Optimizing CoMSIA Field Combinations for Robust 3D-QSAR Models in Drug Discovery
This article provides a comprehensive guide for computational chemists and drug development professionals on optimizing Comparative Molecular Similarity Indices Analysis (CoMSIA) field combinations to enhance model performance.
Optimizing CoMSIA Field Combinations for Robust 3D-QSAR Models in Drug Discovery
Abstract
This article provides a comprehensive guide for computational chemists and drug development professionals on optimizing Comparative Molecular Similarity Indices Analysis (CoMSIA) field combinations to enhance model performance. We explore the foundational principles of CoMSIA's five molecular fields—steric, electrostatic, hydrophobic, and hydrogen bond donor/acceptor—and their biophysical significance in molecular recognition. The content covers methodological approaches for field selection, advanced optimization strategies including machine learning integration, and rigorous validation techniques using benchmark datasets. By synthesizing recent advancements, including open-source implementations and novel algorithmic integrations, this resource offers practical frameworks for constructing predictive and interpretable 3D-QSAR models that accelerate rational drug design.
Understanding CoMSIA Field Fundamentals: The Five Pillars of Molecular Interaction
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between CoMFA and CoMSIA?
CoMFA (Comparative Molecular Field Analysis) and CoMSIA (Comparative Molecular Similarity Indices Analysis) are both ligand-based, alignment-dependent 3D-QSAR methods. However, they differ fundamentally in how they calculate molecular fields and the types of fields they incorporate [1] [2].
CoMFA relies on Lennard-Jones and Coulomb potentials to compute steric and electrostatic fields. This approach can lead to abrupt changes in grid-based probe-atom interactions and is sensitive to molecular alignment [1]. In contrast, CoMSIA employs a Gaussian-type distance-dependent function to calculate similarity indices. This "softer" potential avoids singularities near atomic nuclei and eliminates the need for arbitrary energy cut-offs, making the results less sensitive to small changes in molecular orientation and placement [1] [3] [2].
Furthermore, while classic CoMFA is often limited to steric and electrostatic fields, CoMSIA typically incorporates a broader set of physicochemical properties, including hydrophobic, and hydrogen bond donor and acceptor fields, providing a more holistic view of interactions relevant to biological activity [1] [3].
Q2: Why is molecular alignment so critical in CoMSIA, and what are the common strategies?
CoMSIA is an alignment-dependent technique. The underlying assumption is that the molecules under study bind to the same biological target in a similar conformation and orientation [1]. The quality of the molecular alignment directly impacts the statistical significance and predictive power of the model, as an incorrect alignment will lead to field descriptors that do not correlate meaningfully with the biological response.
Common alignment strategies include:
- Pharmacophore-Based Alignment: Molecules are superimposed based on a common set of chemical features (pharmacophore) believed to be essential for biological activity. Tools like GALAHAD can be used to generate these pharmacophore hypotheses [4].
- Template-Based Alignment: The most active compound is often used as a template, and all other molecules are aligned to it based on a common substructure [1].
- Database Alignment: Using pre-aligned datasets from published studies, such as the steroid benchmark set, to validate new methodologies [3].
Q3: My CoMSIA model has a high fitted correlation coefficient (r²) but a low cross-validated coefficient (q²). What does this indicate?
A high r² value indicates that your model fits the training data well. However, a low q² (typically obtained through leave-one-out cross-validation) suggests that the model lacks predictive power for new, unseen compounds. This discrepancy is often a sign of overfitting, where the model has learned the noise in the training set rather than the underlying structure-activity relationship [4].
To address this, consider the following:
- Re-evaluate Molecular Alignment: An improper alignment is a common source of poor predictability.
- Check for Outliers: Identify and investigate compounds that are poorly predicted by the model. They might be misaligned or have a different binding mode.
- Optimize Field Parameters: Experiment with different combinations of the five CoMSIA fields (steric, electrostatic, hydrophobic, donor, acceptor) and grid spacing to find the most robust descriptor set [1] [3].
- Review Dataset Composition: Ensure your training set is diverse and representative, and that your test set is appropriate for validation [4].
Q4: What is the role of the attenuation factor in CoMSIA calculations?
The attenuation factor (often denoted as α) is a parameter in the Gaussian function used by CoMSIA to calculate similarity indices [3]. It controls the steepness of the Gaussian decay with distance. A lower attenuation factor results in a broader, smoother field, while a higher value makes the field more localized. The default value in many studies is 0.3, but optimizing this parameter for a specific dataset can sometimes improve model performance. The use of this Gaussian function is a key differentiator from CoMFA, as it prevents the fields from becoming infinite when a grid point is very close to an atom [2].
Troubleshooting Common CoMSIA Experimental Issues
Problem 1: Poor Statistical Model Performance (Low q² and r²)
| Potential Cause | Diagnostic Steps | Solution & Resolution |
|---|---|---|
| Incorrect Molecular Alignment [1] | Visually inspect the superimposed molecules in 3D. Check if common functional groups or the pharmacophore are well-aligned. | Re-perform the alignment using a different, well-justified method (e.g., switch from common substructure to a pharmacophore model) [4]. |
| Suboptimal Field Combination [3] | Run CoMSIA with different field combinations (e.g., Steric+Electrostatic vs. all five fields) and compare cross-validated results. | Systematically test field contributions. Exclude fields that do not improve or harm model predictivity. Refer to your thesis context of optimizing field combinations. |
| Presence of Structural Outliers [3] | Calculate the residual values for each compound. Identify structures with much higher prediction errors than the rest. | Investigate the chemical structure of the outlier. If a valid reason is found (e.g., different binding mode), consider removing it from the training set. |
| Improper Grid & Parameter Settings [1] | Check if the grid box extends at least 2.0 Å beyond all molecules in every direction. Test the impact of grid spacing (e.g., 1Å vs 2Å). | Ensure a sufficient grid margin. Use a smaller grid spacing (e.g., 1Å) for finer sampling if computationally feasible, and optimize the attenuation factor [3]. |
Problem 2: Uninterpretable or Counter-Intuitive CoMSIA Contour Maps
| Potential Cause | Diagnostic Steps | Solution & Resolution |
|---|---|---|
| Poor Quality of the Underlying Model | Confirm that the statistical performance (q² and r²) of the model is acceptable. Contour maps from a weak model are not trustworthy. | Focus on improving the model's predictivity first. The interpretability of the maps is directly linked to the model's quality. |
| Inconsistent Biological Data | Review the experimental biological data (e.g., IC₅₀, Kᵢ) for the training set. Look for large errors or inconsistencies in the data source. | If possible, use biological data determined from a single, consistent assay protocol to minimize noise [4]. |
| Incorrect Region Selection | The model might be based on noisy or irrelevant regions of the grid. | Employ variable selection methods like GOLPE or region-focused analyses to isolate the most relevant descriptor regions [2]. |
Problem 3: Software and Technical Implementation Hurdles
| Potential Cause | Diagnostic Steps | Solution & Resolution |
|---|---|---|
| Reliance on Proprietary Software | The discontinuation of commercial platforms like Sybyl creates accessibility issues [3]. | Consider migrating to open-source alternatives. Py-CoMSIA is a validated Python library that replicates the core CoMSIA algorithm and integrates with modern data science workflows [3]. |
| Errors in Preprocessing Steps | Verify each step: structure sketching, energy minimization, and partial charge calculation. | Follow a standardized protocol. Use appropriate force fields (e.g., Tripos Standard) and charge calculation methods (e.g., Gasteiger-Hückel) for consistency [1] [4]. |
Experimental Protocol: Building a Robust CoMSIA Model
The following workflow outlines the key steps for conducting a CoMSIA analysis, incorporating best practices for avoiding common errors.
Step 1: Data Set Preparation
- Select a set of molecules with a common scaffold or pharmacophore, ensuring they are presumed to act via the same mechanism [1].
- Minimize Energy: Generate a low-energy 3D conformation for each molecule. Use a force field like Tripos Standard and a convergence criterion (e.g., energy gradient < 0.01 kcal/mol) [4].
- Calculate Partial Charges: Compute partial atomic charges, for example, using the Gasteiger-Hückel method [1] [4].
Step 2: Molecular Alignment
- This is the most critical step. Superimpose all molecules based on a validated common template or pharmacophore hypothesis [1] [4]. Visually inspect the alignment for consistency.
Step 3: Grid Box Creation
- Construct a 3D grid that encompasses all aligned molecules. The grid should extend at least 2.0 Å beyond the molecular dimensions in all directions to adequately sample the interaction fields [1]. A typical grid spacing of 1.0 or 2.0 Å is used [3].
Step 4: CoMSIA Field Calculation
- Calculate the five similarity index fields using a common probe atom. Standard probe parameters are a charge of +1, hydrophobicity of +1, and hydrogen bond donor and acceptor properties of +1 [1]. The Gaussian function with a default attenuation factor of 0.3 is typically used [3].
Step 5: Statistical Analysis using PLS Regression
- Use Partial Least Squares (PLS) regression to correlate the CoMSIA field descriptors (independent variables) with the biological activity data (dependent variable) [1].
- Perform Leave-One-Out (LOO) cross-validation to determine the optimal number of components (ONC) and the cross-validated correlation coefficient
q². Aq² > 0.5is generally considered statistically significant [4]. - Build the final non-cross-validated model using the ONC to obtain the conventional correlation coefficient
r²and standard error of estimate [3].
Step 6: Model Validation
- Test the predictive power of the model by predicting the activity of an external test set of compounds that were not used in model building. The predictive
r²pred should be reasonably high [4].
Step 7: Visualization and Interpretation
- Interpret the results by visualizing the CoMSIA contour maps. These maps highlight regions where specific physicochemical properties (e.g., steric bulk, negative charge) are favorable or unfavorable for biological activity, providing a direct guide for molecular design [1] [2].
Performance Data from Benchmark Studies
The table below summarizes the results of a CoMSIA study on a benchmark steroid dataset, comparing the performance of a traditional implementation (Sybyl) with the modern open-source alternative (Py-CoMSIA). This data provides a reference for expected model performance metrics [3].
Table 1: Comparison of CoMSIA Models for a Steroid Benchmark Data Set
| Metric / Field Contribution | Published Sybyl (SEH) | Py-CoMSIA (SEH) | Py-CoMSIA (SEHAD) |
|---|---|---|---|
| q² (LOO-CV) | 0.665 | 0.609 | 0.630 |
| r² | 0.937 | 0.917 | 0.898 |
| Standard Error of Estimate (S) | 0.33 | 0.33 | 0.366 |
| Optimal Number of Components | 4 | 3 | 3 |
| Steric Contribution | 0.073 | 0.149 | 0.065 |
| Electrostatic Contribution | 0.513 | 0.534 | 0.258 |
| Hydrophobic Contribution | 0.415 | 0.316 | 0.154 |
| H-Bond Donor Contribution | - | - | 0.274 |
| H-Bond Acceptor Contribution | - | - | 0.248 |
Abbreviations: SEH: Steric, Electrostatic, Hydrophobic fields. SEHAD: Steric, Electrostatic, Hydrophobic, Acceptor, Donor fields. LOO-CV: Leave-One-Out Cross-Validation.
The Scientist's Toolkit: Essential Research Reagents & Software
Table 2: Key Resources for CoMSIA Modeling
| Tool / Resource | Category | Function & Application in CoMSIA |
|---|---|---|
| Py-CoMSIA [3] | Software Library | An open-source Python implementation of CoMSIA, providing a free and flexible alternative to discontinued proprietary software. |
| RDKit [3] | Cheminformatics Toolkit | An open-source toolkit used for cheminformatics and molecular modeling; often integrated for tasks like structure manipulation and descriptor calculation. |
| GALAHAD [4] | Pharmacophore Generation | A tool for generating pharmacophore hypotheses and molecular alignments, which are critical for the CoMSIA pre-processing step. |
| PLSR Algorithm | Statistical Tool | Partial Least Squares Regression is the core statistical method used to derive the relationship between CoMSIA fields and biological activity [1]. |
| Tripos Force Field | Molecular Mechanics | A standard force field used for energy minimization and geometry optimization of molecular structures prior to alignment [4]. |
| Gasteiger-Hückel Charges | Partial Charge Model | A method for calculating partial atomic charges, which are essential for defining the electrostatic field in CoMSIA [1] [4]. |
Frequently Asked Questions (FAQs) on CoMSIA Molecular Fields
Q1: What is the core difference between the molecular fields in CoMFA and CoMSIA?
The fundamental difference lies in how the fields are calculated and the types of interactions they represent.
- CoMFA (Comparative Molecular Field Analysis) primarily uses steric (Lennard-Jones potential) and electrostatic (Coulombic potential) fields. These fields can show sharp energy changes near the molecular surface, making the results sensitive to molecular alignment and requiring energy cut-offs [5] [1].
- CoMSIA (Comparative Molecular Similarity Indices Analysis) introduces a Gaussian-type distance-dependent function to calculate its fields. This "softer" potential avoids singularities and drastic changes, making the model less sensitive to alignment and removing the need for arbitrary cut-offs. Furthermore, CoMSIA expands the descriptor set to include hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields, providing a more holistic view of ligand-receptor interactions [3] [1].
Q2: Which combination of CoMSIA fields typically yields the most predictive model?
While the optimal combination can be project-dependent, systematic studies suggest that using more fields generally leads to better model predictivity. A statistical comparison of 23 data sets concluded that model predictive ability varied significantly depending on the set of CoMSIA fields used, with a general trend of improved predictivity as more molecular fields are included [6]. The study also found that when all five fields are used, the hydrophobic and electrostatic fields often contribute the most, while the steric field tends to contribute the least [6]. It is therefore recommended to start with an all-five-field model and then refine based on statistical significance and field contribution plots.
Q3: How can I troubleshoot a CoMSIA model with a low cross-validated correlation coefficient (q²)?
A low q² value often points to issues with the molecular alignment or the chosen conformation. Below is a troubleshooting guide for this common problem.
| Potential Issue | Diagnostic Steps | Recommended Solution |
|---|---|---|
| Poor Molecular Alignment | Check if pharmacophore features or key structural scaffolds are misaligned. | Switch from a simple common substructure alignment to a more sophisticated pharmacophore-based alignment (e.g., using tools like GALAHAD) or a protein-binding site guided alignment if structural data is available [7]. |
| Suboptimal Bioactive Conformation | The chosen low-energy conformation might not represent the binding mode. | If crystal structures of ligand complexes are available, use them as a template to generate the theoretical active conformers for the entire dataset [8]. |
| Incorrect Field Parameters | The Gaussian attenuation factor or grid spacing might be unsuitable. | Systematically test different attenuation factors (default is 0.3) and reduce grid spacing (e.g., from 2.0 Å to 1.0 Å) to improve the model's resolution [7] [9]. |
Q4: What are the critical steps in the CoMSIA methodology to ensure a robust model?
A robust CoMSIA model relies on a rigorous workflow, from compound preparation to statistical validation. The diagram below outlines the key procedural stages.
Q5: How do I interpret a CoMSIA hydrophobic contour map compared to a CoMFA steric map?
This is a crucial distinction for understanding design implications.
- A CoMFA steric map (e.g., green contours) indicates regions in space around the molecules where bulky groups are favored or disfavored by a hypothetical receptor environment [5] [1].
- A CoMSIA hydrophobic map (e.g., yellow and white contours) indicates areas within the region occupied by the ligands themselves that favor or dislike hydrophobic groups. A yellow contour signifies where hydrophobic substituents enhance activity, while a white contour suggests hydrophobic groups are detrimental [1]. This provides a more direct guide for modifying the ligand's physicochemical properties.
Quantitative Data on CoMSIA Field Performance
The following table summarizes quantitative data from published CoMSIA studies, illustrating the performance achievable with different field combinations.
Table 1: Performance Metrics from Benchmark CoMSIA Studies
| Study Compound Series / Target | Field Combination | Cross-validated q² | Non-cross-validated r² | Field Contributions (Excerpt) |
|---|---|---|---|---|
| Steroid Benchmark Dataset [3] | SEH (Steric, Electrostatic, Hydrophobic) | 0.609 | 0.917 | Steric: 0.149, Electrostatic: 0.534, Hydrophobic: 0.316 |
| Steroid Benchmark Dataset [3] | SEHAD (All Five Fields) | 0.630 | 0.898 | Steric: 0.065, Electrostatic: 0.258, Hydrophobic: 0.154, H-Bond Donor: 0.274, H-Bond Acceptor: 0.248 |
| α1A-Adrenergic Receptor Antagonists [7] | All Five Fields | 0.840 | 0.940 (for CoMSIA) | Information obtained from 3D contour maps. |
| 1,2-dihydropyridine Anticancer Agents [9] | Not Specified | 0.639 | Not Reported | Model used to design a new compound with submicromolar activity. |
| Thiazolone HCV Inhibitors [10] | Not Specified | 0.685 | 0.940 | Model validated with a test set (r²pred = 0.822). |
The Scientist's Toolkit: Essential Reagents & Software for CoMSIA
Table 2: Key Research Reagent Solutions for CoMSIA Studies
| Item / Resource | Function / Application in CoMSIA |
|---|---|
| Molecular Modeling Software (e.g., SYBYL, Schrödinger, MOE) | Provides the integrated computational environment for molecule sketching, conformational analysis, energy minimization, molecular alignment, CoMSIA field calculation, and Partial Least Squares (PLS) regression [3] [8] [7]. |
| Open-Source Python Libraries (e.g., Py-CoMSIA, RDKit, NumPy) | Offers a non-proprietary alternative for implementing the core CoMSIA algorithm, calculating similarity indices, and generating 3D contour maps, enhancing accessibility and customization [3]. |
| Semi-Empirical Quantum Mechanics Programs (e.g., MOPAC/AM1) | Used for geometry optimization and partial charge calculation (e.g., VESPA charges) to ensure high-quality and comparable 3D molecular structures before alignment and field calculation [9]. |
| Pharmacophore Generation Tools (e.g., GALAHAD) | Assists in deriving the critical molecular alignment rule by identifying common pharmacophoric features across active molecules, which is often superior to simple common substructure alignment [7]. |
| Partial Atomic Charges | Assigned to each atom to define the molecular electrostatic potential, which is critical for calculating the electrostatic field. Common methods include Gasteiger-Hückel or Gasteiger-Marsili [7] [9]. |
Advanced Protocol: Optimizing CoMSIA Field Combinations
A systematic approach to selecting the best CoMSIA field combination can significantly enhance model performance. The following diagram and protocol outline this process.
Detailed Protocol:
Initial Model Construction: Begin by constructing a CoMSIA model using all five molecular fields (steric, electrostatic, hydrophobic, hydrogen bond donor, hydrogen bond acceptor) on your training set. Use a consistent and well-validated molecular alignment [7].
Statistical Analysis and Field Contribution Assessment: Run a Partial Least Squares (PLS) analysis with leave-one-out (LOO) cross-validation. Record the cross-validated correlation coefficient (q²), the optimal number of components, the non-cross-validated correlation coefficient (r²), and the relative contribution of each molecular field to the model [3] [6].
Iterative Field Elimination: Systematically create new models by removing the field with the lowest contribution from the previous model. For example, if the steric field contribution is the lowest in the all-five-field model, build a new four-field model (electrostatic, hydrophobic, HBD, HBA) and record all statistical parameters [6].
Model Comparison and Selection: Compare the predictive ability of all generated models. The model with the highest q² and the highest predictive r² (r²pred) for a test set of compounds is generally preferred. A study on 23 datasets found that predictive ability varied significantly with the field set used, and often, models with more fields performed better [6].
Validation and Application: Use the selected optimal model to predict the activity of external test set compounds that were not used in model building. The final model, with its contour maps, can then guide the rational design of new compounds with improved potency [10] [9].
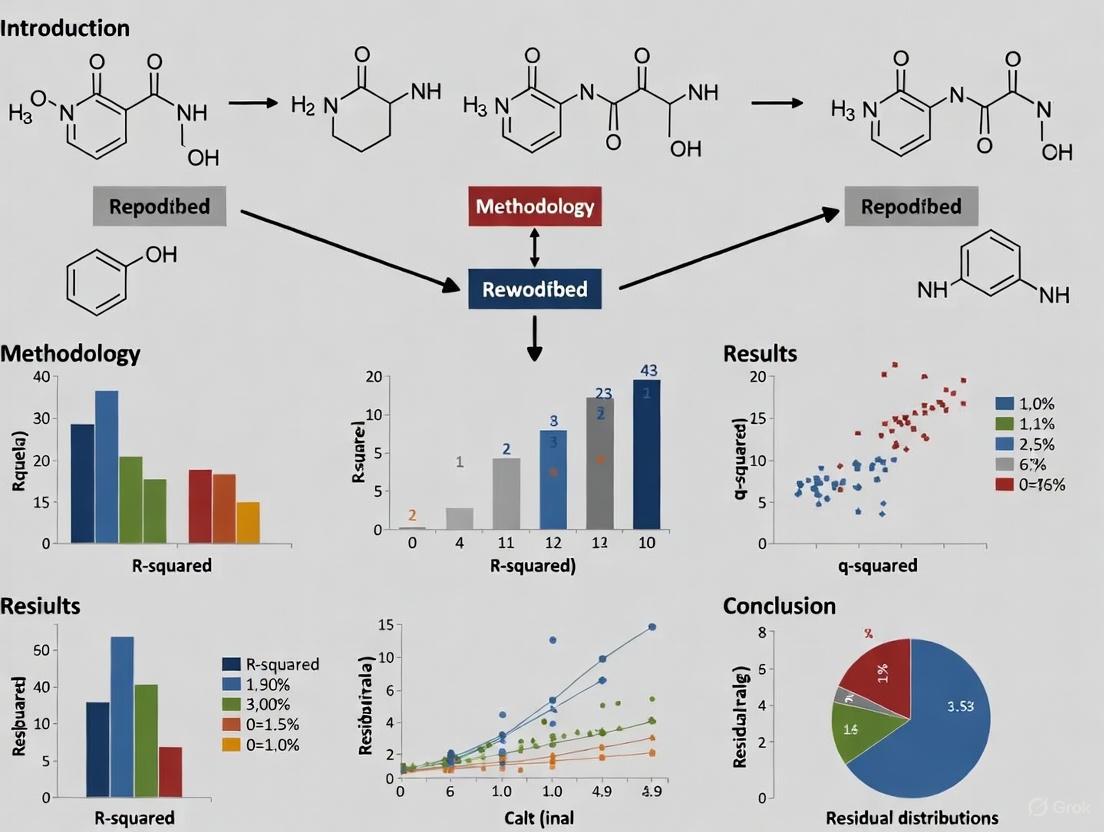
Comparative Molecular Similarity Indices Analysis (CoMSIA) is an advanced 3D-QSAR technique that maps key molecular forces governing biological interactions. Unlike its predecessor CoMFA, CoMSIA employs a Gaussian function to calculate molecular similarity indices, generating continuous molecular similarity maps that avoid the sharp, non-physical cutoffs observed in CoMFA models. This approach provides a more holistic view of the molecular determinants underlying biological activity by incorporating five distinct physicochemical fields: steric (S), electrostatic (E), hydrophobic (H), hydrogen bond donor (D), and hydrogen bond acceptor (A). The selection of appropriate field combinations is crucial for creating predictive models that accurately map to specific biological recognition events in drug discovery.
Troubleshooting Guide: CoMSIA Field Combinations
FAQ 1: What is the scientific basis for selecting specific CoMSIA field combinations?
Answer: The selection of CoMSIA fields should be guided by the specific nature of the target receptor-ligand interaction. Each field represents a distinct physicochemical force that drives molecular recognition:
- Steric (S) and Electrostatic (E) Fields: These fundamental fields correspond to van der Waals and Coulombic interactions, respectively. They are essential for modeling most receptor-ligand interactions where shape complementarity and charge distribution play critical roles [3] [1].
- Hydrophobic (H) Field: This field incorporates solvent-reliant molecular entropic terms, making it crucial for modeling interactions where desolvation and hydrophobic effects significantly contribute to binding affinity [1].
- Hydrogen Bond Donor (D) and Acceptor (A) Fields: These fields specifically model directional hydrogen bonding interactions, which are often critical for ligand specificity and potency [3] [1].
Systematic statistical comparisons across 23 datasets have demonstrated that models incorporating greater numbers of CoMSIA fields generally show improved predictivity, with hydrophobic and electrostatic fields typically contributing most significantly to model performance [6].
FAQ 2: How do different field combinations affect model performance and biological interpretation?
Answer: Different field combinations directly impact both statistical performance and the biological relevance of CoMSIA models. The table below summarizes findings from systematic studies:
Table 1: Performance Characteristics of Common CoMSIA Field Combinations
| Field Combination | Typical Application Context | Statistical Performance | Biological Mapping |
|---|---|---|---|
| SEH | Standard combination for general QSAR | High predictivity (q² = 0.609 in steroid benchmark) [3] | Maps steric complementarity, electrostatic attraction/repulsion, and hydrophobic binding pockets |
| SEHAD | Comprehensive modeling of complex interactions | Good predictivity (q² = 0.630 in steroid benchmark) [3] | Adds specific mapping of hydrogen bonding networks to receptor interactions |
| EAH | Polar interactions-dominated systems | Varies by system; may outperform in specific cases [11] | Focuses on charge-based, hydrophobic, and hydrogen acceptor interactions |
| All Five Fields | Maximum descriptor information | Generally highest predictivity [6] | Provides complete mapping of multiple interaction types simultaneously |
Research indicates that including all five fields typically yields the most predictive models, with hydrophobic and electrostatic fields generally contributing most significantly, while the steric field often shows the smallest contribution [6]. However, field redundancy should be considered, as some fields may contain overlapping information.
FAQ 3: What methodology should I follow to systematically evaluate different field combinations?
Answer: Follow this established experimental protocol to optimize field combinations for your specific dataset:
Table 2: Essential Research Reagents and Computational Tools for CoMSIA
| Reagent/Software Tool | Function in CoMSIA Analysis | Implementation Example |
|---|---|---|
| Molecular Dataset | Training and test compounds with known biological activities | 21 steroid training + 10 test molecules [3] |
| Alignment Tool | Structural superposition of molecules based on pharmacophore | SYBYL-X 2.1, GALAHAD [12] [7] |
| Grid Generation | Creates 3D lattice for field calculation | 1-2 Å spacing, 4 Å padding beyond molecular dimensions [3] |
| Partial Least Squares (PLS) | Correlates field descriptors with biological activity | Leave-one-out cross-validation to determine optimal components [3] [7] |
| Visualization Software | Interprets contour maps for structural optimization | PyVista, SYBYL [3] |
Experimental Protocol:
- Dataset Preparation: Select a congeneric series of molecules with known biological activities and divide them into training and test sets (typically 70-80% for training) [7].
- Molecular Alignment: Superimpose molecules using a common pharmacophore or structural template. Alignment is critical as CoMSIA is alignment-dependent [1] [7].
- Grid Generation: Create a 3D grid around the aligned molecules with recommended spacing of 1-2 Å and extension of 2-4 Å beyond molecular dimensions in all directions [3].
- Field Calculation: Calculate all five CoMSIA fields using a common probe atom (typically an sp³ carbon with +1 charge). The Gaussian function with attenuation factor of 0.3 is standard [3].
- Model Construction & Validation: Build separate CoMSIA models using different field combinations. Validate using leave-one-out cross-validation to obtain q² values and predict test set compounds to obtain r²pred values [3] [7].
- Contour Map Analysis: Interpret the resulting contour maps to identify regions where specific molecular properties enhance or diminish biological activity.
FAQ 4: What are the common pitfalls in field selection, and how can I address them?
Answer: Common issues and their solutions include:
Problem: Overfitting with Too Many Fields Solution: Use cross-validation statistics (q²) and external test set prediction (r²pred) to identify truly predictive models. If adding fields doesn't improve test set prediction, the model may be overfit.
Problem: Low Predictive Power (q² < 0.3) Solution: Verify molecular alignment, which significantly impacts results. Consider alternative alignment methods such as pharmacophore-based alignment or docking-based alignment [7].
Problem: Biologically Implausible Contour Maps Solution: Ensure field combinations match the expected interaction chemistry of your target. For example, if your target has known hydrogen bonding residues, include D and A fields in your analysis.
Problem: Inconsistent Field Contributions Solution: Systematically test different field combinations as shown in Table 1. Research demonstrates that different field combinations work best for different biological targets [6].
Advanced Applications: Field Combinations in Practice
Case Study: Anti-Gout Inhibitor Development
In a study on triazole derivatives as xanthine oxidase inhibitors, researchers successfully developed CoMFA and CoMSIA models to identify key structural features enhancing biological activity. The models revealed that modifying substituents played a critical role in enhancing anti-gout inhibitory activity. Molecular docking complemented the CoMSIA analysis by showing specific interactions with enzyme residues, including hydrogen bonds with SER 69 and ASN 71, and hydrophobic interactions with ALA 70, LEU 74, and ALA 75 [12].
Extended Applications: Protein Mutations and Binding Affinity
The CoMSIA paradigm has been extended beyond small molecules to model the effects of protein mutations in SARS-CoV-2 variants. The MB-QSAR approach treats mutations as perturbations to physicochemical fields at protein interaction interfaces, successfully predicting changes in binding affinity to human ACE2 receptor and antibody escape potential. This demonstrates how field-based analysis can map to complex biological recognition events, achieving correlation coefficients (r²) exceeding 0.8 for hACE2 binding affinity [13].
Scientist's Toolkit: Essential Research Reagents and Software
The following table details the key computational tools and their functions required to perform a Py-CoMSIA analysis.
Table: Essential Components for a Py-CoMSIA Workflow
| Component Name | Type | Primary Function |
|---|---|---|
| Py-CoMSIA | Core Library | Pythonic implementation of the CoMSIA algorithm for calculating molecular similarity fields and building 3D-QSAR models [14]. |
| RDKit | Dependency (Chemistry) | Handles core cheminformatics tasks, including molecular structure manipulation, conformational analysis, and descriptor calculation [3]. |
| NumPy | Dependency (Computation) | Provides support for large, multi-dimensional arrays and matrices, enabling the high-performance mathematical operations required for field calculations [3]. |
| PyVista | Dependency (Visualization) | Generates 3D visualizations and molecular field maps for interpreting the results of the CoMSIA analysis [3]. |
| Partial Least Squares (PLS) | Statistical Method | The core regression technique used to correlate the molecular similarity fields with biological activity data [3]. |
Experimental Protocol: Benchmarking Py-CoMSIA with the Steroid Dataset
This protocol outlines the methodology for validating Py-CoMSIA and optimizing field combinations, as demonstrated in the foundational research [3].
Dataset Preparation and Molecular Alignment
- Dataset: Use the benchmark steroid dataset, which includes 31 molecules (21 for training, 10 for testing) and their associated binding affinities [3].
- Molecular Alignment: Employ a pre-aligned dataset or perform a common substructure alignment to ensure all molecules are superimposed in 3D space based on their shared pharmacophoric features. Consistent alignment is critical for a meaningful comparison of molecular fields [3].
CoMSIA Field Calculation
- Grid Setup: Define a 3D grid that encompasses all aligned molecules. Use a grid spacing of 1 Å and a padding of 4 Å beyond the molecular dimensions [3].
- Field Types: Calculate up to five distinct molecular similarity fields using a Gaussian function with a default attenuation factor (
α) of 0.3 [3] [15]:- Steric
- Electrostatic
- Hydrophobic
- Hydrogen bond Donor
- Hydrogen bond Acceptor
- Field Combinations: For model optimization, systematically test different field combinations, such as SEH (steric, electrostatic, hydrophobic) and SEHAD (all five fields) [3].
Model Building and Validation
- Partial Least Squares (PLS) Regression: Use PLS to build a model correlating the CoMSIA field descriptors with the biological activity values.
- Leave-One-Out Cross-Validation (LOOCV): Perform LOOCV on the training set to determine the optimal number of PLS components. Select the component count that yields the highest cross-validated correlation coefficient (
q²) [3]. - Model Evaluation: Assess the model's performance using the following key metrics:
q²: The cross-validated correlation coefficient, indicating model predictivity.r²: The non-cross-validated correlation coefficient, indicating the model's goodness-of-fit for the training data.SPRESS: The Standard Error of Prediction from the cross-validation.r²pred: The predictiver²for the external test set, which is a crucial measure of the model's external validity [3].
Performance Data: Quantitative Analysis of Field Combinations
The performance of Py-CoMSIA was quantitatively validated against proprietary software (Sybyl) using the steroid benchmark dataset. The table below compares key statistical metrics for different field combinations.
Table: Performance Comparison of CoMSIA Field Combinations on the Steroid Dataset [3]
| Metric | Published (SEH) | Py-CoMSIA (SEH) | Py-CoMSIA (SEHAD) |
|---|---|---|---|
| q² | 0.665 | 0.609 | 0.630 |
| r² | 0.937 | 0.917 | 0.898 |
| SPRESS | 0.759 | 0.718 | 0.698 |
| Standard Error (S) | 0.33 | 0.33 | 0.366 |
| No. of Components | 4 | 3 | 3 |
| Field Contributions | |||
| Steric | 0.073 | 0.149 | 0.065 |
| Electrostatic | 0.513 | 0.534 | 0.258 |
| Hydrophobic | 0.415 | 0.316 | 0.154 |
| Hydrogen Bond Donor | - | - | 0.274 |
| Hydrogen Bond Acceptor | - | - | 0.248 |
Technical Support Center: Troubleshooting Guides and FAQs
Frequently Asked Questions (FAQs)
Q1: What are the key advantages of using Py-CoMSIA over traditional CoMFA? Py-CoMSIA offers several key improvements. It uses a Gaussian function to calculate molecular similarity indices, which eliminates the abrupt, non-physical cutoffs seen in CoMFA and results in smoother, more interpretable contour maps. Furthermore, Py-CoMSIA is less sensitive to molecular alignment and grid spacing parameters. Crucially, it incorporates five different molecular fields (steric, electrostatic, hydrophobic, and hydrogen bond donor/acceptor), providing a more holistic view of ligand-target interactions compared to CoMFA's primary focus on steric and electrostatic fields [3] [15].
Q2: My model shows a high r² for the training set but a low r²pred for the test set. What does this indicate and how can I address it?
This is a classic sign of model overfitting, meaning your model has memorized the training data noise instead of learning the generalizable structure-activity relationship. To address this:
- Re-evaluate Field Selection: The SEHAD model in the benchmark study showed a lower predictive
r²(0.186) compared to the SEH model (0.319). Systematically test different field combinations to find the most robust set for your specific data [3]. - Check for Outliers: Identify and investigate potential outlier molecules that may be unduly influencing the model. The benchmark study correctly identified compound 10 as an outlier [3].
- Validate Alignment: Ensure the molecular alignment is biologically relevant, as poor alignment is a major source of poor external predictivity [3].
Q3: How do I choose the optimal number of components in the PLS analysis?
The optimal number of components is determined through cross-validation. You should use Leave-One-Out Cross-Validation (LOOCV) on your training set and select the number of components that yields the highest q² value. The benchmark analysis, for instance, found that three components were optimal for both SEH and SEHAD models, whereas the original proprietary software used four [3].
Troubleshooting Guide
| Problem | Potential Cause | Solution |
|---|---|---|
Low q² value |
1. Incorrect molecular alignment.2. Suboptimal field combination.3. Excessive noise in the activity data. | 1. Re-examine and refine the molecular superposition strategy.2. Test different field combinations (e.g., SE, SEH, SEHAD).3. Review the experimental data for inconsistencies. |
High r² but low r²pred (Overfitting) |
1. Too many PLS components.2. The model includes non-predictive fields for the specific activity.3. Test set is not well-represented by the training set. | 1. Use LOOCV to find the optimal number of components.2. Systematically remove fields with low contribution and re-evaluate prediction.3. Ensure the training and test sets cover similar chemical space. |
| Uninterpretable or noisy contour maps | 1. Poor molecular alignment.2. Grid spacing is too coarse or too fine. | 1. Verify the alignment is based on a common, relevant scaffold or pharmacophore.2. Adjust the grid spacing (e.g., try 1.0 Å or 2.0 Å) and observe the impact on map clarity. |
Workflow and Field Contribution Diagrams
Py-CoMSIA Analysis Workflow
CoMSIA Molecular Field Contributions
Strategic Field Selection and Implementation for Diverse Molecular Targets
A technical guide for optimizing your CoMSIA models
This resource provides targeted troubleshooting guides and FAQs to help researchers navigate the critical decisions involved in selecting and optimizing Comparative Molecular Similarity Indices Analysis (CoMSIA) field combinations for robust 3D-QSAR models.
Frequently Asked Questions
1. Which field combination should I use for a new target: SEH or SEHAD?
Start with the SEH (Steric, Electrostatic, Hydrophobic) combination. This trio covers the most fundamental intermolecular interactions. A benchmark study on a steroid dataset demonstrated that a model with SEH fields produced a better predictive r² (0.319) compared to a full SEHAD model (0.186) [3]. The SEH model also showed a more robust performance with lower residuals and a comparable cross-validated q² (0.609 for SEH vs. 0.630 for SEHAD) [3]. Use the full SEHAD set when your biological target is known to be heavily dependent on hydrogen bonding, or if the SEH model shows poor performance and you suspect these interactions are critical.
2. Why does my CoMSIA model have poor predictive power even with the SEHAD field set?
Poor predictive power often stems from molecular alignment errors, not just the field selection. The alignment of your molecules is a cornerstone of CoMSIA; even the best field set will fail if the spatial arrangement is incorrect. One study on α1A-AR antagonists achieved highly predictive models (q² = 0.840) by using a pharmacophore-based alignment generated by GALAHAD, which optimally superposed key molecular features [4] [7]. Before adjusting fields, re-investigate your alignment method. Pharmacophore-based alignments are often superior to simple common scaffold overlays, especially for structurally diverse compounds [4].
3. The contour maps from my model are noisy and hard to interpret. How can I improve them?
This is a common issue. First, ensure you are using the Gaussian function inherent to CoMSIA, which naturally produces smoother and more interpretable maps than the potential functions used in older methods like CoMFA [2]. The Gaussian function avoids abrupt changes in field values, leading to less fragmented contours [16] [2]. If maps remain noisy, review your attenuation factor (α), which has a default value of 0.3. This parameter controls the slope of the Gaussian function; a larger value results in a steeper function and stronger attenuation of effects with distance, which can help average local features and simplify the maps [16].
4. How do I know the individual contribution of each field in my model?
After running the Partial Least Squares (PLS) analysis in your CoMSIA software, the model output will provide a table of field contributions. This table shows the relative contribution (often as a proportion) of each field (steric, electrostatic, hydrophobic, donor, acceptor) to the final model. For example, in the steroid benchmark, the SEH model showed contributions of 14.9% (steric), 53.4% (electrostatic), and 31.6% (hydrophobic) [3]. Analyzing these values helps you understand which physicochemical forces are most critical for your dataset's biological activity.
Troubleshooting Guide
| Problem Area | Common Symptoms | Probable Causes & Solutions |
|---|---|---|
| Field Selection | • Low q² & r² values with SEH• Model fails to explain known SAR | • Cause: Missing key interactions (e.g., H-bonding).• Solution: Switch from SEH to SEHAD or a custom set. |
| Model Overfitting | • High q² but very low predictive r² (r²ᵩᵣₑ𝒹)• Excessively high number of optimal components | • Cause: Too many fields/descriptors for a small dataset.• Solution: Use cross-validation to find optimal components; prefer simpler SEH model if performance is comparable. |
| Contour Map Interpretation | • Maps are noisy, fragmented, and lack clear regions | • Cause: Suboptimal alignment or incorrect attenuation factor.• Solution: Verify molecular alignment; adjust the Gaussian attenuation factor (default is 0.3) [16]. |
| Statistical Significance | • Poor cross-validated correlation coefficient (q²) | • Cause: Incorrect data set division or spatial alignment.• Solution: Ensure a representative training/test set split; re-check the alignment of all molecules. |
Performance Comparison of Standard Field Sets
The following table summarizes quantitative performance metrics from a benchmark CoMSIA study on a steroid dataset, comparing the SEH and SEHAD field combinations [3].
| Performance Metric | SEH Field Set | SEHAD Field Set |
|---|---|---|
| Cross-validated q² | 0.609 | 0.630 |
| Non-cross-validated r² | 0.917 | 0.898 |
| Standard Error (S) | 0.33 | 0.366 |
| Optimal Number of Components | 3 | 3 |
| Predictive r² (on test set) | 0.319 | 0.186 |
| Field Contributions | • Steric: 14.9%• Electrostatic: 53.4%• Hydrophobic: 31.6% | • Steric: 6.5%• Electrostatic: 25.8%• Hydrophobic: 15.4%• Donor: 27.4%• Acceptor: 24.8% |
Source: Py-CoMSIA validation study (2025) [3]
Experimental Protocol: Building a Robust CoMSIA Model
Below is a generalized workflow for developing a CoMSIA model, from data preparation to validation, highlighting steps critical for field combination strategy.
Step-by-Step Methodology:
- Dataset Curation and Preparation: A set of 32 N-aryl and N-heteroaryl piperazine α1A-AR antagonists with known binding affinity (pKi) was collected. Their 3D structures were sketched and energy-minimized using the Tripos standard force field with Gasteiger-Hückel atomic partial charges [4] [7].
- Critical Molecular Alignment: All compounds were aligned to a common pharmacophore model using GALAHAD, a superior method for aligning structurally diverse compounds that share few commonalities [4] [7].
- Training/Test Set Division: The dataset was divided into a training set of 32 compounds to build the model and a test set of 12 compounds to validate its predictive power, ensuring both sets covered a wide range of biological activity and structural diversity [7].
- Grid Generation and Field Calculation: A 3D cubic lattice with a grid spacing of 1.0 Å was generated to enclose the aligned molecules. A probe atom with a charge of +1, hydrophobicity of +1, and H-bond donor/acceptor properties of +1 was placed at each grid point. The CoMSIA similarity indices for the selected fields (e.g., SEH or SEHAD) were calculated using a Gaussian function with a default attenuation factor of 0.3 [16] [7].
- Model Building and Validation: Partial Least Squares (PLS) regression was applied. The optimal number of components was determined via leave-one-out (LOO) cross-validation, yielding the cross-validated correlation coefficient, q². A final model was then developed using the optimal number of components and validated by predicting the activity of the external test set, giving the predictive r²ₚᵣₑ𝒹 [3] [7].
Research Reagent Solutions
| Essential Material / Software | Function in CoMSIA Workflow |
|---|---|
| SYBYL (Tripos) | The classic, proprietary software that originally implemented CoMSIA; used for molecular modeling, alignment, and analysis [16] [7]. |
| Py-CoMSIA | An open-source Python library providing a functional alternative to proprietary CoMSIA software, implementing the core algorithm and visualization [3]. |
| RDKit & NumPy | Open-source Python libraries used by Py-CoMSIA for fundamental chemical calculations and numerical operations [3]. |
| GALAHAD (Tripos) | A tool used to generate pharmacophore-based molecular alignments, which are crucial for robust 3D-QSAR models [4] [7]. |
| Gaussian Function | The mathematical function used in CoMSIA (as opposed to Lennard-Jones/Coulomb in CoMFA) to calculate similarity indices, preventing singularities and producing smoother contour maps [16] [2]. |
| Probe Atom | A conceptual atom (typically an sp³ carbon with specific properties) placed at grid points to measure interaction fields with the molecules [16] [7]. |
Dataset Fundamentals and Historical Context
What is the Steroid Benchmark Dataset and why is it a cornerstone for 3D-QSAR validation?
The Steroid Benchmark Dataset is a extensively curated collection of steroids with known affinity for Sex Hormone-Binding Globulin (SHBG). It has been widely used for decades to validate popular molecular field-based QSAR techniques, including CoMFA and CoMSIA [17] [18]. Its longevity as a benchmark stems from its well-characterized biological activities and structural diversity, providing a standard for comparing the performance and predictive power of new computational models and methodologies [18]. For instance, it was central to the original CoMSIA analysis paper and continues to be used in modern implementations, such as the validation of the open-source Py-CoMSIA software [3] [15].
What does the "updated steroid benchmark set" include?
Research has expanded the classic dataset by incorporating nonsteroidal SHBG ligands identified from the literature and experimental studies. This updated molecular set helps develop more robust QSAR models and provides deeper insight into protein-ligand interactions. Surprisingly, alignments generated by docking active compounds into the SHBG active site have contradicted classical ligand-based alignments yet yielded models with higher statistical significance and predictive power [17].
Field Combination Optimization
Which CoMSIA field combinations are most effective for the steroid dataset?
Performance varies by dataset, but analyses on the steroid benchmark provide clear guidance. The table below summarizes a comparative performance analysis of different field combinations [3] [15]:
Table 1: Performance Metrics of CoMSIA Field Combinations on a Steroid Benchmark Dataset
| Field Combination | q² (LOOCV) | r² | Optimal Components | Key Field Contributions |
|---|---|---|---|---|
| SEH (Steric, Electrostatic, Hydrophobic) | 0.609 | 0.917 | 3 | Electrostatic (53.4%), Hydrophobic (31.6%), Steric (14.9%) |
| SEHAD (All Five Fields) | 0.630 | 0.898 | 3 | Electrostatic (25.8%), H-Bond Acceptor (24.8%), H-Bond Donor (27.4%), Hydrophobic (15.4%), Steric (6.5%) |
| Published SEH (Sybyl) | 0.665 | 0.937 | 4 | Electrostatic (51.3%), Hydrophobic (41.5%), Steric (7.3%) |
How do I interpret these results to select the best fields for my model?
The SEH model often provides a robust and predictive baseline, with electrostatic and hydrophobic interactions being dominant drivers for steroid-SHBG binding [3] [15]. While including all five fields (SEHAD) can yield a good cross-validated q², it may sometimes lead to a less robust model with lower predictive r², potentially due to overparameterization or increased model complexity [3] [15]. The workflow for this optimization process is systematic:
Troubleshooting Common Model Performance Issues
My CoMSIA model has a high r² but a low q². What does this indicate and how can I fix it?
A high goodness-of-fit (r²) coupled with a low cross-validated correlation coefficient (q²) is a classic sign of overfitting. This means your model fits the training data well but lacks predictive power for new compounds. To address this:
- Apply Variable Selection: Use algorithms like the Enhanced Replacement Method (ERM) or Genetic Algorithms (GA) to filter out noisy, non-informative variables from the thousands of calculated interaction fields. This can significantly improve the model's predictivity [19].
- Re-evaluate Molecular Alignment: The alignment of molecules is a very sensitive step in CoMSIA. Ensure your alignment strategy is sound and physiologically relevant. Consider using docking-based alignment if ligand-based alignment yields poor results [17] [20].
- Validate with an External Test Set: An independent test set is necessary to truly judge the predictivity of a model. Robust models must demonstrate accurate predictions on an external set that was not used in model building [19].
My model's predictive power is highly sensitive to small changes in molecular orientation within the grid. What can I do?
This was a known challenge in older methods like CoMFA. A key advantage of CoMSIA is that it uses a Gaussian-type function to calculate molecular similarity indices, which makes the model less sensitive to factors like molecular alignment, grid spacing, and probe atom selection compared to CoMFA [3] [15]. If you are using CoMSIA and still experience high sensitivity, you can employ an All-Orientation Search (AOS) strategy, which systematically tests rotations and translations of the molecular aggregate within the grid to find the sampling with the highest q² value [19].
Essential Research Reagents and Computational Tools
Table 2: Research Reagent Solutions for CoMSIA Studies
| Tool / Reagent | Category | Function in Analysis | Example / Note |
|---|---|---|---|
| Py-CoMSIA | Software Library | An open-source Python implementation of CoMSIA, increasing accessibility and flexibility for researchers. | Replicates core CoMSIA algorithm; allows integration with advanced ML techniques [3] [15]. |
| Aligned Molecular Dataset | Data | A pre-aligned set of molecules is the foundational input for any 3D-QSAR study. | The Sybyl pre-aligned steroid dataset from Coats' study is a classic example [3] [15]. |
| Partial Least Squares (PLS) Regression | Statistical Algorithm | The primary method for correlating the CoMSIA fields (independent variables) with biological activity (dependent variable). | Often coupled with Leave-One-Out Cross-Validation (LOOCV) to determine optimal components [3] [15]. |
| Variable Selection Algorithms (e.g., ERM, GA) | Computational Method | Identify and select the most informative variables from the CoMSIA fields, improving model predictivity and robustness. | Enhanced Replacement Method (ERM) has shown noticeable improvement on statistical parameters [19]. |
| Docking Software | Computational Tool | Generates structure-based molecular alignments by placing compounds into the target's active site. | Can provide alternative, sometimes superior, alignments compared to ligand-based methods [17]. |
Comparative Molecular Similarity Indices Analysis (CoMSIA) is an advanced three-dimensional quantitative structure-activity relationship (3D-QSAR) technique that significantly contributes to medicinal chemistry and pharmaceutical discovery [3]. Unlike earlier methodologies, CoMSIA incorporates a broader range of molecular descriptors encompassing five distinct field types: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields [3]. This comprehensive approach addresses key interactions often overlooked by previous methods, particularly in cases where hydrophobic forces or hydrogen bonding dominate receptor-ligand recognition.
A critical advancement in CoMSIA is its use of a Gaussian function to calculate molecular similarity indices, which generates continuous molecular similarity maps and eliminates the sharp, non-physical cutoffs that complicated earlier models like CoMFA [3]. This methodological enhancement makes CoMSIA models less sensitive to molecular alignment, grid spacing, and probe atom selection, providing more robust and interpretable results for drug discovery professionals [3].
The optimization of CoMSIA field combinations represents a crucial research focus for improving model performance and predictive capability. By systematically evaluating different field combinations, researchers can identify the most relevant molecular interaction fields for specific target classes, leading to more accurate activity predictions and better-informed molecular design strategies.
Troubleshooting Common CoMSIA Model Performance Issues
FAQ: Why does my CoMSIA model show high cross-validated q² but poor external predictive ability?
Issue: Discrepancy between internal validation metrics and external prediction performance.
Solutions:
- Re-evaluate molecular alignment: Ensure proper superposition of pharmacophoric elements across all compounds, as CoMSIA is sensitive to alignment quality [3].
- Analyze field contributions: Check if field contributions align with known target binding requirements. For GPCR targets, hydrophobic and hydrogen bond fields often prove critical [21].
- Validate test set selection: Confirm your test set represents the same chemical space as the training set. Use PCA or other diversity analysis methods to verify representativeness.
- Adjust field combinations: Systematically test different field combinations (SEH, SEHAD, etc.) to identify the optimal set for your target class, as performance varies significantly [3].
FAQ: How can I determine the optimal number of components for my CoMSIA model?
Issue: Selection of appropriate components to avoid overfitting or underfitting.
Solutions:
- Use cross-validation: Perform leave-one-out (LOO) or leave-many-out cross-validation to determine the optimal number of components corresponding to the highest q² value [3].
- Monitor standard error: Select components where the standard error of prediction is minimized.
- Validate with external set: Confirm component selection by evaluating predictive r² on an external test set.
- Reference benchmarks: Consult literature for similar target classes; optimal components typically range from 3-6 for well-behaved datasets [3].
FAQ: What should I do when my CoMSIA model shows unexpected field contributions?
Issue: Field contribution patterns contradict established structure-activity relationships.
Solutions:
- Verify alignment: Check for inconsistencies in molecular alignment, particularly for flexible molecules.
- Assess conformation selection: Ensure biologically relevant conformations were used, especially for peptide targets [22].
- Evaluate steric outliers: Identify compounds with extreme steric properties that may disproportionately influence the model.
- Test alternative field combinations: Systematically exclude or include different field types to identify contribution patterns that align with mechanistic understanding [3].
Experimental Protocols for CoMSIA Modeling
Standard CoMSIA Protocol for Protease Inhibitors
Objective: Develop predictive CoMSIA models for protease inhibitor activity prediction.
Methodology:
- Dataset Preparation:
- Curate 20-50 compounds with consistent inhibitory activity data (IC50 or Ki values)
- Ensure structural diversity while maintaining common scaffold elements
- Apply log transformation to activity data: pIC50 = -log(IC50)
Molecular Modeling and Alignment:
- Generate low-energy conformations using molecular mechanics (MMFF94 or similar)
- Align molecules using common pharmacophore elements or database alignment methods
- Verify alignment visually and through RMSD quantification
CoMSIA Field Calculation:
- Set grid spacing to 1.0-2.0 Å with sufficient padding (≥4 Å beyond molecules)
- Use standard probe atom with charge +1, radius 1.0 Å, and hydrophobicity +1
- Calculate all five field types: steric, electrostatic, hydrophobic, H-bond donor, H-bond acceptor
- Apply default attenuation factor (0.3) for the Gaussian function
Statistical Analysis and Validation:
- Perform PLS regression with cross-validation to determine optimal components
- Validate model using external test set (20-30% of total compounds)
- Calculate standard validation metrics: q², r², standard error of estimate
- Generate contour maps for intuitive interpretation
Troubleshooting Notes:
- For peptide-based protease inhibitors, pay special attention to hydrogen bond field calculations [22]
- If model performance is inadequate, test subsets of field combinations focusing on steric, electrostatic, and hydrophobic fields initially
- Verify that activity cliffs are properly handled in the model
GPCR Antagonist-Specific CoMSIA Protocol
Objective: Create targeted CoMSIA models for GPCR antagonist optimization.
Methodology:
- Dataset Curation:
- Select compounds with binding affinity (Ki) or functional antagonist activity (IC50)
- Include diverse chemotypes targeting the same GPCR binding pocket
- Convert activities to pKi or pIC50 values for analysis
Structure-Based Alignment (When Possible):
- Utilize available GPCR crystal structures for alignment guidance [21]
- Focus on key binding pocket residues for orientation reference
- For class A GPCRs, align to transmembrane domain conserved features
Field Calculation Parameters:
- Emphasize hydrophobic and hydrogen bond fields for GPCR targets [21]
- Consider using finer grid spacing (1.0 Å) around key binding regions
- Adjust attenuation factor based on binding pocket size characteristics
Model Validation:
- Implement rigorous external validation with structurally diverse test compounds
- Apply domain of applicability analysis to identify reliable prediction boundaries
- Validate against known mutagenesis data for binding site residues [21]
GPCR-Specific Considerations:
- GPCR binding pockets often require greater emphasis on hydrophobic fields [21]
- For peptide GPCR antagonists, ensure proper handling of flexible terminal regions [22]
- Consider extracellular loop interactions when modeling class B GPCR antagonists [21]
Optimizing Field Combinations for Different Target Classes
Field Combination Performance Across Target Classes
Table 1: Recommended Field Combinations for Different Target Classes
| Target Class | Optimal Field Combination | Key Fields | Typical q² Range | Performance Notes |
|---|---|---|---|---|
| Protease Inhibitors | SEHAD | Electrostatic, H-bond | 0.6-0.8 | Hydrogen bond fields critical for catalytic residue interactions |
| GPCR Antagonists | SEH | Hydrophobic, Steric | 0.5-0.7 | Hydrophobic fields dominate for transmembrane binding pockets |
| Antioxidant Peptides | EHAD | Hydrophobic, H-bond | 0.4-0.6 | Electronic properties less critical for radical scavenging |
Field Contribution Patterns by Target Class
Table 2: Characteristic Field Contribution Patterns for Different Target Classes
| Target Class | Steric | Electrostatic | Hydrophobic | H-Bond Donor | H-Bond Acceptor |
|---|---|---|---|---|---|
| Protease Inhibitors | 15-25% | 25-35% | 10-20% | 15-25% | 10-20% |
| GPCR Antagonists | 20-30% | 20-30% | 30-40% | 5-15% | 5-15% |
| Antioxidant Peptides | 10-20% | 10-20% | 30-40% | 15-25% | 10-20% |
Advanced Troubleshooting: Case Studies and Solutions
Case Study: Steroid Benchmark Dataset Analysis
The steroid benchmark dataset demonstrates the importance of field selection in CoMSIA modeling [3]. Using the standard steric, electrostatic, and hydrophobic (SEH) fields produced a model with q² = 0.609 and r² = 0.917, while adding hydrogen bond donor and acceptor fields (SEHAD) altered performance to q² = 0.630 and r² = 0.898 [3]. This case highlights that while additional fields may improve cross-validation metrics, they don't necessarily enhance external predictive capability.
Key troubleshooting insights from this case:
- Field contribution analysis showed electrostatic fields dominated (53.4%) for steroid binding, aligning with known steroid receptor interactions [3]
- The SEH model demonstrated better predictive r² (0.40) compared to the SEHAD model (0.186), suggesting simpler field combinations sometimes outperform comprehensive ones [3]
- Optimal components differed between field combinations (3 for SEH vs. 3 for SEHAD), emphasizing the need for systematic component optimization [3]
Case Study: GPCR Peptide Antagonist Modeling
For GPCR-targeting peptides, specific considerations apply due to their flexible nature and complex binding modes [21] [22]. Peptide-binding GPCRs exhibit distinctive structural features, with key characteristics being the involvement of extracellular loops and the N-terminal tail in ligand binding [21]. This extended binding interface requires careful attention in CoMSIA modeling.
GPCR-specific troubleshooting strategies:
- Implement multiple alignment strategies based on different binding hypotheses
- Focus on hydrophobic field optimization for class A GPCR transmembrane domains [21]
- For class B GPCRs, emphasize both hydrophobic and hydrogen bond fields to capture peptide hormone interactions [21]
- Consider constrained conformations for peptide ligands based on experimental data
Research Reagent Solutions for CoMSIA Studies
Table 3: Essential Research Reagents and Tools for CoMSIA Modeling
| Reagent/Tool | Function | Application Notes | Representative Examples |
|---|---|---|---|
| Molecular Modeling Software | Structure preparation, alignment, and visualization | Critical for pre-processing and post-analysis | Py-CoMSIA [3], RDKit [3], Schrödinger Suite |
| PLS Analysis Tools | Statistical analysis and model building | Enables correlation of fields with activity | SIMCA, R/Python with PLS packages |
| Grid Computing Resources | Field calculation and resource-intensive computations | Accelerates model development for large datasets | University HPC clusters, cloud computing services |
| Benchmark Datasets | Method validation and performance comparison | Provides reference points for model quality | Steroid dataset [3], GPCR antagonist datasets [21] |
| Chemical Databases | Source of structural and activity data | Provides input for model development | ChEMBL, PubChem, proprietary corporate databases |
Workflow and Signaling Pathway Visualizations
CoMSIA Model Development Workflow
GPCR Antagonist Signaling Blockade
Frequently Asked Questions (FAQs)
Q1: Why does my CoMSIA model show poor predictive power even with a high r² value?
This discrepancy often arises from improper molecular alignment or inadequate conformational sampling. A high non-cross-validated r² indicates the model fits the training data well but does not guarantee its ability to predict new compounds. The predictive power is primarily assessed through the cross-validated q² and r²pred values. For a reliable model, ensure your alignment is based on a pharmacophore hypothesis or the bioactive conformation, and validate with a sufficiently large external test set (typically 25-33% of your data) [7] [1]. Over-reliance on a single, potentially non-bioactive conformation during alignment is a common source of this problem.
Q2: What is the impact of using different molecular alignment methods on CoMSIA field contours?
The choice of alignment method directly and significantly influences the resulting CoMSIA field contours and, consequently, the model's interpretation and predictive accuracy. Different protocols can lead to different contour maps, suggesting alternative structural requirements for activity [7] [1].
- Common Feature Alignment: Relies on a maximum common substructure (MCS), which can be subjective for structurally diverse sets [23].
- Pharmacophore-Based Alignment: Uses tools like GALAHAD to generate an alignment based on common pharmacophoric features, often leading to more robust models for diverse datasets [7].
- Database Alignment: Aligns molecules to a template from a structural database (e.g., PDB), useful when the bioactive conformation of a lead compound is known [23] [24].
Q3: How can I determine the optimal combination of CoMSIA fields for my dataset?
There is no universal "best" combination; it depends on the specific ligand-receptor interactions in your system. A systematic approach is recommended:
- Begin with the standard Steric and Electrostatic (SE) fields.
- Progressively add Hydrophobic (H), Hydrogen Bond Donor (D), and Acceptor (A) fields.
- Construct models for different field combinations and compare their statistical parameters (q², r²pred, Standard Error) [3] [7].
The following table summarizes the interpretation of CoMSIA fields:
Table: Guide to CoMSIA Field Contributions
| Field | Physical Chemical Meaning | Implied Interaction with Receptor |
|---|---|---|
| Steric | Molecular size and shape | Favors or disfavors bulky substituents in specific regions. |
| Electrostatic | Charge distribution | Favors complementary positive or negative charges. |
| Hydrophobic | Lipophilicity | Favors non-polar, water-excluding groups. |
| H-Bond Donor | Presence of donor groups (e.g., OH, NH) | Favors regions where the receptor can accept a hydrogen bond. |
| H-Bond Acceptor | Presence of acceptor atoms (e.g., O, N) | Favors regions where the receptor can donate a hydrogen bond. |
Q4: My CoMSIA model is highly sensitive to small changes in grid spacing and alignment. How can I stabilize it?
This sensitivity is a known challenge. To enhance model stability:
- Employ a Gaussian Function: Unlike CoMFA, CoMSIA uses a Gaussian function to calculate similarity indices, which avoids abrupt changes in potential energy and makes the model less sensitive to grid placement and small alignment variations [2] [3] [1].
- Optimize Grid Parameters: Systematically test grid spacings (e.g., 1.0 Å vs. 2.0 Å) and ensure the grid extends sufficiently (e.g., 4.0 Å beyond all molecules) to capture relevant interactions without introducing excessive noise [3].
- Validate Alignment Robustness: Slightly perturb your chosen alignment and rebuild the model. A robust model should not show drastic statistical changes with minor alignment adjustments.
Troubleshooting Common Experimental Issues
Problem: Low Cross-Validated Correlation Coefficient (q²)
Potential Causes and Solutions:
- Cause 1: Poor Molecular Alignment.
- Solution: Re-evaluate your alignment strategy. For sets with a common core, use MCS-based alignment. For structurally diverse compounds, switch to a pharmacophore-based alignment using tools like GALAHAD or field-fit methods [7]. Verify that all molecules are aligned in a biologically relevant orientation.
- Cause 2: Incorrect Bioactive Conformation.
- Solution: If the crystal structure of the target-ligand complex is unavailable, consider using multi-conformational alignment or conformers generated from molecular dynamics simulations to account for flexibility [25] [24]. The use of a single, potentially incorrect, low-energy conformation is a major source of error.
- Cause 3: Suboptimal Field Combination.
Problem: Uninterpretable or Chemically Illogical CoMSIA Contour Maps
Potential Causes and Solutions:
- Cause: Misalignment of Molecules.
- Solution: This is the most common cause. The contour maps are generated relative to the aligned molecules. If the alignment does not reflect the true binding mode, the maps will be meaningless. Return to the alignment stage and ensure it is pharmacologically sensible [7] [1]. Using a known active compound as a template for alignment can often rectify this issue.
Problem: Large Difference Between Model Prediction and Experimental Activity for New Compounds
Potential Causes and Solutions:
- Cause 1: The new compound explores chemical space not covered by the training set.
- Cause 2: The model lacks a critical molecular field to describe the new compound's activity.
Experimental Protocols & Workflows
Protocol 1: Robust Molecular Alignment for CoMSIA
Principle: A consistent and biologically relevant alignment of all molecules is the most critical step for a successful CoMSIA model [7] [1].
Materials:
- Molecular dataset with known biological activities (e.g., IC50, Ki).
- Molecular modeling software (e.g., Sybyl, Schrodinger, Open-Source Py-CoMSIA [3]).
- Hardware: Standard computer workstation.
Methodology:
- Data Preparation: Sketch 2D structures of all compounds and convert them to 3D. Perform energy minimization using a standard force field (e.g., Tripos or MMFF94) and assign partial atomic charges (e.g., Gasteiger-Hückel) [23] [7].
- Conformational Analysis: For flexible molecules, generate a set of low-energy conformers. If the bioactive conformation is unknown (from a crystal structure), the global minimum or a conformation consistent with a common pharmacophore is often selected.
- Alignment:
- For congeneric series: Identify the Maximum Common Substructure (MCS). Use the MCS to superimpose the molecules, often using a potent and rigid compound as the template [23].
- For diverse series: Use a pharmacophore-based alignment. Tools like GALAHAD can generate a pharmacophore hypothesis and align molecules based on shared steric and electronic features [7].
- Validation: Visually inspect the alignment from multiple angles to ensure all molecules are meaningfully superimposed in 3D space.
Protocol 2: Systematic Construction and Validation of a CoMSIA Model
Principle: To build a statistically robust and predictive CoMSIA model through a structured workflow that includes internal and external validation [23] [24] [7].
Materials:
- An aligned set of molecules.
- Software capable of performing CoMSIA and Partial Least Squares (PLS) regression.
Methodology:
- Dataset Division: Randomly split the dataset into a training set (~70-80% of compounds) for model building and a test set (~20-30%) for external validation [23] [7].
- Field Calculation: Define a 3D grid that encompasses all aligned molecules. Calculate the five CoMSIA similarity indices (steric, electrostatic, hydrophobic, H-bond donor, H-bond acceptor) using a probe atom at each grid point. A typical attenuation factor of 0.3 is used for the Gaussian function [23] [3] [1].
- Partial Least Squares (PLS) Analysis:
- Cross-Validation: Perform Leave-One-Out (LOO) cross-validation on the training set to determine the optimal number of components (ONC) that gives the highest q² value.
- Non-Cross-Validated Analysis: Using the ONC, derive the final PLS model to obtain the conventional correlation coefficient (r²) and standard error of estimate [23] [7].
- Model Validation: Predict the activity of the external test set compounds using the built model. Calculate the predictive r² (r²pred) to evaluate the model's true predictive power [24].
- Contour Map Generation: Visualize the results as 3D contour maps to identify regions where specific physicochemical properties favor or disfavor biological activity.
The following diagram illustrates the logical workflow for building and validating a CoMSIA model:
The Scientist's Toolkit: Research Reagent Solutions
This table details key computational tools and materials essential for conducting CoMSIA studies.
Table: Essential Resources for CoMSIA Modeling
| Tool / Resource | Function / Description | Application in CoMSIA |
|---|---|---|
| Molecular Modeling Suite(e.g., SYBYL, Schrödinger, MOE) | Integrated software platforms providing tools for molecule building, simulation, and QSAR analysis. | Used for the entire workflow: structure sketching, energy minimization, conformational analysis, molecular alignment, and performing CoMSIA/PLS calculations [23] [7]. |
| Open-Source Tools(e.g., Py-CoMSIA [3], RDKit) | Programmable libraries (often in Python) for cheminformatics and molecular modeling. | Provides an accessible alternative for implementing CoMSIA algorithms, offering flexibility and customization for advanced users [3]. |
| Structural Database(e.g., Protein Data Bank, PDB) | A repository for 3D structural data of biological macromolecules. | Source of target protein structures and protein-ligand co-crystals. Used to guide molecular alignment by providing a known bioactive conformation [23] [24]. |
| Partial Least Squares (PLS) Algorithm | A statistical method for modeling relationships between independent variables (fields) and a dependent variable (activity). | The core algorithm for correlating CoMSIA field values with biological activity and deriving the quantitative model [7] [1]. |
| Gaussian Function | A mathematical function that decreases smoothly and gradually. | Used in CoMSIA to calculate similarity indices, avoiding the abrupt energy changes of CoMFA and producing more interpretable contour maps [2] [3] [1]. |
Advanced Optimization Strategies and Machine Learning Integration
Core Concepts: Understanding Model Weaknesses
What are overfitting, noise, and predictive failures in the context of a CoMSIA model?
In 3D-QSAR CoMSIA (Comparative Molecular Similarity Indices Analysis), these issues arise from the method's fundamental structure. A CoMSIA model calculates thousands of similarity indices (steric, electrostatic, hydrophobic, etc.) for each molecule placed in a grid [26] [19]. Among these, many descriptors are uninformative and irrelevant to the biological activity; these are considered noise [26] [19]. When a model, often built using the Partial Least Squares (PLS) algorithm, is overly influenced by this noise instead of the true underlying structure-activity relationship, it becomes too complex and learns the training data's random fluctuations. This is overfitting [26]. An overfit model will exhibit high statistical performance for the training set but will fail to make accurate predictions for new, external compounds, leading to predictive failures [26] [19].
Why is the high number of CoMSIA descriptors a problem?
CoMSIA typically generates several thousand field descriptors for a set of aligned molecules [26] [19]. The core problem is that a significant portion of these variables are uninformative "noise" that do not correlate with biological activity [26] [19]. This excessive number of descriptors, many of which are irrelevant, can introduce noise and compromise the model's efficacy, especially if no feature-selection techniques are applied [26]. Furthermore, the standard linear PLS estimator may not adequately capture non-linear relationships in the data, leading to subpar predictive power [26].
Troubleshooting Guides
FAQ 1: My CoMSIA model has excellent R² for the training set but poor predictive power for the test set. Is this overfitting and how can I fix it?
Diagnosis: This is a classic symptom of an overfit model. The model has likely learned the noise in the training data rather than the genuine structure-activity relationship.
Solutions:
- Implement Feature Selection: Use algorithms to identify and retain only the most informative CoMSIA fields. Proven methods include:
- Recursive Feature Elimination (RFE) and SelectFromModel: These techniques can significantly improve model fitting and predictivity (R², RCV², and R²_test) across numerous estimators [26].
- Enhanced Replacement Method (ERM): This algorithm has shown noticeable improvement in statistical parameters for models built from CoMSIA fields, leading to more predictive and robust models [19].
- Apply Hyperparameter Tuning: For tree-based machine learning models, carefully tuning hyperparameters is crucial. For instance, a Gradient Boosting (GB) model with RFE, when tuned with a specific learning rate, max depth, and number of estimators, has been shown to effectively mitigate overfitting and demonstrate superior performance compared to a standard linear PLS model [26].
- Use a Non-Linear Algorithm: The default PLS algorithm is linear. Employing non-linear machine learning techniques like Support Vector Machines (SVM) or Random Forest (RF) can sometimes better capture the underlying relationships in the data [26] [27].
Experimental Protocol: Mitigating Overfitting with Feature Selection and Machine Learning
- Objective: To build a predictive CoMSIA model by integrating feature selection and non-linear algorithms to overcome overfitting.
- Methodology:
- Generate CoMSIA Fields: Calculate standard CoMSIA fields (steric, electrostatic, hydrophobic, hydrogen bond donor, and acceptor) for a pre-aligned set of molecules.
- Data Splitting: Divide the dataset into a training set (for model building), a test set (for internal validation), and a completely blind evaluation set (for final model assessment) [19].
- Feature Selection: Apply a feature selection algorithm (e.g., GB-RFE, ERM) to the training set descriptors to identify the most relevant variables.
- Model Building & Tuning: Use the selected features to build a model. If using a tree-based method (like Gradient Boosting), perform hyperparameter tuning via GridSearchCV on the training set [26].
- Validation: The final model's performance must be evaluated primarily on the external test set and the blind evaluation set to confirm its predictive power [19].
FAQ 2: How can I determine if my model's poor performance is due to noisy descriptors?
Diagnosis: Noisy, uninformative descriptors can distort the model and lead to poor generalization.
Solutions:
- Variable Selection Algorithms: Employ genetic algorithms (GA) or the successive projection algorithm (SPA) to filter out noisy variables from the large pool of calculated CoMSIA interactions [19].
- Region Focusing: Use techniques like CoMFA Region Focusing (CoMFA-RF) to weight the lattice points in the model, enhancing the contribution of informative regions and attenuating the contribution of noisy ones [19].
- Visual Inspection: A key advantage of CoMSIA and related MFA methods is the ability to visualize the 3D contour maps. If the maps appear chaotic, nonsensical, or do not align with known chemical intuition or receptor-ligand interaction data, it is a strong indicator that the model is being driven by noise rather than true effects.
Experimental Protocol: Identifying and Reducing Noise with Variable Selection
- Objective: To extract meaningful variables from CoMSIA fields and generate a simple, predictive model.
- Methodology:
- Extract Fields: Export the raw CoMSIA field values from the modeling software and remove any columns with zero variance.
- Apply Selectors: Apply variable selection algorithms (e.g., GA, ERM, SPA) to the remaining thousands of CoMSIA fields [19].
- Build MLR Model: Use the selected variables to build a multiple linear regression (MLR) model. This creates a simpler, more interpretable model that relies on a smaller subset of relevant descriptors [19].
- Validate Rigorously: Compare the statistical parameters (R², Q², R²_pred) of the MLR model built with selected variables against the full PLS model. A more robust and predictive model with fewer variables indicates successful noise reduction [19].
FAQ 3: What are the best practices for validating a CoMSIA model to ensure its predictions are reliable?
Diagnosis: A model that is not rigorously validated may appear good during development but fail in real-world applications.
Solutions:
- Use a True External Test Set: The most critical step is to validate the model on a set of compounds that were not used in any part of the model building process, including feature selection [19]. This set should be selected blindly, without visual inspection or pre-treatment that could introduce bias.
- Go Beyond Internal Validation: A high leave-one-out cross-validated correlation coefficient (q²) is necessary but not sufficient to prove a model's predictive power [19]. A model with a high q² can still fail to predict an external test set accurately.
- Follow OECD Principles: For regulatory acceptability, QSAR models should be built according to OECD principles, which include using a defined endpoint, an unambiguous algorithm, and appropriate measures of goodness-of-fit, robustness, and predictivity [19]. The use of an external test set is a separate and critical principle for an ideal model.
Performance Data and Reagent Solutions
Table 1: Comparison of Modeling Approaches for Improving CoMSIA Performance
| Modeling Approach | Key Technique(s) | Reported Performance Outcomes | Key Advantage |
|---|---|---|---|
| Traditional PLS on CoMSIA | Partial Least Squares on all field descriptors | Statistically underperforming models in some cases; can suffer from low R²_test and overfitting [26]. | Standard, widely implemented approach. |
| Feature Selection + PLS/ML | Recursive Feature Elimination (RFE), SelectFromModel, Enhanced Replacement Method (ERM) [26] [19] | Significant improvement in model fitting and predictivity (R², RCV², R²test) for 24 estimators [26]. ERM improved R²test to 0.852 and 0.908 in CoMSIA models [19]. | Reduces noise and model complexity; improves generalizability. |
| Hyperparameter-Tuned ML | Gradient Boosting with RFE (GB-RFE) and tuned hyperparameters (learningrate=0.01, maxdepth=2, etc.) [26] | Superior performance (R²: 0.872, RCV²: 0.690, R²test: 0.759) compared to PLS (R²test: 0.575) [26]. | Effectively mitigates overfitting; handles non-linear relationships. |
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Computational Tools and Materials
| Item / Reagent | Function in CoMSIA Modeling |
|---|---|
| Aligned Molecular Dataset | A set of molecules with known activity, structurally aligned based on a common scaffold or pharmacophore. This is the foundational input for any 3D-QSAR model [27]. |
| Molecular Modeling Software (e.g., Sybyl) | Software used to build, optimize, and align molecular structures, calculate CoMSIA fields, and generate initial PLS models [26] [19]. |
| Python / R with ML Libraries (e.g., scikit-learn) | Programming environments used to implement advanced feature selection (RFE, SelectFromModel), hyperparameter tuning (GridSearchCV), and non-linear machine learning algorithms (Gradient Boosting, SVM) [26]. |
| Variable Selection Algorithm (e.g., ERM, GA) | Computational methods designed to identify and select the most relevant descriptors from thousands of CoMSIA fields, thereby reducing noise and improving model robustness [19]. |
| External Test / Evaluation Set | A set of compounds, strictly withheld from the model building and feature selection process, used for the final, unbiased assessment of the model's true predictive power [19]. |
FAQs and Troubleshooting Guide
This guide addresses common challenges researchers face when using feature selection in CoMSIA studies, helping you refine field descriptors to build more robust and interpretable 3D-QSAR models.
Q1: Why does my model performance drop significantly after using SelectFromModel for CoMSIA field selection?
A sudden performance drop often occurs when the importance threshold is set too high, filtering out relevant field descriptors.
- Diagnosis: Check the number of features selected. Compare it with your baseline model using all fields (e.g., Steric, Electrostatic, Hydrophobic, Hydrogen Bond Donor, Acceptor). A drastic reduction suggests an overly aggressive threshold [28].
- Solution:
- Use the
max_featuresparameter to guarantee a minimum number of features are retained [29]. - Instead of a fixed threshold, use a heuristic string like
threshold="0.1*mean"to dynamically adjust the cutoff based on the computed feature importances [29]. - Validate the selected features against known biological mechanisms to ensure critical field descriptors have not been inadvertently removed [30].
- Use the
Q2: My RFE is taking too long to run on the molecular field data. How can I speed it up?
RFE is computationally intensive because it trains a model multiple times. This is especially pronounced with large grid-based CoMSIA fields [31].
- Diagnosis: The computational load scales with the number of features and the chosen model.
- Solution:
- Increase the
stepparameter. Instead of removing one feature per iteration (step=1), trystep=10orstep=50to eliminate multiple features at once, significantly reducing the number of iterations required [28]. - Use a faster estimator. For the
RFEprocess itself, a less complex model likeLinearSVCorLogisticRegression(with an L1 penalty) can provide a good feature ranking much faster than an ensemble method [29]. - Start with a pre-filtered feature set using a variance threshold or prior knowledge to reduce the initial number of descriptors [29].
- Increase the
Q3: How do I choose between RFE and SelectFromModel for my CoMSIA analysis?
The choice depends on your primary goal: model robustness versus computational efficiency and interpretability.
- Use
RFEwhen you need the most robust set of features and have sufficient computational resources.RFErecursively re-evaluates feature importance, which can lead to a more optimal subset, especially when features are correlated [28] [31]. - Use
SelectFromModelfor a faster, more straightforward approach that provides a baseline understanding of feature importance. It is less robust thanRFEbut highly efficient for initial experiments and high-dimensional data [31].
Q4: Can I use RFECV to optimize for multiple metrics like Precision and Recall simultaneously?
No, RFECV requires a single metric to determine the optimal number of features. It cannot natively optimize for multiple metrics at once [32].
- Diagnosis: Passing a list of metrics to the
scoringparameter will result in an error [32]. - Solution:
- Choose the single most critical metric for your study (e.g.,
scoring='precision'). - Use a composite metric like
'f1'which balances precision and recall. - Run
RFECVseparately for each metric of interest and compare the resulting feature sets to find a consensus [32].
- Choose the single most critical metric for your study (e.g.,
Q5: The feature importance rankings from my tree-based model seem unreliable. What could be wrong?
Tree-based models use "impurity-based" feature importance (Mean Decrease in Impurity, MDI), which can be misleading, especially with high-cardinality features [28].
- Diagnosis: The importance scores may be biased and not reflect the true relevance of the CoMSIA field descriptors.
- Solution:
- Use Permutation Importance instead. This method measures the performance drop when a single feature's values are randomly shuffled, providing a more reliable estimate of its contribution. You can calculate this using
sklearn.inspection.permutation_importance[28]. - Always pair data-driven selection with biological prior knowledge. If a field known to be critical for ligand-receptor binding (e.g., a hydrophobic patch) is ranked low, it warrants further investigation [30].
- Use Permutation Importance instead. This method measures the performance drop when a single feature's values are randomly shuffled, providing a more reliable estimate of its contribution. You can calculate this using
Experimental Protocols for Feature Selection in CoMSIA
These detailed methodologies are designed for integration into a CoMSIA modeling workflow, helping you systematically refine field descriptors.
Protocol 1: Implementing Recursive Feature Elimination (RFE)
RFE is a wrapper method that recursively prunes the least important features to find the optimal subset [33].
- Objective: To identify the smallest set of CoMSIA field descriptors that yields the best or near-best predictive performance for a given model.
- Materials:
- A curated dataset of aligned molecules with biological activity data.
- Computed CoMSIA fields (Steric, Electrostatic, Hydrophobic, H-bond Donor, H-bond Acceptor).
- A machine learning estimator (e.g.,
ExtraTreesClassifier,LinearSVC).
- Methodology:
- Data Preparation: Generate your CoMSIA field matrices and split data into training and test sets.
- Estimator Selection: Choose an estimator. Tree-based models are common, but linear models can be faster.
- Initialize RFE: Create an
RFEobject, specifying theestimator,n_features_to_select(if known), and thestepsize. - Fit and Transform: Fit the
RFEobject on the training data and transform the training and test sets to the selected features. - Validation: Train your final model on the reduced training set and evaluate its performance on the held-out test set.
Protocol 2: Knowledge-Guided Feature Selection with SelectFromModel
This protocol combines the efficiency of SelectFromModel with domain expertise to create highly interpretable models [30].
- Objective: To leverage prior knowledge of drug targets and biological pathways to constrain and guide feature selection.
- Materials: Same as Protocol 1, plus a list of known drug targets or pathway genes relevant to your compound series.
- Methodology:
- Define Feature Subset: Instead of starting with all CoMSIA fields, pre-select fields associated with your drug's known target or its pathway.
- Model Training: Train an estimator (e.g.,
ExtraTreesClassifier) on this biologically constrained feature set. - Apply SelectFromModel: Use
SelectFromModelwith a heuristic threshold (e.g.,"mean") to further refine the subset. - Evaluate and Interpret: Assess model performance and, crucially, interpret the selected features in the context of the drug's mechanism of action [30].
Protocol 3: Systematic Comparison of Feature Selection Methods
A comparative analysis ensures you select the most appropriate feature selection strategy for your specific dataset [34].
- Objective: To empirically compare the performance of different feature selection methods (
RFE,SelectFromModel, knowledge-driven) when applied to your CoMSIA data. - Materials: A standardized dataset and evaluation framework.
- Methodology:
- Define Methods: Choose the feature selection methods and model types to evaluate.
- Set Evaluation Metric: Decide on a primary performance metric (e.g., Pearson's Correlation Coefficient, RMSE).
- Run Experiments: For each drug/compound series, run the modeling workflow with each feature selection method.
- Analyze Results: Compare the performance, number of features, and interpretability of the resulting models.
Table 1: Hypothetical Results from a Comparative Study on a CoMSIA Dataset
| Feature Selection Method | Avg. Number of Features Selected | Mean Pearson's Correlation (CV) | Key Advantage |
|---|---|---|---|
| All Features (Baseline) | ~17,000 (All Fields) | 0.55 | Comprehensive, no information loss |
| RFE (LinearSVC) | ~45 | 0.62 | Robust, handles correlated features well |
| SelectFromModel (ExtraTrees) | ~120 | 0.59 | Very fast, good for initial screening |
| Knowledge-Based (Target Genes) | ~5 | 0.65 | High interpretability, direct biological link |
The Scientist's Toolkit: Research Reagent Solutions
This table lists key computational tools and their functions for implementing feature selection in CoMSIA studies.
Table 2: Essential Computational Tools for Feature Selection in CoMSIA
| Tool / Reagent | Function in the Workflow | Example / Reference |
|---|---|---|
| scikit-learn | Provides the core implementation for RFE, SelectFromModel, and various machine learning estimators. |
sklearn.feature_selection.RFE [29] |
| Py-CoMSIA | An open-source Python implementation for generating CoMSIA fields, overcoming reliance on discontinued proprietary software. | [3] |
| ExtraTreesClassifier | An "Extremely Randomized Trees" ensemble model often used to compute robust, impurity-based feature importances. | sklearn.ensemble.ExtraTreesClassifier [28] [31] |
| Permutation Importance | A model inspection technique that provides a more reliable ranking of feature importance than impurity-based methods. | sklearn.inspection.permutation_importance [28] |
| Stability Selection | A method designed to improve the stability of feature selection, especially with high-dimensional data. | Can be implemented with SelectFromModel and randomized algorithms [30]. |
Workflow and Relationship Visualizations
The following diagrams, generated with Graphviz, illustrate the logical flow and decision points for integrating feature selection into a CoMSIA research project.
CoMSIA Feature Selection Workflow
RFE Process Loop
Troubleshooting Guides & FAQs
This section addresses common challenges encountered when integrating Gradient Boosting Machines (GBMs) with 3D-QSAR CoMSIA studies to optimize field combinations for better model performance.
Common Error: Model Overfitting
Problem: Your CoMSIA-GBM model performs excellently on training data but poorly on the test set or new molecular validation sets. Solution:
- Adjust Tree-Specific Parameters: Increase
min_samples_splitandmin_samples_leafto prevent the tree from learning overly specific patterns [35]. A value ofmin_samples_leaf=50can be a good starting point [36]. - Use Subsampling: Set
subsampleto a value less than 1.0 (e.g., 0.8) to train each tree on a random fraction of the data, introducing robustness [35]. - Tune Learning Rate and Estimators: Employ a lower
learning_rate(e.g., 0.01) coupled with a higher number ofn_estimatorsto make the model more robust [35] [37]. A successful combination reported waslearning_rate=0.01withn_estimators=500[26] [38]. - Apply Regularization: Utilize
max_depthto limit the complexity of individual trees; amax_depthof 3 is often effective [35] [39].
Supporting Protocol: A study on the FTC dataset for antioxidant peptides successfully mitigated overfitting using a GBM with learning_rate=0.01, max_depth=2, n_estimators=500, and subsample=0.5 [26] [38].
Common Error: Poor Predictive Performance (Low R²_test)
Problem: The model's predictive accuracy on external test sets is unsatisfactory, even with what seems like a good internal fit. Solution:
- Conduct Systematic Hyperparameter Tuning: Do not rely on default parameters. Use methods like
GridSearchCVorRandomizedSearchCVto explore the hyperparameter space systematically [35]. - Feature Selection for CoMSIA Fields: The high number of CoMSIA descriptors can introduce noise. Apply feature selection techniques like Recursive Feature Elimination (RFE) or
SelectFromModelbefore GBM fitting to identify the most relevant molecular fields [26] [38]. - Check Data Alignment: In 3D-QSAR, the predictive performance is highly sensitive to molecular alignment. Ensure your alignment protocol is optimal and consistent [15].
Supporting Protocol: Research shows that coupling GB-RFE for feature selection with a tuned Gradient Boosting Regressor (GBR) resulted in a superior model (R²test of 0.759) compared to the traditional PLS model (R²test of 0.575) for an FTC CoMSIA model [26] [38].
Common Error: Handling Imbalanced Datasets in Classification
Problem: When modeling categorical biological activities (e.g., active/inactive), an imbalance between classes can bias the model. Solution:
- Balance the Training Set: Create a subsampled dataset with an equal number of observations from each class for training [36].
- Utilize
class_weightParameter: Use theclass_weightparameter in theGradientBoostingClassifierto automatically adjust weights inversely proportional to class frequencies. A common approach is to set it to'balanced'or calculate a specific ratio (e.g.,ratio_background_to_signal) [36].
Common Error: Long Training Times
Problem: Hyperparameter tuning with GBMs on large CoMSIA datasets is computationally expensive. Solution:
- Use Faster Tuning Methods: For a large hyperparameter space, prefer
RandomizedSearchCVoverGridSearchCV[35]. For even greater efficiency, consider advanced frameworks like Optuna or Bayesian Optimization [35] [40] [41]. - Leverage Warm Start: Use the
warm_startparameter to fit additional trees on a previously fitted model, which can save time during iterative tuning [37].
The following table summarizes the core hyperparameter tuning methods applicable to GBM-CoMSIA workflows.
Table 1: Comparison of Hyperparameter Optimization (HPO) Methods
| Method | Core Principle | Best For | Advantages | Disadvantages |
|---|---|---|---|---|
| GridSearchCV [35] | Exhaustively searches over all predefined combinations in a parameter grid. | Small, well-defined parameter spaces. | Guarantees finding the best combination within the grid. | Computationally prohibitive for large parameter spaces. |
| RandomizedSearchCV [35] | Randomly samples a fixed number of parameter settings from specified distributions. | Larger parameter spaces where an approximate optimum is sufficient. | Faster than grid search; often finds good parameters with fewer iterations. | Does not guarantee a global optimum; result depends on number of iterations. |
| Bayesian Optimization (e.g., via Tree-Parzen Estimator) [40] | Builds a probabilistic model of the objective function to direct the search towards promising parameters. | Complex, high-dimensional spaces where function evaluations are expensive. | More efficient than random search; requires fewer evaluations to find good parameters. | Higher computational overhead per iteration; can be more complex to implement. |
| Optuna [35] | Define-by-run API that can efficiently search over complex spaces with pruning of unpromising trials. | Large, complex search spaces requiring high efficiency. | Flexible and efficient; can handle conditional parameters. | Requires familiarity with the framework. |
A 2025 study comparing HPO methods for XGBoost found that while all methods improved upon default parameters, their performance was similar for datasets with a large sample size, a small number of features, and a strong signal-to-noise ratio [40].
Detailed Experimental Protocol: Tuning a GBM for a CoMSIA Model
This protocol outlines the steps to tune a Gradient Boosting Classifier for a CoMSIA-based classification problem (e.g., predicting active vs. inactive compounds) using the Titanic dataset as an illustrative example [35].
Objective: To identify the optimal set of hyperparameters that maximize the predictive accuracy of a GBM model on an external test set.
Workflow Overview:
Materials & Code Implementation:
Data Preparation and Splitting:
Hyperparameter Tuning with GridSearchCV:
Final Model Training and Evaluation:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software Tools for GBM-CoMSIA Integration
| Tool Name | Type | Primary Function in Workflow |
|---|---|---|
| Scikit-learn [35] [39] [37] | Python Library | Provides the GradientBoostingClassifier/Regressor, GridSearchCV, RandomizedSearchCV, and other essential ML utilities. |
| XGBoost [40] [41] | Optimized Library | An optimized implementation of gradient boosting designed for speed and performance, often a top performer. |
| Optuna [35] | HPO Framework | An automatic hyperparameter optimization software framework, particularly suited for large and complex search spaces. |
| Py-CoMSIA [15] | Python Library | An open-source implementation of CoMSIA, enabling the entire 3D-QSAR pipeline within Python and facilitating integration with ML libraries. |
| SHAP [42] | Python Library | Explains the output of any ML model, including GBMs, which is crucial for interpreting the model's decisions in a medicinal chemistry context. |
Advanced Workflow: Integrating CoMSIA with Machine Learning
The following diagram illustrates a modernized workflow for building predictive 3D-QSAR models by integrating the traditional CoMSIA method with advanced machine learning techniques like Gradient Boosting.
Frequently Asked Questions (FAQs)
1. What are the most common CoMSIA field combinations, and how do I choose? The five standard CoMSIA fields are Steric (S), Electrostatic (E), Hydrophobic (H), Hydrogen Bond Donor (D), and Hydrogen Bond Acceptor (A). The choice of combination is not universal and depends on your specific dataset and the dominant interactions of your biological target. A common and often effective starting point is the SEH combination (Steric, Electrostatic, Hydrophobic). You should systematically test different field combinations and validate their predictive power using a test set of compounds. For example, one study on steroids found that an SEH model provided better predictive performance ((r^2{pred} = 0.40)) than a model using all five fields (SEHAD, (r^2{pred} = 0.186)) [3].
2. My model performs well on training data but poorly on new compounds. What should I check? This is a classic sign of overfitting, often caused by having too many descriptors relative to the number of compounds in your dataset. To address this:
- Apply Dimensionality Reduction: Use techniques like Principal Component Analysis (PCA) to compress your descriptor set into a smaller number of informative components [43] [44].
- Implement Robust Validation: Always use an external test set that is not involved in model training. Employ cross-validation and monitor metrics like (q^2) (cross-validated (R^2)) and (r^2_{pred}) (predictive (R^2) on the test set) to gauge real-world performance [3] [45].
- Conduct Feature Selection: Use methods like Random Forest feature importance or regularization techniques (e.g., LASSO) to identify and retain only the most relevant descriptors [43] [45].
3. How can I capture complex, nonlinear relationships in my data? Classical partial least squares (PLS) regression used in CoMSIA is a linear method. For nonlinear relationships, consider these advanced approaches:
- Nonlinear Machine Learning Models: Replace PLS with algorithms like Random Forests, Support Vector Machines (SVM), or Gradient Boosting, which can model complex patterns [43] [45].
- Gene Expression Programming (GEP): This nonlinear method has been shown to successfully model complex QSAR data, with one study reporting a significant improvement in (R^2) (0.839 for GEP vs. 0.603 for a linear method) on the training set [46].
- Deep Learning and Autoencoders: Autoencoders are powerful neural networks for nonlinear dimensionality reduction and feature learning, often leading to highly predictive models [47] [44].
Troubleshooting Guides
Problem: Model is Highly Sensitive to Molecular Alignment Description: Small changes in the alignment of your compound set lead to large fluctuations in model statistics and contour maps.
| Troubleshooting Step | Action and Rationale |
|---|---|
| Review Alignment Rule | Confirm that all molecules are aligned to a common, rigid scaffold or to the putative pharmacophore. The most active molecule is often chosen as a template [20]. |
| Verify Grid Parameters | Ensure consistent grid spacing and dimensions across all analyses. Py-CoMSIA has demonstrated reduced sensitivity to such parameters compared to older methods [3]. |
| Explore Alignment-Independent Descriptors | If alignment remains problematic, consider supplementing your analysis with alignment-independent 3D descriptors or transitioning to a 2D-QSAR approach for initial insights [46]. |
Problem: Low Predictive (r^2_{pred}) Despite High (q^2) Description: The cross-validated (q^2) from the training set is high, indicating good internal consistency, but the model fails to predict the activity of the external test set accurately.
| Troubleshooting Step | Action and Rationale |
|---|---|
| Check Applicability Domain | Ensure the test set compounds are structurally similar to the training set and fall within the model's "applicability domain." Predictions for outliers are unreliable [48]. |
| Re-evaluate Field Contributions | Analyze the contribution of each CoMSIA field. A field with an unusually low contribution might be adding noise. Try rebuilding the model with different field combinations [3]. |
| Avoid Over-parameterization | Using too many fields or principal components can lead to overfitting. Use statistical criteria (e.g., lowest (SPRESS)) to determine the optimal number of components [3]. |
Experimental Protocol: Systematic Optimization of CoMSIA Field Combinations
This protocol provides a step-by-step methodology for evaluating different CoMSIA field combinations to enhance model performance, directly supporting thesis research.
1. Hypothesis: Systematic testing of CoMSIA field combinations will identify an optimal set of molecular fields that maximizes the predictive accuracy for a given dataset.
2. Materials and Reagents (The Scientist's Toolkit)
| Item / Reagent | Function in the Experiment |
|---|---|
| Molecular Dataset | A curated set of compounds with known 3D structures and consistent biological activity data (e.g., IC50, Ki). |
| Software (e.g., Py-CoMSIA) | Open-source Python implementation for performing CoMSIA calculations, generating fields, and statistical modeling [3]. |
| Partial Least Squares (PLS) Regression | The core statistical algorithm for building the linear relationship between molecular fields and biological activity [3] [43]. |
| Dimensionality Reduction (PCA) | A technique to reduce the number of correlated variables (descriptors) into a smaller set of uncorrelated components, mitigating overfitting [43] [44]. |
3. Procedure: 1. Data Preparation: Prepare and optimize the 3D structures of all compounds. Divide the dataset into a training set (typically ~80%) for model building and a test set (~20%) for external validation [46] [20]. 2. Molecular Alignment: Align all molecules using a common, robust method, such as superimposition on a core scaffold or the most active compound [20]. 3. Define Field Combinations: Prepare a list of field combinations to test. Example combinations include: SE, SH, SEH, SEHD, SEHA, SEHAD. 4. Model Construction & Internal Validation: For each field combination: * Calculate the CoMSIA fields for the training set. * Perform PLS regression to build the QSAR model. * Conduct leave-one-out (LOO) cross-validation to determine the optimal number of components and calculate the cross-validated correlation coefficient ((q^2)) [3]. 5. External Validation & Model Selection: For each model, predict the activity of the test set compounds. Calculate the predictive (r^2{pred}). The model with the highest (r^2{pred}) and a low standard error of estimate (S) is considered the most robust and predictive [3]. 6. Contour Map Analysis: Generate and interpret the CoMSIA contour maps for the optimal model to gain insights into the structural features influencing biological activity [3] [20].
4. Expected Outcomes: A comparative table of performance metrics for all tested field combinations, allowing for the identification of the most predictive model. The steroid benchmark test case, for instance, showed that an SEH model ((q^2 = 0.609), (r^2{pred} = 0.40)) outperformed a full SEHAD model ((q^2 = 0.630), (r^2{pred} = 0.186)) on predictive power [3].
Table: Example Results from a Hypothetical Field Combination Study
| Field Combination | Optimal N.C. | (q^2) | (r^2) | (r^2_{pred}) | Standard Error (S) |
|---|---|---|---|---|---|
| SE | 3 | 0.55 | 0.89 | 0.35 | 0.38 |
| SEH | 3 | 0.61 | 0.92 | 0.40 | 0.33 |
| SEHAD | 3 | 0.63 | 0.90 | 0.19 | 0.37 |
Workflow Visualization
CoMSIA Field Optimization Workflow
Managing Descriptor Dimensionality
Rigorous Validation and Benchmarking Against Established Standards
Within the framework of thesis research focused on optimizing Comparative Molecular Similarity Indices Analysis (CoMSIA) field combinations, robust model validation is not merely a final step but a guiding principle. Reliable 3D-QSAR models are fundamental for rational drug design, as they connect a compound's physicochemical properties to its biological activity. The interpretation of three core metrics—the cross-validated correlation coefficient ((q^2)), the predicted correlation coefficient for an external test set ((r^2_{pred})), and the Standard Error of Prediction (SPRESS)—forms the bedrock of this process. These metrics collectively assess a model's internal stability, predictive power, and reliability, ensuring that the insights gained from optimized steric, electrostatic, hydrophobic, and hydrogen-bonding field combinations are both statistically sound and scientifically valid [3] [26] [49].
Metric Definitions and Core Interpretive Frameworks
The Cross-Validated Correlation Coefficient ((q^2))
What it Measures: (q^2) estimates the model's internal stability and predictive ability using its own training dataset. It is calculated via Leave-One-Out (LOO) or other cross-validation techniques, where one or more compounds are systematically omitted from the model-building process and then predicted by the model derived from the remaining compounds [49].
How to Interpret it:
- (q^2 > 0.5): Generally indicates a statistically robust model with good predictive potential [3] [49].
- (q^2 > 0.6)-(0.7): Suggests a highly reliable model. For instance, a CoMSIA model for 2-Phenylindole derivatives demonstrated high reliability with a (q^2) of 0.814 [49].
- Low (q^2): A value below 0.5 signals that the model is unstable and has poor predictive power. This often arises from issues in molecular alignment, the presence of structural outliers, or the selection of non-informative CoMSIA fields.
Troubleshooting a Low (q^2):
- Verify Molecular Alignment: Ensure all molecules are correctly superimposed based on a common scaffold or pharmacophore. Misalignment is a primary source of poor (q^2).
- Re-evaluate Field Combinations: Test different combinations of CoMSIA fields (e.g., Steric, Electrostatic, Hydrophobic, Hydrogen Bond Donor, Hydrogen Bond Acceptor). An optimized field set can significantly improve (q^2) [3].
- Check for Outliers: Identify and investigate compounds with large prediction errors during cross-validation. These may be structurally unique or have erroneous activity data.
The Predicted Correlation Coefficient for an External Test Set ((r^2_{pred}))
What it Measures: (r^2_{pred}) is the most stringent test, evaluating the model's ability to predict the activity of novel, unseen compounds that were not part of the model training process. It is calculated on a pre-selected test set [3].
How to Interpret it:
- (r^2{pred} > 0.6): Indicates a model with excellent and trustworthy external predictive power. In the steroid benchmark case, an (r^2{pred}) of 0.40 was considered comparable to the original Sybyl implementation, showing acceptable predictive capability [3].
- Low (r^2_{pred}) with High (q^2): This is a classic sign of model overfitting. The model has memorized the training set noise rather than learning the general structure-activity relationship.
Troubleshooting a Low (r^2_{pred}):
- Avoid Overfitting: Use variable selection methods like Recursive Feature Elimination (RFE) to reduce the number of CoMSIA descriptors and build a simpler, more generalizable model [26].
- Review Test/Train Set Division: Ensure the test set is representative of the chemical space covered by the training set. A flawed division can lead to poor external predictions.
- Consider Machine Learning Algorithms: For non-linear datasets, replacing traditional Partial Least Squares (PLS) with algorithms like Gradient Boosting Regression (GBR) has been shown to yield superior (r^2_{pred}) values and mitigate overfitting [26].
The Standard Error of Prediction (SPRESS)
What it Measures: SPRESS represents the average uncertainty or error associated with the model's predictions, typically reported in the same units as the biological activity (e.g., pIC50). It is derived from the cross-validation process [3].
How to Interpret it:
- A lower SPRESS value indicates a more precise model with smaller prediction errors.
- There is no universal "good" value; it must be interpreted in the context of the activity range of your dataset. A SPRESS of 0.33, as seen in a validated steroid CoMSIA model, indicates high precision for a pIC50 scale [3].
- Compare SPRESS with the standard error of the model without cross-validation (S). If they are similar, it suggests a robust model.
Troubleshooting a High SPRESS:
- Increase Data Quality: Review the experimental biological data for high uncertainty or errors.
- Optimize Model Parameters: Adjust grid spacing and the attenuation factor in the CoMSIA calculation. A common grid spacing is 2 Å, and a typical attenuation factor is 0.3 [3] [49].
- Expand the Training Set: If possible, incorporating more high-quality data points can help reduce the overall prediction error.
Table 1: Benchmark Values for CoMSIA Validation Metrics from Case Studies
| Metric | Threshold for a "Good" Model | Exemplary Value from Literature | Context |
|---|---|---|---|
| (q^2) | > 0.5 | 0.814 [49] | 2-Phenylindole derivatives model |
| (r^2_{pred}) | > 0.6 | 0.722 [49] | External test set for the same model |
| SPRESS | As low as possible | 0.33 [3] | Steroid benchmark CoMSIA model |
Advanced FAQ and Troubleshooting Guide
FAQ 1: My (q^2) is acceptable (>0.5), but my (r^2_{pred}) is poor (<0.3). What is the most likely cause and how can I fix it?
This discrepancy is a strong indicator of model overfitting. Your model has learned the training data too well, including its noise, and fails to generalize.
- Solution: Implement feature selection to identify and use only the most relevant CoMSIA descriptors. One study on antioxidant peptides used GB-RFE (Gradient Boosting with Recursive Feature Elimination) to successfully address overfitting. The final model, with tuned hyperparameters (learningrate=0.01, maxdepth=2, nestimators=500, subsample=0.5), achieved a superior (r^2{pred}) of 0.759 compared to 0.575 for the standard PLS model [26].
- Actionable Protocol:
- Use
scikit-learn'sRFEorSelectFromModelin Python. - Build a preliminary model with all CoMSIA fields.
- Apply the feature selection method to rank the importance of each grid-point descriptor.
- Rebuild the model using only the top-performing features.
- Re-evaluate (r^2_{pred}) on your external test set.
- Use
FAQ 2: How do the contributions of different CoMSIA fields (S, E, H, D, A) influence these validation metrics?
The choice of fields directly impacts the model's ability to capture the true structure-activity relationship. An uninformative field adds noise, degrading all validation metrics.
- Interpretation: A significant drop in (q^2) or (r^2_{pred}) when a particular field is added suggests it is not relevant for the specific biological interaction. Conversely, a large increase confirms its importance.
- Case Study Example: In the steroid benchmark, a model using only Steric, Electrostatic, and Hydrophobic (SEH) fields had a higher predictive (r^2{pred}) (0.40) than a model using all five fields (SEHAD, (r^2{pred}) = 0.186). This indicated that for this particular dataset, the hydrogen bond fields introduced noise or were redundant, reducing predictive power [3].
Table 2: Troubleshooting Guide for Common Validation Metric Problems
| Problem | Potential Causes | Corrective Actions |
|---|---|---|
| Low (q^2) (< 0.5) | Poor molecular alignment, uninformative field selection, structural outliers in training set. | Re-check alignment methodology; test different field combinations; identify and remove outliers. |
| Low (r^2_{pred}) (< 0.6) but High (q^2) | Model overfitting, non-representative test/train split, inadequate field descriptors. | Apply feature selection (e.g., RFE); re-partition dataset ensuring chemical space coverage; try non-linear ML algorithms. |
| High SPRESS | Noisy biological data, suboptimal model parameters (grid spacing, attenuation). | Review experimental data sources; optimize grid spacing (e.g., 1-2 Å) and attenuation factor (default 0.3). |
Experimental Protocol for a Validated CoMSIA Study
The following workflow, implemented using software like Sybyl or open-source alternatives like Py-CoMSIA [3], ensures a rigorous validation process.
Diagram 1: CoMSIA Model Validation Workflow. This chart outlines the iterative process of building and validating a 3D-QSAR model, highlighting key checkpoints for (q^2) and (r^2_{pred}).
Step-by-Step Methodology:
Dataset Preparation and Curation:
- Collect a homogeneous set of compounds with consistent biological activity data (e.g., IC50).
- Convert activity to pIC50 (-logIC50) for analysis.
- Divide the dataset into a training set (~80%) for model building and a test set (~20%) for external validation. Ensure both sets represent a similar and broad chemical space [12] [49].
Molecular Modeling and Alignment:
- Sketch 3D molecular structures using a module like the sketch module in SYBYL.
- Optimize geometries using a force field (e.g., Tripos or OPLS_2005) and assign charges (e.g., Gasteiger-Hückel).
- Perform molecular alignment, a critical step. Use the most active compound or a common core scaffold as a template for database alignment [49].
CoMSIA Field Calculation and PLS Analysis:
- Calculate CoMSIA descriptor fields (S, E, H, D, A) using a 3D grid with a spacing of 1-2 Å and a probe atom with a standard attenuation factor of 0.3 [3] [49].
- Use Partial Least Squares (PLS) regression to correlate the field descriptors with biological activity.
- Perform Leave-One-Out (LOO) cross-validation to determine the optimal number of components (N) that gives the highest (q^2).
- Build the final model with this optimal N and analyze the statistical results ((R^2), (S), field contributions) [49].
Model Validation and Interpretation:
- Apply the finalized model to predict the activities of the external test set compounds.
- Calculate the key validation metrics: (r^2_{pred}) and analyze prediction residuals.
- Generate and interpret CoMSIA contour maps to understand the structural features enhancing or diminishing biological activity [50].
Table 3: Key Computational Tools for CoMSIA Model Development and Validation
| Tool / Resource | Function / Description | Application in CoMSIA Workflow |
|---|---|---|
| SYBYL (Tripos) | A comprehensive molecular modeling software suite. | The traditional commercial platform for performing CoMSIA, including alignment, field calculation, and PLS analysis [50] [49]. |
| Py-CoMSIA | An open-source Python implementation of CoMSIA. | Provides a free, flexible alternative to proprietary software, using RDKit and NumPy for calculations [3]. |
| RDKit | Open-source cheminformatics software. | Used within Py-CoMSIA for handling molecular structures and calculations [3]. |
| scikit-learn | A core Python library for machine learning. | Essential for implementing advanced feature selection (RFE) and alternative ML algorithms like Gradient Boosting to combat overfitting [26]. |
| PLS Regression | Partial Least Squares regression algorithm. | The standard linear algorithm for establishing the relationship between CoMSIA descriptors and biological activity [3] [49]. |
| Gradient Boosting (GBR) | A powerful machine learning technique based on ensemble trees. | A non-linear algorithm that can be applied to CoMSIA descriptors to improve predictive performance ((r^2_{pred})) on complex datasets [26]. |
This technical guide supports researchers in performing Comparative Molecular Similarity Indices Analysis (CoMSIA) benchmarking studies, specifically focusing on the validation of the open-source Py-CoMSIA implementation against the traditional, proprietary Sybyl software. The core of this validation utilizes the classic steroid benchmark dataset, a standard in 3D-QSAR methodology development [3] [51]. The experiments are designed to determine whether Py-CoMSIA, developed in Python using libraries like RDKit and NumPy, can generate CoMSIA models with predictive performance and statistical robustness comparable to those produced by the established Sybyl platform [3] [52]. This is critical for enabling accessible, flexible, and reproducible grid-based 3D-QSAR analyses in modern computational drug discovery.
Performance Benchmarking & Data Interpretation
Key Performance Metrics Comparison
The following table summarizes the key statistical outcomes from the CoMSIA analysis of the steroid dataset, comparing models built with different field combinations in Py-CoMSIA against published results from Sybyl.
Table 1: Performance Metrics for CoMSIA Models on the Steroid Benchmark Dataset
| Metric | Published Sybyl (SEH) | Py-CoMSIA (SEH) | Py-CoMSIA (SEHAD) |
|---|---|---|---|
| q² (LOOCV) | 0.665 | 0.609 | 0.630 |
| r² (Non-cross-validated) | 0.937 | 0.917 | 0.898 |
| Standard Error (S) | 0.33 | 0.33 | 0.366 |
| SPRESS | 0.759 | 0.718 | 0.698 |
| Optimal Number of Components | 4 | 3 | 3 |
| Predictive r² (r²pred) | 0.318 | 0.40 | 0.186 |
Field Contributions | | Steric | 0.073 | 0.149 | 0.065 | | Electrostatic | 0.513 | 0.534 | 0.258 | | Hydrophobic | 0.415 | 0.316 | 0.154 | | Hydrogen Bond Donor | - | - | 0.274 | | Hydrogen Bond Acceptor | - | - | 0.248 |
Data adapted from Py-CoMSIA validation study [3].
Troubleshooting Guide: Interpreting Benchmarking Results
Q1: The cross-validated correlation coefficient (q²) of my Py-CoMSIA model is slightly lower than the reported Sybyl value. Does this mean my model is invalid?
A: Not necessarily. A q² value of 0.609 for the primary Py-CoMSIA (SEH) model is considered statistically acceptable and indicates a model with good predictive robustness [3]. Minor variations from the Sybyl benchmark (q² = 0.665) are expected and can be attributed to differences in underlying molecular alignment or slight variations in algorithmic implementation. Focus on the overall statistical profile: your model's strong non-cross-validated r² (0.917) and acceptable predictive r²pred (0.40) confirm its validity.
Q2: Why are the field contributions in my Py-CoMSIA model different from the Sybyl benchmark?
A: Observed differences in field contributions, such as the higher steric contribution in Py-CoMSIA (0.149 vs. 0.073), are a known phenomenon in cross-platform comparisons [3]. This can be influenced by the optimal number of components selected by the Partial Least Squares (PLS) regression algorithm. As long as the relative importance of the fields is logically consistent (e.g., electrostatic and hydrophobic fields dominate in both models), the model should be considered functionally correct.
Q3: Should I use the 3-field (SEH) or 5-field (SEHAD) model for my research?
A: For initial benchmarking and direct comparison with classic studies, the 3-field (SEH) model is recommended. The 5-field (SEHAD) model demonstrates Py-CoMSIA's comprehensive field-handling capability but exhibited a lower predictive r²pred (0.186) in the steroid benchmark, suggesting it may be less robust for this specific dataset [3]. The choice of field combination is a key aspect of model optimization and should be guided by the biological context of your specific system.
Detailed Experimental Protocol
Workflow for Benchmarking Analysis
The following diagram illustrates the end-to-end workflow for conducting a Py-CoMSIA benchmarking study, from data preparation to model validation.
Diagram 1: CoMSIA Benchmarking Workflow. A sequential workflow for performing and validating a Py-CoMSIA analysis against traditional benchmarks.
Step-by-Step Methodology
Dataset Preparation
- Source: Obtain the classic steroid benchmark dataset, which includes 31 steroids (21 in the training set and 10 in the test set) and their associated binding affinity data [3] [51].
- Format: For consistent and reproducible benchmarking, it is highly recommended to use the pre-aligned molecular structures from sources like the Coats' steroid benchmarking study [3]. This minimizes variability introduced by the alignment step, allowing for a direct comparison of the CoMSIA algorithms.
Molecular Alignment
- Method: The database alignment method is used, where all molecules are superimposed onto a common template or a pre-defined alignment [53].
- Technical Note: Molecular alignment is a critical and sensitive step in 3D-QSAR. Using a pre-validated, pre-aligned dataset ensures that any performance differences observed are due to the CoMSIA implementation itself and not alignment artifacts.
Grid Generation and Field Calculation
- Grid Setup: A 3D grid is generated to encompass all aligned molecules. The standard parameters from the original benchmark are a grid spacing of 1.0 Å and a grid padding of 4.0 Å beyond the molecular dimensions [3].
- Field Descriptors: Py-CoMSIA calculates similarity indices using a Gaussian function for different physicochemical properties. The standard field combinations for benchmarking are:
- SEH: Steric, Electrostatic, Hydrophobic.
- SEHAD: Steric, Electrostatic, Hydrophobic, Hydrogen Bond Acceptor, Hydrogen Bond Donor.
- Attenuation Factor: Use the default value of 0.3 for the Gaussian function [3].
Statistical Analysis and Validation
- Partial Least Squares (PLS) Regression: This technique is used to correlate the CoMSIA field descriptors with the biological activity values (e.g., pIC50, pKi) [3] [53].
- Model Validation:
- Internal Validation: Perform Leave-One-Out Cross-Validation (LOOCV) to determine the optimal number of components and calculate the cross-validated correlation coefficient, q².
- External Validation: Use the test set of molecules, which were not used in model building, to calculate the predictive r²pred, which is a crucial metric for assessing the model's true predictive power on new data [3].
Table 2: Key Computational Tools and Resources for CoMSIA Benchmarking
| Item / Resource | Type | Function / Purpose in Benchmarking |
|---|---|---|
| Steroid Benchmark Dataset | Dataset | The gold-standard set of 31 steroids with binding affinities for validating 3D-QSAR methods [3] [51]. |
| Py-CoMSIA Python Library | Software | The open-source implementation of CoMSIA being validated; provides core algorithm and visualization [3] [52]. |
| RDKit | Software/Chemoinformatics | Open-source toolkit for cheminformatics; used by Py-CoMSIA for handling molecules and calculations [3]. |
| NumPy | Software/Library | Fundamental package for scientific computing in Python; handles numerical operations [3]. |
| Sybyl (Tripos) | Software (Proprietary) | Traditional molecular modeling platform that originally hosted CoMSIA; serves as the benchmark for performance comparison [3] [2]. |
| Pre-aligned Molecular Coordinates | Data | Provides a consistent molecular alignment, removing a major source of variability and allowing direct comparison of CoMSIA performance [3]. |
| Partial Least Squares (PLS) | Statistical Method | The regression technique used to build the relationship between molecular fields and biological activity [3] [2]. |
Advanced Technical Notes & FAQs
Q4: What are the fundamental methodological advantages of CoMSIA over CoMFA?
A: CoMSIA introduces several key improvements over its predecessor, Comparative Molecular Field Analysis (CoMFA):
- Smoother Fields: It uses a Gaussian function to calculate similarity indices, which avoids the abrupt energy cut-offs seen in CoMFA's Lennard-Jones and Coulomb potentials. This results in more interpretable and continuous contour maps [3] [2].
- Broader Descriptor Set: While CoMFA typically relies on steric and electrostatic fields, CoMSIA can incorporate additional fields like hydrophobic, and hydrogen bond donor and acceptor properties, providing a more holistic view of molecular interactions [3] [2].
- Reduced Sensitivity: The approach is less sensitive to molecular alignment, grid spacing, and the choice of probe atom, often leading to more robust models [3].
Q5: My model identifies an outlier compound (e.g., steroid #10). How should I proceed?
A: The identification of compound #10 as a predictive outlier in both the original Sybyl study and the Py-CoMSIA replication is a sign that your model is behaving as expected [3]. This consistency actually validates Py-CoMSIA's performance. You should:
- Investigate the outlier structurally. It may possess unique structural or physicochemical features not adequately captured by the model for the standard steroid scaffold.
- Report the outlier transparently in your results, as it highlights the model's applicability domain.
- Avoid simply removing the outlier to improve statistics unless a solid scientific rationale exists.
Q6: Can I integrate Py-CoMSIA with advanced machine learning techniques?
A: Yes, this is a significant advantage of an open-source Python implementation. The Py-CoMSIA library is designed to be a flexible platform, making it easier to integrate the generated 3D field descriptors with advanced statistical or machine learning techniques available in the Python ecosystem (e.g., scikit-learn) for potentially improved model performance [3].
Troubleshooting Guides & FAQs
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between SEH and SEHAD field combinations in CoMSIA? The SEH model uses three molecular fields: Steric, Electrostatic, and Hydrophobic. The SEHAD model incorporates two additional fields: hydrogen bond Acceptor and Donor. These additional fields provide a more comprehensive description of molecular interactions, particularly important for biological systems where hydrogen bonding plays a critical role in receptor-ligand recognition [3].
Q2: My SEHAD model shows a lower predictive r² (r²pred) than my SEH model. Is this expected? Yes, this can occur. In benchmark studies, a SEHAD model demonstrated a lower predictive r² (0.186) compared to a SEH model (0.319) on the same steroid dataset. This does not necessarily mean the model has failed. The SEHAD model may be capturing more complex, nuanced interactions. It remains a statistically acceptable model for CoMSIA analysis, and its interpretative value regarding specific interactions can be greater [3].
Q3: How does the choice of fields impact the number of optimal components in a PLS analysis? The complexity introduced by additional fields can influence the model's optimal dimensionality. For instance, in a benchmark test, both SEH and SEHAD models identified an optimal of 3 components during cross-validation. In contrast, a published Sybyl analysis of the same data with SEH fields found 4 components to be optimal. This highlights that field selection is a key parameter in model optimization [3].
Q4: When should I prioritize using a SEHAD model over a simpler SEH model? Prioritize the SEHAD model when the biological activity you are modeling is known or suspected to be heavily influenced by hydrogen bonding. This is often the case for targets with polar active sites. If your primary interest is in steric, electrostatic, and hydrophobic drivers, or if you are working with a congeneric series where hydrogen bonding is consistent, the SEH model may be sufficient and more robust [3].
Troubleshooting Common Experimental Issues
Problem: High prediction residuals for specific compounds in the test set. Solution: This is a common occurrence and can be part of model validation. For example, in the Py-CoMSIA validation study, both the new implementation and the classic Sybyl analysis correctly identified the same compound (compound 10) as a predictive outlier. Investigate the structural features of the outlier compound; it may possess unique characteristics not well-represented in the training set, offering valuable insights for the next cycle of compound design [3].
Problem: The model shows good statistical fit but poor predictive capability. Solution:
- Verify Molecular Alignment: Confirm that the 3D alignment of your molecules is correct. This is a critical and sensitive step in 3D-QSAR model development. Misalignment is a major source of poor predictive performance [20].
- Check Field Contributions: Analyze the contribution of each field. In the SEHAD benchmark, the electrostatic field had the highest contribution (0.534 for SEH; 0.258 for SEHAD), followed by hydrophobic (0.316 for SEH; 0.154 for SEHAD). If the contributions of the hydrogen bond fields are exceptionally low, they might be adding noise. Consider refining your alignment or testing the SEH model [3].
- Review Training/Test Set Division: Ensure your test set molecules are representative of the chemical space covered by the training set. A random but stratified division is recommended [20].
The following tables consolidate quantitative performance data from a benchmark CoMSIA study on a steroid dataset, providing a clear comparison between SEH and SEHAD models [3].
Table 1: Overall Model Performance Metrics
| Metric | Published SEH (Sybyl) | Py-CoMSIA SEH | Py-CoMSIA SEHAD |
|---|---|---|---|
| q² (LOOCV) | 0.665 | 0.609 | 0.630 |
| SPRESS | 0.759 | 0.718 | 0.698 |
| r² (non-cross-validated) | 0.937 | 0.917 | 0.898 |
| Standard Error (S) | 0.33 | 0.33 | 0.366 |
| Optimal Number of Components | 4 | 3 | 3 |
| Predictive r² (r²pred) | 0.318 | 0.40 | 0.186 |
Table 2: Field Contribution Breakdown
| Field | Published SEH | Py-CoMSIA SEH | Py-CoMSIA SEHAD |
|---|---|---|---|
| Steric | 0.073 | 0.149 | 0.065 |
| Electrostatic | 0.513 | 0.534 | 0.258 |
| Hydrophobic | 0.415 | 0.316 | 0.154 |
| Hydrogen Bond Donor | - | - | 0.274 |
| Hydrogen Bond Acceptor | - | - | 0.248 |
Experimental Protocols
Protocol 1: Standard CoMSIA Model Development and Validation
This protocol outlines the core methodology for developing SEH and SEHAD models, as used in the benchmark study [3].
1. Molecule Preparation and Alignment:
- Construct 3D molecular structures using a molecular sketching tool.
- Energy minimization is performed using a standard force field (e.g., TRIPOS) [20].
- Alignment is the most sensitive step. Select the most active molecule as a template and align all other molecules to it using common substructures or pharmacophores. The benchmark study used a pre-aligned dataset from a previous publication [3].
2. Grid Generation and Field Calculation:
- Create a 3D grid that encompasses all aligned molecules.
- Set grid spacing (typically 1-2 Å) and padding (e.g., 4 Å beyond the molecular dimensions).
- Calculate molecular similarity indices using a Gaussian function with a defined attenuation factor (commonly 0.3) [3]. The five CoMSIA fields are:
- Steric (S)
- Electrostatic (E)
- Hydrophobic (H)
- Hydrogen Bond Donor (D)
- Hydrogen Bond Acceptor (A)
3. Partial Least Squares (PLS) Analysis:
- Split the dataset into a training set (~70-80%) for model building and a test set (~20-30%) for validation [20].
- Perform Leave-One-Out Cross-Validation (LOOCV) on the training set to determine the optimal number of components (latent variables) that gives the highest cross-validated correlation coefficient (q²).
- Use the optimal number of components to derive the final non-cross-validated model and calculate the conventional correlation coefficient (r²) and standard error (S).
4. Model Validation and Prediction:
- Use the final model to predict the activity of the external test set compounds.
- Calculate the predictive r² (r²pred) to evaluate the model's true external predictive power [3].
Workflow for CoMSIA Model Development and Validation
Protocol 2: Systematic Field Combination Comparison
This protocol provides a step-by-step method for directly comparing SEH and SEHAD models to inform field selection.
1. Baseline SEH Model Construction:
- Follow Protocol 1 to build and validate a model using only Steric, Electrostatic, and Hydrophobic fields.
- Record all performance metrics (q², r², r²pred, optimal components, field contributions).
2. Extended SEHAD Model Construction:
- Using the exact same molecular alignment and grid parameters, construct a second model that includes the Hydrogen Bond Donor and Acceptor fields.
- Record the same set of performance metrics.
3. Comparative Analysis:
- Contribution Analysis: Compare the relative contributions of the steric, electrostatic, and hydrophobic fields in both models. A significant shift in contributions when hydrogen bond fields are added indicates these interactions are important and were being partially accounted for by other fields in the SEH model [3].
- Residual Analysis: Compare the prediction residuals for both models across the training and test sets. Identify if the SEHAD model improves predictions for specific compounds known to engage in hydrogen bonding.
- Statistical Comparison: Use the r²pred and the distribution of residuals as the primary indicators of which model generalizes better to new data.
Systematic Comparison of SEH and SEHAD Models
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for CoMSIA Research
| Item | Function & Application in CoMSIA |
|---|---|
| Py-CoMSIA Library | An open-source Python implementation of CoMSIA. It provides a free, flexible alternative to discontinued proprietary software (e.g., Sybyl) and enables integration with modern machine learning libraries [3]. |
| RDKit | An open-source cheminformatics toolkit. Used for fundamental tasks like reading molecules, generating 3D coordinates, optimizing structures, and calculating molecular descriptors [3]. |
| NumPy | A fundamental package for scientific computing in Python. Essential for performing efficient numerical calculations required for grid-based field computations and linear algebra in PLS analysis [3]. |
| Pre-Aligned Benchmark Datasets | Publicly available datasets (e.g., the steroid dataset) with molecules already aligned. Crucial for validating new CoMSIA implementations and methodologies [3]. |
| Partial Least Squares (PLS) Implementation | A statistical method used to relate the CoMSIA fields (X-block) to the biological activity data (Y-block). It is the core algorithm for building the 3D-QSAR model and is available in various scientific computing libraries [3]. |
Within the broader scope of research on optimizing Comparative Molecular Similarity Indices Analysis (CoMSIA) field combinations, prospective validation stands as the definitive test of model utility and predictive power. While internal validation (e.g., cross-validated q²) and statistical goodness-of-fit (e.g., r²) are essential first steps, they merely assess a model's self-consistency and interpolative capability. True prospective validation extends beyond this by using the CoMSIA model to predict the activity of novel, untested compounds before their synthesis and biological evaluation. This process creates a closed loop of rational drug design: prediction → synthesis → experimental testing → model refinement. Successful prospective validation provides unambiguous evidence that the model captures genuine structure-activity relationships rather than statistical artifacts, thereby enabling genuine molecular design.
The optimization of CoMSIA field combinations is particularly crucial for this process. Unlike its predecessor CoMFA, which primarily utilizes steric and electrostatic fields, CoMSIA incorporates up to five molecular field types: steric (S), electrostatic (E), hydrophobic (H), hydrogen bond donor (D), and hydrogen bond acceptor (A) [3] [15]. Selecting the optimal combination of these fields is not trivial; an over-specified model may fit training data well but fail to predict new compounds, while an under-specified model may miss critical interactions governing biological activity. This technical support document addresses the specific challenges researchers face during the prospective validation of CoMSIA models, with a focus on troubleshooting failed predictions and optimizing field selections to enhance model predictivity for experimental confirmation.
Technical Support Center
Frequently Asked Questions (FAQs)
FAQ 1: What constitutes a successful prospective validation for a CoMSIA model? A successful prospective validation requires that the model's predictions correlate well with experimental results for a set of newly designed and synthesized compounds that were not part of the original training set. Key indicators include:
- The model correctly predicts the trend of activity for the new compounds (e.g., high, medium, low potency).
- The quantitative predictions of activity (pIC₅₀, pEC₅₀, etc.) fall within an acceptable error margin of the experimentally measured values.
- The designed compounds, based on the model's contour maps, show improved potency compared to previous leads, confirming the hypothesized structure-activity relationship.
FAQ 2: How do I select the optimal CoMSIA field combination for my dataset to ensure better predictive performance? There is no single best combination that applies to all targets. The optimal field set must be determined empirically through a systematic screening process [54]:
- Construct multiple CoMSIA models using different combinations of the five fields (S, E, H, D, A).
- Evaluate each model based on robust statistical criteria: a cross-validated coefficient q² > 0.5 and a conventional coefficient r² > 0.8 are common thresholds for a reliable model [12] [55].
- The model with the highest predictive q² and the lowest standard error, typically using a minimal number of principal components to avoid overfitting, should be selected for prospective testing. For example, a robust CoMSIA model combining steric and hydrogen-bond acceptor fields (SA) was successfully used to design active antileishmanial compounds [54].
FAQ 3: My model showed excellent statistical parameters (high q² and r²) but failed prospectively. What are the most likely causes? This is a common challenge, often stemming from one or more of the following issues:
- Inadequate Molecular Alignment: The bioactive conformation and alignment of your training set molecules may be incorrect. Re-evaluate your alignment rules, considering multiple low-energy conformations or using a receptor-based alignment if the protein structure is available.
- Overfitting: The model may be too complex, describing noise rather than the true SAR. This occurs with too many components relative to the number of training molecules. Use the lowest number of components that gives a high q².
- Limited Chemical Space: The training set may not cover the structural features of the newly designed compounds, making extrapolation unreliable. Ensure your training set is diverse and representative.
- Incorrect Field Combination: The selected fields may not accurately represent the key interactions driving binding to your specific biological target. Re-screening field combinations or incorporating additional physicochemical properties may be necessary.
FAQ 4: What is the recommended workflow for taking a CoMSIA model from prediction to experimental confirmation? The following diagram outlines the critical steps for a robust prospective validation workflow, incorporating key decision points to troubleshoot and refine the model.
Troubleshooting Guides
Problem: Poor Correlation Between Predicted and Experimental Activities for New Compounds
| Symptom | Possible Cause | Diagnostic Steps | Solution |
|---|---|---|---|
| New compounds are consistently less active than predicted. | The model is overfitted to the training set. | Check the number of components used in the PLS analysis. A high number of components relative to the number of training molecules is a red flag. | Rebuild the model using fewer PLS components and prioritize models with a lower standard error of estimate. |
| Specific chemical scaffolds are poorly predicted. | The training set lacks diversity and does not adequately represent the chemical space of the new scaffolds. | Analyze the structural similarity between the new scaffolds and the training set. | Expand the training set with representative compounds that bridge the chemical space or develop a separate, more localized model for the new scaffold. |
| Predictions are insensitive to specific molecular modifications. | The CoMSIA field combination may be missing a key interaction field (e.g., Hydrophobic or H-bond). | Review the contour maps. Do they highlight regions known from crystallography or docking to be important? | Rebuild models with different field combinations. For example, if hydrophobic interactions are critical, ensure the hydrophobic (H) field is included [3]. |
Problem: Uninterpretable or Chemically Illogical Contour Maps
| Symptom | Possible Cause | Diagnostic Steps | Solution |
|---|---|---|---|
| Contour maps show favorable regions in sterically impossible locations. | Incorrect molecular alignment is the most probable cause. | Superimpose your aligned molecules and check if key pharmacophore features are consistently overlayed. | Re-perform the alignment using a different method (e.g., receptor-based alignment if the protein structure is available, or pharmacophore-based alignment). |
| Maps are noisy and lack clear, contiguous regions of favorable/unfavorable effects. | The model may be based on a suboptimal field combination or the grid spacing may be too fine. | Test different field combinations and check if the maps become more interpretable. | Switch to a CoMSIA field combination that yields smoother, more interpretable maps (e.g., SE, SEH, SHD). Increase the grid spacing slightly (e.g., from 1Å to 2Å). |
Case Studies in Prospective Validation
The following case studies illustrate the successful application of optimized CoMSIA models to design new bioactive compounds, followed by experimental confirmation.
Case Study 1: Design of Bcr-Abl Inhibitors for Chronic Myeloid Leukemia
- Objective: To design new purine-based Bcr-Abl inhibitors to combat imatinib resistance, particularly the T315I mutation [56].
- CoMSIA Model Implementation: A 3D-QSAR model was constructed using a database of 58 purine derivatives with known inhibitory activity (pIC₅₀). The model correlated biological activity with steric and electrostatic potentials.
- Prospective Validation & Outcome: Based on the model's insights, a series of new 2,6,9-trisubstituted purines were designed and synthesized. Experimental testing confirmed high potency, with compounds 7a and 7c exhibiting IC₅₀ values of 0.13 and 0.19 μM, respectively, surpassing the potency of imatinib (IC₅₀ = 0.33 μM). Crucially, some analogs (7e and 7f) showed greater efficacy than imatinib against resistant KCL22-B8 cells expressing the Bcr-Abl[T315I] mutation [56].
Experimental vs. Predicted Activity for Selected Bcr-Abl Inhibitors
| Compound ID | Predicted pIC₅₀ | Experimental pIC₅₀ | Experimental IC₅₀ (μM) | Key Outcome |
|---|---|---|---|---|
| 7a | - | 6.89 | 0.13 | Surpassed imatinib potency |
| 7c | - | 6.72 | 0.19 | Surpassed imatinib potency |
| 7e | - | - | - | Active against T315I mutant |
| 7f | - | - | - | Active against T315I mutant |
| Imatinib | - | 6.48 | 0.33 | Reference drug |
Case Study 2: Design of Hybrid Quinoline–Chalcone Anti-Leishmanial Compounds
- Objective: To design novel quinoline–chalcone hybrids with inhibitory activity against Leishmania amazonensis [54].
- CoMSIA Model Implementation: A robust CoMSIA model was developed using a dataset of chalcone derivatives. The best model combined steric and hydrogen-bond acceptor fields (SA) (q² = 0.664), highlighting the importance of these interactions for antileishmanial activity.
- Prospective Validation & Outcome: The contour maps from the CoMSIA-SA model guided the design of 12 new hybrid compounds. Four of the synthesized compounds (E003, E005, E006, E011) demonstrated inhibitory activity comparable to the standard drug amphotericin B, successfully validating the model's predictive capability [54].
Summary of Prospectively Designed Anti-Leishmanial Compounds
| Compound ID | CoMSIA Field Guidance | Experimental Outcome |
|---|---|---|
| E003 | Designed to fit steric and H-bond acceptor favorable regions | Activity comparable to Amphotericin B |
| E005 | Designed to fit steric and H-bond acceptor favorable regions | Activity comparable to Amphotericin B |
| E006 | Designed to fit steric and H-bond acceptor favorable regions | Activity comparable to Amphotericin B |
| E011 | Designed to fit steric and H-bond acceptor favorable regions | Activity comparable to Amphotericin B |
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table details key software, computational tools, and resources essential for conducting CoMSIA studies and prospective validation.
Key Research Reagent Solutions for CoMSIA Modeling
| Item Name | Function/Application | Example in Search Results |
|---|---|---|
| Py-CoMSIA | An open-source Python implementation of CoMSIA, providing a free alternative to proprietary software. It uses RDKit for cheminformatics and NumPy for calculations [3] [15]. | Provides a functional open-source alternative to proprietary software, validated on benchmark datasets [15]. |
| Proprietary Modeling Suites | Software like SYBYL/Tripos, Schrödinger, or MOE offer integrated, GUI-driven environments for performing CoMSIA and molecular docking. | Classical CoMSIA analysis was conducted using the Sybyl platform [3]. |
| Partial Least Squares (PLS) Regression | The core statistical method used in 3D-QSAR to correlate the CoMSIA field descriptors (independent variables) with biological activity (dependent variable). | Used to determine the optimal number of components and build the final predictive model [3] [12]. |
| Deep Mutational Scanning (DMS) Datasets | Large-scale experimental data on the effects of mutations on protein function and binding. Can be used to validate advanced QSAR approaches for biologics and protein engineering. | Used to train predictive models for SARS-CoV-2 RBD variant binding and antibody escape [13]. |
Standard Experimental Protocol for Prospective Validation
This section provides a detailed methodology for the experimental phase of prospective validation, as referenced in the case studies.
Title: Experimental Protocol for In Vitro Kinase Inhibition Assay Background: This protocol describes a standard method for determining the half-maximal inhibitory concentration (IC₅₀) of novel compounds against a target kinase, such as Bcr-Abl [56]. Materials:
- Test Compounds: Newly synthesized compounds, dissolved in DMSO at a high concentration (e.g., 10 mM stock).
- Target Kinase: Purified recombinant kinase protein (e.g., Bcr-Abl).
- Substrate and Cofactor: Specific peptide substrate and ATP.
- Detection Reagents: ELISA-based kinase kit or ADP-Glo Kinase Assay kit.
- Equipment: Multi-channel pipettes, 96-well or 384-well assay plates, microplate reader or luminescence detector.
Procedure:
- Compound Dilution: Prepare a serial dilution of each test compound in a suitable buffer, typically creating 8-12 concentrations covering a range around the expected IC₅₀ (e.g., from 0.001 μM to 100 μM). Include a control with only DMSO (no inhibitor).
- Reaction Setup: In each well of the assay plate, add the kinase, substrate, ATP, and the diluted compound. The reaction is typically initiated by adding ATP or the kinase.
- Incubation: Incubate the reaction plate at 30°C for a predetermined time (e.g., 60 minutes) to allow the kinase reaction to proceed.
- Detection: Stop the reaction and detect the amount of phosphorylated product formed. With an ELISA kit, this may involve adding a detection antibody and a colorimetric substrate. With the ADP-Glo assay, it involves converting remaining ADP to ATP and generating a luminescent signal.
- Data Analysis: Measure the signal (absorbance or luminescence) for each well. Normalize the data relative to the DMSO control (100% activity) and blank (0% activity). Plot the inhibitor concentration versus the percentage of kinase activity and fit the data to a sigmoidal dose-response curve to calculate the IC₅₀ value. Convert IC₅₀ to pIC₅₀ (-logIC₅₀) for QSAR analysis.
The logical flow of this protocol, from setup to data analysis, is visualized below.
Conclusion
Optimizing CoMSIA field combinations is a critical determinant of model success, moving beyond default settings to strategic, problem-specific configurations. The integration of machine learning for feature selection and model building addresses fundamental limitations of traditional PLS regression, significantly enhancing predictive performance for complex biological endpoints. The emergence of open-source tools like Py-CoMSIA democratizes access to these advanced methodologies while ensuring reproducibility. Future directions point toward dynamic field selection protocols, deeper integration with structural biology data from complexes, and expanded applications in challenging areas like predicting protein mutation effects and polypharmacology. These advancements will further solidify CoMSIA's role in accelerating the design of novel therapeutic agents with optimized binding and selectivity profiles.
References
- 1. Comparative Molecular Similarity Indices Analysis [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/comparative-molecular-similarity-indices-analysis]
- 2. Comparative Molecular Similarity Indices Analysis [https://www.sciencedirect.com/topics/immunology-and-microbiology/comparative-molecular-similarity-indices-analysis]
- 3. Py-CoMSIA: An Open-Source Implementation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11945924/]
- 4. Comparative Molecular Field Analysis (CoMFA) and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3211024/]
- 5. Comparative Molecular Field Analysis - an overview [https://www.sciencedirect.com/topics/nursing-and-health-professions/comparative-molecular-field-analysis]
- 6. Systematic Statistical Comparison of Comparative ... [https://pubmed.ncbi.nlm.nih.gov/16995732/]
- 7. Comparative Molecular Field Analysis (CoMFA) and ... [https://www.mdpi.com/1422-0067/12/10/7022]
- 8. 3D-QSAR CoMFA/CoMSIA models based on theoretical ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523409003080]
- 9. CoMFA and CoMSIA Studies of 1,2-dihydropyridine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4980841/]
- 10. Comparative molecular field analysis (CoMFA) and ... [https://pubmed.ncbi.nlm.nih.gov/18716715/]
- 11. Statistical results of CoMSIA models with different combinations of ... [https://plos.figshare.com/articles/dataset/Statistical_results_of_CoMSIA_models_with_different_combinations_of_molecular_fields_/29342495]
- 12. Design and evaluation of novel triazole derivatives as ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1518777/full]
- 13. Modeling the Mutational Effects on Biochemical ... [https://www.mdpi.com/2218-273X/15/11/1538]
- 14. clhaga/pycomsia: A pythonic implementation of CoMSIA [https://github.com/clhaga/pycomsia/]
- 15. Py-CoMSIA: An Open-Source Implementation of ... [https://www.mdpi.com/1424-8247/18/3/440]
- 16. CoMSIA Field Descriptions [https://life.nthu.edu.tw/~lslth/QSARandCoMFA/sybyl/sybyl/qsar/qsar_theory18.html]
- 17. An updated steroid benchmark set and its application in the ... [https://pubmed.ncbi.nlm.nih.gov/18330978/]
- 18. The CoMFA Steroids as a Benchmark Dataset for ... [https://link.springer.com/chapter/10.1007/0-306-46858-1_13]
- 19. Improvement of the Prediction Power of the CoMFA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3447613/]
- 20. 3D-QSAR, molecular docking and ADMET studies of ... [https://www.sciencedirect.com/science/article/abs/pii/S0019452222003375]
- 21. Structural insights into GPCR signaling activated by peptide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322139/]
- 22. Therapeutic peptides: current applications and future ... [https://www.nature.com/articles/s41392-022-00904-4]
- 23. Design and screening of novel 1,2,4-Triazole-3-thione ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1590711/full]
- 24. Insights into anti-tuberculosis drug design on the scaffold of ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra01362c?page=search]
- 25. Principles and Overview of Sampling Methods for Modeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4849799/]
- 26. Integration of machine learning in 3D-QSAR CoMSIA models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10654693/]
- 27. Molecular field analysis for data-driven molecular design in ... [https://pubs.rsc.org/en/content/articlehtml/2022/ob/d2ob00228k]
- 28. vs SelectFromModel - huge difference in model performance RFE [https://datascience.stackexchange.com/questions/84793/selectfrommodel-vs-rfe-huge-difference-in-model-performance]
- 29. 1.13. Feature selection [https://scikit-learn.org/stable/modules/feature_selection.html]
- 30. Feature selection strategies for drug sensitivity prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC7287073/]
- 31. Techniques | Pier Paolo Ippolito Feature Selection [https://ppiconsulting.dev/blog/blog27/]
- 32. Performing recursive feature elimination with multiple ... [https://stackoverflow.com/questions/78420559/performing-recursive-feature-elimination-with-multiple-performance-metrics]
- 33. Recursive Feature Elimination: A Powerful Technique for ... [https://medium.com/the-modern-scientist/recursive-feature-elimination-a-powerful-technique-for-feature-selection-in-machine-learning-89b3c2f3c26a]
- 34. Comparative evaluation of feature reduction methods for ... [https://www.nature.com/articles/s41598-024-81866-1]
- 35. How to Tune in Hyperparameters Algorithm Gradient Boosting [https://www.geeksforgeeks.org/machine-learning/how-to-tune-hyperparameters-in-gradient-boosting-algorithm/]
- 36. Classifier's hyperparametrs and balancing it Tuning Gradient Boosted [https://datascience.stackexchange.com/questions/14377/tuning-gradient-boosted-classifiers-hyperparametrs-and-balancing-it]
- 37. | Gradient Python Boosting Hyperparameter Tuning [https://www.analyticsvidhya.com/blog/2016/02/complete-guide-parameter-tuning-gradient-boosting-gbm-python/]
- 38. Integration of machine learning in 3D-QSAR CoMSIA ... [https://pubs.rsc.org/en/content/articlelanding/2023/ra/d3ra06690h]
- 39. : Classifier Example Gradient Boosting Hyperparameters Tuning [https://www.datasciencelearner.com/machine-learning/gradient-boosting-hyperparameters-tuning/]
- 40. Comparison of methods for tuning machine learning model ... [https://bmcmedresmethodol.biomedcentral.com/articles/10.1186/s12874-025-02561-x]
- 41. Machine learning with hyperparameter optimization ... [https://www.nature.com/articles/s41598-025-95490-0]
- 42. Why Gradient Boosting Machines Still Matter in 2025 [https://www.linkedin.com/posts/shawnbhatti_an-introduction-to-gradient-boosted-trees-activity-7365105825259991040-uXap]
- 43. AI-Integrated QSAR Modeling for Enhanced Drug Discovery [https://www.mdpi.com/1422-0067/26/19/9384]
- 44. Comprehensive review of dimensionality reduction algorithms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12453773/]
- 45. Quantitative Structure-Activity Relationship Models [https://www.emergentmind.com/topics/quantitative-structure-activity-relationship-qsar-models]
- 46. A novel non-linear approach for establishing a QSAR model of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10565811/]
- 47. A Comparative Study of Dimensionality Reduction Methods ... [https://www.mdpi.com/2079-9292/14/14/2916]
- 48. A review of quantitative structure-activity relationship [https://www.sciencedirect.com/science/article/abs/pii/S0169743924002181]
- 49. Multitarget inhibition of CDK2, EGFR, and tubulin by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12233951/]
- 50. Analyzing the CoMSIA Models [https://life.nthu.edu.tw/~lslth/QSARandCoMFA/sybyl/sybyl/qsar/qsar_tut39.html]
- 51. The CoMFA Steroids as a Benchmark Dataset for ... [https://link.springer.com/content/pdf/10.1007/0-306-46858-1_13]
- 52. Py-CoMSIA: An Open-Source Implementation of ... [https://pubmed.ncbi.nlm.nih.gov/40143216/]
- 53. Receptor-guided 3D-QSAR studies, molecular dynamics ... [https://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-017-0385-5]
- 54. Design of Hybrid Quinoline–Chalcone Compounds Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566680/]
- 55. Structural optimization for pyrimidine analogues inhibitors ... [https://www.sciencedirect.com/science/article/abs/pii/S0022286021008218]
- 56. 3D-QSAR Design of New Bcr-Abl Inhibitors Based on Purine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12195648/]