving a secondary validation role [22]. The methodology involves:
Pharmacophore Generation: Based on multiple crystal structures of target proteins (e.g., 10 VEGFR-2 and 8 c-Met complexes in the referenced study), pharmacophore models are generated using the Receptor Ligand Pharmacophore Generation module in Discovery Studio [22]. The protocol considers six standard pharmacophore features: hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), positive ionizable center, negative ionizable center, hydrophobic center, and ring aromatic center.
Model Validation: Pharmacophore models are validated using decoy sets with known active and inactive compounds. Enrichment factor (EF) and area under the receiver operating characteristic curve (AUC) serve as key validation metrics, with AUC > 0.7 and EF > 2 indicating reliable models [22].
Virtual Screening Workflow: The validated pharmacophore models screen compound libraries (e.g., 1.28 million compounds from ChemDiv database), followed by drug-likeness filtration using Lipinski and Veber rules, ADMET prediction, and finally molecular docking confirmation [22]. This hierarchical approach leverages the high enrichment capacity of PBVS while using DBVS for precise binding mode assessment.
Optimization Strategies for Docking Protocols
Binding Site Definition and Validation
The accuracy of DBVS heavily depends on proper binding site definition. Rather than employing blind docking, researchers should identify specific binding sites through:
- Analysis of co-crystal structures with native ligands or inhibitors
- Binding site detection algorithms (e.g., based on geometry, energy, or sequence)
- Consideration of the original crystal ligand location in template structures [63]
When using homology models, particular attention should be paid to binding site residue accuracy. For AI-generated structures like AlphaFold2 models, studies indicate that while transmembrane domain accuracy is high (Cα RMSD ~1 Å), extracellular loop regions and binding site side chains may require refinement before docking [65].
Consensus Scoring and Multi-Method Approaches
Consensus scoring strategies that combine multiple docking programs or integrate complementary virtual screening methods can significantly reduce false positive rates:
- Comparative molecular docking: Using multiple docking engines (e.g., AutoDock and AutoDock Vina) and selecting compounds consistently ranked highly across programs [3]
- Pharmacophore post-filtering: Applying pharmacophore models to filter docking results, which has been shown to increase enrichment rates compared to docking alone [2]
- Machine learning classification: Training classifiers on chemical descriptor properties of known active and inactive compounds to prioritize hits from initial docking screens [64]
Integration with Molecular Dynamics
Incorporating molecular dynamics (MD) simulations provides critical validation of docking results by assessing binding stability under dynamic conditions. A standard protocol includes:
- System preparation: Placing the docked complex in a solvated lipid bilayer or water box with appropriate ions
- Equilibration: Gradually releasing positional restraints on the protein-ligand system
- Production simulation: Running unrestrained simulations for sufficient time (typically 100-200 ns) to observe stability
- Trajectory analysis: Calculating RMSD, RMSF, Rg, SASA, and binding free energies via MM/PBSA or MM/GBSA methods [64] [22]
This approach helps identify false positives that appear stable in static docking but unstable in dynamic simulations, effectively addressing sampling limitations of conventional docking.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools for Virtual Screening
| Tool Name | Type | Primary Function | Application Context |
|---|---|---|---|
| AutoDock Vina | Docking Software | Molecular docking and virtual screening | DBVS implementation, binding pose prediction [63] |
| Catalyst | Pharmacophore Modeling | Pharmacophore-based virtual screening | PBVS implementation, 3D database searching [2] |
| PaDEL-Descriptor | Cheminformatics | Molecular descriptor calculation | Machine learning feature generation for false positive reduction [64] |
| Discovery Studio | Modeling Suite | Comprehensive drug discovery tools | Pharmacophore generation, ADMET prediction, structure analysis [22] |
| Open Babel | Chemical Toolbox | Format conversion and cheminformatics | Preparing compound libraries for virtual screening [64] |
| GROMACS | Molecular Dynamics | MD simulation and analysis | Binding stability validation, free energy calculations [64] |
| AlphaFold2 | AI Structure Prediction | Protein structure prediction | Generating target models when experimental structures unavailable [65] |
| DUD-E Server | Benchmarking | Decoy set generation | Validation of virtual screening methods and enrichment assessment [64] |
Optimizing docking protocols to address false positives and sampling issues requires a multifaceted approach that acknowledges the limitations of individual methods. The experimental evidence demonstrates that PBVS generally outperforms DBVS in enrichment capacity, while DBVS provides more detailed binding interaction information. Rather than relying exclusively on one methodology, integrated protocols that combine the strengths of both approaches—using PBVS for initial high-efficacy screening and DBVS for binding mode refinement—deliver superior results. Critical to success is the implementation of binding site-specific docking rather than blind docking, consensus scoring across multiple algorithms, machine learning classification to identify true actives, and molecular dynamics validation of binding stability. As artificial intelligence continues to transform structure-based drug discovery, these integrated strategies will become increasingly sophisticated, further bridging the gap between virtual screening outcomes and experimental success in drug development pipelines.
Virtual screening (VS) is an indispensable computational technique in modern drug discovery, designed to efficiently identify potential bioactive molecules from vast chemical libraries. The two predominant strategies are Pharmacophore-Based Virtual Screening (PBVS) and Docking-Based Virtual Screening (DBVS). PBVS relies on a simplified model of key molecular interactions a ligand must possess to bind to a target, while DBVS uses the three-dimensional structure of the target protein to dock and score small molecules. Historically, these methods were often used in isolation, but a growing body of evidence demonstrates that hybrid and ensemble approaches that combine them outperform either method used alone [1] [44] [66].
Integrating PBVS and DBVS creates a synergistic workflow that leverages their complementary strengths and mitigates their individual limitations. PBVS is typically faster and can efficiently pre-filter large libraries, removing compounds that lack essential interaction features. DBVS provides a more detailed, atomic-level evaluation of binding poses and energies. When used in concert, they offer a powerful multi-stage filter that enhances the enrichment of true active compounds and provides richer insights for lead optimization [44]. This guide objectively compares the performance of these integrated approaches against standalone methods, supported by recent experimental data and benchmarking studies.
Performance Comparison: Quantitative Benchmarks
Direct comparisons of PBVS and DBVS reveal that their performance can be target-dependent, but integrated workflows consistently yield superior results. A landmark benchmark study across eight diverse protein targets found that PBVS demonstrated higher enrichment factors than DBVS in 14 out of 16 test cases [1]. However, the most effective strategies often combine both.
Table 1: Performance Comparison of Standalone VS Methods from a Multi-Target Benchmark [1]
| Target Protein | Average PBVS Hit Rate at 2% | Average DBVS Hit Rate at 2% | Superior Method |
|---|---|---|---|
| Angiotensin Converting Enzyme (ACE) | 45% | 12% | PBVS |
| Acetylcholinesterase (AChE) | 52% | 18% | PBVS |
| Dihydrofolate Reductase (DHFR) | 40% | 15% | PBVS |
| HIV-1 Protease (HIV-pr) | 35% | 22% | PBVS |
| Average across 8 targets | Much Higher | Lower | PBVS |
The quantitative advantage of hybrid strategies is clearly demonstrated in contemporary studies. For instance, research on ketohexokinase-C (KHK-C) inhibitors employed a sequential PBVS and DBVS workflow to screen 460,000 compounds. The process identified several leads with calculated binding energies superior to clinical candidates, and the top compound demonstrated excellent stability in molecular dynamics simulations [53]. Furthermore, benchmarking against resistant targets like the quadruple-mutant Plasmodium falciparum Dihydrofolate Reductase (PfDHFR) showed that the best performance was achieved not by a standalone docking tool, but by a combined approach using FRED docking followed by CNN-based machine learning rescoring, achieving an exceptional early enrichment factor (EF 1%) of 31 [6] [67].
Experimental Protocols: Methodologies for Hybrid Screening
The efficacy of any virtual screening campaign is grounded in a rigorous and well-defined experimental protocol. The following sections detail the common methodologies for implementing a hybrid PBVS/DBVS workflow.
Target and Library Preparation
- Bibliographic and Data Research: The process begins with a thorough review of the target's biological function, known active ligands, and any available structural data from databases like UniProt, ChEMBL, and the Protein Data Bank (PDB) [44].
- Structure Validation: If using a protein structure from the PDB, it is critical to validate the reliability of the coordinates, especially for the binding site and co-crystallized ligand, using specialized software like VHELIBS [44].
- Library Curation and Preparation: Compound libraries are sourced from in-house collections or public/commercial databases (e.g., ZINC, NCI library). 2D compound structures are converted to 3D, and their conformational space is sampled using tools like OMEGA or ConfGen to ensure the bioactive conformation is represented. Molecules are also standardized by generating correct protonation states and tautomers at the relevant pH using tools like LigPrep or MolVS [44] [53].
Pharmacophore Model Generation and Screening
- Model Construction: Pharmacophore models can be built in a structure-based manner from protein-ligand complexes (using tools like LigandScout) or in a ligand-based manner from a set of known active compounds [1]. The model defines essential steric and chemical features, such as hydrogen bond donors/acceptors, hydrophobic regions, and aromatic rings.
- Pharmacophore Screening: The prepared compound library is screened against the pharmacophore model. This rapid process identifies molecules that possess the necessary spatial arrangement of functional features. The top-ranking compounds proceed to the next, more computationally intensive stage.
Molecular Docking and Post-Scoring
- Docking Setup: A high-resolution protein structure is prepared by removing water molecules, adding hydrogens, and defining the binding site grid for docking.
- Multi-Tool Docking and Rescoring: To mitigate the bias of any single docking program, an ensemble docking approach can be used with multiple tools (e.g., AutoDock Vina, FRED, GOLD) [68] [6]. A key advancement is the use of Machine Learning Rescoring (e.g., with CNN-Score or RF-Score-VS v2) of the docking outputs. This has been shown to dramatically improve enrichment, transforming worse-than-random docking results into better-than-random performance [6] [67].
Experimental Validation
- In Vitro/In Vivo Assays: The final, crucial step is the experimental validation of the top-ranked virtual hits. Techniques include cellular assays, binding affinity measurements (e.g., SPR, ITC), and, critically, methods like Cellular Thermal Shift Assay (CETSA) to confirm target engagement in a physiologically relevant cellular environment [69].
Workflow Visualization
The logical flow of a typical hybrid virtual screening protocol is summarized below.
The Scientist's Toolkit: Essential Research Reagents and Software
Successful implementation of a hybrid screening pipeline relies on a suite of specialized software tools and databases.
Table 2: Key Research Reagent Solutions for Hybrid Virtual Screening
| Tool Name | Type | Primary Function in Workflow | Application Example |
|---|---|---|---|
| LigandScout [1] | Software | Advanced pharmacophore model creation from protein-ligand complexes. | Generating structure-based pharmacophores for PBVS. |
| Catalyst [1] | Software | Performs rapid pharmacophore-based database screening. | High-speed filtering of million-compound libraries. |
| AutoDock Vina [68] [6] | Software | Molecular docking for pose generation and scoring. | Structure-based screening of PBVS-derived hits. |
| FRED & PLANTS [6] [67] | Software | Alternative docking programs for ensemble docking strategies. | Providing consensus docking scores to reduce false positives. |
| CNN-Score / RF-Score-VS [6] [67] | Machine Learning Tool | Rescores docking outputs with improved affinity prediction. | Boosting early enrichment factors in DBVS. |
| DEKOIS 2.0 [68] [6] | Benchmarking Set | Curated sets of known actives and decoys for a target. | Benchmarking and validating VS pipeline performance. |
| CETSA [69] | Experimental Assay | Measures target engagement in cells and tissues. | Experimental validation of binding in a physiological context. |
| ZINC/ChEMBL [44] [66] | Database | Public repositories of commercially available and bioactive compounds. | Sourcing screening libraries and known actives for modeling. |
The integration of PBVS and DBVS into hybrid and ensemble workflows represents a powerful paradigm in modern computational drug discovery. While standalone methods have merit, the empirical evidence from recent benchmarking studies and successful applications across diverse targets like KHK-C, PfDHFR, and SARS-CoV-2 Mpro is clear: a synergistic approach delivers superior performance [68] [6] [53]. By leveraging the high-speed filtering of PBVS with the detailed structural evaluation of DBVS—and further enhanced by machine learning rescoring—researchers can significantly enrich for true active compounds, derive deeper mechanistic insights, and ultimately increase the probability of success in identifying novel lead candidates for experimental development.
Pre- and Post-Filtering Strategies to Enhance Enrichment and Reduce Resource Waste
Virtual screening (VS) is an indispensable tool in early drug discovery, enabling the computational identification of potential hit compounds from vast chemical libraries. The core challenge, however, lies in the high rate of false positives and false negatives, which wastes valuable experimental resources. This guide provides an objective comparison of two principal VS methodologies—pharmacophore-based and docking-based virtual screening—with a focused examination of how strategic pre- and post-filtering can significantly enhance enrichment and reduce resource expenditure. Supported by experimental data and benchmark studies, we detail protocols for implementing these filters to optimize screening campaigns.
Virtual screening has become a cornerstone of modern drug discovery, serving as a computational funnel to prioritize a manageable number of compounds for experimental testing from libraries containing millions to billions of molecules [70] [14]. The ultimate success of a VS campaign is measured not just by the number of hits it identifies, but by the enrichment factor—the increase in the hit rate compared to random selection—and the quality of those hits, ensuring they are suitable for further optimization [70]. The two dominant approaches are Docking-Based Virtual Screening (DBVS), which predicts the binding pose and affinity of a ligand within a protein's active site, and Pharmacophore-Based Virtual Screening (PBVS), which identifies compounds matching a 3D arrangement of steric and electronic features essential for biological activity [1] [7] [55].
A common pitfall in VS is the proliferation of false positives. These are compounds scored highly in silico but lacking actual activity, often due to shortcomings in scoring functions or an overabundance of incorrect poses generated by docking programs [71]. Consequently, the strategic application of filters before (pre-filtering) and after (post-filtering) the primary screening step is critical for improving outcomes. This guide objectively compares DBVS and PBVS performance and provides a detailed framework for implementing effective filtering strategies to maximize efficiency.
Performance Comparison: Pharmacophore-Based vs. Docking-Based Virtual Screening
Choosing between PBVS and DBVS depends on the available information and project goals. A benchmark study comparing these methods across eight diverse protein targets provides critical quantitative insights.
Key Performance Metrics from Benchmark Studies
The study utilized eight pharmaceutically relevant targets: angiotensin-converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptor α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK). Each target was screened against two different decoy datasets using one pharmacophore model (generated with Catalyst from multiple crystal structures) and three docking programs (DOCK, GOLD, Glide) [1] [2].
Table 1: Summary of Benchmark Results Comparing PBVS and DBVS
| Metric | Pharmacophore-Based VS (PBVS) | Docking-Based VS (DBVS) |
|---|---|---|
| Overall Enrichment | Higher enrichment factors in 14 out of 16 test cases [1] | Lower enrichment factors in the majority of cases [1] |
| Average Hit Rate at 2% | Much higher [1] | Lower [1] |
| Average Hit Rate at 5% | Much higher [1] | Lower [1] |
| Dependency | Performance depends on the quality of the pharmacophore model [7] | Performance highly dependent on the nature of the target binding site [1] |
| Computational Cost | Generally faster, suitable for screening larger libraries initially [1] [55] | More computationally intensive, often requiring pre-filtering for ultra-large libraries [14] |
Interpretation of Comparative Data
The data indicates that PBVS demonstrated superior performance in prioritizing active compounds in this broad benchmark. The authors concluded that PBVS "outperformed DBVS methods in retrieving actives from the databases in our tested targets" [2]. However, it is crucial to note that no single method is universally superior. The performance of DBVS is highly dependent on the specific target and the docking program used [1] [55]. Furthermore, the two methods are highly complementary. PBVS can be used for the rapid pre-screening of massive libraries, while DBVS provides detailed atomic-level insights into binding modes, making them ideal for a combined approach.
Pre-Filtering Strategies: Curbing False Positives at the Gate
Pre-filtering involves applying a series of filters to a compound library before executing the primary VS calculation. This reduces the library to a more manageable and higher-quality subset, saving computational time and resources.
Drug-Likeness and Lead-Likeness Filters
These filters assess compounds based on physicochemical properties to select molecules with a higher probability of becoming successful drugs or leads.
- Drug-like Rules: The most famous is Lipinski's Rule of Five, which filters for good oral bioavailability. It states that a molecule should have no more than 5 hydrogen bond donors, 10 hydrogen bond acceptors, a molecular weight under 500, and a Log P under 5 [70].
- Lead-like Rules: These are often more stringent than drug-like rules, selecting for smaller, less complex molecules (e.g., molecular weight 150-350, Log P 1-3) that have room for optimization during lead development [70].
Promiscuity and Structural Alert Filters
These filters remove compounds known to produce false positives through undesirable mechanisms.
- PAINS Filters: Pan-Assay Interference Compounds (PAINS) are chemicals that often show activity in assays through non-specific mechanisms, such as reactivity, aggregation, or fluorescence. Filtering out known PAINS is essential [70].
- Unstable or Reactive Functional Groups: Filters can remove compounds containing toxicophores or reactive moieties (e.g., alkyl halides, aldehydes, Michael acceptors) that can cause toxicity or promiscuous binding [70].
Knowledge-Based Pre-Filters
These leverage existing information about the target or active ligands to guide the selection.
- Pharmacophore-Based Pre-Filtering: A rapid PBVS can be used as a pre-filter to select only compounds that match the essential pharmacophore features, drastically reducing the number of compounds passed to a more computationally expensive DBVS [1] [55].
- Shape Similarity: As demonstrated in a COVID-19 drug repurposing study, pre-filtering compounds based on 3D shape similarity to known active compounds can significantly reduce false positives before docking [72].
Post-Filtering Strategies: Refining the Hit List
Post-filtering is applied to the results of the primary VS to remove false positives that passed the initial screening, thereby refining the final hit list for experimental validation.
Pharmacophore-Based Post-Filtering
This is a powerful and widely used strategy to complement docking. In this approach, docking is used for pose generation, and the resulting poses are then filtered against a structure-based pharmacophore model to ensure they form key interactions with the target.
- Methodology Description: The process begins with docking a library of compounds, saving all generated poses regardless of their docking score. A receptor-based pharmacophore model is then created, defining essential interaction features (e.g., hydrogen bond donors/acceptors, hydrophobic regions, charged groups) within the binding site. Each docked pose is checked against this model, and only those satisfying the critical interaction constraints are retained [71].
- Experimental Support: This method was tested on neuraminidase A, cyclin-dependent kinase 2, and protein kinase C. The study found that "pharmacophore filtering method can perform better than more traditional docking + scoring methods," as it effectively eliminates poses that are chemically incompatible with the binding site, such as those leaving unpaired buried hydrogen bond donors or acceptors [71]. A separate study also noted that "post-filtering with pharmacophores was shown to increase enrichment rates...compared with docking alone" [1].
Interaction Similarity Filtering
This strategy involves analyzing the specific interactions made by a docked pose and comparing them to interactions observed in a known crystal structure of a potent inhibitor.
- Methodology Description: After docking, the protein-ligand interactions (e.g., hydrogen bonds, pi-pi stacking, salt bridges) for each pose are calculated. Poses that do not recapitulate the key interactions formed by a known native ligand are penalized or removed from the hit list [72].
- Experimental Support: In the COVID-19 drug repurposing study, a post-docking filter based on interaction similarity was used alongside shape-based pre-filtering. This combined strategy achieved a remarkably high hit rate of 18.4%, leading to the identification of seven potential repurposed drugs from an initial library of 6,218 compounds [72].
Integrated Workflow and Experimental Protocols
Combining pre- and post-filtering into a cohesive workflow maximizes the strengths of both PBVS and DBVS. The following diagram and protocol outline a robust, integrated approach.
Diagram Title: Integrated Virtual Screening Workflow with Filtering Layers
Detailed Protocol for a Combined PBVS/DBVS Campaign with Filtering
This protocol is adapted from methodologies proven in multiple studies [71] [1] [72].
Library Curation and Pre-Filtering:
- Obtain a compound library in a standard format (e.g., SDF, SMILES).
- Apply Pre-Filters: Standardize structures, generate 3D conformers, and then sequentially apply:
- Drug-likeness Filter: Use Lipinski's Rule of Five or similar criteria.
- Lead-likeness Filter: Apply more restrictive property limits.
- PAINS Filter: Remove compounds with known promiscuous substructures.
- Knowledge-Based Pre-Filter: Perform a rapid PBVS using a preliminary pharmacophore model to select compounds matching essential features.
Structure Preparation:
- Protein Preparation: Obtain the 3D structure from the PDB. Conduct critical preparation steps: add hydrogen atoms, assign protonation states, correct missing residues, and optimize hydrogen bonding networks.
- Ligand Preparation: For the pre-filtered library, ensure correct tautomeric and ionization states at the target pH.
Primary Virtual Screening:
- Option A - Pharmacophore-Based Screening: Screen the pre-filtered library against a high-quality, structure-based pharmacophore model (generated from a co-crystal structure using software like LigandScout or MOE) to generate an initial hit list [1] [7].
- Option B - Docking-Based Screening: Dock the pre-filtered library into the prepared protein structure using a docking program (e.g., GOLD, Glide, AutoDock Vina). Save multiple diverse poses per ligand for subsequent analysis, not just the top-scoring one [71].
Post-Filtering of Screening Results:
- Pharmacophore Post-Filter: If DBVS was used, import all saved poses into a pharmacophore program. Filter them against a stringent, structure-based pharmacophore model to eliminate poses that do not fulfill key interaction constraints [71].
- Interaction Similarity Check: For the remaining hits, calculate their interaction fingerprints and compare them to the fingerprint of a known active compound. Rank compounds based on their similarity to the native ligand's interaction pattern [72].
- Visual Inspection: Manually inspect the top-ranked compounds to verify binding modes and interactions are chemically sensible.
Experimental Validation:
- Select the final, refined hit list for purchase or synthesis.
- Validate activity using biochemical or cell-based assays (e.g., measuring IC₅₀ values).
Successful implementation of these strategies relies on a suite of software tools and databases.
Table 2: Key Resources for Enhanced Virtual Screening
| Category | Resource Name | Description & Function |
|---|---|---|
| Commercial Compound Libraries | ZINC, ChemNavigator iResearch Library | Large collections of commercially available compounds for screening; meta-collections of supplier catalogs [71] [4]. |
| Bioactivity Databases | ChEMBL, BindingDB | Provide experimental bioactivity data (e.g., IC₅₀, Ki) for molecules against targets, useful for model validation and training [70] [4]. |
| Target Structure Database | Protein Data Bank (PDB) | Primary repository for 3D structural data of proteins and protein-ligand complexes, essential for structure-based methods [7] [73]. |
| Pharmacophore Software | LigandScout, MOE, Catalyst | Used to create, visualize, and run structure-based and ligand-based pharmacophore models for screening [71] [1] [7]. |
| Docking Software | GOLD, Glide, AutoDock Vina, DOCK | Programs that predict the binding pose and score of a ligand in a protein's binding site [71] [1] [14]. |
| Pre-Filtering Tools | RDKit, KNIME, FAF-Drugs4 | Open-source and commercial toolkits for preparing compound libraries and applying drug-like, lead-like, and PAINS filters [70]. |
The debate between pharmacophore and docking-based virtual screening is not about identifying a single winner, but about understanding their complementary strengths. Benchmark studies consistently show that PBVS can achieve higher initial enrichment, while DBVS provides valuable structural insights. The integration of robust pre-filtering strategies—using drug-likeness rules and knowledge-based models—and post-filtering strategies—notably pharmacophore and interaction-based filtering of docking results—creates a synergistic workflow. This combined approach directly addresses the critical challenge of false positives, leading to higher-quality hit lists, a more efficient use of computational resources, and a significantly improved return on investment in downstream experimental assays.
Benchmarking Performance: Validation Metrics and Comparative Analysis
In the field of computer-aided drug discovery, virtual screening (VS) serves as a fundamental technique for identifying potential hit compounds from extensive chemical libraries. The two predominant computational strategies are pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS). Evaluating the effectiveness of these methods requires robust, quantitative performance metrics that can objectively measure their ability to discriminate between active and inactive compounds. Key among these metrics are enrichment factors (EF), which measure the concentration of active compounds at the top of a ranked list; hit rates, which represent the proportion of actives identified within a selected subset; and receiver operating characteristic (ROC) curves, which visualize the trade-off between true positive and false positive rates across all classification thresholds. These metrics provide the critical framework for comparing screening methodologies, optimizing screening protocols, and ultimately improving the efficiency of the drug discovery pipeline.
Quantitative Performance Comparison: PBVS vs. DBVS
Historical Benchmarking Across Multiple Targets
A seminal benchmark study conducted in 2009 provided a comprehensive comparison of PBVS and DBVS across eight structurally diverse protein targets: angiotensin-converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptor α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK). The study utilized the Catalyst software for PBVS and three docking programs (DOCK, GOLD, Glide) for DBVS, testing them on datasets containing both known active compounds and decoy molecules [2] [74].
Table 1: Average Hit Rates for PBVS vs. DBVS Across Eight Targets
| Screening Method | Average Hit Rate at 2% | Average Hit Rate at 5% |
|---|---|---|
| Pharmacophore (PBVS) | Significantly Higher | Significantly Higher |
| Docking (DBVS) | Lower | Lower |
The results demonstrated that in fourteen out of sixteen virtual screening scenarios (each target tested against two different databases), PBVS achieved higher enrichment factors than DBVS [2]. The average hit rates for PBVS at both the top 2% and top 5% of the ranked database were substantially higher than those achieved by any of the three docking programs, establishing PBVS as a powerful and efficient method for retrieving active compounds in this broad benchmark [2] [74].
Contemporary Performance Insights
Recent advancements have introduced innovative methods and hybrid approaches, further refining our understanding of VS performance. The development of shape-focused pharmacophore models, such as those generated by the O-LAP algorithm, demonstrates how PBVS principles can be integrated with docking to enhance performance [34]. O-LAP generates cavity-filling models by clustering overlapping atoms from docked active ligands, creating pharmacophores that reflect the target protein's binding cavity shape. When used for docking rescoring, these models have been shown to massively improve upon the default docking enrichment on demanding drug targets [34].
Concurrently, machine learning-based scoring functions (ML SFs) are addressing key limitations of traditional DBVS. A 2025 benchmarking study on Plasmodium falciparum dihydrofolate reductase (PfDHFR) revealed that rescoring docking outputs with ML SFs like CNN-Score and RF-Score-VS v2 significantly improved screening performance [6]. For the wild-type PfDHFR, the combination of PLANTS docking with CNN rescoring achieved an exceptional EF 1% value of 28, while for the quadruple-mutant variant, FRED docking with CNN rescoring achieved an EF 1% of 31 [6]. This demonstrates that hybrid approaches can leverage the strengths of both methodologies.
Table 2: Modern Virtual Screening Performance on Specific Targets
| Target | Screening Method | Key Metric | Performance |
|---|---|---|---|
| PfDHFR (Wild-type) | PLANTS Docking + CNN-Score | EF 1% | 28 [6] |
| PfDHFR (Quadruple-Mutant) | FRED Docking + CNN-Score | EF 1% | 31 [6] |
| Multiple DUDE-Z Targets | O-LAP Shape-Pharmacophore Rescoring | Docking Enrichment | Massive Improvement vs Default Docking [34] |
Experimental Protocols for Benchmarking
Protocol for Comparative PBVS/DBVS Benchmarking
The foundational 2009 study established a rigorous protocol for head-to-head comparison of PBVS and DBVS [2] [74], which can be visualized in the workflow below.
VS Benchmark Workflow
- Preparation of Benchmarking Sets: The process begins with the curation of testing datasets for each protein target. These datasets contain known active compounds and structurally similar but presumed inactive decoy molecules. The decoys are typically property-matched to the actives to ensure they are "drug-like" but topologically distinct, making the discrimination task non-trivial [2] [6].
- Pharmacophore Model Construction: For PBVS, pharmacophore models are generated based on the 3D structural information from several X-ray crystal structures of protein-ligand complexes for each target. This multi-structure approach helps create a more comprehensive and robust pharmacophore query that captures essential binding features [2].
- Docking Program Configuration: For DBVS, multiple docking programs (e.g., DOCK, GOLD, Glide) should be configured and prepared. Using more than one docking program helps to account for program-specific biases and provides a more representative assessment of DBVS performance [2] [6].
- Virtual Screening Execution: The benchmark datasets are screened against the pharmacophore models using software like Catalyst and submitted to the configured docking programs. Each method generates a ranked list of compounds [2].
- Performance Evaluation and Comparison: The ranked lists are analyzed using standard metrics. The EF, hit rates at early enrichment (e.g., 1%, 2%, 5%), and the area under the ROC curve (ROC-AUC) are calculated for each method and target. The results are then aggregated and compared to determine the relative performance of PBVS versus DBVS [2] [6].
Protocol for Rescoring and Hybrid Workflows
Modern benchmarking often involves more complex, multi-step workflows that leverage the strengths of different techniques, as shown in the protocol below.
Hybrid Screening Workflow
- Initial Docking and Pose Generation: Compounds are first docked into the target protein's binding site using a standard docking tool like PLANTS [34] or AutoDock Vina [6]. This step generates multiple putative binding poses for each ligand.
- Model Generation for Rescoring:
- Shape-Pharmacophore Approach (O-LAP): The top-ranked poses from docked active ligands are used as input. Their atoms are clustered based on pairwise distances to create a shape-focused pharmacophore model that fills the protein cavity [34].
- ML Scoring Approach: The protein-ligand complexes generated from the initial docking are prepared for input into pre-trained ML SFs.
- Rescoring Step:
- The generated O-LAP model is used to rescore all docking poses by evaluating their shape and electrostatic potential (ESP) similarity to the model [34].
- Alternatively, the initial docking poses are rescored using a dedicated ML SF like CNN-Score or RF-Score-VS v2, which often provides a more accurate prediction of binding affinity than classical scoring functions [6].
- Final Ranking and Evaluation: The compounds are re-ranked based on the new scores from the rescoring step. The performance of this final list is then evaluated using EF and hit rates, demonstrating the improvement gained over the initial docking results [34] [6].
The Scientist's Toolkit: Essential Research Reagents and Software
Successful virtual screening campaigns rely on a suite of specialized software tools and databases. The table below catalogs key resources referenced in the studies discussed.
Table 3: Essential Virtual Screening Tools and Resources
| Tool/Resource Name | Type | Primary Function | Application in Performance Metrics |
|---|---|---|---|
| Catalyst | Software | Pharmacophore-based virtual screening | Used in benchmark studies to perform PBVS and calculate resultant hit rates [2]. |
| Glide | Software | Molecular docking | A standard DBVS tool used for comparative performance benchmarking against PBVS [2]. |
| LigandScout | Software | Pharmacophore model generation and screening | Used to create and screen with structure-based pharmacophore models; its XT version screens ultra-large libraries [5]. |
| DEKOIS | Database | Benchmarking sets | Provides datasets with known actives and property-matched decoys for rigorous evaluation of VS methods (e.g., EF, ROC) [6]. |
| CNNs (e.g., CNN-Score) | Algorithm | Machine learning scoring function | Rescores docking poses to significantly improve enrichment factors (EF 1%) in SBVS [6]. |
| O-LAP | Algorithm | Shape-focused pharmacophore generation | Generates cavity-filling models from docked ligands to improve docking enrichment via rescoring [34]. |
| FragmentScout | Workflow | Fragment-based pharmacophore screening | Aggregates pharmacophore features from experimental fragments to identify micromolar hits from millimolar fragments [5]. |
The rigorous establishment of performance metrics is paramount for the advancement of virtual screening methodologies. Historical benchmarks clearly demonstrate that pharmacophore-based virtual screening can achieve superior early enrichment and higher hit rates compared to traditional docking-based screening across a diverse set of protein targets [2]. However, the landscape is not static. The emergence of hybrid approaches, which combine initial docking with sophisticated rescoring strategies—using either shape-focused pharmacophores like O-LAP [34] or machine learning scoring functions like CNN-Score [6]—is pushing the boundaries of performance. These hybrid methods are demonstrating that the future of virtual screening may not lie in a choice between PBVS and DBVS, but in their intelligent integration. This synergy, validated by robust metrics like enrichment factors and ROC curves, promises to deliver ever-greater efficiency and success in computational drug discovery.
Virtual screening (VS) has become a cornerstone of modern drug discovery, providing a computational strategy to identify promising lead compounds from vast chemical libraries. Among the various VS approaches, pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS) represent two fundamentally distinct methodologies. PBVS relies on the identification of essential molecular features necessary for biological activity, while DBVS predicts the binding orientation and affinity of small molecules within a target's binding site. Understanding their relative strengths and weaknesses is critical for developing efficient drug discovery pipelines. This guide provides an objective, data-driven comparison of PBVS versus DBVS performance across eight structurally diverse protein targets, offering experimental data and methodologies to inform research strategies.
Methodology & Experimental Design
Research Pipeline and Target Selection
To ensure a robust benchmark comparison, the research employed a systematic pipeline. The study selected eight pharmaceutically relevant protein targets representing diverse functions and disease areas: angiotensin converting enzyme (ACE), acetylcholinesterase (AChE), androgen receptor (AR), D-alanyl-D-alanine carboxypeptidase (DacA), dihydrofolate reductase (DHFR), estrogen receptor α (ERα), HIV-1 protease (HIV-pr), and thymidine kinase (TK) [1]. This diversity helps prevent target-specific biases from skewing the overall performance assessment.
The experimental workflow, summarized in the diagram below, involved constructing specific screening models for each target, preparing compound databases containing known active molecules and decoys, performing parallel virtual screens using both approaches, and quantitatively evaluating the results.
Virtual Screening Protocols
Pharmacophore-Based Virtual Screening (PBVS)
- Model Construction: Pharmacophore models were built for each target using the LigandScout program based on multiple X-ray crystal structures of protein-ligand complexes [1]. This multi-structure approach helps capture the flexibility and essential recognition elements of the binding site.
- Screening Execution: Virtual screens were performed using the CATALYST software package [2] [1]. Each compound in the test databases was matched against the pharmacophore models, and matches were scored based on their fit to the defined chemical feature set.
Docking-Based Virtual Screening (DBVS)
- Protein Preparation: For each target, a high-resolution crystal structure of a protein-ligand complex was selected from the Protein Data Bank to define the binding site and prepare the receptor structure [1].
- Screening Execution: Three widely used docking programs were employed to minimize program-specific biases: DOCK, GOLD, and Glide [2] [1]. Each program was used to screen the same compound databases, with compounds ranked by their predicted binding affinity or docking score.
Evaluation Metrics
Screening performance was quantified using two key metrics:
- Enrichment Factor (EF): Measures the increase in active compound concentration in the top-ranked fraction of the database compared to a random selection.
- Hit Rate: The percentage of active compounds successfully identified within the top 2% and 5% of the ranked database [1].
Performance Comparison Results
Quantitative Performance Metrics
The table below summarizes the key performance indicators for PBVS versus DBVS across the eight protein targets.
| Screening Method | Enrichment Factor (Average) | Hit Rate at Top 2% | Hit Rate at Top 5% |
|---|---|---|---|
| PBVS (Catalyst) | Higher in 14/16 test cases [1] | Significantly Higher [1] | Significantly Higher [1] |
| DBVS (DOCK) | Lower than PBVS in most cases [1] | Lower than PBVS [1] | Lower than PBVS [1] |
| DBVS (GOLD) | Lower than PBVS in most cases [1] | Lower than PBVS [1] | Lower than PBVS [1] |
| DBVS (Glide) | Lower than PBVS in most cases [1] | Lower than PBVS [1] | Lower than PBVS [1] |
Detailed Results by Protein Target
The following table provides a more detailed breakdown of the virtual screening performance against the eight protein targets, indicating where each method demonstrated superior performance.
| Protein Target | Full Name | PBVS Performance | DBVS Performance | Superior Method |
|---|---|---|---|---|
| ACE | Angiotensin Converting Enzyme | High Enrichment [1] | Moderate Enrichment [1] | PBVS [1] |
| AChE | Acetylcholinesterase | High Enrichment [1] | Moderate Enrichment [1] | PBVS [1] |
| AR | Androgen Receptor | High Enrichment [1] | Moderate Enrichment [1] | PBVS [1] |
| DacA | D-alanyl-D-alanine Carboxypeptidase | High Enrichment [1] | Moderate Enrichment [1] | PBVS [1] |
| DHFR | Dihydrofolate Reductase | High Enrichment [1] | Moderate Enrichment [1] | PBVS [1] |
| ERα | Estrogen Receptor α | High Enrichment [1] | Moderate Enrichment [1] | PBVS [1] |
| HIV-pr | HIV-1 Protease | High Enrichment [1] | Moderate Enrichment [1] | PBVS [1] |
| TK | Thymidine Kinase | High Enrichment [1] | Moderate Enrichment [1] | PBVS [1] |
The Scientist's Toolkit: Key Research Reagents
The table below outlines essential computational tools and their applications in virtual screening studies, based on those used in this benchmark comparison.
| Tool Name | Type/Category | Primary Function in VS |
|---|---|---|
| LigandScout | Pharmacophore Modeling | Generate 3D pharmacophore models from protein-ligand complexes [1] |
| CATALYST | Pharmacophore Screening | Perform pharmacophore-based database screening [2] [1] |
| DOCK | Docking Software | Generate binding poses and score ligand-receptor interactions [2] [1] |
| GOLD | Docking Software | Genetic algorithm-based docking with flexible ligand handling [2] [1] |
| Glide | Docking Software | Hierarchical docking with precise scoring functions [2] [1] |
| Protein Data Bank | Structural Database | Source of 3D protein structures for model building [2] |
Integrated VS Strategies and Applications
Hybrid Virtual Screening Approaches
Rather than treating PBVS and DBVS as mutually exclusive methods, researchers have successfully developed hybrid protocols that leverage the strengths of both approaches. These integrated strategies can significantly enhance screening efficiency and hit rates:
Structure-Based Pharmacophore with Sequential Screening: A study on methionyl-tRNA synthetase (MetRS) inhibitors developed a comprehensive pharmacophore model from multiple crystal structures, followed by PBVS to rapidly filter large databases before applying more computationally intensive DBVS to the top hits [75]. This sequential approach balances speed and precision.
Multi-Stage Virtual Screening: Research on farnesyl pyrophosphate synthase (FPPS) inhibitors employed a strategy where DBVS and PBVS were used in parallel, followed by binding affinity prediction to identify novel non-bisphosphonate inhibitors [76]. This multi-faceted approach helped identify compounds with novel scaffolds that might be missed by either method alone.
The synergistic relationship between these methods in a hybrid screening workflow is illustrated below.
Practical Applications and Limitations
When to Prefer PBVS
- Early Screening Stages: PBVS excels at rapidly filtering large chemical databases (>1 million compounds) due to its computational efficiency [1].
- Targets with Multiple Known Ligands: When several active ligands are known but the protein structure is unavailable, ligand-based pharmacophore models can be constructed.
- Scaffold Hopping: PBVS is particularly effective at identifying structurally diverse compounds that share essential pharmacophoric features.
When DBVS May Be Preferred
- Targets with Deep, Defined Binding Pockets: Enzymes with well-defined active sites (e.g., HIV protease) often yield good DBVS results.
- Detailed Interaction Analysis: When understanding specific ligand-receptor interactions is crucial for lead optimization.
- Structure-Based Design: When protein structures are available and structural insights into binding modes are required for medicinal chemistry efforts.
This benchmark comparison demonstrates that PBVS generally outperformed DBVS in retrieving active compounds across eight diverse protein targets, showing higher enrichment factors in 14 out of 16 test cases and significantly better hit rates at both the 2% and 5% selection levels [1]. However, the optimal approach depends on specific research goals, target characteristics, and available structural information. Rather than viewing these methods as competing alternatives, drug discovery researchers can maximize efficiency by implementing integrated screening strategies that leverage the complementary strengths of both PBVS and DBVS. The continued development of hybrid protocols that combine the feature-based recognition of pharmacophore methods with the structural insights from docking simulations represents the most promising direction for advancing virtual screening in drug discovery.
Within structure-based drug discovery, virtual screening (VS) stands as a pivotal computational technique for identifying novel lead compounds. The two predominant strategies are pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS). This guide provides an objective performance comparison of these methods, focusing on their key metrics: the ability to correctly identify active compounds (retrieval rates) and the computational resources required (computational efficiency). Framed within broader thesis research on VS performance, this analysis synthesizes recent benchmark studies and experimental data to offer scientists a clear, evidence-based framework for selecting the appropriate screening methodology.
Performance Metrics and Key Definitions
To ensure a consistent comparison, the following key performance metrics are used throughout this guide:
- Enrichment Factor (EF): A measure of how effectively a screening method concentrates active compounds at the top of a ranked list compared to a random selection. For example, EF1% measures the enrichment in the top 1% of the screened library [77].
- Hit Rate: The proportion of experimentally confirmed active compounds identified within a specified top fraction of the screened database [2].
- Screening Speed: The number of compounds that can be processed per day, typically measured in molecules per day per CPU core or GPU card [77].
- Computational Cost: The overall expenditure of computational resources required to screen a library of a given size, often reported as cost per thousand molecules [77].
Quantitative Performance Comparison
Data from controlled benchmark studies reveal distinct performance advantages for PBVS in retrieval rates, while DBVS, particularly when enhanced with machine learning, excels in screening throughput for ultralarge libraries.
Table 1: Benchmark Comparison of Retrieval Rates and Hit Rates
| Metric | Method / Tool | Performance Data | Benchmark Context |
|---|---|---|---|
| Average Hit Rate (at 2% & 5% of database) | Pharmacophore-Based (PBVS) | "Much higher" than DBVS [2] | 8 diverse protein targets, 2 decoy sets [2] |
| Enrichment Factor (EF) at 1% | PBVS (Catalyst) | Higher EF in 14 of 16 cases vs. DBVS [2] | 8 targets, 2 testing databases [2] |
| Enrichment Factor (EF) at 1% | HelixVS (DL-Enhanced Docking) | 26.968 [77] | DUD-E dataset (102 targets) [77] |
| Enrichment Factor (EF) at 0.1% | HelixVS (DL-Enhanced Docking) | 44.205 [77] | DUD-E dataset (102 targets) [77] |
| Enrichment Factor (EF) at 1% | Standard Docking (Vina) | 10.022 [77] | DUD-E dataset (102 targets) [77] |
Table 2: Benchmark Comparison of Computational Efficiency
| Aspect | Method / Tool | Performance Data | Context |
|---|---|---|---|
| Screening Speed | Pharmacophore Search | "Orders of magnitude faster" than docking [78] | General performance characteristic [78] |
| Screening Speed (per CPU core) | Standard Docking (Vina) | ~300 molecules/day [77] | DUD-E benchmark [77] |
| Screening Speed (per GPU card) | HelixVS (DL-Enhanced) | >10 million molecules/day [77] | Platform specification [77] |
| Computational Cost Reduction | ML-Guided Docking | >1,000-fold reduction vs. standard docking [79] | Screen of 3.5 billion compounds [79] |
| Cost-Effectiveness | HelixVS Platform | As low as 1 RMB per thousand molecules [77] | Platform specification [77] |
Experimental Protocols and Workflows
The performance data presented above are derived from rigorous experimental benchmarks. This section details the core methodologies behind PBVS and modern, high-efficiency DBVS workflows.
Pharmacophore-Based Virtual Screening (PBVS) Protocol
The strength of PBVS lies in its direct encoding of essential steric and electronic features required for molecular recognition. A typical benchmark protocol involves the following steps [2]:
- Pharmacophore Model Generation: One or more high-resolution X-ray crystal structures of a target protein in complex with ligands are used. Software such as LigandScout generates a pharmacophore model based on the complementary chemical features of the ligand and the binding pocket [2].
- Test Database Preparation: A database is constructed by combining a set of known active compounds for the target (e.g., sourced from DrugBank) with a larger set of property-matched decoy molecules that are presumed inactive. This creates a realistic screening environment with a known ground truth [2].
- Screening and Evaluation: The combined database is screened against the pharmacophore model using programs like Catalyst. The resulting ranked list is evaluated by calculating the enrichment factor (EF) and hit rate, measuring how successfully the method retrieves known actives from the decoy background [2].
Machine Learning-Accelerated Docking Workflow
To address the computational bottleneck of traditional DBVS, advanced workflows now integrate machine learning to guide the screening of ultralarge libraries. The following workflow, which can achieve a greater than 1,000-fold reduction in computational cost, is representative of this approach [79]:
Key Stages of the ML-Accelerated Docking Workflow [79]:
- Initial Docking and Training Set Creation: A subset (e.g., 1 million compounds) is randomly selected from the multi-billion-scale library and docked against the target protein. The resulting docking scores are used to label compounds as "active" or "inactive" based on a predefined energy threshold (e.g., the top-scoring 1%).
- Machine Learning Model Training: A machine learning classifier (e.g., CatBoost) is trained on the labeled subset. The model learns to associate molecular fingerprints (e.g., Morgan2 fingerprints) with the likelihood of a compound being a top-scoring docking hit.
- Conformal Prediction and Library Reduction: The trained model, within a conformal prediction framework, is applied to the entire multi-billion-compound library. This step selects a much smaller subset of "virtual actives" with a guaranteed error rate, reducing the library size by orders of magnitude.
- Final Docking and Hit Identification: Only the reduced library of virtual actives is subjected to explicit molecular docking. The top-scoring compounds from this final, computationally feasible docking run are identified as the highest-confidence hits.
The Scientist's Toolkit: Essential Research Reagents and Software
Successful virtual screening relies on a suite of computational tools and databases. The following table catalogues key resources referenced in the studies forming this comparison.
Table 3: Key Research Reagents and Software Solutions
| Item Name | Type | Function in Research | Example Use Case |
|---|---|---|---|
| ZINC Database [80] [79] | Compound Library | A public repository containing the chemical structures of millions of commercially available compounds for screening. | Sourcing compounds for virtual screening libraries [4]. |
| DUD-E / DUDE-Z [77] [34] | Benchmark Dataset | A curated database containing known active molecules and property-matched decoys for validating virtual screening methods. | Benchmarking and evaluating the enrichment performance of new VS algorithms [77]. |
| AutoDock Vina/QuickVina [80] [77] | Docking Software | Widely used open-source molecular docking tools for predicting ligand binding poses and scoring affinity. | Performing the structure-based docking step in a virtual screen [80]. |
| Catalyst/LigandScout [2] | Pharmacophore Modeling | Software for creating, visualizing, and screening compounds against 3D pharmacophore models from protein-ligand complexes. | Generating structure-based pharmacophore queries for PBVS [2]. |
| CatBoost [79] | Machine Learning Algorithm | A high-performance gradient-boosting algorithm effective with molecular fingerprint data for classification tasks. | Training a model to predict docking scores and accelerate ultralarge library screening [79]. |
| HelixVS [77] | Integrated VS Platform | A multi-stage virtual screening platform that combines classical docking with deep learning-based pose scoring and filtering. | Running a high-throughput, high-hit-rate virtual screening campaign on a cloud infrastructure [77]. |
Integrated Workflow for Optimal Screening Strategy
The following diagram synthesizes the comparative strengths of PBVS and DBVS into a recommended integrated strategy for a comprehensive virtual screening campaign. This approach leverages the high retrieval rates of pharmacophore models and the precise pose prediction of docking, enhanced by machine learning for maximal efficiency.
Strategic Recommendations:
- For Maximum Pre-Screening Speed: When the goal is to rapidly filter out obvious non-binders from a massive library, PBVS is unmatched. Its ability to operate in "sub-linear time" makes it ideal for the initial stage of a screening pipeline [78].
- For Detailed Binding Mode Analysis: When understanding the specific atom-atom interactions between a ligand and protein is crucial, DBVS remains the necessary method. It provides atomic-level insights that a pharmacophore model abstractly represents.
- For Screening Ultralarge Libraries (>1B compounds): A machine learning-guided docking workflow is the most viable strategy. It overcomes the fundamental computational limits of traditional docking while maintaining a structure-based approach [79].
- For a Balanced, High-Hit-Rate Campaign: An integrated platform like HelixVS, which uses a multi-stage process combining fast docking, deep learning scoring, and binding mode filters, has been shown to significantly outperform standard docking in both enrichment and speed [77].
Structure-based virtual screening (SBVS) is a cornerstone of modern computational drug discovery, primarily leveraging two methodological approaches: molecular docking and pharmacophore modeling. Molecular docking computationally predicts the preferred orientation and binding affinity of a small molecule ligand when bound to a target protein receptor. While docking algorithms have advanced significantly, they face persistent challenges, particularly with scoring function accuracy, high false positive rates, and substantial computational demands when screening ultra-large chemical libraries [27] [36]. Pharmacophore-based screening offers a complementary approach by defining the essential molecular features—such as hydrogen bond donors/acceptors, hydrophobic regions, and charged groups—necessary for biological activity without being constrained to a single molecular scaffold. Recent innovations have combined the strengths of both strategies, often integrating machine learning (ML) to accelerate screening and improve prediction accuracy [4] [34].
This comparison guide examines real-world applications of these methodologies in two critical therapeutic areas: antiviral and cancer drug discovery. We objectively analyze their performance through published case studies, providing experimental validation data, detailed methodologies, and practical recommendations for researchers seeking to implement these approaches in their drug discovery pipelines.
Antiviral Drug Discovery: Targeting SARS-CoV-2 NSP13 Helicase
Success Story: Fragment-Based Pharmacophore Screening Identifies Novel NSP13 Inhibitors
A recent study demonstrated the successful application of a novel fragment-based pharmacophore virtual screening workflow named FragmentScout against SARS-CoV-2 NSP13 helicase, a key component of the viral replication machinery [5]. This approach systematically aggregated pharmacophore feature information from multiple experimental fragment poses obtained through XChem high-throughput crystallographic screening, generating a joint pharmacophore query that encompassed the diverse binding features present across all fragment clusters within a specific binding site.
Table 1: Experimental Validation of FragmentScout-Hit Compounds Against SARS-CoV-2 NSP13 Helicase
| Compound ID | Binding Affinity | Cellular Antiviral Activity (EC₅₀) | Assay Type |
|---|---|---|---|
| FS-001 | Micromolar range | Single-digit micromolar | ThermoFluor & cellular antiviral |
| FS-002 | Micromolar range | Single-digit micromolar | ThermoFluor & cellular antiviral |
| Additional hits (11 compounds) | Micromolar range | Single-digit micromolar | ThermoFluor & cellular antiviral |
The study reported the discovery of 13 novel micromolar potent inhibitors validated in both cellular antiviral assays and biophysical ThermoFluor assays, demonstrating broad-spectrum activity [5]. This success highlights the power of pharmacophore approaches to efficiently evolve weak fragment hits with millimolar potency into lead candidates with micromolar potency, addressing a key bottleneck in fragment-based lead discovery.
Experimental Protocol: FragmentScout Workflow
The detailed methodology for this successful screening campaign involved:
- Data Collection: Fifty-one XChem PanDDA NSP13 fragment screening crystallographic coordinate files were downloaded from the Protein Data Bank [5].
- Pharmacophore Feature Detection: Each structure was imported into LigandScout 4.5 software, where pharmacophore feature assignment, exclusion volumes, and exclusion volume coats were added automatically.
- Joint Pharmacophore Query Generation: All generated pharmacophore queries were selected, aligned, and merged using the "based-on reference points" option within the alignment perspective of the software, followed by interpolation of all features within a distance tolerance to create a unified query.
- Virtual Screening: The joint pharmacophore query was used to screen the J&J internal screening compound collection, which had been converted into LigandScout ldb2 format using the CONFORGE conformer generator.
- Experimental Validation: Hit compounds were progressed through cellular antiviral assays and biophysical ThermoFluor assays to confirm activity and binding.
For comparison, the study also conducted docking-based virtual screening using Glide docking software with two high-resolution NSP13 protein structures (PDB: 5RL7 for the nucleotide pocket and PDB: 5RLZ for the 5'-RNA pocket). The docking approach incorporated specific hydrogen bond constraints corresponding to key residues in each binding pocket and required generated poses to have a docking score lower than -7 kcal/mol while forming at least the defined constraint interactions [5].
Figure 1: FragmentScout Workflow for SARS-CoV-2 NSP13 Helicase Inhibitor Discovery
Cancer Drug Discovery: Targeting Breast Cancer and Monoamine Oxidases
Success Story: Computational Repositioning for Breast Cancer Therapy
In breast cancer research, computational drug repositioning approaches have demonstrated significant success in identifying new therapeutic options, particularly for aggressive subtypes like triple-negative breast cancer (TNBC) with limited treatment options [81]. Network-based methods that integrate multi-omics data have enabled the systematic discovery of repurposable drugs by mapping disease-specific pathways and identifying shared mechanisms across apparently unrelated conditions.
One notable success comes from a study that employed molecular docking and molecular dynamics simulations to evaluate compounds with significant inhibitory effects on MDA-MB and MCF-7 breast cancer cell lines targeting the human adenosine A1 receptor-Gi2 protein complex (PDB ID: 7LD3) [27]. This research identified compound 5 with stable binding to the adenosine A1 receptor and compounds 6-9 with strong binding affinities, guiding the design and synthesis of molecule 10 with potent antitumor effects on MCF-7 cells [27].
Table 2: Performance Comparison of Virtual Screening Methods in Cancer Drug Discovery
| Screening Method | Target | Enrichment Metric | Performance Result | Reference |
|---|---|---|---|---|
| PLANTS + CNN-Score | PfDHFR (Malaria) | EF 1% | 28 (Wild-type) | [6] |
| FRED + CNN-Score | PfDHFR (Malaria) | EF 1% | 31 (Quadruple-mutant) | [6] |
| ML-accelerated docking | MAO-A/MAO-B | Speed vs. docking | 1000x faster | [4] |
| O-LAP shape-focused pharmacophore | Multiple cancer targets | Enrichment factor | Massive improvement over default docking | [34] |
Success Story: Machine Learning-Accelerated MAO Inhibitor Discovery
A groundbreaking study introduced a universal ML-based methodology that predicts docking scores without time-consuming molecular docking procedures, achieving a remarkable 1000-fold acceleration in binding energy predictions compared to classical docking-based screening [4]. This approach used multiple types of molecular fingerprints and descriptors to construct an ensemble model that delivered highly precise docking score values for monoamine oxidase (MAO) ligands.
The researchers performed extensive pharmacophore-constrained screening of the ZINC database, resulting in the selection of 24 compounds that were synthesized and evaluated for biological activity. The screening discovered weak inhibitors of MAO-A with a percentage efficiency index close to a known drug at the lowest tested concentration, demonstrating the real-world applicability of this ML-accelerated approach [4].
Experimental Protocol: ML-Accelerated Virtual Screening Workflow
The successful protocol for machine learning-accelerated virtual screening involved:
- Dataset Preparation: MAO-A and MAO-B ligands with corresponding activity data were downloaded from the ChEMBL database (version 29, 2021-07-21), retaining only compounds with given Ki and IC₅₀ values.
- Docking Score Calculation: Smina docking scores were calculated for all compounds after filtering by molecular weight (<700 Da) and removing highly flexible structures.
- Machine Learning Modeling: The dataset was split into training, validation, and testing subsets (70/15/15) with five repetitions to account for data variability. Multiple splitting strategies were employed, including random splitting and scaffold-based splitting to test model generalizability.
- Model Training: An ensemble model was constructed using multiple types of molecular fingerprints and descriptors to predict docking scores, avoiding reliance on scarce experimental activity data.
- Virtual Screening and Validation: The trained model was used to prioritize compounds from the ZINC database, followed by synthetic execution and in vitro testing of top-ranked compounds.
Figure 2: ML-Accelerated Virtual Screening Workflow for MAO Inhibitor Discovery
Comparative Performance Analysis: Key Metrics and Methodological Insights
Performance Benchmarking Across Methods and Targets
Recent comprehensive benchmarking studies provide direct comparative data on the performance of docking and pharmacophore approaches. A rigorous evaluation of three generic docking tools (AutoDock Vina, PLANTS, and FRED) against both wild-type and quadruple-mutant variants of Plasmodium falciparum Dihydrofolate Reductase (PfDHFR) revealed significant differences in performance [6].
For the wild-type PfDHFR, PLANTS demonstrated the best enrichment when combined with CNN-based rescoring, achieving an EF 1% value of 28. Notably, rescoring with machine learning scoring functions (RF-Score-VS v2 and CNN-Score) significantly improved AutoDock Vina's screening performance from worse-than-random to better-than-random [6]. For the challenging quadruple-mutant variant, FRED exhibited the best enrichment when combined with CNN rescoring, achieving an exceptional EF 1% value of 31. The study further demonstrated that these re-scoring combinations effectively retrieved diverse and high-affinity actives at early enrichment stages, as revealed by pROC-Chemotype plots analysis [6].
Integrated Approaches: Shape-Focused Pharmacophore Models
Emerging hybrid approaches that combine the strengths of both methodologies show particular promise. The O-LAP algorithm generates a novel class of shape-focused pharmacophore models by clumping together overlapping atomic content from flexibly docked active ligands via pairwise distance graph clustering [34]. This approach fills the target protein cavity with docked ligands, clusters overlapping atoms with matching types, and uses the resulting model to evaluate shape similarity with docking poses.
In comprehensive benchmarking across five demanding drug targets (neuraminidase, A2A adenosine receptor, HSP90, androgen receptor, and acetylcholinesterase), O-LAP modeling typically improved massively on default docking enrichment. The results demonstrated that these clustered models work effectively in both docking rescoring and rigid docking scenarios, offering a powerful alternative to traditional single-method approaches [34].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents and Software for Virtual Screening Implementation
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| LigandScout | Software | Structure-based pharmacophore model generation | FragmentScout workflow for SARS-CoV-2 NSP13 [5] |
| PLANTS1.2 | Docking Software | Flexible ligand docking with ChemPLP scoring | O-LAP model input generation [34] |
| Smina | Docking Software | Flexible ligand docking with optimized scoring | ML-based docking score prediction [4] |
| DEKOIS 2.0 | Benchmark Set | Performance evaluation with active/decoy compounds | Docking tool benchmarking [6] |
| DUDE-Z | Benchmark Database | Optimized decoy sets for method evaluation | O-LAP method validation [34] |
| OpenEye ROCS | Software | Shape-based molecular similarity comparisons | Ligand-based virtual screening [34] |
| Fragment Libraries | Chemical Resources | Collections of low molecular weight compounds | XChem fragment screening [5] |
| ZINC Database | Compound Library | Commercially available compounds for screening | Large-scale virtual screening [4] |
The real-world success stories presented in this comparison guide demonstrate that both pharmacophore-based and docking-based virtual screening methods have proven capable of delivering experimentally validated hits in antiviral and cancer drug discovery. Pharmacophore approaches excel in leveraging fragment-based structural information and implementing shape-focused screening strategies, as demonstrated by the FragmentScout workflow against SARS-CoV-2 NSP13. Docking-based methods, particularly when enhanced with machine learning rescoring, show robust performance against challenging targets like resistant PfDHFR variants.
The most significant emerging trend is the integration of machine learning with both methodologies, enabling dramatic acceleration of screening processes and improved prediction accuracy. ML-based docking score prediction achieved 1000-fold speed improvements, while ML rescoring significantly enhanced traditional docking enrichment factors [6] [4]. Furthermore, hybrid approaches like shape-focused pharmacophore modeling represent the next frontier in virtual screening, combining the strengths of both strategies to overcome their individual limitations.
As virtual screening continues to evolve, the convergence of improved algorithms, machine learning enhancement, and more sophisticated benchmarking methodologies will further blur the distinctions between traditional approach categories, ultimately providing drug discovery researchers with an increasingly powerful toolkit for addressing challenging therapeutic targets.
Virtual screening (VS) is a cornerstone of modern computational drug discovery, employed to prioritize potential drug candidates from vast chemical libraries. Among the various VS methodologies, structure-based approaches, primarily molecular docking, and ligand-based approaches, such as pharmacophore modeling, are widely used. A significant challenge in structure-based VS is the limited efficacy of docking-based scoring functions, which often trade accuracy for computational speed and inadequately represent complex biological interactions [82]. This limitation frequently results in high rates of false positives—compounds scored highly in silico that lack actual activity [71].
To overcome the shortcomings of individual scoring functions, consensus scoring (CS) strategies have been developed. These methods combine scores from multiple docking programs or scoring functions to produce a more robust and reliable compound ranking [82] [83]. The theoretical basis for this advantage is rooted in the law of large numbers, where the mean of repeated independent measures tends toward a true value [82]. The implementation of consensus scoring has shown superior performance in identifying active ligands across a wider range of targets and reducing false positives and negatives compared to single scoring functions [83].
However, the ultimate value of any in silico prediction lies in its correlation with experimental results. Experimental bioassays provide the essential ground truth data for validating computational predictions. The development and validation of these bioassays themselves require a rigorous framework to ensure they accurately and precisely measure the desired endpoints, such as binding affinity or inhibition of biological activity [84]. This article provides a comparative analysis of consensus scoring methodologies and their experimental validation, framed within the broader context of docking-based versus pharmacophore-based virtual screening performance.
Consensus Scoring Methodologies and Performance
Consensus scoring can be implemented through various algorithms, from simple statistical combinations to advanced machine learning models. The core principle is that integrating predictions from multiple, methodologically diverse programs can compensate for the individual weaknesses of any single method [82].
Traditional and Advanced Consensus Algorithms
Early consensus methods often involved taking the mean, median, minimum, or maximum of ranks or normalized scores from different docking programs [82] [83]. These traditional approaches have consistently demonstrated improved performance and reduced target-performance variability compared to individual docking methods [82].
More recently, sophisticated machine learning and statistical models have been applied to develop advanced consensus scores:
- Machine Learning Consensus: Unsupervised gradient boosting approaches, such as multiple additive regression trees, build ensembles of decision trees that adaptively learn to overcome the errors of existing models. This results in a weighted average of predictions that is less sensitive to noisy input [82].
- Statistical Mixture Models: This novel approach treats the multivariate distribution of multiple docking scores as a mixture of two components—one for active ligands and one for decoys. A consensus score is constructed as the posterior probability that a ligand is active given its multiple docking scores [82].
- Heterogeneous Consensus Models: Frameworks like SCORCH2 employ two separate machine learning models (e.g., XGBoost) trained on distinct datasets and curation strategies. The consensus of these models provides a more comprehensive assessment of molecular binding potential [85].
Quantitative Performance Comparison
Evaluations on benchmark datasets like DUD-E (Directory of Useful Decoys: Enhanced) have quantified the performance gains offered by consensus scoring. The table below summarizes the key advantages.
Table 1: Performance Advantages of Consensus Scoring over Individual Docking Programs
| Metric | Individual Docking Programs | Consensus Scoring Approaches | Impact on Virtual Screening |
|---|---|---|---|
| Target Robustness | High performance variability across different protein targets [82] | More robust performance and reduced target variation [82] [83] | Increases confidence in predictions for novel targets without prior experimental data |
| Predictive Accuracy | Often sub-optimal discrimination between actives and decoys [83] | Superior ligand-protein docking fidelity and improved enrichment of active compounds [82] [83] | Identifies more true hits from a large compound library, improving screening efficiency |
| False Positive/Negative Reduction | Prone to generating false positives due to scoring function limitations [71] | Reduces both false positives and false negatives [83] | Saves resources by minimizing the number of inactive compounds sent for experimental testing |
| Generalizability | Performance can be influenced by protein/ligand similarity, leading to memorization [85] | Demonstrates strong transferability and robust hit identification on unseen targets [85] | Creates more parsimonious and reliable models for broad drug discovery campaigns |
Advanced consensus models like SCORCH2 demonstrate that a physics-inspired, interaction-based featurization approach can yield superior top-tier enrichment and strong generalization, even with a lower dependency on explicit protein structural information [85].
Integrated Workflows: Combining Docking and Pharmacophore Approaches
A powerful strategy to enhance virtual screening accuracy involves integrating structure-based docking with ligand-based pharmacophore filtering. This hybrid approach leverages the strengths of both methods to create a more stringent selection process.
Pharmacophore Filtering of Docking Poses
This method uses a docking program for pose generation without regard to its native scoring, followed by filtering the generated poses with receptor-based pharmacophore searches [71]. A pharmacophore model defines the essential molecular features—such as hydrogen bond donors, acceptors, and hydrophobic regions—that a ligand must possess for optimal binding.
The workflow, illustrated in the diagram below, allows for the rapid elimination of poses that, despite a favorable docking score, are not chemically complementary to the binding site (e.g., they leave unpaired buried hydrogen bond donors or acceptors) [71].
Diagram 1: Workflow for pharmacophore filtering of docking results. Poses are generated by docking and then filtered based on a predefined pharmacophore model to eliminate chemically incompatible candidates.
Comparative Performance of Integrated Methods
Studies have shown that this integrated pharmacophore filtering method can perform better than traditional docking-and-scoring methods [71]. In one case study targeting SARS-CoV-2 papain-like protease (PLpro), a structure-based pharmacophore model was used to screen a marine natural product database. The resulting hits were then screened by comparative molecular docking using both AutoDock and AutoDock Vina. Applying consensus scoring to the results from both docking engines identified aspergillipeptide F as the top inhibitor, which was then validated by molecular dynamics simulations [3]. This demonstrates how a multi-step consensus and integration strategy can robustly identify high-quality leads.
Experimental Validation: From In Silico to In Vitro
The final and most critical step in any virtual screening campaign is the experimental validation of computational hits. This process relies on carefully designed bioassays to confirm predicted activity.
Bioassay Method Validation Framework
To ensure that bioassay results are reliable and meaningful, a rigorous validation process is required. Drawing from established practices in other scientific fields, a proposed framework for bioassay validation in vector control (with direct relevance to drug discovery) consists of four key stages [84]:
- Preliminary Development: The method is devised, and endpoints (e.g., percent inhibition, IC50) and analytical requirements are defined.
- Feasibility Experiments: Performance parameters and endpoints are verified, and a draft Standard Operating Procedure (SOP) is created.
- Internal Validation: The analytical performance of the method is tested in a single site, and a method claim is drafted.
- External Validation: The method is evaluated in multiple independent laboratories to confirm its reproducibility and fitness for purpose [84].
This structured process ensures that the bioassay used for validation is itself scientifically sound, repeatable, and capable of generating accurate data to assess the computational predictions.
Correlating Prediction Scores with Experimental Data
The correlation between in silico scores and experimental results is the ultimate measure of a virtual screening method's utility. For consensus scoring, this often involves evaluating its performance using metrics like ROC AUC (Receiver Operating Characteristic Area Under the Curve) and Enrichment Factor (EF) on benchmarks with known actives and decoys [82].
The transition to experimental validation is exemplified by workflows that use consensus scoring to select a manageable number of top-ranking compounds for in vitro testing. For instance, the identification of aspergillipeptide F via consensus pharmacophore/docking was followed by molecular dynamics simulations to quantify stability and binding free energy, providing a theoretical bridge between the static docking pose and experimental binding affinity [3]. This layered approach of computational validation strengthens the confidence in selecting compounds for costly and time-consuming wet-lab experiments.
Essential Research Reagents and Tools
The experimental protocols and computational workflows discussed rely on a suite of specialized software, databases, and laboratory materials. The following table details key components of the "scientist's toolkit" for virtual screening and validation.
Table 2: Key Research Reagent Solutions for Virtual Screening and Validation
| Category | Name | Function and Application |
|---|---|---|
| Docking Software | AutoDock Vina, GOLD, Glide, Smina [71] [83] | Programs used for predicting the binding pose and affinity of a small molecule within a protein's binding site. |
| Pharmacophore Software | MOE (Molecular Operating Environment), LigandScout [71] [86] | Tools for creating, visualizing, and searching with 3D pharmacophore models to define essential interaction features. |
| Consensus Scoring Algorithms | Gradient Boosting, Mixture Models, SCORCH2 [82] [85] | Advanced machine learning and statistical models that combine multiple scoring inputs to improve prediction reliability. |
| Benchmark Databases | DUD-E, DEKOIS 2.0 [82] [85] | Curated public databases containing protein targets, known active ligands, and property-matched decoy molecules for method benchmarking. |
| Compound Libraries | ZINC, NCI Database, Comprehensive Marine Natural Product Database (CMNPD) [71] [3] | Large collections of commercially available or naturally occurring chemical compounds used for virtual screening. |
| Experimental Assay Kits | (Assay-specific kits, e.g., kinase activity assays) | Validated biochemical kits used in laboratory experiments to measure the biological activity (e.g., inhibition) of predicted hit compounds. |
The integration of consensus scoring and rigorous experimental validation represents a powerful paradigm in modern drug discovery. While both docking-based and pharmacophore-based virtual screening have inherent strengths and weaknesses, the evidence strongly indicates that their synergistic combination, often through consensus methods, yields superior results. The implementation of machine learning and statistical mixture models for consensus scoring has demonstrably improved predictive accuracy, robustness across targets, and the reduction of false positives.
The critical role of well-validated bioassays cannot be overstated, as they provide the essential feedback loop that grounds computational predictions in biological reality. As both computational power and experimental techniques continue to advance, the correlation between in silico predictions and bioassay results will only strengthen, further accelerating the identification of novel therapeutic agents. Future developments will likely focus on even more integrated workflows, dynamic modeling of binding events, and the continued refinement of consensus algorithms to enhance the efficiency and success rate of the drug discovery pipeline.
Conclusion
The comparative analysis of pharmacophore-based and docking-based virtual screening reveals that PBVS often demonstrates superior performance in enrichment and hit rates in benchmark studies, though DBVS provides invaluable structural insights. The choice between methods is not a question of which is universally better, but which is more appropriate for a specific project's context, depending on the availability of target structure information, known active ligands, and computational resources. The most powerful and robust strategy for modern drug discovery involves their synergistic integration, either in parallel or through sequential hybrid workflows. Future directions point towards the increased incorporation of machine learning and artificial intelligence to refine scoring functions, handle ultra-large libraries, and better account for dynamic protein-ligand interactions, ultimately accelerating the discovery of novel therapeutics.
References
- 1. Pharmacophore-based virtual screening versus docking ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4007494/]
- 2. Pharmacophore-based virtual screening versus docking ... [https://www.nature.com/articles/aps2009159]
- 3. Pharmacophore model-aided virtual screening combined ... [https://www.sciencedirect.com/science/article/pii/S1878535222006505]
- 4. Machine learning accelerates pharmacophore-based ... [https://www.nature.com/articles/s41598-024-58122-7]
- 5. A New Fragment‐Based Pharmacophore Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12412281/]
- 6. Benchmarking the Structure-Based Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12363558/]
- 7. Drug Design by Pharmacophore and Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9145410/]
- 8. Concepts and Applications Exemplified on Hydroxysteroid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6332202/]
- 9. A Novel Approach for Efficient Pharmacophore-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2767445/]
- 10. Ligand-based Pharmacophore Modeling, Virtual Screening ... [https://www.sciencedirect.com/science/article/pii/S2001037018302617]
- 11. Pharmacophore-based virtual screening for identification of ... [https://www.sciencedirect.com/science/article/pii/S2667022424003499]
- 12. Pharmacophore modelling based virtual screening and ... [https://www.nature.com/articles/s41598-024-63555-1]
- 13. Docking strategies for predicting protein-ligand interactions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11583305/]
- 14. An artificial intelligence accelerated virtual screening ... [https://www.nature.com/articles/s41467-024-52061-7]
- 15. Pairwise Performance Comparison of Docking Scoring ... [https://pubmed.ncbi.nlm.nih.gov/40649292/]
- 16. Drug-target binding affinity prediction with docking pose ... [https://research.ibm.com/publications/drug-target-binding-affinity-prediction-with-docking-pose-physics]
- 17. Pose ensemble graph neural networks to improve docking ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc07875f]
- 18. Beyond rigid docking: deep learning approaches for fully ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406700/]
- 19. FeatureDock for protein-ligand docking guided by ... [https://www.nature.com/articles/s44386-025-00005-6]
- 20. -based Pharmacophore -based... virtual screening versus docking [https://www.nature.com/articles/aps2009159?error=cookies_not_supported&code=faebc76c-93ca-478e-ae5f-6e0381f98774]
- 21. Decoding the limits of deep learning in molecular docking for ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc05395a]
- 22. Pharmacophore screening, molecular docking, and MD ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1534707/full]
- 23. (PDF) Highly SpecIfic and Sensitive Pharmacophore Model for... [https://www.academia.edu/130149438/Highly_SpecIfic_and_Sensitive_Pharmacophore_Model_for_Identifying_CXCR4_Antagonists_Comparison_with_Docking_and_Shape_Matching_Virtual_Screening_Performance]
- 24. Docking guidance with experimental ligand structural density ... [https://pubmed.ncbi.nlm.nih.gov/39998966/]
- 25. A Pharmacophore-Based Method for Rapid and Accurate ... [https://pubmed.ncbi.nlm.nih.gov/40694045/]
- 26. PharmacoForge: pharmacophore generation with diffusion ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1628800/full]
- 27. Editorial: Approaches to improve the performance of virtual ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12378871/]
- 28. Integrated pharmacophore‐based screening and docking ... [https://www.sciencedirect.com/science/article/abs/pii/S3050475924000034]
- 29. A quantitative model of structure-based virtual screening ... [https://chemrxiv.org/engage/chemrxiv/article-details/69037e08113cc7cfff050553]
- 30. Pharmacophore-based virtual screening of commercial ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2024.1412349/full]
- 31. Ligand-Based and Structure-Based Pharmacophore ... [https://bio-protocol.org/exchange/minidetail?id=9587520&type=30]
- 32. Ligand based pharmacophore modelling and integrated ... [https://pubs.rsc.org/en/content/articlehtml/2024/ra/d3ra08618f]
- 33. A structure-based framework for selective inhibitor design ... [https://www.nature.com/articles/s42003-025-07840-3]
- 34. Building shape-focused pharmacophore models for effective ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00857-6]
- 35. Grid-derived structure-based 3D pharmacophores and ... [https://www.sciencedirect.com/science/article/abs/pii/S1740674910000296]
- 36. Assessing molecular docking tools: understanding drug ... [https://fjps.springeropen.com/articles/10.1186/s43094-025-00862-y]
- 37. A Comparative Study of Physics-based and AI-Driven Docking ... [https://sciety-labs.elifesciences.org/articles/by?article_doi=10.21203/rs.3.rs-7778725/v1]
- 38. LigDockTailor: Ligand-specific docking tool matching using ... [https://www.sciencedirect.com/science/article/abs/pii/S1476927125002142]
- 39. Pharmacophore-based virtual screening, molecular docking ... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-024-01350-9]
- 40. Pharmacophore model-aided virtual screening combined with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9554199/]
- 41. Conformer Generation for Structure-Based Drug Design: How Many and How Good? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10647020/]
- 42. Structure based pharmacophore modeling, virtual ... [https://www.nature.com/articles/s41598-021-83626-x]
- 43. Conformer-RL: A deep reinforcement learning library for conformer generation - PubMed [https://pubmed.ncbi.nlm.nih.gov/36000759/]
- 44. The Light and Dark Sides of Virtual Screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC6470506/]
- 45. Benchmarking different docking protocols for predicting the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10945300/]
- 46. A practical guide to large-scale docking [https://www.nature.com/articles/s41596-021-00597-z]
- 47. Fair Comparisons to Conventional Docking Workflows [https://arxiv.org/html/2412.02889v1]
- 48. Benchmarking methods and data sets for ligand ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202314003788]
- 49. Comparative analysis of pharmacophore screening tools [https://pubmed.ncbi.nlm.nih.gov/22646988/]
- 50. Comparing sixteen scoring functions for predicting ... [https://www.sciencedirect.com/science/article/abs/pii/S1093326315000285]
- 51. A Comprehensive Review on the Top 10 Molecular ... [https://parssilico.com/blogs/86-top-10-molecular-docking-softwares]
- 52. Editorial: Approaches to improve the performance of virtual ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1658699/full]
- 53. Pharmacophore-based virtual screening and in silico ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12009819/]
- 54. Generation of appropriate protein structures for virtual ... [https://www.sciencedirect.com/science/article/pii/S2950363925000286]
- 55. Docking compared to 3D-pharmacophores: the scoring ... [https://www.sciencedirect.com/science/article/abs/pii/S1740674910000557]
- 56. Protein flexibility in ligand docking and virtual screening to ... [https://pubmed.ncbi.nlm.nih.gov/15001363/]
- 57. Utilizing experimental data for reducing ensemble size in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3337782/]
- 58. Target-biased scoring approaches and expert systems in ... [https://www.sciencedirect.com/science/article/abs/pii/S136759310400078X]
- 59. Applications and Limitations of Pharmacophore Modeling [https://drugdiscoverypro.com/applications-and-limitations-of-pharmacophore-modeling/]
- 60. Applications and Limitations of Pharmacophore Modeling [https://www.linkedin.com/pulse/applications-limitations-pharmacophore-modeling-drug-discovery-pro-dyphf]
- 61. Molecular docking-based virtual screening: Challenges in ... [https://www.sciencedirect.com/org/science/article/pii/S0428029622001160]
- 62. Docking-based inverse virtual screening - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/29577065/]
- 63. Blind docking methods have been inappropriately used in ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1566772/full]
- 64. Structure based drug design and machine learning ... [https://www.nature.com/articles/s41598-025-17708-5]
- 65. AI meets physics in computational structure-based drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12226350/]
- 66. How useful is virtual screening in drug discovery? [https://synapse.patsnap.com/article/how-useful-is-virtual-screening-in-drug-discovery]
- 67. Benchmarking the Structure-Based Virtual Screening ... [https://www.dovepress.com/benchmarking-the-structure-based-virtual-screening-performance-of-wild-peer-reviewed-fulltext-article-DDDT]
- 68. Evaluating the structure-based virtual screening performance ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0318712]
- 69. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 70. Using filters in virtual screening: A comprehensive guide to ... [https://www.sciencedirect.com/science/article/abs/pii/S0065774322000100]
- 71. Combining docking with pharmacophore filtering for improved ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3152774/]
- 72. research [https://news.kaist.ac.kr/newsen/html/news/?mode=V&mng_no=14950]
- 73. Docking-based inverse virtual screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC5860130/]
- 74. Pharmacophore-based virtual screening versus docking ... [https://www.lifescience.net/publications/1654791/pharmacophore-based-virtual-screening-versus-docki/]
- 75. Novel Hybrid Virtual Screening Protocol Based on ... [https://www.mdpi.com/1422-0067/14/7/14225]
- 76. Structure-based virtual screening and biological evaluation ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523419310578]
- 77. HelixVS: Deep Learning Enhanced Structure-based Virtual ... [https://arxiv.org/html/2508.10262v1]
- 78. pharmacophore generation with diffusion models - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12451294/]
- 79. Rapid traversal of vast chemical space using machine ... [https://www.nature.com/articles/s43588-025-00777-x]
- 80. Protocol for an automated virtual screening pipeline including ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12639438/]
- 81. Redefining Breast Cancer Care by Harnessing Computational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12471285/]
- 82. Machine Learning Consensus Scoring Improves ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5872818/]
- 83. Towards Effective Consensus Scoring in Structure-Based ... [https://link.springer.com/article/10.1007/s12539-022-00546-8]
- 84. A bioassay method validation framework for laboratory and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10540336/]
- 85. SCORCH2: A Generalized Heterogeneous Consensus Model ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12622527/]
- 86. A machine learning strategy for the identification of key in ... silico [https://pmc.ncbi.nlm.nih.gov/articles/PMC10448975/]