High-Throughput qPCR in Cancer Biomarker Screening: A Comprehensive Guide for Translational Research
This article provides a comprehensive overview of high-throughput quantitative PCR (HT-qPCR) and its transformative role in cancer biomarker screening and validation.
High-Throughput qPCR in Cancer Biomarker Screening: A Comprehensive Guide for Translational Research
Abstract
This article provides a comprehensive overview of high-throughput quantitative PCR (HT-qPCR) and its transformative role in cancer biomarker screening and validation. Tailored for researchers, scientists, and drug development professionals, it explores the foundational principles of cancer biomarkers, including circulating tumor DNA (ctDNA), microRNAs (miRNAs), and mRNA. It details methodological workflows, from automated liquid handling and nanofluidics to data analysis pipelines. The scope extends to practical troubleshooting, optimization strategies for challenging samples, and rigorous validation protocols. Finally, it presents a comparative assessment of HT-qPCR against other genomic technologies like next-generation sequencing (NGS), offering insights to guide platform selection for precision oncology applications.
The Rise of Precision Oncology and the Critical Role of Biomarkers
The Global Cancer Burden and the Imperative for Early Detection
The global burden of cancer is substantial and growing, underscoring an urgent need for scalable early detection technologies. In 2022, there were an estimated 20 million new cancer cases and 9.7 million deaths worldwide according to the World Health Organization (WHO) [1]. The most common cancers include lung (2.5 million new cases), breast (2.3 million), and colorectal cancer (1.9 million) [1]. Projections indicate a dramatic increase to 35 million new annual cases by 2050, a 77% rise from 2022 figures, with the most significant proportional increases expected in low and medium-resource countries [1].
This escalating burden creates a pressing imperative for diagnostic tools that are not only accurate but also cost-effective, rapid, and deployable at scale. High-throughput quantitative PCR (qPCR) represents a foundational technology in this effort, combining high analytical sensitivity, rapid turnaround time, and cost-efficiency essential for informing therapeutic decision-making at scale [2]. Its utility is particularly pronounced in time-sensitive or resource-constrained settings where complex infrastructure is unavailable.
Quantitative Analysis of the Global Cancer Landscape
Table 1: Global Incidence and Mortality for Major Cancers (2022)
| Cancer Type | New Cases (Millions) | Proportion of Total Cases | Deaths (Millions) | Proportion of Total Deaths |
|---|---|---|---|---|
| Lung | 2.5 | 12.4% | 1.8 | 18.7% |
| Breast | 2.3 | 11.6% | 0.67 | 6.9% |
| Colorectal | 1.9 | 9.6% | 0.9 | 9.3% |
| Prostate | 1.5 | 7.3% | - | - |
| Stomach | 0.97 | 4.9% | 0.66 | 6.8% |
| Liver | - | - | 0.76 | 7.8% |
| Total (All Cancers) | 20.0 | 100% | 9.7 | 100% |
Table 2: Projected Cancer Incidence Growth and Health System Preparedness
| Development Index | Projected Increase in Incidence by 2050 | Countries Adequately Financing Cancer Services |
|---|---|---|
| High HDI Countries | +4.8 million cases (Greatest absolute increase) | Higher likelihood of service coverage |
| Medium HDI Countries | 99% increase | Limited data available |
| Low HDI Countries | 142% increase (Largest proportional increase) | Significant gaps in service financing |
| Global Average | 77% increase (35 million total cases) | 39% of countries cover basic cancer management |
Alarming disparities exist in cancer burden and care accessibility. For example, women in lower Human Development Index (HDI) countries are 50% less likely to be diagnosed with breast cancer than women in high HDI countries, yet they face a much higher risk of mortality due to late diagnosis and inadequate treatment access [1]. These inequities highlight the critical need for accessible, affordable, and scalable diagnostic technologies like qPCR that can function effectively in diverse healthcare environments.
High-Throughput qPCR as a Solution for Early Detection
Technical Advantages in Oncology Diagnostics
qPCR offers several distinct advantages that make it particularly suitable for addressing the global cancer detection challenge:
Multiplexing Capability: Modern qPCR platforms can simultaneously detect multiple clinically relevant mutations in a single reaction without compromising sensitivity or speed [2]. For non-small cell lung cancer (NSCLC), multiplexed qPCR panels can simultaneously assess alterations in EGFR, KRAS, BRAF and ALK, delivering results faster with less input material than sequential sequencing approaches [2].
Rapid Turnaround and Minimal Infrastructure: Unlike sequencing platforms that can take days to generate data, qPCR delivers clinically actionable results within hours, which is critical for selecting targeted therapies or enrolling patients in mutation-driven clinical trials [2]. The technology is highly scalable and automation-friendly, supporting high-throughput testing in 96- or 384-well formats without significant capital investment [2].
Cost-Effectiveness for Population-Scale Screening: qPCR remains significantly more cost-effective than next-generation sequencing (NGS), with test costs typically ranging from $50 to $200 compared to $300 to $3,000 for NGS [2]. This affordability makes it particularly well-suited for large-scale screening initiatives and routine clinical diagnostics in resource-conscious healthcare systems.
Innovations in qPCR Chemistry
Recent advancements in qPCR chemistry have further enhanced its utility for cancer biomarker detection:
- Inhibitor Resistance: Next-generation polymerases and buffers are engineered to tolerate PCR inhibitors commonly found in clinical matrices such as heparinized plasma, whole blood, or FFPE-derived nucleic acids [2].
- Thermal Stability: Enzymes now withstand higher-temperature, faster-cycling protocols without loss of activity, enabling faster run times and greater assay reliability [2].
- Lyophilization Compatibility: Ambient-temperature stable formulations support cold chain-independent transport and storage, ideal for decentralized testing environments [2].
- Multiplexing Efficiency: Advanced master mixes and probe systems enable detection of multiple mutations in a single reaction, critical for cancers with complex mutational profiles [2].
Application Note: Multiplex RT-qPCR for Breast Cancer Subtyping
Background and Rationale
Breast cancer remains a leading cause of cancer mortality globally, with approximately 2.3 million new cases anticipated worldwide [3]. Current diagnostic standards rely heavily on Immunohistochemistry (IHC), which can be slow, expensive, and dependent on proficient pathologists [3]. We developed a novel multiplex Reverse Transcription quantitative PCR (RT-qPCR) approach with touch-down methods to enhance the accuracy, speed, and cost-effectiveness of BC diagnosis and subtyping.
Experimental Protocol
Sample Preparation and RNA Extraction
- Sample Collection: Collect Formalin-Fixed Paraffin-Embedded (FFPE) tumor block samples. In our validation study, we used 61 tumor samples from different breast cancer groups (Luminal type, TN, HER2 positive, and TP subtypes) and 9 benign samples [3].
- RNA Extraction: Extract total RNA from FFPE samples using the Quick-DNA/RNATM FFPE Kit (or equivalent), following manufacturer instructions. Evaluate RNA concentration and purity using a Nano-Drop One C spectrophotometer [3].
- Storage: Store isolated RNA at -80°C until use.
Primer and Probe Design
- Target Genes: Design primers and probes for ESR, PGR, HER-2, Ki67, HIF1A, ANG, and VEGF genes, using RPL13A as the endogenous control gene [3].
- Preparation: Resuspend lyophilized primers and probes in PCR-grade water to 100 μM final concentration, then prepare 1:10 dilution to achieve 10 μM working concentration [3].
Multiplex RT-qPCR Workflow
Diagram 1: Multiplex RT-qPCR workflow for breast cancer subtyping
- cDNA Formation: 50°C for 10 minutes [3]
- Initial Denaturation: 95°C for 2 minutes [3]
- Touch-Down PCR Cycling:
- 3 cycles: 95°C for 10s, 70°C for 15s
- 3 cycles: 95°C for 10s, 67°C for 15s
- 3 cycles: 95°C for 10s, 63°C for 15s
- Final Amplification: 40 cycles: 95°C for 5s, 60°C for 30s (with data collection) [3]
Data Analysis and Interpretation
- ΔCT Calculation: Calculate ΔCT values for target genes using the reference gene (RPL13A) as control: ΔCT = CT(target) - CT(reference) [3]
- Inverted ΔCT: Subtract ΔCT values from the maximum PCR cycle number to obtain inverted ΔCT values [3]
- Normalization: Mathematically normalize inverted ΔCT values to the scale of IHC for direct comparison with immunohistochemistry results [3]
- Fold Change Calculation: Use the ΔΔCT method to calculate gene expression fold changes: Fold Change = 2^(-ΔΔCT) [3]
Results and Validation
This methodology demonstrated remarkable precision, nearly equivalent to IHC, in detecting gene expressions vital for BC diagnosis and subtyping [3]. The touch-down PCR approach consistently yielded significantly lower Cycle Threshold (CT) values, enhancing detection sensitivity [3]. Additionally, the protocol enabled exploration of angiogenesis gene expression (Hif1A, ANG, and VEGFR), shedding light on the metastatic potential of the tested BC tumours [3].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for High-Throughput qPCR Cancer Biomarker Screening
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Nucleic Acid Extraction Kits | Quick-DNA/RNATM FFPE Kit | Extracts high-quality RNA/DNA from challenging FFPE samples; critical for working with archival clinical specimens [3]. |
| qPCR Master Mixes | Inhibitor-resistant master mixes | Engineered to tolerate PCR inhibitors in clinical matrices (heparinized plasma, whole blood); enables direct amplification without purification [2]. |
| Reference Genes | RPL13A, GAPDH | Endogenous controls for data normalization; RPL13A demonstrated stable expression across breast cancer subtypes [3]. |
| Primers & Probes | Target-specific primers with dual hybridization probes | Enable multiplex detection of cancer biomarkers (ESR, PGR, HER2, Ki67); designed for specific amplification and reliable detection [3]. |
| Ambient-Stable Formulations | Lyophilized qPCR reagents | Reduce cold chain requirements; ideal for decentralized testing or global distribution to resource-limited settings [2]. |
| Targeted Biomarker Panels | Aspyre Lung Reagents, AmoyDx Pan Lung Cancer PCR Panel | Multiplexed panels for specific cancers; simultaneously assess alterations in EGFR, KRAS, BRAF, ALK in NSCLC [2]. |
Biomarker Development Pipeline: From Discovery to Clinical Application
The translation of cancer biomarkers from initial discovery to clinical application follows a structured pathway with distinct stages:
Diagram 2: Cancer biomarker development and validation pipeline
Key Considerations at Each Stage
Biomarker Discovery: Study design quality is paramount; "samples of convenience" can introduce confounding factors and contribute to false positive associations [4]. Large-scale, well-predefined prospective studies provide the most reliable evidence [4].
Assay Development and Analytical Validation: Following discovery, candidate biomarkers must be adapted to robust, clinically applicable platforms like qPCR [4]. This requires careful optimization of sensitivity, specificity, and reproducibility under standardized conditions.
Clinical Validation and Implementation: Biomarkers must demonstrate clinical utility in improving patient outcomes [4]. Only an estimated 0.1% of initially discovered biomarkers successfully complete this translation process [4].
The growing global cancer burden, projected to reach 35 million new cases annually by 2050, demands urgent implementation of accessible, scalable, and cost-effective early detection technologies [1]. High-throughput qPCR represents a foundational technology in this effort, with demonstrated utility across multiple cancer types including lung, breast, and hepatocellular carcinomas [2] [3] [5].
The multiplex RT-qPCR protocol detailed herein for breast cancer subtyping exemplifies how this technology can deliver precision nearly equivalent to IHC while offering advantages in speed, cost-effectiveness, and objectivity [3]. As the field advances, ongoing innovations in qPCR chemistry, multiplexing efficiency, and ambient-stable formulations will further enhance the deployability of these assays in diverse healthcare settings worldwide [2].
For researchers and drug development professionals, focusing on robust assay design, careful validation, and attention to the specific requirements of the biomarker development pipeline will be essential to translating promising discoveries into clinically impactful tools that can reduce the global cancer burden.
Cancer biomarkers have revolutionized oncology, shifting the paradigm from a one-size-fits-all approach to personalized precision medicine. A biomarker refers to any biological marker found in blood, body fluids, or tissues that signals the presence of normal or abnormal biological processes, conditions, or diseases [6]. In oncology, these biomarkers provide critical insights into cancer type, likely disease progression, recurrence chances, and expected treatment outcomes [6]. They are broadly classified as prognostic biomarkers, which indicate the natural course of the disease independent of treatment, and predictive biomarkers, which forecast how a cancer will respond to a specific therapy [6].
The ideal cancer biomarker should possess attributes that facilitate easy, reliable, and cost-effective assessment, coupled with high sensitivity and specificity [6]. It should demonstrate remarkable detectability at early stages and accurately reflect tumor burden, enabling continuous monitoring of disease evolution during treatments [6]. The rapid expansion of biological sciences has markedly driven technological advancements in biomarker discovery, from early immunological techniques to contemporary sophisticated analytical methodologies including mass spectrometry, protein and DNA arrays, and next-generation sequencing [6].
Classes of Cancer Biomarkers: From Coding to Non-Coding RNAs
DNA and mRNA Biomarkers
Traditional cancer biomarkers primarily focused on DNA alterations and protein-coding mRNA. These include specific genetic alterations such as mutations, amplifications, and translocations at the single gene level, as well as comprehensive genetic profiles created through microarrays [6]. For example, identifying activating epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer patients enables clinicians to select EGFR inhibitors for those most likely to respond [7]. Similarly, multi-gene expression patterns have been exploited as biomarkers for clinical outcomes in numerous cancer studies, such as the PAM50 50-gene panel effectively used for breast cancer classification [8].
Table 1: DNA and mRNA Biomarkers in Cancer
| Biomarker Type | Example | Cancer Type | Clinical Utility |
|---|---|---|---|
| Gene Mutation | EGFR mutations | Non-Small Cell Lung Cancer | Predicts response to EGFR inhibitors |
| Gene Amplification | HER2 amplification | Breast, Gastric Cancer | Guides HER2-targeted therapies |
| Gene Fusion | ALK rearrangements | Non-Small Cell Lung Cancer | Predicts response to ALK inhibitors |
| mRNA Signature | PAM50 | Breast Cancer | Molecular subtyping and prognosis |
| DNA Methylation | F12 gene CpG site | Hepatocellular Carcinoma | Early detection in liquid biopsies |
The Emerging Role of Non-Coding RNAs
Non-coding RNAs represent a significant proportion of the human genome and have established crucial roles in cancer biology. Among these, microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have attracted substantial research interest as potential biomarkers and therapeutic targets [9].
MicroRNAs are short RNA transcripts of 18-24 nucleotides that regulate gene expression at the translational level [9]. According to the canonical view, miRNAs function as negative regulators of gene expression that upon binding to the 3'-untranslated region of target messenger RNA (mRNA) cause a block of translation and/or degradation of the transcript [9]. A single miRNA can target several mRNAs, enabling simultaneous regulation of multiple target genes both within a single pathway or across different pathways [9]. MiRNA expression patterns are tissue specific and often define the physiological nature of the cell, with altered expression occurring in numerous diseases including cancer [9].
Long non-coding RNAs are non-coding RNAs exceeding 200 nucleotides in length that play important roles as transcriptional or post-transcriptional regulators [9]. These molecules demonstrate high stability in the bloodstream due to extensive secondary structures, transport by protective exosomes, and stabilizing post-translational modifications, making them particularly attractive as circulating biomarkers [10]. Many lncRNAs function as oncogenes or tumor suppressors, influencing tumour growth, invasion, and metastasis by regulating key genes involved in cancer development [9] [10].
Table 2: Non-Coding RNA Biomarkers in Cancer Diagnostics
| RNA Type | Example | Cancer Type | Diagnostic Performance |
|---|---|---|---|
| miRNA | miR-21 | Various Cancers | Suppresses p53, TGF-β and mitochondrial apoptosis pathways |
| miRNA | miR-155 | Various Cancers | Binds to coding sequence of target mRNAs; oncogenic |
| lncRNA | PCA3 | Prostate Cancer | FDA-approved for prostate cancer diagnosis (Progensa) |
| lncRNA | HOTAIR | Colorectal Cancer | 92.5% specificity for identifying colorectal cancer |
| lncRNA | MALAT-1 | Non-Small Cell Lung Cancer | 96% specificity for detecting NSCLC |
| lncRNA | LINC00152 | Gastric Cancer | 85.2% specificity for detecting gastric cancer |
| lncRNA | UCA1 | Hepatocellular Carcinoma | 82.1% specificity for detecting HCC |
High-Throughput qPCR: A Cornerstone Technology in Biomarker Screening
Advantages of qPCR in Oncology Diagnostics
Quantitative PCR (qPCR) remains a foundational tool in oncology diagnostics despite the emergence of newer technologies like next-generation sequencing [2]. Its combination of high analytical sensitivity, rapid turnaround time, and cost-efficiency makes it uniquely suited for informing therapeutic decision-making at scale, especially in time-sensitive or resource-constrained settings [2]. Key advantages include:
Multiplexing Capability: qPCR allows multiple clinically relevant mutations to be detected in a single reaction without compromising sensitivity or speed. For example, in non-small cell lung cancer (NSCLC), multiplexed qPCR panels can simultaneously assess alterations in EGFR, KRAS, BRAF and ALK – delivering results faster and using less input material than sequential or panel-based NGS approaches [2].
Rapid Turnaround: Unlike sequencing platforms, which can take days to generate and analyze data, qPCR delivers clinically actionable results within hours. This rapid turnaround is especially valuable in time-sensitive scenarios such as selecting targeted therapies or enrolling patients into mutation-driven clinical trials [2].
Cost-Effectiveness: Test costs typically range from $50 to $200 – substantially less than the $300 to $3,000 price range of NGS. This affordability makes qPCR especially well-suited for large-scale screening initiatives and routine clinical diagnostics [2].
Innovations in qPCR Chemistry and Applications
Recent innovations have significantly enhanced qPCR's clinical utility for cancer biomarker detection:
Inhibitor Resistance: Next-generation polymerases and buffers are engineered to tolerate PCR inhibitors commonly found in clinical matrices, such as heparinized plasma, whole blood, or FFPE-derived nucleic acids [2].
Thermal Stability: Enzymes now withstand higher-temperature, faster-cycling protocols without loss of activity – enabling faster run times and greater assay reliability [2].
Multiplexing Efficiency: Advanced master mixes and probe systems enable detection of multiple mutations in a single reaction – critical for cancers with complex mutational profiles [2].
Sensitivity Optimization: Methodologies have been developed to quantify surrogate markers of immunity from very low numbers of PBMCs, reducing costs by almost 90% compared to standard practice while maintaining single-cell analytical sensitivity [11].
Experimental Protocols for Biomarker Detection
High-Throughput, Cost-Optimized RT-qPCR for Immune Marker Profiling
This protocol enables sensitive and specific quantification of surrogate transcriptional markers of immunity from low numbers of PBMCs, optimized for high-throughput screening [11].
Sample Preparation and Stimulation
- Isolate PBMCs from healthy donors by standard density gradient centrifugation and cryopreserve in 90% FBS/10% DMSO.
- Before use, thaw samples rapidly at 37°C, treat with DNase I (100μg/mL), and rest for 18 hours at 2x10^6 cells/mL in complete R10 media at 37°C and 5% CO2.
- Stimulate with synthetic peptides (10μg/mL) representing well-defined CD4+ or CD8+ T cell epitopes alongside PMA/Iono mitogen positive-control (50ng/mL PMA, 1,000ng/mL Ionomycin) and media-only negative-control.
- For RT-qPCR analysis, stimulate PBMCs in 200μL R10 media in 96-well U-bottom plates.
RNA Extraction and Reverse Transcription
- Extract RNA using MagMAX mirVana Total RNA Isolation Kit following manufacturer's instructions.
- Convert extracted RNA to cDNA with SuperScript IV First-Strand Synthesis System.
- For cost-optimized "Half Volume" or "Quarter Volume" protocols, use reagents at 50% or 25% of manufacturer-recommended volumes, maintaining equal reaction volume with DEPC-Treated H2O.
Quantitative PCR (qPCR)
- Determine mRNA copies/reaction with absolute quantification based on a standard curve.
- Use IFN-γ, TNF-α and IL-2 specific desalt-grade primers at 500nM with SYBR Green Master-Mix.
- Run reactions in technical triplicate at either 10μL or 5μL total volume.
- For 5μL reaction volumes, add 1μL of reverse transcription eluent diluted 1:4 in Ultra-Pure H2O.
- Acquire data using a QuantStudio5 Real-Time PCR system.
- Calculate primer reaction efficiency by amplification of logarithmically diluted cDNA.
Circulating lncRNA Detection from Blood Samples
This protocol outlines the recommended approach for investigating circulating lncRNAs in blood samples from cancer patients [10].
Sample Collection and Processing
- Collect peripheral blood in EDTA tubes and process within 2 hours of collection.
- Centrifuge at 1,200 × g for 10 minutes at 4°C to separate plasma.
- Transfer supernatant to a fresh tube and centrifuge at 12,000 × g for 10 minutes at 4°C to remove residual cells.
- Aliquot cleared plasma and store at -80°C until RNA extraction.
RNA Extraction and Quality Control
- Extract RNA from plasma using commercial kits specifically designed for liquid biopsies.
- Include synthetic spike-in RNAs during extraction to monitor efficiency and potential degradation.
- Assess RNA quality and quantity using spectrophotometry or fluorometry.
Reverse Transcription and qPCR
- Perform reverse transcription using gene-specific primers or random hexamers.
- Use stem-loop RT primers for specific miRNA detection if analyzing multiple RNA species.
- Conduct qPCR reactions using SYBR Green or TaqMan chemistry.
- Normalize data using stable reference genes or spike-in controls.
- Analyze using the 2^(-ΔΔCt) method for relative quantification.
Visualization of Experimental Workflows and Biomarker Classification
High-Throughput qPCR Workflow for Cancer Biomarker Screening
Cancer Biomarker Classification and Clinical Applications
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for High-Throughput qPCR Biomarker Studies
| Reagent/Material | Function | Example Products | Key Features |
|---|---|---|---|
| Nucleic Acid Extraction Kits | Isolation of high-quality RNA/DNA from various sample types | MagMAX mirVana Total RNA Isolation Kit | Optimized for challenging samples including liquid biopsies |
| Reverse Transcription Kits | Conversion of RNA to cDNA for qPCR analysis | SuperScript IV First-Strand Synthesis System | High efficiency and robustness for limited samples |
| qPCR Master Mixes | Enzymes and buffers for efficient amplification | Luna Universal qPCR Master Mix, ssoAdvanced Universal SYBR Green Master-Mix | Inhibitor-resistant, thermal stability, multiplexing compatibility |
| qPCR Primers & Probes | Target-specific amplification | PrimerBank primers, Custom TaqMan assays | High specificity and efficiency for biomarker targets |
| Reference Genes | Normalization of qPCR data | GAPDH, β-actin, 18S rRNA | Stable expression across sample types and conditions |
| Automation Equipment | High-throughput sample processing | Liquid handlers, automated pipetting systems | Enables miniaturization and reproducible results |
| Quality Control Tools | Assessment of RNA/DNA quality | Bioanalyzer, spectrophotometers | Ensures input material quality for reliable results |
The future of cancer biomarker research lies in shifting towards multiparameter approaches that incorporate diverse molecular classes including DNA, mRNA, miRNAs, and lncRNAs, alongside dynamic processes and immune signatures [6]. The integration of artificial intelligence and machine learning is further revolutionizing biomarker analysis by identifying subtle patterns in large datasets that human observers might miss [8]. These technologies enable the integration and analysis of various molecular data types with imaging to provide a comprehensive picture of cancer, consequently enhancing diagnostic accuracy and therapeutic recommendations [12] [8].
High-throughput qPCR maintains its pivotal role in this evolving landscape due to its unique combination of speed, cost-efficiency, and scalability [2]. As biomarker discovery continues to advance, qPCR technologies have kept pace through innovations in chemistry, automation, and miniaturization, ensuring their continued relevance in both research and clinical settings [11] [2]. By embracing technological sophistication without compromising practical utility, the field moves closer to realizing the full potential of personalized cancer diagnosis and treatment through comprehensive biomarker profiling.
The global molecular diagnostics market is demonstrating robust growth, propelled by its indispensable role in modern healthcare. This expansion is quantified by several key reports, all pointing towards a consistent upward trajectory, though with varying projections based on different methodological assumptions. The market was valued at approximately USD 27 billion in 2024 and is projected to grow to between USD 40.4 billion and USD 63.86 billion by 2034, reflecting compound annual growth rates (CAGRs) of 4.2% to 3.87% over the forecast period [13] [14]. This growth is primarily fueled by the rising global prevalence of infectious diseases, technological advancements in diagnostic techniques, an increasing focus on early disease diagnosis, and the growing demand for point-of-care (POC) testing solutions [13]. Furthermore, the global geriatric population, which is more susceptible to chronic diseases, is a significant demographic driver; the population aged 60 and above is projected to rise from 1.1 billion in 2023 to 1.4 billion by 2030, necessitating more frequent health monitoring [13].
Table 1: Global Molecular Diagnostics Market Size Projections
| Report Source | Base Year Value | Projected Year Value | Forecast Period | CAGR |
|---|---|---|---|---|
| GM Insights [13] | USD 27 Billion (2024) | USD 40.4 Billion (2034) | 2025-2034 | 4.2% |
| Precedence Research [14] | USD 45.11 Billion (2025) | USD 63.86 Billion (2034) | 2025-2034 | 3.87% |
| Research and Markets [15] | USD 15.9 Billion (2025) | USD 30.9 Billion (2035) | 2025-2035 | 6.2% |
The application of molecular diagnostics in oncology is a particularly fast-growing segment, driven by the critical need for early detection and the development of precision medicine [14]. Molecular diagnostics enable the identification of cancer-related biomarkers at very early stages, allowing for treatment initiation before symptoms arise. In 2023, nearly 1.96 million new cancer cases were projected in the United States alone, underscoring the massive demand for accurate diagnostic methods [14]. Techniques such as quantitative PCR (qPCR), next-generation sequencing (NGS), and microarrays are pivotal for detecting genetic mutations, gene expression patterns, and other nucleic acid-based biomarkers that guide treatment decisions and facilitate personalized therapeutic strategies [16] [15].
Table 2: Molecular Diagnostics Market Segmentation (2024 Estimates)
| Segmentation Basis | Leading Segment | Market Share / Key Stat |
|---|---|---|
| Product Type | Reagents & Kits | ~66-70% of market [13] [14] |
| Technology | Polymerase Chain Reaction (PCR) | 70.4% of market [13] |
| Application | Infectious Disease Diagnostics | ~78% of market [14] |
| End User | Hospitals & Central Laboratories | Largest market share [14] |
| Regional Market | North America | >40% revenue share [14] |
| Fastest-Growing Region | Asia Pacific | CAGR of ~4.9% [14] |
Application Note: High-Throughput qPCR for Cancer Biomarker Screening
Quantitative PCR (qPCR) and its derivative, reverse transcription qPCR (RT-qPCR), are cornerstone technologies in molecular diagnostics and biomarker research due to their sensitivity, specificity, and capacity for precise quantification [17]. In the context of cancer, these techniques allow for the rapid, sensitive, and accurate detection of potential biomarkers from a variety of sample sources, including formalin-fixed paraffin-embedded (FFPE) tissue, circulating tumor cells, and liquid biopsies [16]. The primary biomarkers quantified include DNA, mRNA, microRNA (miRNA), and long non-coding RNA (lncRNA). Notably, miRNA is a highly stable biomarker resistant to fragmentation, making it an excellent candidate for precision diagnostics in cancer [16]. The power of qPCR is now being amplified through high-throughput screening (HTS) methodologies, which enable the processing of thousands of reactions simultaneously, dramatically accelerating the pace of biomarker discovery and validation.
Detailed Protocol: A Cost-Optimized HTS qPCR Workflow
The following protocol is adapted from an HTS-optimized RT-qPCR assay designed for quantifying surrogate markers of immunity, with principles directly applicable to cancer biomarker screening from limited cell samples [18]. This protocol is miniaturized to reduce reagent costs by nearly 90% while maintaining high sensitivity and specificity.
Sample Preparation and Stimulation
- Cell Source: Utilize relevant cell samples, such as purified cancer cell lines, patient-derived PBMCs, or other primary cells.
- Resting: Thaw and rest cells (e.g., 2x10^6 cells/mL) in complete media for 18 hours at 37°C and 5% CO2.
- Stimulation: Stimulate cells with the desired oncogenic or therapeutic agents. Include a mitogen-positive control (e.g., PMA/Ionomycin) and a media-only negative control. For kinetic studies, harvest cells at multiple time points (e.g., hourly from 0-12 hours) to identify peak biomarker expression [18].
RNA Extraction
- Method: Extract total RNA using a magnetic bead-based RNA isolation kit, such as the MagMAX mirVana Total RNA Isolation Kit, following the manufacturer's instructions. This method is efficient for 96-well or 384-well format processing [18].
Reverse Transcription (cDNA Synthesis) - Cost Optimized
- Kit: Use a high-efficiency system like the SuperScript IV First-Strand Synthesis System.
- Miniaturization: Scale down the reaction volumes to 25% of the manufacturer's recommended volume to drastically reduce costs while maintaining cDNA yield and quality [18].
- Example: If the standard protocol uses 20 µL, the miniaturized reaction would be 5 µL.
- Priming: Use a combination of oligo(dT) and random primers for comprehensive cDNA synthesis.
Quantitative PCR (qPCR) - High-Throughput Setup
- Reaction Volume: Perform qPCR in a miniaturized 5 µL total reaction volume [18].
- Chemistry: Use a SYBR Green or probe-based master mix, such as the ssoAdvanced Universal SYBR Green Master-Mix.
- Template: Add 1 µL of a 1:4 dilution of the synthesized cDNA from the previous step.
- Primers: Use gene-specific primers at a concentration of 500 nM. For cancer biomarkers, this could include primers for mRNA, miRNA (using a specialized Mir-X miRNA kit), or lncRNA targets [16].
- Platform: Run the reactions on a high-throughput real-time PCR system capable of handling 384-well or higher-density plates, such as the SmartChip Real-Time PCR System, which allows for thousands of nanoscale reactions per chip [16].
- Replication: Conduct all reactions in technical triplicate in accordance with MIQE guidelines to ensure reproducibility [18].
Data Analysis and Interpretation
- Baseline and Threshold Setting: Accurate quantification cycle (Cq) values are critical. The baseline should be set using early cycles (e.g., 5-15) where fluorescence is stable. The threshold must be set within the exponential phase of all amplification curves, sufficiently above the baseline to ensure signal significance [19].
- Relative Quantification (ΔΔCq Method): This is the most common method for gene expression analysis [17] [20].
- Normalization: Normalize the Cq of the target gene (ΔCq) to a stable endogenous control (reference gene) [17].
- Calibration: Compare the ΔCq of the test sample to the ΔCq of a control sample (e.g., untreated cells) to calculate the ΔΔCq.
- Fold Change: Calculate the fold change in gene expression using the formula: 2^(-ΔΔCq) [20].
- PCR Efficiency: For the ΔΔCq method to be valid, the amplification efficiency of the target and reference genes must be approximately equal and near 100% (90-110%). Efficiency (E) is calculated from a standard curve of serial dilutions using the formula: E (%) = (10^(-1/slope) - 1) × 100 [20]. If efficiencies are not equivalent, alternative models like the Pfaffl method should be used [19] [20].
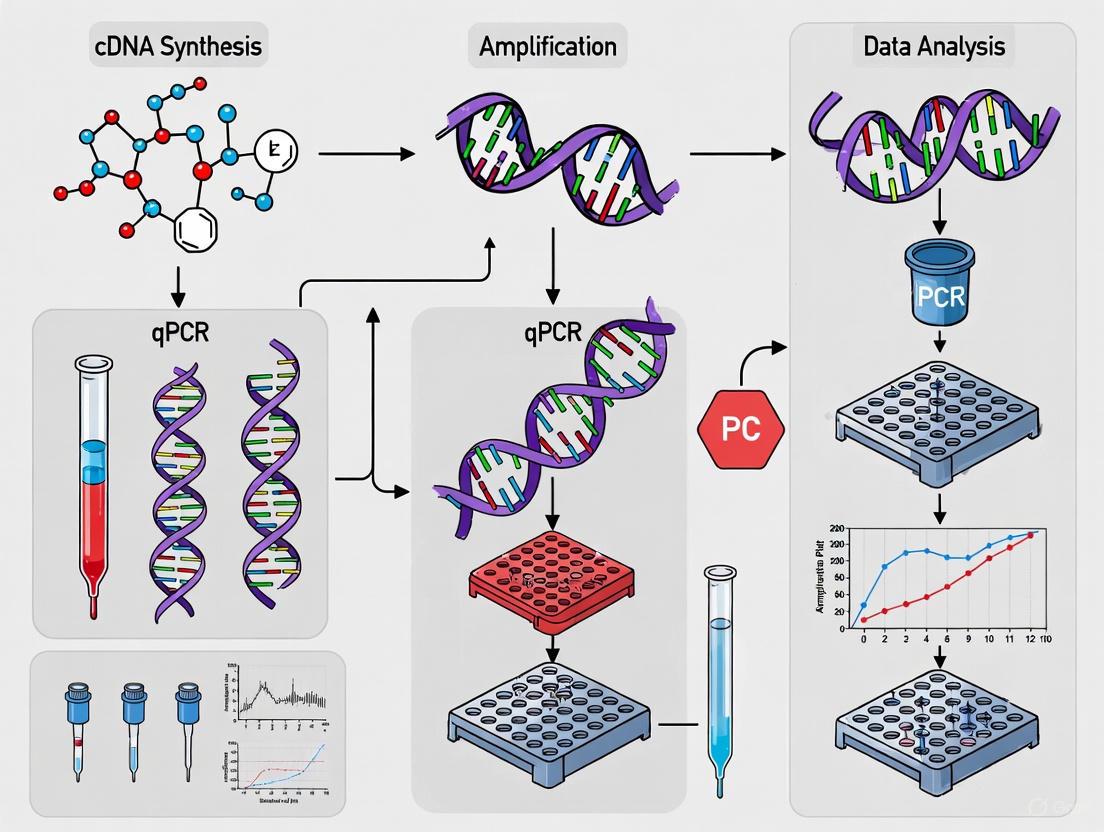
The workflow for this high-throughput qPCR protocol, from sample preparation to data analysis, is summarized in the diagram below.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of high-throughput qPCR for cancer biomarker discovery relies on a suite of specialized reagents and instruments. The following table details key solutions for constructing a robust screening pipeline.
Table 3: Essential Research Reagent Solutions for High-Throughput qPCR Biomarker Screening
| Item / Solution | Function / Application | Example Products / Notes |
|---|---|---|
| High-Throughput RNA Isolation Kits | Efficient, automated-friendly total RNA extraction from multiple sample types (cells, FFPE tissue). | MagMAX mirVana Total RNA Isolation Kit [18] |
| cDNA Synthesis Kits | High-efficiency reverse transcription of RNA to cDNA, crucial for sensitive detection. | SuperScript IV First-Strand Synthesis System [18] |
| qPCR Master Mixes | Provides enzymes, buffers, and fluorescence chemistry (SYBR Green or Probe-based) for amplification. | ssoAdvanced Universal SYBR Green Master-Mix [18] |
| Pre-Designed qPCR Assays/Panels | Pre-validated primer/probe sets for specific cancer-related genes or pathways; save time and ensure reproducibility. | Lyophilized primer sets for colorectal, lung, prostate cancer etc.; PCR arrays for pathways [16] [17] |
| miRNA Quantification Kits | Specialized single-step, single-tube reaction for accurate and sensitive quantification of stable miRNA biomarkers. | Mir-X miRNA qRT-PCR TB Green Kit [16] |
| High-Throughput qPCR Systems | Automated platforms for running and analyzing thousands of nanoscale qPCR reactions. | SmartChip Real-Time PCR System [16] |
| Endogenous Controls | Stable reference genes (e.g., housekeeping genes) for normalization of qPCR data. | TaqMan Endogenous Controls (e.g., for human, mouse, rat) [17] |
Pathway Visualization: From Biomarker Discovery to Clinical Application
The journey from initial biomarker screening to its integration into clinical diagnostics and personalized treatment strategies involves a logical, multi-stage pathway. This process, underpinned by high-throughput qPCR and other omics technologies, is critical for advancing cancer management [21].
Quantitative PCR (qPCR) remains a foundational technology in clinical oncology, providing a unique combination of high analytical sensitivity, rapid turnaround time, and cost-efficiency that makes it uniquely suited for informing therapeutic decision-making at scale [2]. In the context of high-throughput cancer biomarker screening, qPCR enables the detection of clinically actionable biomarkers at increasingly low concentrations while supporting broader mutational profiling to guide precise treatment decisions [2]. This application note details the specific implementations of qPCR across the cancer care continuum, from initial diagnosis through therapy selection and monitoring.
The advantages of qPCR are particularly evident in time-sensitive or resource-constrained settings. Unlike sequencing platforms that can take days to generate and analyze data, qPCR delivers clinically actionable results within hours, which is especially valuable when selecting targeted therapies or enrolling patients into mutation-driven clinical trials [2]. Furthermore, its strong multiplexing capability allows multiple clinically relevant mutations to be detected in a single reaction without compromising sensitivity or speed, making it ideal for oncology applications where actionable targets span several genes and sample material is scarce [2].
Application Note: qPCR for Cancer Diagnosis and Early Detection
Circulating Tumor DNA (ctDNA) Analysis
Liquid biopsies analyzing ctDNA have transformed cancer diagnosis by providing a minimally invasive method to detect tumor-derived genetic material in blood and other body fluids [22] [23]. qPCR assays can detect cancer-specific mutations and epigenetic alterations in ctDNA with sensitivity sufficient to identify early-stage malignancies. The rapid clearance of ctDNA, with estimated half-lives ranging from minutes up to a few hours, provides a dynamic window into current disease status, though it also presents technical challenges for detection [23].
DNA methylation biomarkers in liquid biopsies offer particular promise for early cancer detection. Methylation patterns often emerge early in tumorigenesis and remain stable throughout tumor evolution, making them ideal biomarker candidates [23]. The inherent stability of the DNA double helix provides additional protection compared to single-stranded nucleic acid-based biomarkers, and methylated DNA appears to have enhanced resistance to degradation during sample collection, storage, and processing [23].
Multiplexed Panels for Comprehensive Screening
Multiplexed qPCR panels exemplify the power of high-throughput applications in cancer diagnostics. For example, in non-small cell lung cancer (NSCLC), multiplexed qPCR panels can simultaneously assess alterations in EGFR, KRAS, BRAF, and ALK – delivering results faster and using less input material than sequential or panel-based next-generation sequencing (NGS) approaches [2]. Solutions such as Biofidelity's Aspyre Lung Reagents and the AmoyDx Pan Lung Cancer PCR Panel showcase the strong clinical potential of multiplexed qPCR for comprehensive molecular profiling [2].
Table 1: Diagnostic qPCR Biomarkers in Common Cancers
| Cancer Type | Key Biomarkers | Sample Source | Clinical Utility |
|---|---|---|---|
| Lung Cancer | EGFR, KRAS, BRAF, ALK fusions | Plasma, FFPE | Diagnosis, mutation profiling [2] |
| Colorectal Cancer | DNA Methylation Markers | Stool, Plasma | Early detection, screening [23] |
| Liver Cancer (HCC) | AFP | Serum | Diagnosis, recurrence monitoring [22] |
| Prostate Cancer | PSA | Serum | Screening, diagnosis [22] |
| Bladder Cancer | DNA Methylation Markers | Urine | Non-invasive detection [23] |
Application Note: qPCR for Prognostic Stratification
Minimal Residual Disease (MRD) Detection
qPCR enables highly sensitive detection of minimal residual disease following curative-intent treatment. By tracking tumor-specific mutations or methylation patterns in ctDNA, qPCR can identify molecular recurrence weeks or months before clinical or radiographic evidence emerges [23]. This early warning system allows clinicians to initiate interventions sooner, potentially improving outcomes. The quantitative nature of qPCR further enables monitoring of disease burden over time, providing dynamic prognostic information.
Gene Expression Profiling
While ctDNA analysis focuses on genetic and epigenetic alterations, qPCR also facilitates gene expression profiling of tumor tissue. By quantifying expression levels of genes associated with aggressive phenotypes or treatment resistance, qPCR panels can stratify patients according to recurrence risk. This application requires careful normalization to appropriate reference genes and validation of expression thresholds with clinical outcomes [24].
Table 2: Prognostic Biomarkers Detectable by qPCR
| Biomarker Category | Specific Examples | Prognostic Significance |
|---|---|---|
| Circulating Tumor DNA | EGFR mutations, methylation markers | Molecular recurrence, tumor burden [23] |
| Gene Expression Signatures | Oncogenes, tumor suppressor genes | Risk stratification, treatment response [24] |
| MicroRNAs | Various cancer-associated miRNAs | Disease progression, metastasis [22] |
| Cancer/Testis Antigens | CT antigens | Aggressive disease course [22] |
Application Note: qPCR for Therapy Selection
Companion Diagnostic Applications
qPCR serves as the technological foundation for numerous companion diagnostics that match patients with targeted therapies. For example, detection of EGFR mutations in NSCLC determines eligibility for EGFR tyrosine kinase inhibitors, while BRAF V600E identification guides use of BRAF inhibitors in melanoma and other cancers [2]. The rapid turnaround time of qPCR (typically within hours) supports timely treatment decisions without compromising accuracy.
Resistance Mutation Monitoring
During treatment with targeted therapies, tumors often develop resistance mutations that can be detected by qPCR before clinical progression occurs. Serial monitoring of known resistance mechanisms (e.g., EGFR T790M in NSCLC) enables timely adaptation of treatment strategies. qPCR's cost-effectiveness and technical accessibility make it ideal for repeated testing throughout a patient's treatment journey [2].
Experimental Protocols
Protocol 1: ctDNA Extraction and Mutation Analysis from Plasma
Principle: Circulating tumor DNA fragments are extracted from blood plasma and analyzed for cancer-specific mutations using allele-specific qPCR assays.
Reagents and Equipment:
- Blood collection tubes (EDTA or specialized ctDNA tubes)
- Plasma separation reagents
- ctDNA extraction kit
- qPCR master mix optimized for low input DNA
- Mutation-specific primers and probes
- Control templates (wild-type and mutant)
- Centrifuge capable of 16,000 × g
- Quantitative PCR instrument
Procedure:
- Sample Collection and Processing: Collect blood in EDTA tubes. Process within 2 hours of collection by centrifugation at 800 × g for 10 minutes to separate plasma. Transfer supernatant to a fresh tube and centrifuge at 16,000 × g for 10 minutes to remove residual cells [23].
- ctDNA Extraction: Extract ctDNA from plasma using a specialized circulating nucleic acid kit according to manufacturer's instructions. Elute in an appropriate buffer and quantify using a fluorometric method sensitive to low DNA concentrations.
- Assay Preparation: Prepare qPCR reactions containing mutation-specific primers and probes. Include appropriate controls: no-template control, wild-type control, and mutant control at known variant allele frequencies.
- qPCR Amplification: Run reactions under the following conditions:
- Initial denaturation: 95°C for 2 minutes
- 45 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 1 minute
- Data Analysis: Determine Cq values using appropriate baseline and threshold settings. For variant allele frequency calculation, use a standard curve generated from control templates with known mutation percentages [24].
Protocol 2: High-Throughput Multiplex qPCR for Cancer Panel Screening
Principle: Simultaneous detection of multiple cancer-associated mutations in a single reaction using a multiplex qPCR approach with target-specific probes differentially labeled with fluorescent reporters.
Reagents and Equipment:
- Multiplex qPCR master mix
- Target-specific primer and probe sets
- DNA template (from tumor tissue, plasma, or other sources)
- 384-well qPCR plates
- High-throughput qPCR instrument with multiple detection channels
Procedure:
- Assay Design: Design primer and probe sets for each target mutation, ensuring similar annealing temperatures and minimal cross-reactivity. Label each probe with a different fluorescent dye compatible with available detection channels.
- Reaction Setup: Prepare multiplex qPCR reactions containing all primer and probe sets. For high-throughput applications, use automated liquid handling systems to dispense reactions into 384-well plates.
- qPCR Amplification:
- Initial denaturation: 95°C for 10 minutes
- 50 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 1 minute (with fluorescence acquisition)
- Data Analysis: Analyze amplification curves for each fluorescence channel separately. Set thresholds within the exponential phase of amplification where all assays display parallel log-linear phases [24]. Determine Cq values and interpret results based on established cutoffs for each target.
Protocol 3: DNA Methylation Analysis Using Bisulfite Conversion
Principle: Bisulfite conversion of unmethylated cytosines to uracils, followed by qPCR with methylation-specific primers to detect promoter hypermethylation of tumor suppressor genes.
Reagents and Equipment:
- Bisulfite conversion kit
- Methylation-specific primers
- qPCR master mix
- Converted DNA controls (fully methylated and unmethylated)
Procedure:
- Bisulfite Conversion: Treat extracted DNA with bisulfite reagent according to manufacturer's instructions. This process deaminates unmethylated cytosines to uracils while leaving methylated cytosines unchanged.
- Purification: Purify bisulfite-converted DNA and elute in an appropriate buffer.
- Methylation-Specific qPCR: Set up reactions with primers specifically designed to amplify either the methylated or unmethylated sequence. Always run both reactions for each sample.
- Amplification:
- Initial denaturation: 95°C for 10 minutes
- 45 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing: Primer-specific temperature for 30 seconds
- Extension: 72°C for 30 seconds
- Data Interpretation: Calculate the percentage of methylated alleles using the ΔΔCq method with reference to standards of known methylation percentage [24].
Workflow Visualization
High-Throughput qPCR Clinical Workflow: This diagram illustrates the standardized workflow for processing samples in cancer biomarker screening, from sample collection through clinical reporting.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for High-Throughput qPCR in Oncology Research
| Reagent/Material | Function | Key Characteristics |
|---|---|---|
| Inhibitor-Resistant Master Mix | Amplification of challenging clinical samples | Engineered polymerases that tolerate PCR inhibitors in plasma, whole blood, or FFPE-derived nucleic acids [2] |
| Multiplex qPCR Reagents | Simultaneous detection of multiple targets | Advanced master mixes and probe systems enabling detection of several mutations in a single reaction [2] |
| Ambient-Stable Formulations | Decentralized testing applications | Lyophilized reagents that maintain stability without cold chain storage [2] |
| Mutation-Specific Assays | Detection of low-frequency variants | Optimized primers and probes capable of detecting variants at <0.1% variant allele frequency [2] |
| Bisulfite Conversion Kits | DNA methylation analysis | Efficient conversion of unmethylated cytosines while preserving methylated sites [23] |
| Reference Gene Assays | Data normalization | Consistently expressed genes for reliable quantification across sample types [24] |
Data Analysis and Quality Control
Baseline and Threshold Setting
Accurate Cq determination requires proper baseline correction and threshold setting. The baseline should be set using early cycles (typically cycles 5-15) where amplification remains linear, avoiding the initial cycles (1-5) which may contain reaction stabilization artifacts [24]. The threshold should be set:
- Sufficiently above background fluorescence to avoid premature threshold crossing
- Within the logarithmic phase of amplification, before plateau effects begin
- At a position where all amplification curves display parallel log-linear phases [24]
Quantitative Analysis Methods
Standard Curve Quantification: Using a dilution series of standards with known concentrations, this method provides absolute quantification of target molecules. The Cq values of standards are plotted against the logarithm of their concentrations to generate a standard curve, from which unknown sample concentrations can be interpolated [24].
Comparative Cq Method (ΔΔCq): This relative quantification approach uses the difference in Cq values between target genes and reference genes compared to a control sample. The formula 2^(-ΔΔCq) provides the relative expression fold change, assuming amplification efficiencies are near 100% [25] [24].
Efficiency-Corrected Model: For more precise quantification when amplification efficiencies differ from 100%, this model incorporates actual PCR efficiencies (determined from standard curves) into the relative quantification calculation, providing more accurate fold-change measurements [24].
Quality Assessment Metrics
Amplification Efficiency: Calculated from standard curves using the formula E = 10^(-1/slope) - 1, with ideal values between 90-110% [24].
Linearity (R²): The correlation coefficient of the standard curve, with values >0.985 indicating acceptable linearity.
Limit of Detection (LOD): The lowest variant allele frequency consistently detectable, typically <0.1% for optimized oncology assays [2].
qPCR maintains a crucial position in clinical oncology workflows due to its unique combination of sensitivity, speed, and practicality. While emerging technologies like next-generation sequencing provide broader genomic coverage, qPCR offers unmatched efficiency for targeted detection of clinically validated biomarkers across the cancer care continuum. The ongoing development of more robust reagents, enhanced multiplexing capabilities, and stable formulations ensures that qPCR will remain an indispensable tool for high-throughput cancer biomarker screening in both centralized and decentralized settings. As the field moves toward earlier detection and more personalized treatment approaches, qPCR's role in providing rapid, cost-effective, and clinically actionable molecular information continues to expand.
Implementing High-Throughput qPCR Workflows: From Sample to Data
High-throughput quantitative polymerase chain reaction (HT-qPCR) represents a cornerstone technology in modern oncology research, enabling the rapid, sensitive, and parallelized detection of nucleic acid biomarkers critical for early cancer detection and personalized medicine. This platform overview examines the core architectures—96-well systems, microfluidics, and emerging nanodispenser technologies—that empower researchers to screen large patient cohorts and multiplexed biomarker panels with high efficiency and reproducibility. The transition from conventional qPCR to HT formats addresses the pressing need in cancer diagnostics to process numerous samples while conserving precious reagents and clinical material, such as cell-free DNA (cfDNA) from liquid biopsies [26]. In the context of cancer biomarker discovery, HT-qPCR facilitates the validation of candidate markers, including circulating tumor DNA (ctDNA), microRNAs (miRNAs), and DNA methylation patterns, across diverse sample types from tissue biopsies to liquid biopsies [22] [23]. The compatibility of these platforms with standardized well plates and automated liquid handling makes them indispensable for translational research aimed at bringing novel biomarkers from concept to clinic.
HT-qPCR Platform Architectures and Technical Specifications
96-Well Plate Systems
The standard 96-well plate format remains the most widely adopted platform for HT-qPCR due to its direct compatibility with established laboratory automation, thermal cyclers, and liquid handling robotics. This format strikes a balance between throughput, reagent consumption, and accessibility, making it a versatile choice for various applications in cancer research.
Key Applications:
- Biomarker Validation: Ideal for profiling the expression of a focused panel of candidate genes (e.g., oncogenes, tumor suppressors, miRNAs) across many patient samples [27].
- Mutation Screening: Multiplexed qPCR panels can simultaneously assess clinically actionable mutations in genes such as EGFR, KRAS, and BRAF from limited samples like fine-needle aspirates or liquid biopsies [26].
- Data Analysis: The R/Bioconductor package
HTqPCRis specifically designed to process, normalize, and perform statistical analysis on the large datasets generated from 96-well (and higher density) formats, handling cycle threshold (Ct) values, quality control, and differential expression [27].
Table 1: Key Characteristics of 96-well HT-qPCR Platforms
| Feature | Specification | Utility in Cancer Research |
|---|---|---|
| Throughput | 96 reactions per run | Efficient for screening 10s-100s of samples against a defined biomarker panel |
| Reaction Volume | Typically 10-25 µL | Balances sensitivity with reagent cost |
| Multiplexing Capability | Moderate (typically 2-4 plex) | Suitable for simultaneous detection of a few key mutations or expression targets |
| Data Analysis | Compatible with specialized software (e.g., HTqPCR package for R) |
Enables quality control, normalization, and statistical testing across multiple conditions [27] |
| Cost per Sample | Low ($50-$200 per test) [26] | Cost-effective for large-scale screening and routine diagnostics |
Microfluidic qPCR Systems
Microfluidic HT-qPCR systems represent a significant advancement in miniaturization and integration. These systems use microfabricated fluidic channels and chambers to partition reactions at nanoliter scales, dramatically increasing throughput while reducing sample and reagent consumption. A prominent example is the TaqMan Low Density Array (TLDA) cards, which are pre-configured with assay primers and probes [27].
Key Applications:
- High-Density Profiling: Enables the parallel analysis of hundreds of targets (e.g., a comprehensive miRNA signature or a large panel of methylation markers) from a single, limited patient sample [27] [23].
- Liquid Biopsy Analysis: Ideal for analyzing ctDNA or other rare nucleic acid species from liquid biopsies where input material is scarce [23] [26].
- Integrated Perfusion Culture: Novel systems like the High-Throughput microfluidics-enabled uninterrupted perfusion system (HT-μUPS) integrate microfluidic perfusion with 96-well plates, allowing for long-term dynamic cell culture and chronic all-optical electrophysiology. This is particularly useful for maintaining highly metabolic cells like patient-derived cardiomyocytes or studying cell responses under controlled conditions, which can support research into cancer cell behavior and cardiotoxicity of chemotherapeutics [28] [29].
Table 2: Key Characteristics of Microfluidic HT-qPCR Platforms
| Feature | Specification | Utility in Cancer Research |
|---|---|---|
| Throughput | 384 wells or more per run | High-density profiling of hundreds of targets from a single sample |
| Reaction Volume | Nanoliter scale (e.g., 1-10 nL) | Drastically reduces reagent costs and conserves precious samples (e.g., liquid biopsies) |
| Multiplexing Capability | High (dozens to hundreds of pre-configured assays) | Comprehensive screening of biomarker panels for molecular stratification |
| System Integration | Can be coupled with perfusion systems for cell culture [28] | Allows for longitudinal study of cell responses and functional assays |
| Shear Stress Management | Designed to keep shear stress low (e.g., <2.4 dyn/cm² for excitable cells) [28] | Maintains viability of sensitive cell cultures for downstream analysis |
Nanodispenser Systems
Nanodispenser systems focus on the front-end of the HT-qPCR workflow: the precise, high-speed dispensing of nanoliter-scale reaction volumes into high-density well plates (e.g., 384- or 1536-well formats). These systems are critical for achieving the miniaturization and automation that define true high-throughput screening.
Key Applications:
- Ultra-High-Throughput Screening: Essential for large-scale drug discovery campaigns, functional genomics screens, and population-scale biomarker validation studies.
- Reagent Miniaturization: Enables the setup of thousands of qPCR reactions with significant savings on costly primers, probes, and master mixes.
- Workflow Automation: Can be integrated with robotic plate handlers to create fully automated, walk-away systems for processing thousands of samples per day.
Table 3: Key Characteristics of Systems with Nanodispenser Technology
| Feature | Specification | Utility in Cancer Research |
|---|---|---|
| Dispensing Volume | Nanoliter to picoliter range | Enables reaction setup in 384- and 1536-well plates, minimizing costs |
| Dispensing Precision | High accuracy and reproducibility | Critical for assay robustness and reliable quantification of low-abundance targets |
| Throughput | Thousands of wells per hour | Facilitates population-scale studies and large compound screens |
| Automation Compatibility | Designed for integration into robotic workstations | Supports complex, multi-step workflows with minimal manual intervention |
Experimental Protocols for Cancer Biomarker Applications
Protocol: Multiplexed Mutation Detection in Liquid Biopsies using 96-well qPCR
This protocol is designed for detecting somatic mutations in ctDNA from plasma, relevant for non-small cell lung cancer (NSCLC) and other solid tumors [26].
1. Sample Preparation and Nucleic Acid Extraction
- Collect whole blood in EDTA or cell-stabilizing tubes. Process within 2 hours to prevent lysis of blood cells.
- Isolate plasma by double centrifugation (e.g., 1,600 x g for 10 min, then 16,000 x g for 10 min).
- Extract cfDNA from 1-5 mL of plasma using a silica-membrane or bead-based kit optimized for low-concentration samples. Elute in a low-EDTA TE buffer or nuclease-free water.
- Quantify cfDNA using a fluorescence-based method sensitive to low DNA concentrations (e.g., Qubit dsDNA HS Assay).
2. Assay Design and qPCR Setup
- Primers/Probes: Use a validated multiplex qPCR panel targeting key mutations (e.g., in EGFR, KRAS, BRAF). Assays should be designed for high specificity to discriminate single-nucleotide variants. Use dual-labeled probes (e.g., TaqMan) with distinct fluorophores for each target.
- Reaction Mix (10 µL final volume in a 96-well plate):
- 5 µL of 2x Multiplex qPCR Master Mix (inhibitor-resistant, with antibody-mediated hot-start) [26]
- 1 µL of primer-probe mix (pre-optimized concentrations)
- 2-4 µL of cfDNA template (up to 50 ng total)
- Nuclease-free water to 10 µL
- Sealing and Centrifugation: Seal the plate with an optical adhesive film and centrifuge briefly to collect contents at the bottom of the wells.
3. qPCR Amplification and Data Acquisition
- Run the plate on a real-time PCR instrument with multicolor detection capability. Use the following typical cycling conditions:
- Initial Denaturation: 95°C for 2 minutes (polymerase activation)
- 45 Cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 1 minute (acquire fluorescence)
- Controls: Include no-template controls (NTCs) for each assay, wild-type control DNA, and positive controls for each mutation if available.
4. Data Analysis with HTqPCR Package in R
- Data Input: Import raw Ct value exports from the qPCR instrument software into the R environment using the
HTqPCRpackage [27]. - Quality Control (QC):
- Use
plotCtDensityandplotCtBoxesto assess the distribution and quality of Ct values across samples. - Flag Ct values above a reliable detection threshold (e.g., Ct > 35) as "Unreliable".
- Use
- Normalization: Normalize data using the
normalizeCtDatafunction. The ΔΔCt method is common, using one or more reference genes (e.g., β-actin) that are stably expressed across samples. Alternatively, use quantile normalization if stable reference genes are not available. - Calling Mutations: Calculate ΔCt values (Ct[target] - Ct[reference]). A ΔCt value beyond a pre-defined threshold (established from wild-type controls) indicates the presence of a mutation.
Diagram 1: Workflow for multiplexed mutation detection in liquid biopsies.
Protocol: DNA Methylation Analysis via Bisulfite Conversion and qPCR
DNA methylation is a stable epigenetic biomarker often altered in cancer. This protocol outlines its detection using bisulfite conversion followed by qPCR, which is highly relevant for analyzing promoter hypermethylation of tumor suppressor genes [23].
1. Bisulfite Conversion of cfDNA
- Use 10-50 ng of input cfDNA.
- Treat DNA with sodium bisulfite using a commercial kit. This process converts unmethylated cytosines to uracils, while methylated cytosines remain unchanged.
- Purify the bisulfite-converted DNA and elute in a small volume.
2. qPCR Assay for Methylated Alleles
- Assay Design: Design primers and probes that are specific to the bisulfite-converted, methylated sequence. The probe typically spans several CpG sites to maximize specificity.
- Reaction Setup:
- Use a qPCR master mix optimized for bisulfite-converted DNA, which is often more fragmented and of lower quality.
- Set up two parallel reactions for each sample: one with primers/probes for the methylated allele and one for a reference gene (to control for DNA input).
- Amplification: Run on a real-time PCR instrument with standard cycling conditions. Methylation-specific amplification will typically yield a lower Ct value if the methylated allele is present.
3. Data Analysis and Quantification
- Calculate the ΔCt value: Ct(methylated assay) - Ct(reference assay).
- The level of methylation can be expressed as 2^(-ΔCt). For absolute quantification, compare to a standard curve of known methylated DNA.
The Scientist's Toolkit: Essential Reagents and Materials
The successful implementation of HT-qPCR in cancer research relies on a suite of specialized reagents and materials designed to ensure sensitivity, specificity, and robustness, particularly when working with challenging clinical samples.
Table 4: Research Reagent Solutions for HT-qPCR in Oncology
| Reagent/Material | Function | Key Characteristics for Cancer Apps |
|---|---|---|
| Inhibitor-Resistant Master Mix | Amplification of nucleic acids | Contains engineered polymerases and buffers to tolerate PCR inhibitors common in clinical samples (e.g., from plasma, FFPE tissue) [26] |
| Multiplex qPCR Assays | Simultaneous detection of multiple targets | Pre-optimized primer-probe sets for mutation panels (e.g., EGFR, KRAS) or biomarker signatures; different fluorophores enable multiplexing [26] |
| cfDNA Extraction Kits | Isolation of cell-free DNA from liquid biopsies | Optimized for low-abundance DNA from large volume plasma/serum inputs; maximizes recovery and minimizes contamination |
| Ambient-Stable/Lyophilized Reagents | Ready-to-use assay formats | Reduces cold-chain dependency, ideal for decentralized testing; simplifies workflow and improves reproducibility [26] |
| Standard 96-/384-Well Plates | Reaction vessel for HT-qPCR | Manufactured to high standards for optimal thermal conductivity and low well-to-well cross-talk; compatible with automation |
| Microfluidic Cartridges (e.g., TLDA) | Integrated, high-density reaction vessel | Pre-configured with assays for specific cancer pathways; minimizes pipetting steps and reduces risk of contamination [27] |
The integrated use of 96-well, microfluidic, and nanodispenser HT-qPCR platforms provides a powerful, scalable toolkit for advancing cancer biomarker research. From validating targeted mutation panels in a standard 96-well format to performing ultra-high-throughput methylation profiling on microfluidic chips, these technologies enable researchers to navigate the complexities of cancer genomics with precision and efficiency. The continued evolution of associated reagents—such as inhibitor-resistant master mixes and ambient-stable formulations—further enhances the reliability of these platforms for analyzing real-world clinical specimens like liquid biopsies. By leveraging the detailed protocols and tools outlined in this overview, researchers and drug development professionals can robustly employ HT-qPCR to accelerate the discovery and translation of molecular diagnostics into clinical practice, ultimately contributing to earlier cancer detection and personalized therapeutic strategies.
The rising global cancer burden, with new cases expected to exceed 35 million annually by 2050, places unprecedented demands on cancer diagnostics and biomarker development [23]. In this context, high-throughput quantitative PCR (qPCR) has emerged as a cornerstone technology for sensitive and specific detection of cancer biomarkers in liquid biopsies, including circulating tumor DNA (ctDNA), microRNAs, and other RNA species. The need to screen large patient cohorts for biomarker discovery and validation, combined with increasing reagent costs and limited sample availability, has driven laboratories toward automation and miniaturization strategies. These approaches enable researchers to maintain high data quality while significantly reducing per-reaction costs and increasing experimental throughput, thereby accelerating the pace of cancer research and precision oncology initiatives.
Miniaturization in qPCR refers to the systematic reduction of reaction volumes from traditional 20-50 µL scales down to 1-10 µL volumes using 384-well plates or higher-density formats. This paradigm shift, when coupled with automated liquid handling systems, allows researchers to achieve substantial cost savings while maximizing output from precious clinical samples. The integration of these approaches is particularly valuable in cancer biomarker research, where sample availability is often limited and the need for robust, reproducible results is paramount for clinical translation.
Quantitative Benefits: Cost and Resource Optimization
The economic advantages of miniaturization in high-throughput qPCR are substantial and directly impact research efficiency. A detailed cost analysis reveals significant savings in reagent consumption and associated expenses when transitioning from 96-well to 384-well formats.
Table 1: Cost Comparison Between 96-Well and 384-Well qPCR Formats
| Parameter | 96-Well Format | 384-Well Format | Savings with 384-Well |
|---|---|---|---|
| Reaction Volume | 20 µL | 10 µL | 50% reduction in reagents |
| Reactions per Plate | 96 | 384 | 4x increase in throughput |
| Cost per Reaction | 0.72 EUR / 0.89 USD | 0.36 EUR / 0.44 USD | 50% reduction per data point |
| Cost per Full Plate | 50 EUR / 85.78 USD | 60 EUR / 167.86 USD | Higher total but lower per reaction |
| Plates to Break Even on Instrument Cost | - | 77 plates (~29,600 reactions) | Initial investment recouped rapidly at scale [30] |
Beyond direct reagent costs, miniaturization preserves valuable clinical samples. A 2.5-fold difference in sample input exists between 20 µL (typically 5 µL sample) and 10 µL (typically 2 µL sample) reactions, with only a minimal impact on Cq values (approximately 0.83 cycles higher in 10 µL reactions) [30]. This allows researchers to perform more replicates, include additional controls, or analyze more biomarkers from the same sample volume, thereby increasing the statistical power and reliability of cancer biomarker studies.
For specialized cell models such as iPSC-derived cells, which can cost over $1,000 per vial of 2 million cells, miniaturization from 96-well to 384-well format reduces cell requirements from approximately 23 million to 4.6 million cells for a 3,000 data-point screen, representing substantial cost savings of approximately $6,900 excluding additional savings on specialized culture media and growth factors [31].
Implementation Strategies: Methodologies and Workflows
Automated Liquid Handling and Miniaturization Protocols
Successful implementation of miniaturized qPCR workflows requires specialized equipment and optimized protocols. Automated liquid handling systems with positive displacement technology enable accurate and precise transfer of volumes from 500 nL to 5 µL, eliminating cross-contamination through pre-sterilized disposable pipettes and maximizing efficiency through rapid plate-to-plate pipetting [32]. The following protocol outlines a standardized approach for miniaturized qPCR setup for cancer biomarker analysis:
Protocol: Miniaturized qPCR Setup for Cancer Biomarker Analysis
Reagents and Equipment:
- Automated liquid handler (e.g., mosquito HV genomics)
- 384-well qPCR plates with white wells and ultra-clear seals
- Low-volume qPCR master mix (e.g., biotechrabbit Capital qPCR Mix)
- Template DNA/cDNA (minimum 2 µL per 10 µL reaction)
- Target-specific primers/probes
- qPCR instrument with 384-well block (e.g., qTOWERiris)
Procedure:
- Reaction Plate Preparation: Program the automated liquid handler to dispense 8 µL of master mix into each well of a 384-well plate.
- Template Addition: Add 2 µL of template DNA/cDNA to each well using the automated system.
- Sealing and Centrifugation: Seal the plate with optical-quality clear seals and centrifuge briefly (1000 × g, 1 minute) to collect reaction mixture at the bottom of wells and eliminate bubbles.
- qPCR Amplification: Run the reaction using the following optimized thermocycling conditions:
- Initial denaturation: 95°C for 30 seconds (for genomic DNA)
- 40 cycles of:
- Denaturation: 95°C for 5-15 seconds (shorter for smaller templates)
- Annealing/Extension: 60°C for 1 minute (combined shuttle PCR)
- Melt curve analysis: As recommended by instrument manufacturer [33]
Critical Optimization Steps:
- Primer Design: Design primers with 40-60% GC content, length of 28 bp or larger, and Tm between 58-65°C with less than 4°C difference between forward and reverse primers.
- Probe Design: For probe-based detection, design probes with Tm approximately 10°C higher than primers, length between 9-40 bp, and avoid G repeats, especially at the 5' end.
- Evaporation Control: Use proper sealing methods and consider plate designs with evaporation barriers to prevent edge effects, particularly critical in low-volume reactions [31] [33].
Integration with Cancer Biomarker Applications
In cancer biomarker research, miniaturized qPCR workflows can be applied to various liquid biopsy analytes. For RNA biomarkers, a two-step RT-qPCR approach is recommended when analyzing multiple transcripts from a single sample. This involves reverse transcription primed with either oligo d(T)16 or random primers, followed by miniaturized qPCR analysis of specific targets [17]. This approach is particularly valuable for analyzing circulating miRNAs, lncRNAs, and other RNA species identified as promising cancer biomarkers in recent studies [8].
For DNA methylation-based cancer biomarkers, which demonstrate enhanced stability in liquid biopsies, targeted qPCR approaches following bisulfite conversion enable sensitive detection of hypermethylated promoter regions of tumor suppressor genes. The minimal sample requirements of miniaturized workflows are especially beneficial for these applications, as they allow for parallel assessment of multiple methylation markers from limited liquid biopsy samples [23].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Essential Research Reagent Solutions for Miniaturized qPCR in Cancer Biomarker Research
| Item | Function | Application Notes |
|---|---|---|
| Automated Liquid Handler | Precise nanoliter-scale dispensing for 384-well plate setup | Positive displacement technology (e.g., mosquito HV) minimizes cross-contamination; enables 10x miniaturization of NGS library prep [32] |
| Low-Volume qPCR Master Mix | Optimized enzyme and buffer system for 10 µL reactions | Antibody-mediated hot-start polymerases eliminate need for 10-15 minute activation step [33] |
| 384-Well qPCR Plates | Optical reaction vessels with minimal well-to-well variability | White wells reduce light distortion; ultra-clear caps optimize signal detection [33] |
| Target-Specific Assays | Pre-designed primer/probe sets for cancer biomarkers | Commercial assays available for known biomarkers; custom designs needed for novel targets; ensure 90-110% amplification efficiency [17] |
| Quality Control Tools | RNA/DNA integrity assessment and quantification | Bioanalyzer systems (e.g., Implen N80) and spectrophotometers essential for input quality control [33] |
Workflow Integration and Pathway Analysis
The implementation of automated, miniaturized qPCR requires careful planning of the complete workflow from sample collection to data analysis. The following diagram illustrates the integrated process for cancer biomarker screening:
The successful implementation of this workflow requires careful consideration of the logical relationships between key experimental parameters. The following diagram illustrates the optimization pathway for miniaturized qPCR experiments:
Automation and miniaturization of qPCR workflows represent a transformative approach in cancer biomarker research, addressing the dual challenges of increasing throughput and reducing costs without compromising data quality. The strategic implementation of 384-well formats, coupled with automated liquid handling systems, enables researchers to achieve significant reagent savings while maximizing the utility of precious liquid biopsy samples. As cancer biomarker discovery evolves toward multi-analyte panels and larger validation cohorts, these approaches will become increasingly essential for accelerating the development of next-generation cancer diagnostics and personalized treatment strategies. The protocols and methodologies outlined in this application note provide a foundation for laboratories seeking to implement these efficient workflows in their cancer research programs.
The shift toward personalized oncology has made the molecular profiling of tumors a prerequisite for effective treatment. Oncogenic drivers such as EGFR, KRAS, and BRAF are critical biomarkers that predict response to targeted therapies, particularly monoclonal antibodies against the epidermal growth factor receptor (EGFR) and small molecule inhibitors [34] [35]. For instance, KRAS mutations are well-established negative predictors of response to anti-EGFR therapies in colorectal cancer (CRC), as they lead to constitutive activation of downstream signaling pathways, rendering EGFR inhibitors ineffective [34] [35]. The National Comprehensive Cancer Network (NCCN), American Society of Clinical Oncology (ASCO), and other regulatory bodies therefore recommend mandatory testing of these biomarkers before initiating treatment [34] [35]. This creates a pressing need for diagnostic methods that are not only accurate and sensitive but also fast, cost-effective, and capable of maximizing information output from often limited patient samples. High-throughput quantitative PCR (qPCR) and its advanced derivatives have emerged as powerful tools to meet this need, enabling the simultaneous detection of multiple actionable mutations in a single, streamlined assay [2] [36].
Multiplex Assay Design Strategy
Core Principles for Multiplexing Oncogenic Drivers
The design of a successful multiplex PCR assay for oncogenic drivers hinges on several key principles that ensure specificity, sensitivity, and robustness. The fundamental mechanism is the amplification refractory mutation system (ARMS), also known as allele-specific PCR. This technique relies on primer design where the 3'-terminal nucleotide is complementary to a specific mutant sequence. When there is a perfect match, amplification proceeds efficiently; a mismatch (as with the wild-type sequence) significantly hinders amplification [35]. To further enhance specificity, the introduction of an additional deliberate mismatch at the penultimate or antepenultimate base of the primer is a common strategy [35]. This design is perfectly suited for detecting single-nucleotide variants (SNVs) like those found in KRAS codons 12/13 and BRAF V600E.
Probe-based detection, typically using hydrolysis (TaqMan) probes, allows for multiplexing in real-time. Each probe is labeled with a different fluorophore (e.g., FAM, HEX, Texas Red) and a quencher, enabling the simultaneous detection and distinction of multiple targets in a single tube [37]. The move from singleplex to multiplex reactions offers substantial benefits, including higher throughput, lower reagent consumption, reduced analysis time, and conservation of precious sample material [2] [37]. This is particularly vital for cancer samples, which are often derived from formalin-fixed paraffin-embedded (FFPE) tissues or liquid biopsies where nucleic acid is limited [2].
The Oncogenic Signaling Pathway and Assay Targets
The EGFR, KRAS, and BRAF proteins form part of a critical signaling network, the Ras/Raf/MAPK pathway, which regulates cell proliferation, differentiation, and survival. Mutations in these genes lead to ligand-independent, constitutive activation of this pathway, driving tumorigenesis. The following diagram illustrates this key pathway and the primary mutation sites targeted by multiplex assays.
Detailed Experimental Protocol
This protocol provides a step-by-step guide for a multiplex allele-specific qPCR assay to detect the seven most common mutations in KRAS codons 12 and 13, adaptable for other oncogenic drivers like EGFR and BRAF [35].
Sample Preparation and DNA Extraction
- Source Material: The assay is validated for FFPE tissue sections. Tumor-rich areas (>60% tumor cells) should be identified by a pathologist and isolated via macrodissection or coring if necessary [34].
- DNA Extraction: Use a dedicated FFPE DNA extraction kit, such as the QIAamp DNA FFPE Tissue Kit.
- DNA Quantification and Quality Control: Quantify DNA using a fluorometric method (e.g., Qubit Fluorometer) for accuracy. Assess DNA integrity via multiplex PCR of control genes (e.g., Tbxas, Rag1) generating 100-400 bp amplicons to confirm amplifiability [34].
Primer and Probe Design
Design is critical for specificity and multiplexing success. The following reagents are needed:
- Allele-Specific Forward Primers: One for each mutation (e.g., G12A, G12C, G12D, G12R, G12S, G12V, G13D). The 3' base must match the mutant sequence. Incorporate an additional artificial mismatch at the second or third base from the 3' end to destabilize wild-type binding [35].
- Common Reverse Primer: A single reverse primer that binds to a conserved region downstream of the mutations.
- TaqMan Probe: A single probe binding to a conserved region within the amplicon. It should be dual-labeled with a fluorophore (e.g., FAM) and a quencher.
Example Primer/Probe Sequences (KRAS Exon 2 Amplicon) [34]:
- Common Reverse Primer: 5'-CAAAGAATGGTCCTGCACCAG-3'
- TaqMan Probe: 5'-[FAM]AGGCACTCTTG[CQ]C-[...] 3' (Sequence abbreviated, quencher not specified)
Multiplex qPCR Reaction Setup
- Reaction Volume: 10 µL
- Master Mix: 1X TaqMan Genotyping Master Mix
- Primers/Probes:
- Allele-Specific Primers: 0.4 µM each
- Common Reverse Primer: 0.55 µM
- TaqMan Probe: 0.5 µM
- DNA Template: 35 ng (3.5 µL) of FFPE-derived genomic DNA.
- Controls:
- No Template Control (NTC): Nuclease-free water.
- Wild-Type Control: DNA from normal tissue or commercial wild-type genomic DNA.
- Positive Mutant Controls: Plasmid DNA or commercially available reference standards for each mutation.
Thermal Cycling Conditions
- Initial Denaturation: 95°C for 10 minutes.
- Amplification (50 cycles):
- Denature: 95°C for 30 seconds.
- Anneal/Extend: 60°C for 30 seconds (with fluorescence data acquisition).
- Hold: 4°C.
Data Analysis
- Threshold Setting: Set the fluorescence threshold in the exponential phase of the amplification plots across all replicates.
- Cq Determination: Record the quantification cycle (Cq) for each reaction.
- ΔCq Calculation: Calculate ΔCq as the difference in Cq between the multiplex allele-specific assay and a reference assay for a non-mutated region (e.g.,
ΔCq = Cq(Multiplex) - Cq(Reference)). - Interpretation: A sample is considered positive for a mutation if the Cq value is below a predetermined cutoff (e.g., 37-40) and shows a characteristic amplification curve. Positive results in the initial multiplex screen should be confirmed with individual allele-specific reactions to identify the specific mutation [35].
The entire experimental workflow, from sample to result, is summarized below.
Performance Validation and Data
Robust validation is essential for clinical application. The following tables summarize key performance metrics for multiplex PCR assays targeting oncogenic drivers, as demonstrated in recent studies.
Table 1: Analytical Performance of Published Multiplex PCR Assays
| Target(s) | Technology | Analytical Sensitivity (LoD) | Reported Clinical Performance | Source |
|---|---|---|---|---|
| KRAS (7 mutations) | Allele-Specific Multiplex qPCR | ≤1% mutant allele frequency | 100% concordance with reference standards | [35] |
| EGFR, KRAS, BRAF, ERBB2 (12 SNVs/indels) | Multiplex Digital PCR (dPCR) | N/A | 100% PPA, 98.5% NPA* vs. NGS | [36] |
| SARS-CoV-2 (E, N, RP genes) | One-Step Multiplex RT-qPCR | 10 copies/reaction | 100% Sensitivity, 96% Specificity vs. commercial kit | [37] |
| KRAS, BRAF | SNaPshot Multiplex Assay | Confirmed by sequencing & HRM | 34.5% KRAS and 10% BRAF mutation prevalence in CRC cohort | [34] |
| *PPA: Positive Percent Agreement | *NPA: Negative Percent Agreement |
Table 2: Cost and Time Efficiency Comparison of Detection Methods [34]
| Method | Approximate Cost per Sample | Typical Turnaround Time | Key Characteristics |
|---|---|---|---|
| Multiplex qPCR/dPCR | \$50 - \$200 [2] | Several hours [2] [36] | Fast, cost-effective, high sensitivity, targeted |
| Sanger Sequencing | N/A | 1-2 days | Low sensitivity (requires 10-30% mutant alleles), considered a gold standard [35] |
| Next-Generation Sequencing (NGS) | \$300 - \$3,000 [2] | Several days [2] | Comprehensive, detects novel variants, higher cost and complexity [36] |
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these protocols relies on specific reagents and tools. The following table details key components for setting up a multiplex qPCR assay for oncogenic drivers.
Table 3: Essential Reagents and Materials for Multiplex qPCR Assay Development
| Item | Function / Role | Example Products / Notes |
|---|---|---|
| FFPE DNA Extraction Kit | Isolation of high-quality, amplifiable DNA from archived clinical samples. | QIAamp DNA FFPE Tissue Kit (Qiagen) [34] [35] |
| qPCR Master Mix | Provides enzymes, dNTPs, and optimized buffer for efficient, specific amplification. | TaqMan Genotyping Master Mix (Applied Biosystems); Luna Universal One-Step RT-qPCR Kit (for RNA) [35] [37] |
| Allele-Specific Primers & TaqMan Probes | Core components for specific amplification and detection of mutant alleles. | Custom-designed oligonucleotides; may include locked nucleic acid (LNA) probes for enhanced specificity [35] [36] |
| Reference DNA Standards | Assay validation, sensitivity determination, and run controls. | Horizon Diagnostics DNA reference standards; cloned plasmid controls [34] [35] |
| qPCR Plates & Seals | Reaction vessel compatible with real-time PCR instruments. | Low-profile, clear 96-well or 384-well plates. |
| Real-Time PCR Instrument | Platform for thermal cycling and fluorescence detection. | Instruments from Bio-Rad, Thermo Fisher, Illumina, etc. [35] [37] |
Multiplex qPCR represents a powerful and practical approach for the high-throughput screening of key oncogenic drivers in cancer research and molecular diagnostics. The protocols detailed herein, centered on the principles of allele-specific amplification and probe-based detection, enable the rapid, sensitive, and cost-effective identification of clinically actionable mutations in EGFR, KRAS, and BRAF. As the field of oncology continues to evolve, the flexibility and scalability of multiplex qPCR and its advanced digital PCR counterparts ensure their continued relevance. Future developments will likely focus on expanding the multiplexing capacity and integrating these assays into fully automated workflows, further solidifying their role in enabling precision medicine and guiding personalized therapeutic strategies.
The advancement of precision oncology is intrinsically linked to the reliable analysis of molecular biomarkers. While high-throughput qPCR has emerged as a powerful tool for cancer biomarker screening, the quality of its results is fundamentally dependent on the integrity of the input RNA. Formalin-fixed, paraffin-embedded (FFPE) tissues and liquid biopsy samples represent two of the most valuable yet challenging sample types in modern cancer research. FFPE archives, encompassing over a billion samples worldwide, provide a vast, morphologically preserved resource for retrospective studies [38]. However, the formalin fixation process induces RNA cross-linking, fragmentation, and chemical modification, compromising nucleic acid quality [39]. Conversely, liquid biopsy—analyzing circulating tumor cells, cell-free DNA, and exosomal RNA from blood—offers a minimally invasive method for real-time monitoring of tumor dynamics but presents challenges in isolating rare, fragile targets against a background of normal cellular material [40] [41]. This application note details standardized, optimized protocols for extracting high-quality RNA from these challenging samples to ensure reliable, reproducible results in high-throughput qPCR-based cancer biomarker screening.
RNA Extraction from FFPE Tissues
Understanding the Challenge
The process of formalin fixation creates methylene bridges between proteins and nucleic acids, resulting in a tight meshwork of crosslinks that stabilizes tissue morphology but fragments and damages RNA [39]. The quality of extracted RNA is further influenced by pre-analytical factors including fixation time, storage duration, and tissue type [38]. Successful extraction requires protocols that effectively reverse these crosslinks while minimizing further RNA degradation.
Comparative Performance of RNA Extraction Kits
A systematic comparison of seven commercial FFPE RNA extraction kits revealed significant disparities in both the quantity and quality of recovered RNA [38]. The performance of different kits, summarized in Table 1, was evaluated based on RNA concentration, RNA Quality Score (RQS), and DV200 value (the percentage of RNA fragments >200 nucleotides).
Table 1: Performance Comparison of Selected FFPE RNA Extraction Kits
| Kit Name | Key Features | Input Limit | Deparaffinization | Digestion Conditions | Performance Summary |
|---|---|---|---|---|---|
| Roche Kit (High Quality Recovery) | Not specified | Not specified | Not specified | Not specified | Superior RNA quality (RQS) [38] |
| ReliaPrep FFPE Total RNA Miniprep (Promega) | Spin-column based | Not specified | Xylene/EtOH series | Not specified | Best balance of high yield and high quality [38] |
| RecoverAll Total Nucleic Acid Isolation Kit (Thermo Fisher) | Glass-fiber filter spin column | 80 µm total | Xylene (or substitute) & EtOH | RNA: 15 min @ 50°C + 15 min @ 80°C [39] | Good yield and purity [39] |
| MagMAX FFPE DNA/RNA Ultra Kit (Thermo Fisher) | Magnetic bead-based, high-throughput | 20 µm total | Not required | RNA: 45 min @ 60°C + 30 min @ 80°C [39] | Good yield and purity; amenable to automation [39] |
Optimized Protocol for RNA Extraction from FFPE Tissues
The following protocol is optimized for maximizing RNA yield and quality for downstream qPCR applications.
- Sectioning: Cut 2-4 sections of 10-20 µm thickness from the FFPE block. To avoid regional bias, use a systematic distribution method where non-consecutive slices are combined into a single sample tube [38].
- Deparaffinization:
- Add 1 mL xylene (or manufacturer's recommended deparaffinization solution) to the tube, vortex, and incubate for 5-10 minutes at room temperature.
- Centrifuge at full speed for 5 minutes. Carefully remove and discard the supernatant.
- Wash with 1 mL of 100% ethanol, vortex, and centrifuge for 5 minutes. Discard the supernatant.
- Air-dry the pellet briefly (5-10 minutes) to ensure complete ethanol evaporation [39].
- Proteinase K Digestion:
- Add the appropriate volume of digestion buffer and Proteinase K (as per kit instructions).
- Incubate using a optimized temperature profile. For example, incubate at 50°C for 15 minutes followed by 80°C for 15 minutes (RecoverAll protocol) or 60°C for 45 minutes followed by 80°C for 30 minutes (MagMAX protocol) [39]. These elevated temperatures are critical for reversing formalin crosslinks.
- RNA Isolation:
- Follow the manufacturer's protocol for binding RNA to a silica membrane (spin-column) or magnetic beads.
- Perform rigorous on-column DNase I treatment to remove genomic DNA contamination.
- Wash with provided wash buffers.
- Elution: Elute RNA in a small volume (e.g., 30-50 µL) of nuclease-free water. Heated elution (e.g., 65°C) may increase yield [39].
RNA Quality Assessment and Library Preparation
- Quality Control: Quantify RNA using a fluorometric method (e.g., Qubit) and assess quality. For FFPE RNA, the DV200 value is a more reliable metric of usability than RIN. A DV200 > 30% is generally considered acceptable for downstream sequencing or qPCR applications [42].
- Library Preparation for Sequencing: When moving towards RNA-seq, select library prep kits designed for degraded RNA. A recent comparison found that the TaKaRa SMARTer Stranded Total RNA-Seq Kit v2 generated high-quality data comparable to the Illumina Stranded Total RNA Prep Ligation kit, but with 20-fold lower RNA input requirements, a significant advantage for limited FFPE samples [42].
The following workflow diagram summarizes the optimized path for obtaining viable RNA from FFPE samples.
RNA and DNA Isolation from Liquid Biopsies
Pre-analytical Considerations
The reliability of liquid biopsy analysis is highly dependent on pre-analytical handling. Key considerations include:
- Collection Tubes: Use blood collection tubes containing preservatives (e.g., CellSave, Streck BCT) that stabilize cells and nucleic acids, allowing processing within up to 96 hours of collection [41].
- Processing Time: Process samples within 6 hours of collection for optimal results [41].
- Plasma Separation: Use differential centrifugation to isolate plasma:
- Centrifuge at 800-1600 × g for 10-20 minutes at room temperature to separate plasma from cells.
- Transfer the supernatant (plasma) to a new tube.
- Perform a second, higher-speed centrifugation (16,000 × g for 10 minutes) to remove remaining cellular debris and platelets [41].
- Storage: Aliquot plasma and store at -80°C to avoid repeated freeze-thaw cycles.
Isolation of Circulating Cell-Free DNA (ccfDNA)
A comparative study of ccfDNA isolation methods for liquid biopsy identified significant performance differences, as summarized in Table 2.
Table 2: Performance of ccfDNA Isolation Kits from Plasma
| Kit Name | Technology | Performance Summary | Key Advantage |
|---|---|---|---|
| QIAamp Circulating Nucleic Acid Kit (Qiagen) | Vacuum-column based | Highest quantities of isolated ccfDNA (P < 0.001) [41] | Optimal Yield |
| QIAamp ccfDNA/RNA Kit (Qiagen) | Spin-column based | Moderate yield, with potential for high molecular weight DNA contamination in some isolates [41] | Simultaneous DNA/RNA |
| NucleoSpin cfDNA XS Kit (Macherey-Nagel) | Spin-column based | Lowest quantities of isolated ccfDNA [41] | Not Recommended |
Isolation of Exosomes and Exosomal RNA
Exosomes are a rich source of stable RNA, including microRNAs and circular RNAs, which are promising cancer biomarkers [43] [41]. A comparison of two common exosome isolation kits revealed trade-offs between exosome size, concentration, and protein content.
Table 3: Performance of Exosome and Exosomal RNA Isolation Kits
| Kit Name | Target | Performance Summary |
|---|---|---|
| Total Exosome Isolation Kit (from plasma) (Invitrogen) | Exosomes | Yields larger exosomes but at a significantly lower concentration and with low exosomal marker (CD9, CD63) content [41]. |
| miRCURY Exosome Serum/Plasma Kit (Qiagen) | Exosomes | Yields a higher concentration of exosomes with strong presence of exosomal markers (CD9, CD63, TSG101, Alix) and higher total protein content [41]. |
| miRNeasy Serum/Plasma Advanced Kit (Qiagen) | Exosomal RNA | No significant difference in miR-19a-3p and miR-92a-3p detection compared to the Invitrogen kit [41]. |
| Total Exosome RNA & Protein Isolation Kit (Invitrogen) | Exosomal RNA | No significant difference in miR-19a-3p and miR-92a-3p detection compared to the Qiagen kit [41]. |
High-Throughput qPCR for Cancer Biomarker Screening
Optimizing the RT-qPCR Workflow
For high-throughput screening of cancer biomarkers from challenging samples, every step of the RT-qPCR process must be optimized for sensitivity, specificity, and cost-effectiveness.
- Reverse Transcription (RT): The choice of reverse transcriptase profoundly impacts detection sensitivity. A comparative analysis demonstrated that SuperScript IV VILO (VILO) has superior reverse transcription efficacy compared to Sensiscript (SS), leading to a significantly higher number of biomarker-positive patients identified in liquid biopsy samples [44].
- qPCR Miniaturization: The reaction volume for the qPCR step can be successfully miniaturized to 5 μL without compromising data quality, using a 1:4 dilution of the cDNA eluent. This "Quarter Volume" protocol, alongside reagent concentration optimization, can reduce costs by nearly 90% while maintaining HTS-quality performance (Z' factor >0.5) and single-cell analytical sensitivity [18].
Application in Monitoring Drug Resistance
Circular RNAs (circRNAs) are exceptionally stable non-coding RNAs abundant in body fluids, making them ideal liquid biopsy biomarkers. They are functionally involved in mediating resistance to chemotherapy, targeted therapy, and immunotherapy through mechanisms like miRNA sponging and regulation of signal transduction pathways [43]. High-throughput qPCR panels can be designed to monitor specific circRNAs, such as those listed in Table 4, providing a dynamic, non-invasive strategy for the early detection of therapeutic resistance.
Table 4: Key circRNAs Implicated in Cancer Drug Resistance
| circRNA Name | Tumor Type | Target Pathway/Gene | Mechanism of Resistance | Clinical Relevance |
|---|---|---|---|---|
| circHIPK3 | Colorectal, Lung, Bladder | miR-124, miR-558 | Sponges tumor-suppressor miRNAs; promotes resistance to 5-FU and cisplatin [43]. | Biomarker for chemotherapy resistance [43]. |
| circFOXO3 | Breast, Lung, Gastric | FOXO3, p21, CDK2 | Interferes with cell cycle regulation and apoptosis pathways [43]. | Prognostic marker; potential therapeutic target [43]. |
| circ_0001946 | NSCLC | miR-135a-5p, STAT6 | Promotes gefitinib resistance by activating STAT6/PI3K/AKT pathway [43]. | Marker for EGFR-TKI resistance monitoring [43]. |
| circ-PVT1 | Gastric Cancer | miR-124-3p, ZEB1 | Promotes paclitaxel resistance and invasive phenotypes [43]. | Potential predictor of treatment response [43]. |
The following diagram illustrates the workflow for applying high-throughput qPCR to liquid biopsy samples for monitoring treatment resistance.
The Scientist's Toolkit: Essential Reagents and Kits
Table 5: Key Research Reagent Solutions
| Product/Kit Name | Function/Application | Key Feature/Benefit |
|---|---|---|
| ReliaPrep FFPE Total RNA Miniprep (Promega) | RNA extraction from FFPE tissues | Provides the best balance of high RNA yield and quality from FFPE samples [38]. |
| MagMAX FFPE DNA/RNA Ultra Kit (Thermo Fisher) | Nucleic acid extraction from FFPE tissues | Bead-based, high-throughput, and amenable to automation on platforms like KingFisher [39]. |
| QIAamp Circulating Nucleic Acid Kit (Qiagen) | ccfDNA isolation from plasma | Provides the highest yield of ccfDNA for liquid biopsy applications [41]. |
| miRCURY Exosome Serum/Plasma Kit (Qiagen) | Exosome isolation from plasma | Yields high concentrations of exosomes with well-preserved surface markers and protein content [41]. |
| SuperScript IV VILO (Thermo Fisher) | Reverse Transcription (RT) | Superior sensitivity for cDNA synthesis from low-abundance targets in challenging samples [44]. |
| ssoAdvanced Universal SYBR Green Master-Mix (Bio-Rad) | Quantitative PCR (qPCR) | Reliable performance for SYBR Green-based detection in miniaturized, high-throughput reactions [18]. |
The robust analysis of RNA from FFPE tissues and liquid biopsies is a cornerstone of modern cancer biomarker discovery and validation. Success hinges on selecting optimized, sample-specific protocols for nucleic acid extraction and library preparation, as detailed in this application note. By implementing these best practices—such as using the Promega ReliaPrep kit for FFPE RNA, the Qiagen QIAamp kit for ccfDNA, and the sensitive SuperScript IV VILO for reverse transcription—researchers can unlock the full potential of high-throughput qPCR. This enables sensitive, specific, and cost-effective screening of critical biomarkers, including the highly stable circRNAs, ultimately accelerating the development of personalized cancer diagnostics and monitoring tools in the era of precision oncology.
Quantitative real-time PCR (qPCR) is a cornerstone technique in molecular biology for detecting and quantifying nucleic acids, playing a critical role in cancer biomarker screening research. In the context of high-throughput studies, where 384 or 1536 reactions can be generated in a single experiment, robust data analysis frameworks are essential for drawing accurate biological conclusions [45]. The journey of a biomarker from discovery to clinical application is long and requires rigorous validation, with molecular biomarkers often consisting of cancer-associated proteins, gene mutations, or gene rearrangements that inform clinical decision-making in precision oncology [46]. The reliability of qPCR data depends heavily on appropriate normalization and accurate quantification strategies to minimize technical variability while maximizing the detection of biologically significant changes in gene expression.
This application note provides detailed methodologies for efficiency-adjusted quantification and normalization strategies specifically framed within cancer biomarker research. We present standardized protocols and analytical frameworks that adhere to MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines, which establish essential qPCR performance metrics including PCR efficiency, dynamic range, limit of detection, target specificity, and assay precision [45]. These protocols are designed to ensure robust assay performance and reproducibility in high-throughput screening environments where the validation of potential cancer biomarkers occurs.
Efficiency-Calibrated Quantification Frameworks
Fundamental Principles of Efficiency Adjustment
The accuracy of qPCR quantification depends critically on accounting for variations in amplification efficiency between different assays and samples. PCR efficiency refers to the rate at which amplification products double during each cycle, with 100% efficiency (represented by an efficiency value of 2) indicating perfect doubling [47]. In practice, efficiency often deviates from this ideal due to factors such as primer characteristics, template purity, amplicon length, and reaction conditions. The efficiency-calibrated model provides a mathematical framework for accounting for these variations, offering a more generalized approach than the simpler ΔΔCt method [47].
The core mathematical relationship for efficiency-calibrated quantification is expressed in Equation 1, where the ratio of target gene expression in treatment versus control samples is derived from the ratio between target gene efficiency (Etarget) raised to the power of target ΔCt (ΔCttarget) and reference gene efficiency (Ereference) raised to the power of reference ΔCt (ΔCtreference) [47]. This model can be simplified to the 2^(-ΔΔCt) method only when both target and reference genes achieve perfect PCR amplification efficiency (E=2) [47]. For cancer biomarker research, where expression differences may be subtle and clinically significant, proper efficiency calibration is essential for accurate quantification.
Equation 1: Efficiency-Calibrated Model
Where:
- E_target = Amplification efficiency of target gene
- E_reference = Amplification efficiency of reference gene
- ΔCttarget = Ctcontrol - Ct_treatment for target gene
- ΔCtreference = Ctcontrol - Ct_treatment for reference gene
Statistical Analysis Models for Robust Quantification
Appropriate statistical treatment of qPCR data provides confidence intervals and significance testing that are essential for reliable biomarker discovery. Several statistical approaches can be implemented for the analysis of real-time PCR data:
- Multiple regression analysis: Develop a model to derive ΔΔCt from estimation of interaction of gene and treatment effects [47]
- ANCOVA (analysis of covariance): Propose a model where ΔΔCt can be derived from analysis of effects of variables [47]
- Pairwise group comparisons: Calculate ΔCt followed by a two-group t-test and non-parametric analogous Wilcoxon test [47]
These statistical methodologies can be adapted to various mathematical models and provide standard deviations, confidence levels, and P values essential for robust interpretation of qPCR data in biomarker research. Without proper statistical modeling, researchers risk drawing false positive conclusions, which is particularly problematic in clinical applications [47].
Data Quality Assessment Using the "Dots in Boxes" Method
The "dots in boxes" method provides a high-throughput visual approach for evaluating qPCR assay performance, capturing key MIQE guidelines metrics as single data points plotted in two dimensions [45]. This method plots PCR efficiency on the y-axis against ΔCq (the difference between Cq values of no-template control and the lowest template dilution) on the x-axis, creating a graphical box where successful experiments should fall (PCR efficiency of 90-110% and ΔCq ≥ 3) [45].
A quality scoring system (1-5) incorporates additional performance criteria including precision, fluorescence signal consistency, curve steepness, and sigmoidal curve shape. Parameters for these criteria differ slightly for probe-based chemistry compared to intercalating dye-based detection (Table 1) [45]. This method enables researchers to quickly visualize multiple targets and conditions on a single graph, facilitating rapid evaluation of overall experimental success in high-throughput biomarker screening workflows.
Table 1: Quality Score Metrics for qPCR Data Assessment
| Criteria | Intercalating Dye Chemistry | Hydrolysis Probe Chemistry |
|---|---|---|
| Linearity | R² ≥ 0.98 | R² ≥ 0.98 |
| Reproducibility | Replicate curves shall not vary by more than 1 Cq* | Replicate curves shall not vary by more than 1 Cq* |
| RFU Consistency | Maximum plateau fluorescence signal for all curves within 20% of mean; signal not jagged | Increase of fluorescence signal consistent for all curves with parallel slopes; signal not jagged |
| Curve Steepness | Curves rise from baseline to plateau within 10 Cq values or less | Curves rise from baseline to 50% maximum RFU within 10 Cq values or less |
| Curve Shape | Curves exhibit sigmoidal shape with fluorescence signal plateau | Curves need not be sigmoidal but should approach horizontal asymptote by final PCR cycle |
*At extremely low input (e.g., single copy), lack of amplification due to Poisson distribution is considered
Normalization Strategies for High-Throughput qPCR
Reference Gene Normalization
The use of reference genes (RGs), also known as housekeeping genes, represents the most common normalization approach for minimizing technical variability introduced during sample processing, RNA extraction, and cDNA synthesis. Ideal reference genes should maintain stable expression across all experimental conditions, tissues, and physiological states [48]. However, classical housekeeping genes like GAPDH, ACTB, and 18S ribosomal RNA may demonstrate variable expression under different pathological conditions, including cancer, making validation of RG stability essential for each experimental setup [48].
RG stability can be assessed using algorithms such as GeNorm and NormFinder, which rank candidate genes based on their expression stability across samples [48]. For canine intestinal tissues with different pathologies, a recent study identified RPS5, RPL8, and HMBS as the most stable reference genes, while noting that ribosomal protein genes tend to be co-regulated and may not represent ideal independent normalizers when used together [48]. For cancer biomarker research, where tumor tissues may exhibit fundamentally different metabolic and transcriptional profiles than healthy tissues, rigorous validation of proposed reference genes across all sample types is essential.
Global Mean Normalization
The global mean (GM) normalization method uses the average expression of all reliably detected genes in the experiment as a normalization factor, representing a data-driven approach that does not require a priori selection of reference genes [48]. This method conventionally finds application in gene expression microarrays and microRNA profiling but can be adapted for high-throughput qPCR studies profiling tens to hundreds of genes [48].
In comparative studies of normalization strategies for canine gastrointestinal tissues with different pathologies, GM normalization demonstrated superior performance in reducing technical variability compared to single or multiple reference gene approaches, particularly when profiling larger gene sets (>55 genes) [48]. The performance of GM normalization improves with the number of genes included, as the average of more genes provides a more stable estimate of the true technical variation. For cancer biomarker research involving comprehensive gene panels, GM normalization offers a robust alternative to traditional reference gene approaches.
Data-Driven Normalization Algorithms
Quantile Normalization
Quantile normalization assumes that the distribution of gene transcript levels remains nearly constant across samples, so that increases in some genes are balanced by decreases in others [49]. This method, widely used in microarray analysis, can be adapted for high-throughput qPCR data, particularly when addressing plate-specific effects in large experiments where samples are distributed across multiple PCR plates [49].
The quantile normalization algorithm involves:
- Storing qPCR data from a single RNA sample in a matrix with genes as rows and plates as columns
- Sorting each column into ascending order
- Calculating the average quantile distribution by taking the average of each row
- Replacing each column with this average quantile distribution
- Repeating the process for each sample and applying between-sample normalization [49]
This approach effectively removes variability associated with plate-specific effects when genes are randomly assigned or assigned based on factors unrelated to expected expression levels.
Rank-Invariant Set Normalization
Rank-invariant set normalization identifies genes that maintain their expression rank order across experimental conditions, using these stable genes to calculate normalization factors [49]. This method does not require a priori assumptions about which genes are stably expressed, instead leveraging the data itself to identify an appropriate set of normalizers [49].
The algorithm involves:
- Storing qPCR data from all samples in a matrix with genes as rows and samples as columns
- Selecting gene sets that are rank-invariant across each sample compared to a common reference
- Taking the intersection of all sets to obtain the final rank-invariant gene set
- Calculating scale factors based on the average expression of rank-invariant genes
- Normalizing raw data by multiplying each sample by its corresponding scale factor [49]
For the PMA time series data of macrophage-like human cells, this approach identified five rank-invariant genes: GAPDH, ENO1, HSP90AB1, ACTB, and EEF1A1 [49].
Table 2: Comparison of Normalization Strategies for High-Throughput qPCR
| Method | Principle | Requirements | Advantages | Limitations |
|---|---|---|---|---|
| Reference Genes | Uses stably expressed internal controls for normalization | Pre-validated stable reference genes | Simple implementation, widely accepted | Reference gene stability must be verified for each condition |
| Global Mean | Uses average expression of all genes as normalizer | Large number of genes (>55 recommended) | No need for reference gene validation; data-driven | Requires profiling many genes; performance depends on gene set size |
| Quantile Normalization | Forces same expression distribution across samples | Multiple plates with random gene assignment | Corrects for plate effects; robust distribution matching | Assumes overall transcript levels are constant across conditions |
| Rank-Invariant Set | Uses genes maintaining rank order across conditions | Large gene set; common reference sample | Data-driven; no prior assumptions about specific genes | Performance depends on finding sufficient rank-invariant genes |
Experimental Protocols
Protocol 1: Efficiency-Calibrated Relative Quantification
Sample Preparation and Experimental Design
- Experimental Design: Include control and treatment samples, with each sample containing both target and reference genes. For cancer biomarker studies, include appropriate positive and negative control samples relevant to the cancer type being investigated.
- Serial Dilutions: Prepare serially diluted aliquots for each sample to derive amplification efficiency. A five-log dilution series is recommended for comprehensive efficiency assessment [45].
- Replication: Include several replicates for each diluted concentration (typically 3-5 technical replicates) to account for technical variability.
- No-Template Controls: Include NTCs in every qPCR run to identify spurious amplification products and contamination [45].
qPCR Run Parameters
- Reaction Setup: Utilize either intercalating dye (e.g., SYBR Green I) or hydrolysis probe (e.g., TaqMan) chemistry based on application requirements. For high-throughput biomarker screening, probe-based chemistry may offer better specificity.
- Thermal Cycling Conditions: Standard cycling conditions typically include:
- Initial denaturation: 95°C for 2-10 minutes
- 40-50 cycles of:
- Denaturation: 95°C for 10-30 seconds
- Annealing/Extension: 60°C for 30-60 seconds
- Melt Curve Analysis: If using intercalating dye chemistry, include a melt curve stage (65°C to 95°C with continuous fluorescence measurement) to verify amplification specificity.
Data Analysis and Efficiency Calculation
- Baseline Correction: Define baseline using early cycles (typically cycles 5-15) to determine background fluorescence. Avoid the initial cycles (1-5) due to potential reaction stabilization artifacts [50].
- Threshold Setting: Set threshold at a point where all amplification curves are in their parallel logarithmic phases. Ensure the threshold is:
- Sufficiently above background fluorescence
- Within the logarithmic phase of amplification, unaffected by plateau effects
- At a position where all amplification plots are parallel [50]
- Efficiency Calculation:
- Plot Ct values against the logarithm of template concentration for each dilution series
- Calculate slope of the standard curve
- Determine PCR efficiency using the equation: Efficiency = 10^(-1/slope) - 1 [45]
- Convert to percentage efficiency: % Efficiency = (Efficiency - 1) × 100%
- Relative Quantification:
- Calculate ΔCt for each sample (Cttarget - Ctreference)
- Calculate ΔΔCt (ΔCttreatment - ΔCtcontrol)
- Compute expression ratio using efficiency-adjusted formula:
Protocol 2: Global Mean Normalization for High-Throughput Data
Experimental Setup and Quality Control
- Gene Panel Design: Select a comprehensive panel of genes representing various biological processes and expression levels. For cancer biomarker studies, include genes relevant to the cancer type and potential reference genes.
- Quality Control:
- Assess RNA quality (RIN > 7.0 recommended)
- Verify cDNA synthesis efficiency
- Perform pre-amplification if necessary for low-abundance targets
- Plate Design: For large gene sets, randomize gene assignment across plates to avoid confounding biological effects with plate position effects.
Data Preprocessing and Curatio
- Data Collection: Extract Cq values for all genes across all samples.
- Data Filtering:
- Remove replicates differing by more than 2 PCR cycles [48]
- Exclude genes with poor PCR efficiency (<80% or >120%)
- Remove genes with non-specific amplification (assessed by melt curve analysis for dye-based chemistry)
- Exclude genes with low amplification signal or technical issues
- Efficiency Correction: Apply efficiency correction to all Cq values using the formula: Alternatively, convert Cq to relative quantities using:
Implementation of Global Mean Normalization
- Calculate Global Mean:
- For each sample, compute the arithmetic mean of efficiency-corrected Cq values (or geometric mean of relative quantities) for all reliably detected genes
- Alternatively, use the mean of log-transformed relative quantities
- Normalization Factor:
- Compute sample-specific normalization factors as the global mean for that sample
- Alternatively, compute as the difference between the sample global mean and the overall global mean across all samples
- Apply Normalization:
- For efficiency-corrected Cq values:
- For relative quantities:
- Quality Assessment:
- Calculate coefficient of variation (CV) for normalized data
- Compare CV before and after normalization to assess effectiveness
- Evaluate reduction in technical variability
Visualization of Data Analysis Workflows
Efficiency-Adjusted Quantification Workflow
qPCR Data Normalization Strategies
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Reagents and Materials for qPCR Cancer Biomarker Research
| Category | Specific Products/Solutions | Function/Application |
|---|---|---|
| qPCR Master Mixes | Luna Universal qPCR Master Mix, SYBR Green-based mixes, Probe-based mixes | Provides optimized reaction components for efficient amplification; selection depends on detection chemistry requirements |
| Reverse Transcription Kits | LunaScript RT SuperMix, High-Capacity cDNA Reverse Transcription Kit | Converts RNA to cDNA for gene expression analysis; critical for RT-qPCR experiments |
| RNA Stabilization Reagents | RNAlater, RNA stabilization tubes | Preserves RNA integrity during sample collection and storage |
| Quality Control Assays | RNA integrity assays, DNA contamination removal kits | Ensures input material quality; critical for reproducible results |
| Reference Gene Assays | Pre-validated human reference gene panels | Provides stable normalizers for data normalization; requires validation for specific cancer types |
| qPCR Plates and Seals | Optical reaction plates, adhesive seals | Ensures optimal thermal conductivity and prevents evaporation |
| Standard Curve Materials | Synthetic oligonucleotides, linearized plasmids, control cDNA | Enables efficiency calculation and absolute quantification |
| Automation Solutions | Liquid handling systems, plate loaders | Facilitates high-throughput processing; reduces technical variability |
Robust data analysis frameworks for efficiency-adjusted quantification and normalization are essential components of reliable cancer biomarker research using high-throughput qPCR. The efficiency-calibrated model provides superior accuracy compared to the simple ΔΔCt method, particularly when amplification efficiencies deviate from the ideal value of 2. For normalization, the global mean method offers a robust data-driven alternative to traditional reference gene approaches, especially when profiling large gene sets. The choice of normalization strategy should be guided by experimental design, number of genes profiled, and thorough validation specific to the cancer type and sample conditions under investigation. Implementation of these frameworks with appropriate quality control measures ensures generation of reliable, reproducible data suitable for informing clinical decision-making in precision oncology.
Overcoming Technical Hurdles for Robust and Reproducible Results
In the field of cancer biomarker research, the accuracy of quantitative PCR (qPCR) is paramount, as it directly influences the reliability of diagnostic and prognostic assays [51]. Dynamic modeling, which involves understanding and applying the mathematical principles of the PCR amplification process, is a critical factor in achieving precise nucleic acid quantification [52]. This approach is especially crucial in high-throughput screening environments where consistent performance and the ability to detect subtle, biologically significant fold changes are required for translating findings into clinical applications [52] [51]. These notes detail the application of dynamic modeling principles to optimize PCR efficiency, thereby enhancing the quantification accuracy essential for robust cancer biomarker research.
The Critical Role of Dynamic Range in Quantification
The linear dynamic range of a qPCR assay defines the span of template concentrations over which the fluorescence signal is directly proportional to the initial amount of DNA [53]. Operating within this range is a prerequisite for accurate quantification, as it ensures that the cycle threshold (Ct) values reliably reflect the starting template concentration.
When template concentrations fall outside the dynamic range, accuracy is compromised. Samples below the range may yield false negatives or imprecise data, while concentrations above the range lead to saturation and an underestimation of the true quantity [52]. Dynamic modeling involves characterizing this range for every assay, providing a clear operational framework for researchers to validate their sample measurements against.
Key Parameters for Dynamic Modeling
The following parameters, derived from dynamic modeling, are fundamental for assessing and ensuring quantification accuracy [53] [51].
Table 1: Key PCR Validation Parameters and Their Definitions
| Parameter | Definition | Impact on Quantification Accuracy |
|---|---|---|
| Linear Dynamic Range | The range of template concentrations where the signal is directly proportional to the input. | Enables reliable relative quantification; operating outside this range causes inaccuracies. |
| Amplification Efficiency | The rate at which a PCR target is amplified per cycle, ideally between 90–110%. | Poor efficiency (outside this range) distorts the calculated initial template concentration. |
| Limit of Detection (LOD) | The lowest concentration of target that can be detected. | Defines the sensitivity of the assay for identifying the presence of a biomarker. |
| Limit of Quantification (LOQ) | The lowest concentration of target that can be quantified with acceptable precision. | Determines the threshold for reliable quantitative measurements, crucial for low-abundance targets. |
Experimental Protocol for Dynamic Range and Efficiency Validation
This protocol provides a step-by-step guide for validating the dynamic range and amplification efficiency of a qPCR assay, which is a critical step for any cancer biomarker study.
Pre-Experiment Design and Preparation
- Primer Design: Design primers to span an exon-exon junction to prevent amplification of genomic DNA. Follow best practices: primer length of 18-24 nucleotides, GC content of 40-60%, and a melting temperature (Tm) of 60-65°C for both forward and reverse primers [54].
- Control Selection: Include a No-Template Control (NTC) to check for reagent contamination and a No-Reverse-Transcriptase Control (-RT control) to detect genomic DNA contamination in RNA workflows [54].
- Reference Gene Selection: For gene expression analysis, select and validate stable reference genes (e.g., ACTB, GAPDH) that are unchanged under your experimental conditions to ensure accurate normalization [54].
Materials and Reagents
Table 2: Essential Research Reagent Solutions
| Item | Function/Description |
|---|---|
| Nucleic Acid Standards | A serial dilution of a sample with known concentration (e.g., synthetic oligonucleotides, plasmid DNA) to establish the standard curve [53]. |
| qPCR Master Mix | A pre-mixed solution containing DNA polymerase, dNTPs, buffer, and salts. For probe-based assays, ensure it is compatible with your chosen chemistry (e.g., TaqMan) [54]. |
| Passive Reference Dye | A dye (e.g., ROX) included at a fixed concentration in the master mix to normalize for non-PCR-related fluorescence fluctuations between wells, improving precision [52]. |
| SYBR Green dye / TaqMan Probes | Fluorescent reporters for detecting amplification. SYBR Green binds double-stranded DNA, while TaqMan probes offer higher specificity through a target-specific, fluorophore-labeled oligonucleotide [54]. |
Step-by-Step Workflow
- Prepare a Standard Curve: Create a 10-fold serial dilution series of your standard (e.g., 10^1 to 10^8 copies/µL), ideally consisting of at least 5 dilution points, each run in triplicate [53].
- Run qPCR: Amplify the standard curve and your experimental samples on the same plate using your optimized qPCR protocol.
- Data Analysis:
- Plot the log of the known starting quantity (SQ) of each standard against its mean Ct value.
- Perform a linear regression analysis on the plot. The R² value indicates the goodness-of-fit and should be ≥ 0.980 [53].
- Calculate the amplification efficiency (E) using the slope of the standard curve with the formula: E = [10^(-1/slope) - 1] × 100%. An efficiency between 90% and 110% is generally acceptable [54].
- Validate Sample Quantification: Ensure that the Ct values of your unknown samples fall within the linear dynamic range defined by your standard curve. Use the regression equation to interpolate the starting quantity of your samples.
Diagram 1: Dynamic Range & Efficiency Workflow
Advanced Application: Digital PCR for Absolute Quantification
Digital PCR (dPCR) represents a powerful application of dynamic modeling through physical partitioning. It enables absolute quantification of nucleic acids without a standard curve, enhancing precision for low-abundance targets like circulating tumor DNA [55].
Table 3: Comparison of dPCR Platform Performance
| Parameter | QIAcuity One (Nanoplate dPCR) | QX200 (Droplet dPCR) |
|---|---|---|
| Partition Type | Nanoscale chambers in a solid chip [55] | Water-in-oil droplets [55] |
| Limit of Detection (LOD) | ~0.39 copies/µL input [56] | ~0.17 copies/µL input [56] |
| Limit of Quantification (LOQ) | ~1.35 copies/µL input [56] | ~4.26 copies/µL input [56] |
| Typical Precision (CV) | < 11% (above LOQ) [56] | < 13% (above LOQ) [56] |
| Impact of Restriction Enzymes | Less affected by enzyme choice [56] | Precision significantly improved with HaeIII vs. EcoRI [56] |
Diagram 2: Digital PCR Workflow
Integrating dynamic modeling into qPCR workflows, from fundamental dynamic range validation to advanced dPCR applications, is indispensable for achieving the high level of quantification accuracy required in cancer biomarker research. Adherence to established validation guidelines like the MIQE guidelines ensures the reproducibility and reliability of data, which is the foundation for robust diagnostic and therapeutic development [57] [51]. By rigorously applying these principles, researchers can confidently utilize qPCR and dPCR to discover and validate novel biomarkers, ultimately supporting the advancement of precision oncology.
The reliable detection of low abundance molecular targets represents a pivotal challenge in modern cancer research and diagnostic development. Targets such as circulating tumor DNA (ctDNA), rare transcriptional biomarkers, and low-frequency mutations often exist at concentrations below 1 copy/μL in complex biological matrices like plasma, urine, or tissue extracts [58] [59]. The accurate quantification of these scarce molecules is crucial for advancing liquid biopsy applications, early cancer detection, minimal residual disease (MRD) monitoring, and profiling of tumor heterogeneity [59] [60]. Traditional quantitative PCR (qPCR) methods, while robust for higher concentration analytes, face significant sensitivity limitations at these ultralow concentrations due to factors including PCR inhibition, suboptimal amplification efficiency, and stochastic sampling effects [58] [51]. This application note synthesizes current methodologies and provides detailed protocols to maximize detection sensitivity for low abundance targets within high-throughput qPCR workflows for cancer biomarker screening.
Technology Comparison: qPCR versus ddPCR for Ultrasensitive Detection
The selection of an appropriate platform is fundamental to the success of low abundance target detection. While both qPCR and droplet digital PCR (ddPCR) are established technologies, their performance characteristics differ significantly at low target concentrations, as detailed in Table 1.
Table 1: Performance Comparison of qPCR and ddPCR for Low Abundance Target Detection
| Parameter | Quantitative PCR (qPCR) | Droplet Digital PCR (ddPCR) |
|---|---|---|
| Fundamental Principle | Relies on standard curves and cycle threshold (Ct) for relative quantification [17] | Partitions sample into nanodroplets for absolute counting via Poisson statistics [58] |
| Limit of Detection | Varies with assay efficiency and standard curve quality [51] | Superior sensitivity, particularly below 1 copy/μL [58] |
| Quantification Precision at Low Concentration | Lower; heavily dependent on standard curve accuracy [58] | Higher; offers improved quantification precision for rare targets [58] |
| Susceptibility to PCR Inhibitors | More susceptible; affects amplification efficiency and Ct values [58] | Less susceptible; partitioning dilutes inhibitors in reaction mix [58] |
| Requirement for Standard Curves | Essential, introducing a source of technical variation [58] [17] | Not required, enabling absolute quantification [58] |
| Ideal Application Context | High-throughput screening where targets are reasonably abundant [2] | Detection of very rare targets (e.g., MRD, early-stage ctDNA) [58] [60] |
Recent studies directly comparing these technologies through Bayesian inference demonstrate that ddPCR provides higher detection sensitivity and quantification precision, particularly critical for accurately measuring fold-changes in rare transcripts or mutant allele frequencies in ctDNA [58]. In oncology applications, this enhanced precision translates to more reliable patient stratification, as evidenced by studies where baseline ctDNA detection in plasma was prognostic for significantly shorter progression-free survival [60].
Experimental Design and Workflow for Maximized Sensitivity
A meticulously planned experimental workflow is paramount for achieving robust, reproducible detection of low abundance targets. The following diagram outlines the critical stages, from sample collection to data analysis, highlighting key decision points that influence sensitivity.
Sample Preparation and Nucleic Acid Extraction
The preanalytical phase is often the greatest source of variability. For liquid biopsies, collect blood into cell-stabilizing tubes (e.g., Streck, PAXgene) to prevent genomic DNA release and preserve ctDNA profile [59]. Process samples within 4-6 hours of collection. Isolate plasma through double centrifugation (e.g., 1600 × g for 10 min, then 16,000 × g for 10 min) to minimize cellular contamination. For ctDNA extraction, use commercially available silica-membrane kits specifically validated for cell-free DNA, typically yielding 3-20 ng cfDNA per 10 mL plasma [60]. For tissue samples, laser capture microdissection can enrich for specific cell populations to reduce background [61]. Always include control samples from healthy donors to establish baseline levels.
Assay Design for Ultrasensitive Detection
Assay design directly dictates the upper limit of sensitivity. Follow these specific criteria:
- Amplicon Length: Design amplicons between 65-120 bp for fragmented samples (e.g., FFPE, ctDNA) to maximize amplification efficiency [17].
- Specificity Verification: Use BLAST analysis against reference databases (NCBI, Ensembl) to ensure target specificity and exclude cross-homology with pseudogenes or related family members [62].
- Efficiency Optimization: Design and test multiple primer pairs to achieve 90-110% amplification efficiency, calculated from a standard curve using the formula: Efficiency = [10^(-1/slope) - 1] × 100% [17] [61].
- Probe Selection: For multiplex qPCR, use hydrolysis probes (e.g., TaqMan) with distinct fluorophores (FAM, VIC, CY5) to minimize spectral overlap. For ddPCR, the same probe chemistry applies but requires validation for partitioning efficiency [58] [17].
Detailed Experimental Protocols
High-Sensitivity qPCR Protocol for ctDNA Detection
This protocol is optimized for detecting low-frequency mutations in circulating tumor DNA from plasma samples [2] [60].
Reagents and Materials:
- Inhibitor-resistant qPCR master mix (e.g., Meridian Bioscience oncology formulations)
- Custom TaqMan SNP genotyping assay (20X concentration)
- DNA from human plasma (1-10 ng input per reaction)
- Nuclease-free water
- 384-well optical reaction plates
- Optical adhesive seals
Procedure:
- Reaction Setup: Prepare a 10 μL reaction mix containing:
- 5 μL of 2X qPCR master mix
- 0.5 μL of 20X TaqMan assay (final 1X)
- 2.5 μL template DNA (1-10 ng)
- 2 μL nuclease-free water
Plate Loading: Dispense 10 μL of reaction mix into 384-well plate in triplicate for each sample. Include negative controls (no-template) and positive controls (synthetic DNA with known mutation) on each plate.
Thermocycling Conditions: Run on a high-precision real-time PCR system using these parameters:
- Hold stage: 50°C for 2 minutes (UNG incubation, if applicable)
- Enzyme activation: 95°C for 10 minutes
- 50-55 cycles of:
- Denature: 95°C for 15 seconds
- Anneal/Extend: 60°C for 1 minute (with fluorescence acquisition)
Data Analysis: Calculate Ct values using the system software with automatic baseline settings and threshold set in the exponential phase of amplification. For variant allele frequency determination, use ΔΔCt method with wild-type and mutant-specific assays [62].
ddPCR Protocol for Absolute Quantification of Rare Targets
This protocol provides absolute quantification of rare targets without standard curves, ideal for MRD monitoring [58] [60].
Reagents and Materials:
- ddPCR supermix for probes (no dUTP)
- Target-specific FAM-labeled probe assay
- Reference gene HEX-labeled probe assay
- DG8 cartridges for droplet generator
- Droplet generation oil
- 96-well PCR plates
- PX1 PCR plate sealer
Procedure:
- Reaction Assembly: Prepare 20 μL reaction mix containing:
- 10 μL of 2X ddPCR supermix
- 1 μL of 20X target assay (FAM-labeled)
- 1 μL of 20X reference assay (HEX-labeled)
- 5-8 μL template DNA (up to 100 ng)
- Nuclease-free water to 20 μL
Droplet Generation: Transfer 20 μL reaction mix to DG8 cartridge well. Add 70 μL droplet generation oil to appropriate well. Place cartridge in droplet generator. After generation (approximately 1 minute), carefully transfer 40 μL emulsified sample to a 96-well PCR plate.
PCR Amplification: Seal plate with PX1 plate sealer (heat: 180°C, 5 seconds). Perform PCR amplification with the following profile:
- Enzyme activation: 95°C for 10 minutes
- 40 cycles of:
- Denature: 94°C for 30 seconds
- Anneal/Extend: 60°C for 1 minute (ramp rate: 2°C/second)
- Enzyme deactivation: 98°C for 10 minutes
- Hold: 4°C (until droplet reading)
Droplet Reading and Analysis: Place plate in droplet reader. Analyze using manufacturer's software with amplitude thresholds set based on negative controls. Apply Poisson statistics to calculate absolute concentration (copies/μL) using the formula: Concentration = -ln(1 - p) / V, where p = fraction of positive droplets, V = droplet volume [58].
Essential Reagents and Research Solutions
Successful implementation of high-sensitivity detection strategies requires carefully selected reagents and tools. Table 2 catalogues key solutions specifically engineered for challenging targets in oncology research.
Table 2: Research Reagent Solutions for High-Sensitivity Detection
| Reagent/Tool | Function | Key Features for Low Abundance Targets |
|---|---|---|
| Inhibitor-Resistant Master Mixes [2] | PCR amplification with enhanced robustness | Engineered polymerases tolerate inhibitors in plasma, FFPE, and blood samples; critical for reliable ctDNA detection |
| Ambient-Stable qPCR Reagents [2] | Enable decentralized testing | Lyophilized formulations maintain stability without cold chain; ideal for multi-site clinical trials |
| Multiplex qPCR Panels [2] | Simultaneous detection of multiple targets | Detect numerous mutations (e.g., EGFR, KRAS, BRAF) from minimal sample input; essential for tissue-limited cases |
| TaqMan-based Assays [17] | Specific target detection with probe-based chemistry | Fluorogenic 5' nuclease chemistry provides specific signal accumulation; compatible with both qPCR and ddPCR |
| Droplet Digital PCR Supermix [58] | Partitioning for absolute quantification | Enables absolute counting of target molecules without standard curves; superior for targets <1 copy/μL |
| Custom Assay Design Tools [17] | Bioinformatics for optimal primer/probe design | Ensures high amplification efficiency (90-110%) and specificity through in silico validation |
Data Analysis and Reporting Standards
Advanced Analytical Approaches
Moving beyond basic 2^(-ΔΔCt) calculations significantly enhances analytical robustness. Analysis of Covariance (ANCOVA) provides greater statistical power and is less affected by variability in qPCR amplification efficiency compared to traditional methods [63]. This approach models Ct values as a function of treatment group while controlling for technical covariates, offering particular advantages in detecting small fold-changes characteristic of low abundance targets.
For ddPCR data, apply Poisson confidence intervals to quantify uncertainty in concentration estimates, especially critical when positive droplet counts are low (<10). Use specialized statistical packages (e.g., ddpcR, digitPCR) for accurate modeling of technical and biological variance components [58].
Adherence to MIQE and FAIR Principles
Compliance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines is non-negotiable for rigorous reporting [62]. Essential items include:
- Detailed description of sample processing and storage conditions
- Complete primer/probe sequences and concentrations
- Amplification efficiency values with confidence intervals
- Evidence of assay specificity (e.g., melt curves, sequencing)
- Limit of detection (LOD) and limit of quantification (LOQ) determined from serial dilutions
- Raw data deposition in public repositories (e.g., Figshare, GEO)
Implement FAIR (Findable, Accessible, Interoperable, Reproducible) principles by sharing complete analysis scripts (e.g., in R or Python) that transparently document all data transformation and statistical testing steps [63].
The reliable detection of low abundance targets in cancer research demands an integrated strategy spanning careful experimental design, appropriate technology selection, and rigorous analytical practices. While qPCR remains a powerful, cost-effective tool for high-throughput applications, ddPCR offers superior sensitivity and precision for the most challenging targets existing at concentrations below 1 copy/μL. By implementing the detailed protocols and best practices outlined in this application note, researchers can significantly enhance the sensitivity, reproducibility, and clinical utility of their biomarker detection workflows, ultimately accelerating the development of next-generation cancer diagnostics and monitoring tools.
Managing PCR Inhibitors and Sample Purity in Complex Matrices
In the pursuit of reliable high-throughput qPCR for cancer biomarker screening, researchers consistently face two major technical hurdles: the presence of PCR inhibitors in complex sample matrices and the variable purity of tumor samples. Inhibitory substances can lead to false-negative results or significant underestimation of target concentrations, directly impacting the accuracy of biomarker quantification [64]. Simultaneously, the presence of non-cancerous cells in tumor specimens—including immune cells, fibroblasts, and normal epithelial cells—confounds genomic analysis and can alter the biological interpretation of results [65]. This application note provides detailed protocols and data-driven strategies to overcome these challenges, enabling more robust and reproducible cancer biomarker detection.
Understanding and Managing PCR Inhibition
In complex matrices such as wastewater, clinical samples, and soil extracts, a variety of substances can inhibit PCR amplification. These include complex polysaccharides, lipids, proteins, metal ions, RNases, and other compounds that interfere with molecular detection through multiple mechanisms: inhibition of DNA polymerase activity, interaction with or degradation of template nucleic acids, or chelation of essential metal ions [64]. In the context of cancer research, common inhibitors may originate from blood components, formalin-fixed paraffin-embedded (FFPE) tissue preservatives, or biopsy collection materials.
Quantitative Assessment of PCR Enhancement Strategies
A systematic evaluation of eight different PCR enhancement approaches revealed significant differences in their ability to restore amplification efficiency in inhibited samples [64]. The following table summarizes the performance of these strategies:
Table 1: Comparison of PCR Enhancement Approaches for Inhibitor Management
| Approach | Concentration/Type | Effect on Detection | Inhibition Reduction | Practical Considerations |
|---|---|---|---|---|
| T4 gp32 Protein | 0.2 μg/μL | Most significant improvement | Complete elimination of false negatives | Cost-effective, high-throughput compatible |
| 10-fold Sample Dilution | 1:10 dilution | Improved detection | Eliminated false negatives | Reduces sensitivity, simple implementation |
| Bovine Serum Albumin (BSA) | Various concentrations | Improved detection | Eliminated false negatives | Widely available, cost-effective |
| Inhibitor Removal Kit | Commercial column | Improved detection | Eliminated false negatives | Additional step, increased cost |
| DMSO | Various concentrations | Variable effects | Partial reduction | Concentration-dependent efficacy |
| Formamide | Various concentrations | Variable effects | Partial reduction | Optimization required |
| Tween-20 | Various concentrations | Limited improvement | Minimal reduction | Effective for specific inhibitor types |
| Glycerol | Various concentrations | Limited improvement | Minimal reduction | Primarily enzyme stabilization |
Advanced Polymerase Engineering for Inhibition Resistance
Recent advances in polymerase engineering have yielded novel Taq DNA polymerase variants with superior resistance to diverse PCR inhibitors. A live culture PCR (LC-PCR) screening approach enabled rapid selection of inhibitor-resistant mutants from randomly mutagenized libraries [66]. The following variants demonstrated enhanced performance:
Table 2: Novel Inhibitor-Resistant DNA Polymerase Variants
| Variant | Mutation | Resistance Profile | Performance Characteristics |
|---|---|---|---|
| Taq C-66 | E818V | Chocolate, black pepper, blood, humic acid, plant extracts | Maintains activity in 2-3 μL of 10% chocolate or black pepper extract per 35 μL reaction |
| Klentaq1 H101 | K738R | Blood, plant tissues, humic acid | Intrinsic enzymatic tolerance persisting after purification |
| OmniTaq (Reference) | Previously reported | Broad spectrum | Used as control in screening experiments |
Structural analysis suggests these substitutions may enhance nucleotide binding or stabilize the polymerase-DNA complex, reducing susceptibility to inhibitor interference [66]. These engineered enzymes provide robust solutions for detecting cancer biomarkers in challenging clinical samples without extensive nucleic acid purification.
Assessing and Accounting for Tumor Purity
Impact of Tumor Purity on Genomic Analyses
Tumor purity, defined as the proportion of cancer cells in a tissue sample, has profound implications for the interpretation of qPCR data in cancer biomarker studies. Systematic pan-cancer analysis of over 10,000 samples from The Cancer Genome Atlas (TCGA) revealed substantial variation in purity across cancer types [65]. Purity estimates showed high concordance between DNA, RNA, and methylation-based methods, though correlation with immunohistochemistry (IHC) was lower across all cancer types.
Notably, cancer types with high mutational burden (e.g., lung adenocarcinoma, squamous cell carcinomas) generally exhibited lower purity, while brain-originating tumors (e.g., adrenocortical carcinoma, lower-grade glioma) showed higher purity levels [65]. This relationship between mutational burden and purity has significant implications for immune signature detection and biomarker validation.
Computational Estimation of Tumor Purity
Machine learning approaches have enabled accurate tumor purity prediction using gene expression data. The XGBoost algorithm, applied to 33 TCGA tumor types, achieved median correlations between 0.75 and 0.87 when predicting tumor purity from RNA-seq data [67]. This analysis identified a ten-gene set (CSF2RB, RHOH, C1S, CCDC69, CCL22, CYTIP, POU2AF1, FGR, CCL21, and IL7R) whose expression levels effectively predict tumor purity regardless of tumor type [67]. When applied to an independent dataset, this gene set maintained high correlation (ρ = 0.88) with actual observed tumor purity, confirming its utility as a biomarker for purity assessment.
Detailed Experimental Protocols
Protocol 1: Inhibition-Resistant qPCR Setup
Principle: Enhance PCR reaction tolerance to inhibitors through optimized reaction composition and specialized additives.
Materials:
- Inhibitor-resistant DNA polymerase (e.g., Taq C-66, Klentaq1 H101) [66]
- T4 gene 32 protein (gp32)
- Bovine Serum Albumin (BSA)
- PCR buffer (50 mM Tris-HCl, pH 9.2, 2.5-3.5 mM magnesium chloride, 16 mM ammonium sulfate, 0.025% Brij-58)
- dNTP mix (250 μM each)
- Target-specific primers
- SYBR Green I nucleic acid gel stain (0.5X)
- PEC-1 enhancer (0.5X) [66]
Procedure:
- Prepare master mix on ice according to the following composition:
- 5 μL PCR buffer (10X concentration)
- 1 μL dNTP mix (2.5 mM each)
- 1.5 μL forward primer (10 μM)
- 1.5 μL reverse primer (10 μM)
- 0.5 μL SYBR Green I
- 0.5 μL PEC-1 enhancer
- 1 μL T4 gp32 protein (2 μg/μL stock)
- 1 μL inhibitor-resistant DNA polymerase
- Nuclease-free water to 45 μL total volume
Add 5 μL template DNA (10-100 ng) to each reaction well.
For inhibitor challenge experiments, include 2-3 μL of 10% inhibitor extract (e.g., chocolate, black pepper, blood) per 35 μL reaction [66].
Perform PCR amplification with the following cycling conditions:
- Initial denaturation: 94°C for 10 minutes
- 40-45 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing: 54°C for 40 seconds
- Extension: 70°C for 2 minutes
- Final extension: 70°C for 5 minutes
Include appropriate controls:
- No-template control (NTC) to detect contamination
- Positive control with known template quantity
- Inhibition control (positive control spiked with inhibitor)
Troubleshooting:
- If amplification efficiency remains suboptimal, titrate T4 gp32 concentration (0.1-0.5 μg/μL final concentration) [64].
- For highly inhibited samples, combine 10-fold template dilution with T4 gp32 supplementation.
- Optimize magnesium concentration (2.0-4.0 mM) based on inhibitor type and concentration.
Protocol 2: Tumor Purity Assessment Using Gene Expression Signature
Principle: Quantify tumor purity computationally using expression levels of a validated ten-gene signature.
Materials:
- RNA extracted from tumor tissue
- RNA quality assessment tools (e.g., Bioanalyzer)
- cDNA synthesis kit
- qPCR reagents and platform
- Primers for ten-gene purity signature (CSF2RB, RHOH, C1S, CCDC69, CCL22, CYTIP, POU2AF1, FGR, CCL21, IL7R)
- Reference genes for normalization
- Computational resources with XGBoost implementation [67]
Procedure:
- Sample Preparation and QC:
- Extract high-quality RNA from tumor tissue specimens using standardized protocols.
- Assess RNA integrity number (RIN) – accept only samples with RIN >7.0.
- Synthesize cDNA using reverse transcriptase with uniform input RNA amounts.
qPCR Profiling:
- Perform qPCR amplification for the ten-gene purity signature and reference genes.
- Use three technical replicates per gene to ensure measurement precision.
- Include standards for efficiency calculation if using absolute quantification.
Data Preprocessing:
- Calculate average Cq values for each gene across replicates.
- Transform Cq values to linear scale using efficiency-corrected calculations [68].
- Normalize data using reference genes with stable expression.
Purity Estimation:
- Input normalized expression values into pre-trained XGBoost model.
- Apply logit transformation to handle bounded [0,1] purity values.
- Generate purity estimates for each sample.
- Apply inverse logit transformation to express predictions in original scale [67].
Validation:
- Compare computational purity estimates with histopathological assessment when available.
- Correlate purity estimates with immune cell marker expression.
- Establish sample-specific quality thresholds based on model confidence scores.
Workflow Visualization
Diagram 1: Integrated workflow for managing PCR inhibitors and tumor purity in cancer biomarker studies. The red pathway addresses inhibitor management, while the green pathway handles tumor purity assessment. Both streams converge for corrected data interpretation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Inhibition-Resistant qPCR and Purity Assessment
| Reagent/Category | Specific Examples | Function/Application | Performance Characteristics |
|---|---|---|---|
| Inhibitor-Resistant Polymerases | Taq C-66 (E818V), Klentaq1 H101 (K738R) [66] | Amplification in inhibitor-rich samples | Tolerant to blood, plant extracts, humic acid, food derivatives |
| PCR Enhancers | T4 gene 32 protein (gp32) [64] | Binds inhibitory substances and stabilizes nucleic acids | 0.2 μg/μL final concentration eliminates false negatives |
| Protein Additives | Bovine Serum Albumin (BSA) [64] | Binds inhibitors and stabilizes polymerase | Cost-effective enhancement for moderate inhibition |
| Sample Processing | Inhibitor removal columns [64] | Physical removal of inhibitory compounds | Additional processing step with variable recovery |
| qPCR Master Mixes | Oncology-optimized formulations [2] | Enhanced sensitivity for low-abundance targets | Compatible with FFPE, plasma, cfDNA, low-input samples |
| Purity Assessment | Ten-gene signature panel [67] | Computational tumor purity estimation | XGBoost model with ρ = 0.88 correlation to actual purity |
| Reference Standards | Pre-quantified control templates | Quality control and run validation | Essential for inter-laboratory reproducibility |
Data Analysis and Interpretation Considerations
Proper Cq Value Interpretation in Inhibited Samples
Accurate interpretation of quantification cycle (Cq) values is essential for reliable biomarker quantification. The Cq value depends not only on target concentration but also on PCR efficiency and quantification threshold level [68]. Inhibition typically manifests as increased Cq values or complete amplification failure. When comparing samples, efficiency-corrected analysis is essential, as small differences in efficiency can substantially impact Cq values and resulting concentration estimates [68].
The relationship between Cq, target concentration, and PCR efficiency is described by:
Cq = log(Nq) - log(N₀) / log(E)
Where Nq is the threshold template quantity, N₀ is the initial template quantity, and E is the amplification efficiency [68]. This equation highlights why efficiency differences between samples—potentially caused by varying inhibitor concentrations—can invalidate direct Cq comparisons.
Tumor Pity Adjustment in Biomarker Quantification
When reporting cancer biomarker levels, correction for tumor purity provides more accurate representation of true molecular alterations. The consensus measurement of purity estimations (CPE), derived from multiple genomic methods, offers a robust approach for this correction [65]. For high-throughput applications, the ten-gene expression signature provides a practical balance between accuracy and throughput [67].
Differential expression analysis that accounts for tumor purity can reveal biologically significant signatures that remain undetected in traditional analyses. For example, after purity adjustment, immunotherapy-related T-cell activation pathways may become apparent in cancer types where they were previously masked by purity variation [65].
Effective management of PCR inhibitors and tumor sample purity is essential for robust cancer biomarker detection using high-throughput qPCR. The integrated strategies presented here—employing inhibitor-resistant polymerase variants, T4 gp32 protein supplementation, and computational purity estimation—provide a comprehensive framework for enhancing assay reliability. By implementing these protocols and accounting for both inhibition and purity effects, researchers can significantly improve the accuracy and reproducibility of their cancer biomarker studies, ultimately supporting more precise molecular diagnostics and therapeutic decision-making.
Within high-throughput qPCR campaigns for cancer biomarker discovery, the pressure to reduce reagent costs and increase sample throughput often drives research toward assay miniaturization. While this strategy can yield significant economic benefits, it introduces unique technical challenges that can compromise data fidelity if not meticulously managed. The drive to miniaturize must be carefully balanced against the stringent requirements for reproducibility and accuracy in clinical research. This application note details the major pitfalls encountered during miniaturization and provides validated protocols to navigate these challenges, ensuring that volume reduction does not come at the cost of data reliability in cancer biomarker screening.
The Core Challenges of Miniaturized qPCR
Miniaturization of qPCR reactions is not merely a matter of scaling down volumes. It introduces specific, measurable challenges that can impact key assay performance metrics. The following table summarizes the primary pitfalls and their potential effects on data quality.
Table 1: Key Pitfalls in qPCR Miniaturization and Their Impact on Data Fidelity
| Pitfall | Underlying Cause | Impact on Assay Performance |
|---|---|---|
| Evaporation and Reaction Integrity | Increased surface-area-to-volume ratio, leading to greater proportional loss of liquid [69]. | Altered reagent concentrations, increased replicate variability, and failed amplification [70]. |
| Liquid Handling Inaccuracy | Significant volumetric errors when pipetting sub-microliter volumes with standard liquid handlers [71]. | Decreased precision, higher coefficients of variation (CV), and compromised PCR efficiency [18]. |
| Increased Impact of Surface Interactions | Adsorption of enzymes or nucleic acids to tube walls becomes more significant with lower analyte concentrations [69]. | Reduced effective concentration of critical reagents, leading to lower sensitivity and a higher limit of detection. |
| Suboptimal Signal Intensity | Reduced total number of fluorescent molecules in smaller reaction volumes [72]. | Lower signal-to-noise ratio, making accurate Cq determination more difficult and increasing data variability. |
Establishing Miniaturization Limits: A Case Study
Empirical testing is critical for defining the lower boundary of viable miniaturization. One systematic study evaluated the success of an immunology gene expression panel under different volume-reduction conditions. The results provide a clear framework for determining miniaturization thresholds [70].
Table 2: Experimental Outcomes of Volume Reduction in a Gene Expression Panel [70]
| Miniaturization Condition | Total Reaction Volume | Amplification Success Rate | Data Quality Outcome |
|---|---|---|---|
| Full Reaction (FR) | Standard Volume (e.g., 20 µL) | High | Robust and reproducible data. |
| 1.5x Concentration | ~67% of FR Volume | High | Successful amplification; strong correlation with FR data. |
| 2.5x Concentration | ~40% of FR Volume | Suboptimal | Lower success rate; not recommended. |
| 5x Concentration | ~20% of FR Volume | None | No amplification; complete failure. |
This study concluded that a 1.5x concentration factor (e.g., a ~67% reduction in volume) maintained data quality and reproducibility, while more aggressive miniaturization led to rapid performance degradation [70]. This highlights that miniaturization has a firm limit, and pushing beyond it renders the assay non-functional.
Optimized Protocols for High-Fidelity Miniaturized qPCR
The following protocols are designed to mitigate the pitfalls outlined above, providing a roadmap for implementing robust miniaturized qPCR in a high-throughput setting.
Protocol 1: Automated, Miniaturized RT-qPCR for Surrogate Markers of Immunity
This protocol, adapted from a study on profiling immune responses from PBMCs, demonstrates a cost-effective and miniaturized workflow achieving 90% cost reduction while maintaining diagnostic sensitivity [18].
Application in Cancer Research: Ideal for screening cytokine and immune checkpoint biomarkers from limited patient PBMC samples in immuno-oncology studies.
Workflow Overview:
Step-by-Step Methodology:
Cell Stimulation and Lysis:
- Plate 50,000-100,000 PBMCs per well in a 96- or 384-well U-bottom plate.
- Stimulate with antigens (e.g., tumor lysates, peptide pools) or controls (PMA/Ionomycin for positive control, media for negative control).
- Incubate for a predetermined optimal period (e.g., 6 hours for IFN-γ).
- Lyse cells directly in the plate using a magnetic bead-based RNA extraction kit.
Miniaturized RNA Extraction and Reverse Transcription:
- Perform RNA extraction following kit instructions, eluting in a minimal volume (e.g., 10-15 µL).
- Set up reverse transcription reactions at a quarter of the manufacturer's recommended volume.
- Master Mix per Reaction:
- 4 µL of 5x RT Buffer
- 1 µL of dNTP Mix (10mM)
- 1 µL of Random Hexamers
- 0.5 µL of Reverse Transcriptase
- 0.5 µL of RNase Inhibitor
- 8 µL of RNA template + Nuclease-free Water (to a total reaction volume of 15 µL)
Miniaturized qPCR:
- Use a SYBR Green or probe-based master mix.
- Set up 5 µL reactions in a 384-well plate.
- Master Mix per Reaction:
- 2.5 µL of 2x SYBR Green Master Mix
- 0.5 µL of Primer Mix (10 µM each)
- 1.0 µL of cDNA template (diluted 1:4 from the RT reaction)
- 1.0 µL of Nuclease-free Water
Quality Control and Data Analysis:
- Calculate PCR efficiency and R² from a standard curve.
- Perform HTS validation by calculating the Z' factor (>0.5 is excellent) from positive and negative controls to confirm robust assay performance [18].
Protocol 2: Miniaturization and Automation of NGS Library Prep for qPCR Panel Validation
While not qPCR itself, this protocol for next-generation sequencing (NGS) library preparation is critical for validating the breadth of biomarkers in a qPCR panel. It demonstrates principles of miniaturization that are directly transferable, achieving over 80% cost savings and a 90% reduction in preparation time [71].
Application in Cancer Research: Creating cost-effective, high-throughput NGS libraries from tumor RNA to validate novel biomarker panels intended for downstream qPCR screening.
Workflow Overview:
Step-by-Step Methodology:
Sample Preparation and Dehydration:
- Dilute extracted total RNA in water and transfer to a 384-well PCR plate.
- Dehydrate the samples completely using a vacuum evaporator (e.g., GeneVac EZ-2) at 40°C for ~25 minutes. Studies show this does not compromise RNA Integrity Numbers (RIN) [71].
Automated Miniaturized Library Preparation:
- Use an acoustic liquid handler (e.g., Labcyte Echo 525) to dispense nanoliter volumes of library preparation reagents (fragmentation mix, RT master mix, ligation mix, PCR master mix) directly onto the dehydrated RNA pellets.
- Rehydrate and mix the samples by shaking the plate. This eliminates pipetting error for sub-microliter volumes.
Library Quality Control:
- Quantify the final libraries using a fluorometric method and assess size distribution with a Bioanalyzer or equivalent.
- The libraries should maintain fidelity comparable to full-volume manual preps, as confirmed by spike-in controls (e.g., ERCC RNA) [71].
The Scientist's Toolkit: Essential Reagents and Platforms
Table 3: Key Research Reagent Solutions for Miniaturized qPCR
| Item | Specific Example | Function in Miniaturized Workflow |
|---|---|---|
| Acoustic Liquid Handler | Labcyte Echo 525 [71] | Enables precise, non-contact transfer of nanoliter volumes of reagents, critical for accuracy in miniaturized setups. |
| Positive Displacement Liquid Handler | TTP Labtech Mosquito HV [70] | Automated pipetting system suitable for miniaturizing reaction setup in 384-well formats. |
| RT-qPCR Master Mix | SsoAdvanced Universal SYBR Green Supermix [18] | A robust, efficient master mix tolerant to volume reduction and compatible with low-volume reactions. |
| RNA-to-cDNA Kit | SuperScript IV First-Strand Synthesis System [18] | A high-performance reverse transcriptase that can be used at quarter-volumes with maintained efficiency. |
| High-Density qPCR Plates | 384-well PCR plates [69] [18] | The standard platform for high-throughput, low-volume qPCR reactions. |
| Automated Data Analysis Software | HTqPCR (R/Bioconductor package) [27] | Facilitates quality control (flagging unreliable Cqs), normalization, and statistical analysis of hundreds of qPCR datasets. |
Successful miniaturization of qPCR for cancer biomarker research is a balance between cost efficiency and technical rigor. The pitfalls of evaporation, liquid handling inaccuracy, and reduced signal are real but manageable. By adhering to defined limits of volume reduction—such as the 1.5x concentration threshold—and implementing automated, optimized protocols, researchers can achieve substantial cost savings without sacrificing the data fidelity required for robust, clinically relevant biomarker discovery. The integration of specialized liquid handling platforms and stringent, MIQE-guided quality control is paramount to ensuring that miniaturized data is both reproducible and biologically meaningful.
Primer Design and Hot-Start PCR to Minimize Non-Specific Amplification
In the context of high-throughput qPCR for cancer biomarker screening, the accuracy of molecular diagnostics depends overwhelmingly on the specificity of amplification. Non-specific amplification refers to the amplification of non-target DNA sequences during PCR, leading to false-positive results, reduced sensitivity, and compromised data interpretation [73]. For cancer researchers and drug development professionals, this is particularly critical when analyzing precious clinical samples such as liquid biopsies, fine-needle aspirates, or formalin-fixed paraffin-embedded (FFPE) tissue, where sample material is often limited and maximal information yield is essential [2].
The major forms of non-specific amplification include primer-dimers (amplicons formed by two primers hybridizing to each other, typically 20-60 bp in length), primer multimers (larger complexes that create ladder-like patterns on gels), and general smears caused by random DNA amplification across a wide size range [73]. In cancer biomarker detection, where assays must distinguish single-nucleotide mutations or quantify low-abundance transcripts, these artifacts can obscure legitimate signals, reduce amplification efficiency of true targets, and ultimately lead to erroneous clinical interpretations.
Understanding Hot-Start PCR Mechanisms
Hot-Start PCR represents a fundamental solution to non-specific amplification by maintaining DNA polymerase in an inactive state during reaction setup at room temperature [74]. This technique is particularly valuable in high-throughput screening environments where numerous reactions are assembled, potentially introducing delays that allow non-specific priming events to occur before thermal cycling begins [75].
Comparative Hot-Start Technologies
The table below summarizes the primary Hot-Start technologies used in modern PCR reagents, each with distinct advantages for cancer biomarker applications:
Table 1: Comparison of Hot-Start PCR Technologies
| Technology | Mechanism | Activation | Key Benefits | Considerations | Example Enzymes |
|---|---|---|---|---|---|
| Antibody-Based | Antibody blocks polymerase active site | Short initial denaturation (2-5 min) | Full activity restored; maintains native enzyme properties | Animal-origin components; higher exogenous protein | DreamTaq Hot Start, Platinum II Taq [76] |
| Chemical Modification | Covalent linkage of inhibitory groups | Longer activation (10-12 min) | Stringent inhibition; animal-component-free | Potentially incomplete activation; affects long targets | AmpliTaq Gold DNA Polymerase [76] |
| Affibody-Based | Alpha-helical peptide blockers | Short initial denaturation | Low protein content; animal-component-free | Possibly less stringent than antibody methods | Phire Hot Start II DNA Polymerase [76] |
| Aptamer-Based | Oligonucleotide blockers | Short initial denaturation | Animal-component-free; rapid activation | Potential reversibility at lower temperatures | Various specialized formulations [76] |
The fundamental principle common to all Hot-Start methods is that they prevent enzymatic activity during reaction setup, when primers may bind non-specifically to template DNA or to each other at suboptimal temperatures [74]. When the thermal cycler reaches the initial denaturation temperature (typically 95°C), the inhibitory compound is released or inactivated, restoring full polymerase activity only after potential mispriming sites have been melted [76]. This simple yet effective mechanism explains why Hot-Start PCR typically demonstrates increased sensitivity, higher yields of desired products, and cleaner background compared to conventional PCR [75].
Primer Design Strategies for Specific Amplification
Optimized primer design is equally crucial for minimizing non-specific amplification, particularly in complex applications like cancer biomarker detection where homologous genes and sequence polymorphisms must be distinguished.
Critical Considerations for qPCR Primer Design
Sequence Specificity Based on SNPs: When designing primers for homologous genes, utilize single-nucleotide polymorphisms (SNPs) to ensure specificity. The 3'-end of primers should contain nucleotides unique to the target sequence, as DNA polymerases can differentiate SNPs in the last one or two nucleotides at the 3'-end under optimized conditions [77].
Amplicon Length and Position: Design amplicons between 85-125 bp for optimal qPCR efficiency. Primers should be positioned across intron-exon boundaries where possible, with amplification regions designed at the 3'-end to reduce impacts of RNA degradation [78].
Primer Thermodynamics: Follow these parameters: primer length of 18-25 bp, Tm value of 58-62°C (with forward and reverse primers within 2°C), GC content of 40-65%, and avoidance of self-dimers or hairpin structures [78].
Validation Against Homologous Sequences: Before experimental validation, conduct in silico analysis using tools like primer-BLAST to test off-target binding across all homologous sequences in the genome [77].
Integrated Experimental Protocols
Protocol 1: Stepwise qPCR Optimization for Cancer Biomarker Assays
This protocol combines Hot-Start PCR with systematic primer optimization, specifically adapted for cancer biomarker detection in high-throughput settings.
Table 2: Stepwise qPCR Optimization Protocol
| Step | Parameter | Optimal Conditions | Validation Criteria |
|---|---|---|---|
| 1. Initial Primer Design | Sequence specificity based on SNPs | Design across intron-exon boundaries; 3'-end SNPs for homologous discrimination | In silico specificity confirmation via primer-BLAST [77] |
| 2. Hot-Start Master Mix Preparation | Reaction assembly | Use antibody-based Hot-Start polymerase for rapid activation; assemble on ice | Enzyme remains inactive until thermal activation [76] |
| 3. Thermal Cycling - Initial Activation | Initial denaturation/activation | 95°C for 2-5 min (antibody-based) or 10-12 min (chemical modification) | Complete polymerase activation without damage [74] [76] |
| 4. Annealing Temperature Optimization | Temperature gradient | Test range from 55°C to 65°C in 2°C increments | Single peak in melt curve; no primer-dimer formation [77] |
| 5. Primer Concentration Titration | Primer concentration | Test 50-500 nM in serial dilutions | Maximum efficiency with minimal non-specific products [77] |
| 6. cDNA Concentration Range | Template dilution | 5-point serial dilution (1:10 to 1:10000) | R² ≥ 0.99 in standard curve; efficiency = 100% ± 5% [77] |
| 7. Validation in Biological Matrix | Clinical sample type | Spike controls into plasma, FFPE, or cfDNA samples | Consistent efficiency in clinical matrix vs. controls [2] |
Protocol 2: Hot-Start qPCR Setup for High-Throughput Cancer Biomarker Screening
Materials:
- Hot-Start DNA polymerase (antibody-based for rapid activation)
- Optimized primer pairs (validated against homologous sequences)
- Template cDNA (from clinical samples: plasma, FFPE, tissue)
- qPCR plates compatible with high-throughput systems
- Multichannel pipettes or automated liquid handling system
Procedure:
- Reaction Assembly at Room Temperature: Prepare master mix containing Hot-Start polymerase, buffer, dNTPs, and optimized primer concentrations. Aliquot into qPCR plates.
Template Addition: Add template DNA/cDNA (20-100 ng equivalent per reaction). For high-throughput applications, utilize automated liquid handling systems to minimize setup time variation.
Seal Plates and Centrifuge: Apply optical seals and briefly centrifuge to collect reaction mixture at plate bottom.
Thermal Cycling Protocol:
- Initial denaturation/activation: 95°C for 5 minutes
- 40-45 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing: Optimized temperature (60°C±2) for 30 seconds
- Extension: 72°C for 30 seconds
- Melt curve analysis: 95°C for 15 seconds, 60°C for 1 minute, then gradual increase to 95°C with continuous fluorescence monitoring
Data Analysis: Calculate amplification efficiency using standard curve method. Accept only reactions with efficiency between 95-105% and R² ≥ 0.990 for biomarker quantification [77].
Troubleshooting Non-Specific Amplification
Table 3: Troubleshooting Guide for Non-Specific Amplification
| Problem | Possible Causes | Solutions | Prevention Tips |
|---|---|---|---|
| Primer Dimers (bright bands at 20-60 bp) | Excessive primer concentration; low annealing temperature; slow reaction setup | Reduce primer concentration (50-200 nM); increase annealing temperature; use Hot-Start polymerase | Assemble reactions on ice; use pre-formulated Hot-Start master mixes [73] |
| Multiple Bands (unexpected sizes on gel) | Non-specific priming; homologous gene amplification; low annealing stringency | Redesign primers with 3'-end SNPs; increase annealing temperature; optimize Mg²⁺ concentration | Validate primer specificity against all homologous sequences before use [77] |
| Smear Throughout Lane | Random DNA amplification; degraded templates; contaminated reagents | Use cleaner template DNA; replace reagents; increase annealing temperature | Implement rigorous laboratory practices; use inhibitor-resistant polymerases [73] |
| Reduced Amplification Efficiency | Inhibitors in clinical samples; suboptimal primer design; incomplete Hot-Start activation | Dilute template; use inhibitor-resistant polymerases; ensure complete initial denaturation | Validate assay in relevant clinical matrix (plasma, FFPE) during development [2] |
Application in Cancer Biomarker Research
The combination of optimized primer design and Hot-Start PCR technology enables highly sensitive and specific detection of cancer biomarkers across various sample types. In non-small cell lung cancer (NSCLC), multiplexed qPCR panels simultaneously assess alterations in EGFR, KRAS, BRAF and ALK with rapid turnaround times essential for treatment decisions [2]. For pancreatic cancer research, proper validation of reference genes (such as EIF2B1 and IPO8) using these techniques ensures accurate quantification of gene expression patterns between cancerous and non-neoplastic tissues [78].
Recent advances in liquid biopsy applications highlight the critical importance of these methods. High-performance ctDNA biomarker panels for colorectal cancer detection achieve sensitivities of 91-95% and specificities of 93-98% when properly optimized, enabling non-invasive cancer screening [79]. Similarly, folate receptor-targeted qPCR methods for gastric cancer circulating tumor cell detection demonstrate 104% amplification efficiency with strong linearity (R² = 0.9970), enabling precise disease monitoring [80].
Essential Research Reagent Solutions
Table 4: Key Reagent Solutions for High-Throughput qPCR in Cancer Biomarker Research
| Reagent Category | Specific Examples | Key Features | Optimal Applications |
|---|---|---|---|
| Antibody-Based Hot-Start Polymerases | DreamTaq Hot Start, Platinum II Taq, Platinum SuperFi II | Rapid activation (2-5 min); full enzyme activity; familiar performance characteristics | High-throughput screening; multiplex qPCR; time-sensitive diagnostics [76] |
| Inhibitor-Resistant Master Mixes | Meridian Bioscience oncology reagents | Tolerant to heparin, hemoglobin, FFPE inhibitors; compatible with plasma, whole blood | Liquid biopsy analysis; FFPE-derived nucleic acids; direct blood PCR [2] |
| Ambient-Stable Formulations | Lyophilized qPCR panels | Cold chain-independent; room temperature storage; pre-aliquoted for automation | Decentralized testing; resource-limited settings; OEM applications [2] |
| Multiplex qPCR Reagents | Advanced probe master mixes | Efficient multicolor detection; minimal cross-talk; uniform amplification | Multi-biomarker panels; internal control co-amplification; pathway analysis [2] |
| Rapid Cycling Optimized Enzymes | SolisFAST products | Fast extension rates; reduced cycle times; maintained specificity | High-throughput applications; rapid diagnostic turnaround; large cohort studies [75] |
The integration of sophisticated primer design strategies with Hot-Start PCR technology provides a robust foundation for reliable cancer biomarker detection in high-throughput qPCR applications. The systematic approach outlined in these application notes—combining in silico primer validation, appropriate Hot-Start enzyme selection, and stepwise optimization—enables researchers to achieve the specificity and sensitivity required for modern oncology diagnostics. As cancer biomarker research increasingly focuses on liquid biopsies and early detection, these methodologies will remain essential for extracting maximum information from minimal sample material while maintaining the rigorous quality standards demanded by clinical applications and drug development pipelines.
Benchmarking Performance and Integrating with Multi-Omics Approaches
Within the field of cancer biomarker research, the accurate and reliable detection of molecular signatures is paramount for both early diagnosis and prognostic assessment. High-throughput quantitative polymerase chain reaction (qPCR) platforms represent a cornerstone technology in this endeavor, enabling the screening of numerous potential biomarkers across large sample sets. However, the selection of an appropriate platform necessitates a critical evaluation of its performance characteristics. This application note provides a structured comparison of various high-throughput platforms, focusing on their sensitivity, specificity, and reproducibility in the context of cancer biomarker screening. We summarize quantitative performance data from recent studies, detail standardized experimental protocols for cross-platform validation, and visualize the key workflows and decision pathways to guide researchers and drug development professionals in selecting the optimal technological platform for their specific research objectives.
Comparative Performance Data of High-Throughput Platforms
The choice of platform can significantly influence experimental outcomes. The tables below synthesize key performance metrics from recent comparative studies to inform platform selection.
Table 1: Key Performance Metrics from Platform Comparison Studies
| Platform / Technology | Application Context | Sensitivity & Specificity | Reproducibility & Concordance | Key Findings |
|---|---|---|---|---|
| qPCR-based Platforms (TaqMan OpenArray, miRCURY LNA) [81] | Circulating microRNA (ct-miR) profiling in Non-Small Cell Lung Cancer (NSCLC) plasma | Identified significantly dysregulated miRs (e.g., miR-150-5p, miR-210-3p) with discriminatory power [81] | High intra-platform reproducibility; >80% concordance in miR signature classifier (MSC) risk assignment for 4/5 platforms [81] | qPCR-based platforms showed the highest inter-platform correlations among themselves [81] |
| nCounter NanoString [82] | Copy Number Alteration (CNA) validation in oral cancer | Demonhigh sensitivity for multiplex CNA detection [82] | Moderate to substantial agreement with qPCR for 10/24 genes; Spearman's correlation ranged from weak (r=0.188) to moderate (r=0.517) [82] | Technology influenced prognostic associations; for example, ISG15 was associated with both better (qPCR) and worse (NanoString) survival outcomes [82] |
| High-Resolution Melting (HRM) Analysis [83] | Malaria species differentiation (as a model for mutation detection) | High sensitivity and specificity for identifying Plasmodium species, with complete agreement with sequencing in tested samples [83] | Enabled significant species differentiation based on a Tm difference of 2.73°C, demonstrating high technical reproducibility [83] | A reliable, closed-tube method for species identification and SNP detection when paired with precise primer design [83] |
| Multiple Cross Displacement Amplification (MCDA) with AuNP-LFB [84] | Point-of-care detection of HBV/HCV | 100% sensitivity and specificity, concordant with qPCR; Limit of Detection (LoD) of 10 copies [84] | Rapid (35 min) isothermal amplification with visual readout, offering a reproducible alternative for decentralized settings [84] | Highlights a paradigm shift towards POC technologies that match qPCR accuracy with reduced cost and time [84] |
Table 2: Technical Specifications of Exemplary qPCR Platforms
| Feature / Specification | LightCycler 480 System [85] [86] | QuantStudio 12K Flex System [87] |
|---|---|---|
| Block Formats | Interchangeable 96- and 384-well plates [85] | Five interchangeable blocks: OpenArray, TaqMan Array Card, 384-well, 96-well (0.1 & 0.2 mL) [87] |
| Throughput | 384 wells per run | Up to four 3,072-reaction OpenArray plates (12,000+ data points) per run [87] |
| Thermal Uniformity | Therma-Base technology for optimal well-to-well homogeneity [85] | Standard Peltier-based cycling |
| Detection Capabilities | High-resolution melting (HRM); up to 6 colors for hydrolysis and HybProbe probes [86] | Up to 6-plex targets with enhanced OptiFlex system [87] |
| Dynamic Range | Not explicitly stated | 9-log dynamic range demonstrated [87] |
| Typical Run Time | <40 minutes for 384-well block [85] | ~4 hours for four OpenArray plates [87] |
Experimental Protocols for Platform Assessment
Protocol: Cross-Platform Comparison for microRNA Profiling
This protocol is adapted from a study comparing five high-throughput platforms for ct-miR analysis in NSCLC [81].
1. Sample Preparation and RNA Extraction
- Sample Cohort: Use plasma samples from defined clinical groups (e.g., 10 NSCLC patients and 10 matched healthy donors) [81].
- Blood Processing: Collect blood in specialized tubes (e.g., BD P100). Centrifuge twice at 1,600g for 10 minutes to generate plasma aliquots. Store at -80°C [81].
- RNA Extraction: Extract total RNA from 200 μL plasma using an automated system (e.g., Promega Maxwell 48). Elute in nuclease-free water.
- Spike-in Controls: Add exogenous synthetic miRs (e.g., ath-miR-159a, cel-miR-39-3p) during processing to monitor extraction efficiency and minimize template loss [81].
2. Platform-Specific Profiling
- qPCR-based Platforms (e.g., TaqMan OpenArray):
- Reverse Transcription (RT) and Pre-amplification: Perform using MegaPlex Pools following manufacturer's instructions.
- qPCR Setup: Dilute pre-amplified products, mix with Master Mix, and load onto OpenArray plates [81].
- Sequencing-based Platforms (e.g., EdgeSeq, QiaSeq):
- EdgeSeq: Profile 2,083 human miRs directly from plasma without extraction via quantitative nuclease protection.
- QiaSeq: Use this discovery-oriented platform for whole miRNome capture and library preparation [81].
3. Data Analysis
- Quality Control: Assess extraction efficiency using spike-in controls.
- Differential Expression: Identify dysregulated ct-miRs (e.g., miR-150-5p, miR-210-3p) between patient and control groups.
- Classifier Concordance: Apply a pre-defined biomarker signature (e.g., MSC) and calculate the percentage agreement in risk level assignment across platforms [81].
- Inter-platform Correlation: Calculate correlation coefficients (e.g., Spearman's) between platforms for commonly detected targets [81].
Protocol: Validation of Copy Number Alterations (CNAs)
This protocol is based on a comparison between real-time PCR and nCounter NanoString for validating CNAs in oral cancer [82].
1. Probe and Assay Design
- Gene Selection: Select target genes based on prior genomic studies (e.g., 24 genes associated with clinical outcomes) [82].
- nCounter NanoString: Design 3 probes for genes with amplifications and 5 probes for genes with deletions to ensure comprehensive coverage.
- Real-time PCR: Use commercially available TaqMan assays or custom-designed primers/probes.
2. Experimental Setup
- Sample DNA: Use DNA from patient samples (e.g., 119 oral cancer samples) with a female pooled DNA reference [82].
- nCounter NanoString: Perform reactions in singleton as per manufacturer's guidelines.
- Real-time PCR: Perform reactions in quadruplicate following MIQE guidelines to ensure precision [82].
3. Data Analysis and Validation
- CNA Quantification: Calculate copy numbers for each gene relative to the reference DNA.
- Statistical Correlation: Perform Spearman's rank correlation for each gene across the two techniques.
- Agreement Analysis: Calculate Cohen's Kappa score to evaluate concordance in classifying samples as having a gain or loss of copy number [82].
- Clinical Correlation: Associate CNA data with clinical outcomes (e.g., recurrence-free survival, overall survival) to compare the prognostic value of each platform [82].
Workflow and Technology Selection Diagrams
The following diagrams outline the experimental workflow for platform comparison and a decision tree for selecting the appropriate technology.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Platforms and Reagents for High-Throughput qPCR and Biomarker Validation
| Category / Item | Specific Example | Function and Application Note |
|---|---|---|
| High-Throughput qPCR Systems | QuantStudio 12K Flex System [87] | Offers unmatched flexibility with five interchangeable blocks for scaling from 12K data points to single reactions; ideal for large-scale biomarker screening campaigns. |
| Versatile Real-Time PCR Systems | LightCycler 480 System [85] [86] | Provides exceptional thermal homogeneity and flexible multi-color detection (up to 6 colors), supporting applications from HRM to multiplex hydrolysis probe assays. |
| Multiplex Digital Detection | nCounter NanoString Analysis System [82] | Enables direct, highly multiplexed measurement of gene expression or CNAs without enzymatic reaction, offering high sensitivity and digital readouts. |
| RNA Extraction & QC | Maxwell 48 Instrument (Promega) [81] | Automated nucleic acid extraction ensures high reproducibility for precious clinical samples like plasma. |
| Synthetic spike-in miRs (ath-miR-159a, cel-miR-39-3p) [81] | Critical exogenous controls added during plasma RNA extraction to monitor and correct for efficiency and technical variation. | |
| Specialized Detection Chemistries | SYBR Green I Dye [86] | Intercalating dye for dsDNA quantification and product characterization; cost-effective for amplicon validation. |
| Hydrolysis (TaqMan) Probes [86] | Provide high specificity for target quantification via 5' nuclease assay; essential for multiplexed, specific detection in complex backgrounds. | |
| HybProbe Probes (FRET-based) [86] | Two sequence-specific probes increase specificity and are ideal for SNP genotyping and mutation detection via melting curve analysis. | |
| HRM Dyes (e.g., ResoLight) [86] | Saturating dyes that enable high-resolution melting analysis for mutation discovery, methylation analysis, and species identification. | |
| Emerging POC Technologies | MCDA-AuNP-LFB Assay [84] | Represents the next generation of POC diagnostics, combining isothermal amplification with lateral flow biosensors for rapid, visual, and qPCR-sensitive detection. |
Within cancer biomarker research, the selection of an appropriate analytical platform is critical for obtaining reliable and actionable data. High-throughput quantitative PCR (HT-qPCR) and Next-Generation Sequencing (NGS) represent two powerful yet fundamentally different approaches for genomic analysis. While both methods can detect genetic variants, their technical principles, performance characteristics, and applications differ significantly. This application note provides a systematic concordance analysis between HT-qPCR and NGS methodologies, offering detailed protocols and comparative data to guide researchers in selecting the optimal approach for cancer biomarker screening within drug development pipelines. The analysis presented herein stems from a broader thesis investigating optimized molecular workflows for oncology applications, with particular emphasis on biomarker discovery, validation, and clinical translation.
Technical Comparison: Fundamental Principles and Capabilities
HT-qPCR and NGS employ distinct technical principles that directly influence their application in biomarker research. HT-qPCR utilizes multiplexed primer sets in a nanofluidics platform to simultaneously amplify and quantify hundreds of predefined targets through fluorescence detection [88]. This method relies on prior knowledge of target sequences for primer design and measures amplification kinetics to provide absolute or relative quantification of specific genes. In contrast, NGS is a sequencing-by-synthesis approach that fragments the entire genome, sequences millions of fragments in parallel, and computationally reassembles them to identify variants across targeted or entire genomic regions [89]. Two primary target enrichment strategies are employed in targeted NGS: hybridization capture and amplicon sequencing [90] [91].
Table 1: Core Technical Characteristics of HT-qPCR and NGS Methods
| Feature | HT-qPCR | NGS: Amplicon Sequencing | NGS: Hybridization Capture |
|---|---|---|---|
| Principle | Multiplexed amplification with fluorescence detection | Sequence-specific amplification followed by sequencing | Solution-based hybridization with biotinylated baits to regions of interest |
| Throughput | 100-500 targets per run [88] | Flexible, typically <10,000 amplicons [90] | Virtually unlimited by panel size [90] [91] |
| Sensitivity | High (detects low abundance targets) [88] | Down to 5% variant allele frequency [91] | Down to 1% variant allele frequency [91] |
| Best Applications | Profiling known antibiotic resistance genes, pathogen detection [92] [88] | Smaller gene panels, mutation hotspots, germline SNPs/indels [90] [91] | Larger gene panels, exome sequencing, rare variant detection, oncology [90] [91] |
Concordance Analysis: Comparative Performance Data
Empirical studies directly comparing HT-qPCR and NGS reveal important patterns in detection concordance and performance. A study on antibiotic resistance gene (ARG) profiling demonstrated that HT-qPCR detected 28 out of 31 targeted ARGs, while metagenomic NGS identified a greater diversity of 402 ARGs and associated mobile genetic elements [92] [88]. Despite these differences, both methods effectively captured variations in ARG profiles across environments, with HT-qPCR providing superior absolute quantification and NGS offering comprehensive diversity profiling [92]. In a separate study on Helicobacter pylori detection in pediatric biopsies, real-time PCR methods identified the pathogen in 40% of samples, while NGS detected it in 35% of samples, with PCR showing slightly higher sensitivity for two additional low-abundance samples [93].
Table 2: Quantitative Comparison of Detection Performance from Comparative Studies
| Study Focus | Method | Key Detection Metrics | Strengths | Limitations |
|---|---|---|---|---|
| ARG Profiling in Aquaculture [92] [88] | HT-qPCR | 28/31 targeted ARGs detected | Absolute quantification; Identified dominant ARG-hosts (e.g., Pseudomonas) | Limited to predefined targets |
| Shotgun Metagenomic NGS | 402 ARGs detected; 18 overlapped with HT-qPCR | Comprehensive diversity profiling; Identified plasmid sequences (1567) and integrons (168) | Does not provide absolute quantification | |
| H. pylori Detection [93] | Real-Time PCR | 16/40 samples (40.0%); Cq values: 17.51-32.21 | Slightly higher sensitivity for low-abundance targets | Limited to specific primer targets |
| NGS | 14/40 samples (35.0%); read counts: 7768-42924 | Ability to detect multiple pathogens simultaneously | Higher cost and complexity | |
| Prostate Cancer Mutations [94] | NGS (Tissue) | 100% mutation detection rate (gold standard) | Comprehensive variant profiling | Invasive sampling procedure |
| NGS (Liquid Biopsy) | Plasma: 67.6%; Urine: 65.6%; Semen: 33.3% | Non-invasive alternative; High concordance with tissue | Lower sensitivity for localized disease |
Detailed Experimental Protocols
Protocol 1: HT-qPCR for Targeted Biomarker Profiling
This protocol is adapted from sediment resistome profiling studies [88] and can be modified for cancer biomarker panels.
Reagents and Equipment:
- SmartChipTM Nano-Dispenser (TakaraBio)
- SmartChipTM qPCR system with 5184 wells
- SmartChipTM Green Gene Expression Master Mix
- Custom-designed multiplex primers (typically 296 primers for 268 ARGs, 8 integrons, 20 other genes)
- QIAamp Power Soil Kit (Qiagen) for DNA isolation
- Qubit Fluorometer with HS dsDNA assay kit (Thermo Fisher)
Procedure:
- DNA Isolation: Extract genomic DNA from samples (0.25 mg sediment or tissue) using the QIAamp Power Soil Kit according to manufacturer instructions. Include mechanical lysis with bead beating for 10 minutes.
- Quality Assessment: Quantify DNA using Qubit Fluorometer and verify integrity on 0.8% agarose gel.
- PCR Reaction Setup: Prepare reaction mix containing:
- 1× SmartChipTM Green Gene Expression Master Mix
- 300 nM of each primer
- 2 ng/µL template DNA
- Nuclease-free water to volume
- Nano-Dispensing: Dispense 100 nL reaction volume (in triplicate) into SmartChipTM wells using automated nanodispenser.
- Thermal Cycling: Perform qPCR with the following conditions:
- Initial denaturation: 95°C for 10 min
- 40 cycles of: 95°C for 30 sec, 60°C for 30 sec (data acquisition)
- Melt curve analysis: 60°C to 95°C with 0.5°C increments
- Data Analysis: Process raw Cq data using SmartChipTM software. Normalize to internal controls or 16S rRNA genes for absolute quantification.
Protocol 2: Targeted NGS via Hybridization Capture for Cancer Panels
This protocol leverages NGS methodologies from prostate cancer studies [94] and technical guides on hybridization capture [90] [91].
Reagents and Equipment:
- Illumina HiSeq4000 or equivalent NGS platform
- KAPA Hyper DNA Library Prep Kit (Roche)
- Targeted hybridization capture panels (e.g., 437 cancer-related genes)
- QIAamp Circulating Nucleic Acid Kit (Qiagen)
- Qubit Fluorometer (Life Technologies)
Procedure:
- Library Preparation:
- Fragment genomic DNA (1 μg input recommended) to 200-500 bp via sonication or enzymatic fragmentation.
- Repair ends, add A-tailing, and ligate Illumina sequencing adapters using KAPA Hyper Prep Kit.
- Amplify libraries with 8-10 PCR cycles using indexed primers.
Hybridization Capture:
- Pool libraries (up to 96-plex) and hybridize with biotinylated oligonucleotide baits for 16-24 hours at 65°C.
- Capture bait-bound libraries using streptavidin-coated magnetic beads.
- Wash to remove non-specifically bound DNA.
Post-Capture Amplification:
- Amplify captured libraries with 12-14 PCR cycles.
- Validate library quality and quantity using Qubit and Bioanalyzer.
Sequencing:
- Pool final libraries at equimolar concentrations.
- Sequence on Illumina platform with minimum 100x coverage (recommended 150x for low-frequency variants).
Bioinformatic Analysis:
- Align reads to reference genome (GRCh37/hg19) using Burrows-Wheeler Aligner.
- Call variants using VarScan2 with threshold of:
- Tissue: VAF ≥ 1%, supporting reads ≥ 5
- Plasma: VAF ≥ 0.3%, supporting reads ≥ 3 [94]
- Annotate variants using ANNOVAR and filter against population databases.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for HT-qPCR and NGS Workflows
| Product Category | Specific Examples | Function | Application Context |
|---|---|---|---|
| Nucleic Acid Extraction | QIAamp Power Soil Kit (Qiagen) [88]QIAamp Circulating Nucleic Acid Kit [94] [95] | Isolation of high-quality DNA from challenging samples (soil, sediment, plasma) | Pre-analytical step for all molecular methods; critical for input quality |
| qPCR Master Mixes | SmartChipTM Green Gene Expression Master Mix [88]HTOne Ultra RT-qPCR Probe Master Mix [95] | Optimized enzyme blends for efficient, specific amplification | HT-qPCR and validation assays; provides detection chemistry |
| Target Enrichment | Custom hybridization panels (e.g., 437 cancer-related genes) [94] | Solution-based capture of genomic regions of interest | Targeted NGS; enables focused sequencing on disease-relevant genes |
| Library Preparation | KAPA Hyper DNA Library Prep Kit (Roche) [94] | Fragmentation, end-repair, adapter ligation for NGS | NGS workflow; prepares DNA fragments for sequencing |
| Quantification Tools | Qubit Fluorometer with HS dsDNA assay (Thermo Fisher) [94] [88] | Accurate nucleic acid quantification | Quality control at multiple workflow steps |
Application in Cancer Research: Integration Perspectives
The complementary strengths of HT-qPCR and NGS enable their strategic application across the cancer biomarker development pipeline. NGS excels in discovery phases where comprehensive profiling of 437 cancer-related genes identifies novel biomarkers and mechanisms of therapy resistance [94]. Its ability to detect multiple biomarker classes (SNVs, indels, CNVs, fusions) in a single assay makes it invaluable for comprehensive genomic profiling in oncology [89]. HT-qPCR becomes particularly advantageous in validation and clinical translation phases where high-throughput, cost-effective quantification of established biomarker panels is required across large sample cohorts [92] [88].
Liquid biopsy applications represent a particularly promising area where both technologies contribute significantly. NGS of circulating tumor DNA (ctDNA) enables non-invasive tumor genotyping and monitoring of treatment response [96] [94] [89]. The high sensitivity of NGS (detecting mutations present at as little as 5% allele frequency) makes it suitable for monitoring minimal residual disease and emerging resistance mutations [89]. HT-qPCR complements this approach by offering rapid, economical quantification of specific, clinically-validated mutations in ctDNA, suitable for high-frequency monitoring and routine clinical testing.
From a practical implementation perspective, targeted NGS panels containing 4+ genes have demonstrated cost-effectiveness compared to sequential single-gene tests by reducing turnaround time, healthcare staff requirements, and hospital visits [97]. This economic consideration, coupled with the technical performance data presented herein, supports a hybrid approach where NGS enables comprehensive initial profiling followed by HT-qPCR for focused validation and longitudinal monitoring of priority cancer biomarkers.
The reliability of bioanalytical data is paramount in translational cancer research, particularly in the development of high-throughput screening pipelines for biomarker discovery. Molecular techniques, especially quantitative PCR (qPCR), serve as a cornerstone for quantifying biomarkers critical for diagnosis, prognosis, and therapeutic monitoring [98]. However, without rigorous and standardized validation, the powerful amplification capacity of PCR can lead to misleading results, misdirecting research and drug development efforts [53]. The absence of specific regulatory guidance for molecular assays supporting novel therapeutic modalities like cell and gene therapies has historically led to discussions on a lack of harmonization within the industry [98]. This application note provides a structured framework and a detailed colorectal cancer case example to guide the validation of qPCR assays within high-throughput cancer biomarker screening programs, ensuring the generation of robust, reliable, and actionable data.
Core Principles of qPCR Assay Validation
Validation establishes the performance characteristics of an assay, demonstrating its fitness for a specific purpose. The following parameters are essential for qPCR assays used in regulated bioanalysis [98] [53].
Table 1: Essential Validation Parameters for qPCR Biomarker Assays
| Validation Parameter | Definition and Purpose | Acceptance Criteria |
|---|---|---|
| Inclusivity (Analytical Specificity) | Measures the assay's ability to detect all intended target variants (e.g., genetic mutations across a cancer type) [53]. | The assay should reliably detect all target strains/isolates. Testing with up to 50 well-defined strains is recommended [53]. |
| Exclusivity/Cross-reactivity | Assesses the assay's ability to avoid amplification of genetically similar non-targets or background (e.g., normal host genome) [53]. | No amplification signal from non-target, cross-reactive species. |
| Linear Dynamic Range | The range of template concentrations over which the signal is directly proportional to the input, defining the quantitative range [53]. | A linear range of 6–8 orders of magnitude is typical; linearity (R²) should be ≥ 0.980 [53]. |
| Amplification Efficiency | The rate of PCR product amplification per cycle during the exponential phase. | Efficiency between 90% and 110% is acceptable [53]. |
| Limit of Detection (LOD) | The lowest concentration of the target that can be reliably detected but not necessarily quantified. | Often determined using probit analysis; should meet predefined confidence levels (e.g., 95%) [98]. |
| Limit of Quantification (LOQ) | The lowest concentration of the target that can be reliably quantified with stated precision and accuracy. | Precision (CV%) and accuracy (e.g., 70-130%) must meet pre-defined criteria at the LOQ [98]. |
| Precision | The degree of reproducibility of measurements, assessed as repeatability (intra-run) and intermediate precision (inter-run). | Coefficient of variation (CV%) typically ≤ 25-30% at the LOQ and ≤ 20% at other concentrations [98]. |
| Accuracy | The closeness of the measured value to the true value. | Mean accuracy typically within 70-130% at the LOQ and 80-120% for other concentrations [98]. |
Case Example: Validating a qPCR Assay for a Novel Colorectal Cancer Target
Experimental Context and Objective
Colorectal cancer (CRC) is the third most common cancer worldwide, with rising incidence, particularly in younger populations [99]. The discovery of new therapeutic targets is critical. The 14-3-3ζ protein, an anti-apoptotic molecular scaffold that sequesters pro-apoptotic proteins like BAD, is overexpressed in CRC and implicated in chemoresistance [100]. Disrupting the 14-3-3ζ/BAD interaction presents a promising therapeutic strategy.
In a 2025 high-throughput screening (HTS) study, a Bioluminescence Resonance Energy Transfer (BRET)-based biosensor was developed to identify disruptors of the 14-3-3ζ/BAD protein-protein interaction (PPI) in live cells [100]. The objective was to screen a library of 1971 FDA-approved or orphan drugs to identify compounds that could be repurposed as chemotherapeutics. A validated qPCR assay was integral to the subsequent mechanistic validation, quantifying changes in apoptotic gene expression in CRC cell lines following treatment with hit compounds.
Detailed Experimental Protocol
Stage 1: High-Throughput Screening with a BRET Biosensor
- Biosensor Construction: The BRET sensor was constructed by fusing 14-3-3ζ to Renilla luciferase-8 (Rluc8, the donor) and BAD to mCitrine (the acceptor). Interaction between 14-3-3ζ and BAD brings the donor and acceptor close, enabling energy transfer [100].
- Cell Culture and Transfection: NIH-3T3 fibroblasts were cultured and transfected with the BRET biosensor construct.
- Compound Screening: Cells were treated with compounds from the drug library. A disruptor of the 14-3-3ζ/BAD interaction increases the distance between the donor and acceptor, causing a decrease in the BRET ratio.
- Hit Identification: The Z'-factor for the screening assay was 0.52, indicating a robust and reliable assay for HTS. Compounds causing a statistically significant reduction in the BRET ratio were classified as primary hits (101 identified) [100].
Stage 2: Validation of Pro-Apoptotic Effects in CRC Cells
- Cell Culture: Human colorectal cancer cell lines (HT-29 and Caco-2) were maintained in standard culture conditions.
- Compound Treatment: Cells were treated with top candidate compounds (e.g., Terfenadine, Penfluridol, Lomitapide) identified from the BRET screen and follow-up cytotoxicity assays.
- RNA Extraction: Total RNA was extracted from treated and control cells using a commercial kit, including a DNase digestion step to remove genomic DNA contamination.
- Reverse Transcription: Equal amounts of RNA were reverse transcribed into cDNA using a high-capacity cDNA reverse transcription kit.
- Validated qPCR Analysis:
- Primers/Probes: Validated primer/probe sets for pro-apoptotic genes (e.g., BAX, BAD) and a housekeeping gene (e.g., GAPDH, HPRT1) were used.
- Reaction Setup: Reactions were performed in triplicate using a commercial qPCR master mix on a real-time PCR instrument.
- Quantification: The comparative Cт (ΔΔCт) method was used to calculate relative fold-changes in gene expression, normalized to the housekeeping gene and relative to the untreated control.
The following workflow diagram illustrates the key stages of this case study:
Key Findings and Data Analysis
The integrated screening and validation cascade successfully identified three candidate compounds with potential for repurposing. Terfenadine, penfluridol, and lomitapide were confirmed to disrupt the 14-3-3ζ/BAD PPI, induce cell death in CRC cell lines, and directly bind to 14-3-3ζ as shown by surface plasmon resonance [100]. The validated qPCR assay provided crucial mechanistic evidence by quantifying the upregulation of key pro-apoptotic genes following treatment with these compounds, linking PPI disruption to the activation of the intrinsic apoptotic pathway.
Table 2: Key Data from the CRC Drug Repurposing Screen [100]
| Parameter | Screening Outcome | Downstream Validation |
|---|---|---|
| Screening Assay | BRET-based biosensor in live cells | N/A |
| Assay Quality (Z'-score) | 0.52 (Robust for HTS) | N/A |
| Primary Hits | 101 compounds | N/A |
| Cytotoxicity Hits | 41 compounds | N/A |
| Final Candidates | 13 compounds selected for in-depth study | 3 leads confirmed (Terfenadine, Penfluridol, Lomitapide) |
| qPCR Role | N/A | Quantified induction of pro-apoptotic gene expression in HT-29 and Caco-2 cells. |
| Binding Confirmation | N/A | Direct binding to 14-3-3ζ confirmed by Surface Plasmon Resonance (SPR). |
The Scientist's Toolkit: Research Reagent Solutions
The following table details essential materials and reagents used in the featured CRC case study and high-throughput qPCR workflows.
Table 3: Essential Research Reagents for HTS and qPCR Validation
| Reagent / Material | Function and Importance |
|---|---|
| Validated Primer/Probe Sets | Specifically designed oligonucleotides for the target of interest (e.g., apoptotic genes). Their design and validation for specificity and efficiency are fundamental for assay accuracy [98] [53]. |
| BRET Biosensor Plasmids | Engineered constructs (e.g., 14-3-3ζ-Rluc8 and BAD-mCitrine) that enable the study of protein-protein interactions in a high-throughput, live-cell context [100]. |
| qPCR Master Mix | An optimized buffer containing DNA polymerase, dNTPs, and salts. Probe-based mixes are preferred for specificity in complex biological samples [98]. |
| Nucleic Acid Extraction Kits | For the purification of high-quality, inhibitor-free DNA or RNA from cell lines or tissues, which is critical for robust qPCR performance [98]. |
| Reference DNA Standards | Samples of known concentration used to construct the standard curve for determining the linear dynamic range, amplification efficiency, and for absolute quantification [53]. |
| Cell Line Models | Biologically relevant models (e.g., HT-29, Caco-2 for CRC) used to test compound efficacy and biomarker response in a physiological context [100]. |
The journey from a high-throughput screen to validated mechanistic insights requires a disciplined approach to assay validation. The colorectal cancer case study demonstrates how a validated qPCR assay, embedded within a broader experimental cascade, provides the confirmatory data needed to confidently identify and characterize novel therapeutic candidates. Adherence to community-driven guidelines and a focus on core validation parameters ensure that qPCR data generated in cancer biomarker research are reliable, reproducible, and ultimately, translatable into meaningful advances in oncology drug development.
The Synergy of HT-qPCR and Liquid Biopsy for Minimal Residual Disease (MRD) Monitoring
Minimal Residual Disease (MRD) refers to the persistent presence of a very small number of cancer cells after treatment that remain undetectable by conventional imaging methods, yet pose a high risk for eventual disease recurrence [101]. The detection of MRD represents one of the most promising clinical applications of liquid biopsy, which involves the analysis of tumor-derived components from body fluids such as blood [101]. Liquid biopsy enables the identification of circulating tumor DNA (ctDNA), circulating tumor cells (CTCs), and other biomarkers that carry tumor-specific genetic alterations, providing a minimally invasive window into the molecular status of cancer [96] [102].
High-throughput quantitative PCR (HT-qPCR) has emerged as a powerful technological platform that synergizes with liquid biopsy to address the key challenges in MRD monitoring: the extremely low abundance of tumor-derived material in circulation and the need for highly sensitive, specific, and scalable detection methods [103] [104]. This Application Note details the integrated workflows and protocols that leverage HT-qPCR for sensitive MRD detection, enabling researchers to advance cancer management strategies through precision oncology approaches.
The Rationale for HT-qPCR in MRD Liquid Biopsy
The application of HT-qPCR technologies to liquid biopsy addresses several critical requirements for effective MRD monitoring. Traditional tissue biopsies are ill-suited for repeated assessment of residual disease due to their invasive nature and inability to fully capture tumor heterogeneity [102]. In contrast, liquid biopsy permits serial monitoring through minimally invasive blood collection, providing a more comprehensive molecular profile that reflects contributions from all tumor sites [96] [105].
HT-qPCR platforms offer distinct advantages for MRD detection, including exceptional sensitivity capable of detecting mutant allele frequencies as low as 0.001-0.01% [103] [102], high reproducibility across technical and biological replicates [106] [104], and the capacity to process hundreds of samples in parallel with cost-effectiveness compared to more complex sequencing approaches [106]. The quantitative nature of qPCR provides direct measurement of biomarker levels, enabling dynamic monitoring of treatment response and early detection of recurrence [103].
Table 1: Comparison of MRD Detection Technologies
| Technology | Sensitivity | Throughput | Cost Profile | Key Applications in MRD |
|---|---|---|---|---|
| HT-qPCR | 0.001-0.01% MAF | High | Moderate | Target-specific monitoring, serial assessment |
| ddPCR | 0.01-1.0% MAF | Medium | Low-Moderate | Known mutation tracking, validation |
| NGS-based Approaches | 0.02-0.1% MAF | Variable | High | Comprehensive profiling, novel mutation discovery |
| BEAMing | 0.01% MAF | Medium | Moderate | Known mutation detection, single-molecule analysis |
MAF: Mutant Allele Frequency
Experimental Workflow for HT-qPCR-based MRD Detection
The complete workflow for MRD monitoring integrates sample collection, processing, and HT-qPCR analysis, with stringent quality control at each step to ensure reliable results.
Sample Collection and Processing
Blood Collection Protocol:
- Collect peripheral blood using specialized cell-stabilization tubes (e.g., Streck Cell-Free DNA BCT or PAXgene Blood cDNA tubes) to prevent cellular degradation and preserve nucleic acid integrity [104].
- Process samples within 2-6 hours of collection using a standardized double-centrifugation protocol: initial centrifugation at 1,600×g for 10 minutes at room temperature to separate plasma, followed by transfer of supernatant and a second centrifugation at 16,000×g for 10 minutes to remove residual cellular debris [104].
- Aliquot cleared plasma and store at -80°C until nucleic acid extraction to prevent freeze-thaw degradation.
Cell-Free DNA Extraction:
- Extract cfDNA from 2-10 mL of plasma using automated systems (e.g., Maxwell RSC ccfDNA Plasma Kit, QIAamp Circulating Nucleic Acid Kit, or MagMAX Cell-Free DNA Isolation Kit) to ensure consistent recovery and minimal contamination [101] [102].
- Include synthetic internal control sequences (e.g., cel-miR-39-3p, ath-miR-159a) during extraction to monitor efficiency and potential inhibition [106] [104].
- Quantify cfDNA yield using fluorometric methods (e.g., Qubit dsDNA HS Assay) and assess fragment size distribution (expected peak ~166 bp) via microcapillary electrophoresis (e.g., Agilent 2100 Bioanalyzer) [101].
Assay Design Strategies for MRD Detection
Tumor-Informed Approach:
- For patients with available tumor tissue sequencing data, design patient-specific assays targeting clonal mutations identified through prior whole-exome or targeted sequencing of tumor tissue [107].
- Select 5-16 somatic mutations with high variant allele frequency (VAF >15%) and clonal representation to maximize detection sensitivity [107].
- Design assays across different genomic regions to account for potential heterogeneity and technical variation.
Tumor-Naïve Approach:
- For patients without prior tumor sequencing, utilize predefined panels targeting recurrent mutations in cancer-associated genes relevant to the specific cancer type (e.g., KRAS, NRAS, BRAF for colorectal cancer; EGFR, KRAS, TP53 for non-small cell lung cancer) [107] [108].
- Incorporate epigenetic markers such as cancer-specific DNA methylation patterns that demonstrate high tissue specificity and consistent alteration in malignant cells [101] [102].
qPCR Assay Design Specifications:
- Design amplicons of 60-150 bp to accommodate fragmented cfDNA, with primer melting temperature (Tm) of 60±1°C and GC content between 40-60% [106].
- Position amplicons to avoid known single nucleotide polymorphisms (SNPs) and repetitive regions.
- Incorporate unique molecular identifiers (UMIs) during library preparation to enable correction for PCR amplification biases and accurate quantification of template molecules [103].
HT-qPCR Experimental Protocol
Reaction Setup:
- Utilize 384-well or 1536-well plate formats with automated liquid handling systems to minimize technical variation and enable high-throughput processing [106] [104].
- Prepare master mixes containing hot-start DNA polymerase, SYBR Green or sequence-specific probes (TaqMan), dNTPs, and optimized buffer components [106].
- Include no-template controls (NTCs) and positive controls (synthetic DNA standards with known mutations) in each run to monitor contamination and assay performance.
Amplification Protocol:
- Standard thermal cycling conditions: initial denaturation at 95°C for 10 minutes, followed by 45-50 cycles of denaturation at 95°C for 15 seconds, and combined annealing/extension at 60°C for 60 seconds [106].
- Perform melting curve analysis following amplification when using SYBR Green chemistry to verify amplification specificity and absence of primer-dimer formation [106].
Data Acquisition and Quality Assessment:
- Acquire fluorescence data at the end of each extension phase for quantification analysis.
- Establish baseline and threshold settings consistently across all plates using automated algorithms with manual verification.
- Validate assay performance parameters: amplification efficiency (90-105%), linear dynamic range (5-6 orders of magnitude), and limit of detection (LOD) determined through serial dilution experiments [103] [106].
Table 2: Essential Research Reagent Solutions for HT-qPCR MRD Detection
| Reagent Category | Specific Examples | Function | Quality Control Parameters |
|---|---|---|---|
| Nucleic Acid Extraction Kits | QIAamp Circulating Nucleic Acid Kit, Maxwell RSC ccfDNA Plasma Kit | Isolation of high-quality cfDNA from plasma | Yield ≥5 ng/mL plasma, A260/A280: 1.8-2.0 |
| Reverse Transcription Kits | TaqMan MicroRNA Reverse Transcription Kit, SuperScript III/IV | cDNA synthesis from RNA templates | No-RT controls, spike-in controls |
| qPCR Master Mixes | Power SYBR Green Master Mix, TaqMan PreAmp Master Mix | Amplification with fluorescence detection | Efficiency: 90-105%, R² >0.98 in standard curves |
| Reference Assays | GAPDH, TFRC, ACTB, B2M | Normalization for sample input variation | Stable expression across sample sets |
| Control Templates | Synthetic wild-type and mutant DNA sequences | Assay validation and quantification | Linear response in dilution series |
| Unique Molecular Identifiers | Custom UMI adapter systems | Correction for PCR amplification bias | Minimum of 8 random nucleotides |
Data Analysis and Interpretation
Quantification and Normalization
Efficiency-Corrected Quantification:
- Apply efficiency-corrected ΔΔCq calculation methods that incorporate reaction-specific efficiency values rather than assuming 100% efficiency [103].
- Utilize algorithms such as LinRegPCR to calculate PCR efficiency from the amplification curve of each individual reaction based on the exponential phase of amplification [103] [106].
- For relative quantification, normalize target Cq values to a combination of reference genes (e.g., GAPDH, TFRC) that demonstrate stable expression across all sample types [103] [106].
Absolute Quantification Approach:
- For absolute quantification, generate standard curves using synthetic DNA templates containing the target mutations in known concentrations, spanning 5-6 orders of magnitude [103].
- Calculate mutant allele frequency (MAF) as the ratio of mutant to wild-type alleles, with typical MRD-positive thresholds ranging from 0.01% to 0.1% MAF depending on assay sensitivity and background levels [107].
Statistical Analysis and MRD Calling
Threshold Determination:
- Establish sample-specific background signals using pre-treatment samples when available, or population-based background levels from healthy controls.
- Define positive detection thresholds based on statistical significance above background (typically 3-5 standard deviations above mean background signal) [103].
- Implement replicate-based calling requiring detection in multiple technical replicates (e.g., 2/3 or 3/4 replicates) to enhance specificity.
Longitudinal Analysis:
- For serial monitoring, analyze trends in ctDNA levels rather than single timepoint measurements, with rising levels indicating molecular progression often preceding radiographic evidence of recurrence [101] [107].
- Utilize joint statistical models that incorporate both longitudinal ctDNA trends and time-to-recurrence data to enhance prognostic accuracy [103].
Technical Validation and Quality Assurance
Assay Performance Validation:
- Determine limit of detection (LOD) and limit of quantification (LOQ) through serial dilution experiments using mutant DNA in wild-type background, with target LOD of at least 0.01% MAF for MRD applications [103] [107].
- Assess intra-assay and inter-assay precision through replicate measurements, with coefficient of variation (CV) <15% for Cq values and <25% for quantitative measurements [104].
- Establish analytical specificity by testing against common polymorphisms and related genetic sequences to ensure minimal cross-reactivity.
Sample Quality Metrics:
- Implement quality thresholds for sample inclusion: minimum cfDNA yield (≥5 ng/mL plasma), absorbance ratios (A260/A280 ≥1.8, A260/A230 ≥2.0), and appropriate fragment size distribution [106].
- Monitor extraction efficiency through spike-in controls, with acceptable recovery rates typically >50% [104].
- Verify absence of PCR inhibitors through dilution tests or internal amplification controls.
Applications in Cancer Management and Research
The integration of HT-qPCR with liquid biopsy for MRD monitoring enables multiple advanced applications in clinical research and therapeutic development:
Recurrence Risk Stratification:
- Post-treatment MRD status powerfully stratifies patients into distinct recurrence risk categories [101] [107]. In colorectal cancer, GUCY2C mRNA expression quantification in blood samples via qRT-PCR has demonstrated prognostic value for predicting recurrence risk [103]. Similarly, in non-small cell lung cancer, ctDNA-based MRD detection post-resection identifies patients at highest risk of relapse who might benefit from treatment intensification [107].
Therapeutic Response Monitoring:
- Serial MRD assessment during adjuvant therapy provides early indication of treatment effectiveness, potentially enabling adaptive therapy modifications before clinical or radiographic progression becomes evident [101] [105]. The dynamics of ctDNA clearance during treatment have been shown to correlate with pathological response and long-term outcomes across multiple cancer types [105] [108].
Clinical Trial Applications:
- MRD status serves as a robust enrichment biomarker for clinical trial recruitment, enabling selection of high-risk patients who are most likely to benefit from investigational adjuvant therapies [107] [105].
- ctDNA-based endpoints can accelerate drug development by providing early indicators of drug activity and potentially serving as surrogate endpoints for recurrence-free survival [105].
Troubleshooting and Technical Considerations
Addressing Common Challenges:
- For low cfDNA yield, increase input plasma volume, verify blood collection tube integrity, and minimize processing delays.
- For high background signal, optimize primer specificity, increase annealing temperature, implement UMI-based error correction, and verify reagent quality.
- For poor assay efficiency, redesign suboptimal primers/probes, adjust magnesium concentration, and verify template quality.
Technical Limitations and Mitigation Strategies:
- The stochastic sampling limitation at very low ctDNA concentrations can be addressed by increasing plasma input volume and implementing technical replicates.
- Clonal hematopoiesis of indeterminate potential (CHIP) can generate false positive signals; mitigation strategies include paired white blood cell analysis, bioinformatic filtering of known CHIP mutations, and prioritization of cancer-specific mutation patterns [107] [105].
- Tumor heterogeneity may lead to missed mutations; this can be addressed by targeting multiple genomic regions and incorporating epigenetic markers that are conserved across subclones [101].
The synergy between HT-qPCR and liquid biopsy provides a powerful methodological platform for sensitive, specific, and scalable MRD monitoring in cancer patients. The protocols detailed in this Application Note enable researchers to implement robust MRD detection assays that can inform therapeutic decisions and advance our understanding of cancer dynamics.
Future developments in this field will likely include the integration of multidimensional biomarkers (including methylation patterns, fragmentomics, and protein markers) [101] [102], enhanced computational methods for more accurate quantification at ultra-low allele frequencies [103], and standardized reporting frameworks to facilitate cross-study comparisons and clinical translation. As these technologies continue to evolve, MRD monitoring using HT-qPCR and liquid biopsy is poised to become an integral component of precision oncology, enabling earlier intervention and more personalized cancer management strategies.
Standardization and Quality Control for Clinical Translation
The translation of high-throughput quantitative PCR (qPCR) from a research tool into a clinically validated methodology for cancer biomarker screening demands rigorous standardization and quality control. This process is critical for ensuring that laboratory results are accurate, reproducible, and meaningful for clinical decision-making, such as guiding targeted therapies or monitoring minimal residual disease (MRD) [109]. The inherent complexity of cancer samples, including the low abundance of circulating tumor DNA (ctDNA) against a background of wild-type DNA, amplifies these challenges [110]. This application note outlines a standardized framework and detailed protocols to support the robust clinical translation of qPCR assays in oncology.
Pre-Analytical Quality Control
The pre-analytical phase is a critical determinant of assay success, encompassing all procedures from sample collection to nucleic acid extraction.
Sample Collection and Handling
Standardized collection protocols are essential to prevent analyte degradation and ensure sample integrity. Key parameters are summarized in Table 1.
Table 1: Pre-Analytical Sample Handling Standards for Blood-Based Liquid Biopsies
| Parameter | Standardized Protocol | Quality Control Metric |
|---|---|---|
| Blood Collection | Use of specific cell-stabilizing blood collection tubes. | Record tube type and lot number. |
| Cold Ischemic Time | Minimize time from sample draw to processing; ideally <2 hours [111]. | Document time of draw and processing. |
| Plasma Separation | Two-step centrifugation (e.g., 1600 × g for 10 min, then 16,000 × g for 10 min at 4°C). | Assess plasma for hemolysis (visually or by spectrophotometry). |
| Storage | Plasma stored at -80°C in low-binding tubes. | Avoid multiple freeze-thaw cycles. |
Nucleic Acid Extraction and Quantification
Consistent extraction methods are vital for yield and purity. The College of American Pathologists (CAP) has emphasized the importance of documenting pre-analytical variables, including fixative and fixation time for tissue samples, underscoring the need for detailed record-keeping [111].
- Extraction: Use of automated, bead-based extraction systems is recommended for plasma cell-free DNA (cfDNA) to improve reproducibility and yield.
- Quantification: Utilize fluorometric methods (e.g., Qubit) over spectrophotometry for accurate quantification of cfDNA, as the latter is less sensitive for low-concentration samples and cannot distinguish between DNA and RNA.
Analytical Phase: qPCR Assay Validation
For a qPCR assay to be clinically applicable, it must undergo a formal validation process to establish its performance characteristics. The following protocol and metrics are adapted from rigorous methodologies, such as the TEAM-PCR approach used for detecting the EGFR T790M mutation in non-small cell lung cancer (NSCLC) [110].
Experimental Protocol: Validation of an Ultrasensitive qPCR Assay
Objective: To establish the analytical sensitivity, specificity, and precision of a qPCR assay for a specific cancer point mutation (e.g., EGFR T790M) in cfDNA.
Materials:
- Research Reagent Solutions: Key materials and their functions are listed in Table 2.
- Equipment: qPCR instrument, spectrophotometer/fluorometer, microcentrifuge.
Table 2: Research Reagent Solutions for qPCR Biomarker Assay Validation
| Reagent/Material | Function | Example/Note |
|---|---|---|
| Plasma-derived cfDNA | The target analyte containing potential tumor-derived DNA. | Extract from patient plasma using a validated kit. |
| Mutation-specific TaqMan Assay | Selective amplification and detection of the target mutation. | Includes primers and a FAM-labeled probe. |
| Wild-type Genomic DNA | Serves as a negative control and background for LOD studies. | Used to assess assay specificity. |
| Synthetic Mutation Control | A quantifiable positive control for the target mutation. | Used for creating standard curves and determining LOD. |
| qPCR Master Mix | Provides enzymes, dNTPs, and buffer for efficient amplification. | Use a robust, commercially available mix. |
| Reference Gene Assay | Control for DNA input quantity and PCR inhibition. | e.g., assay for a stable genomic region; VIC-labeled. |
Procedure:
- Assay Design: Design primers and probes to specifically discriminate the single-nucleotide variant. In silico specificity analysis is recommended.
- Standard Curve and Dynamic Range: Serially dilute the synthetic mutation control in a background of wild-type DNA (e.g., 10 ng/µL) to simulate the clinical scenario. A validated assay should demonstrate a linear quantitative range over several orders of magnitude (e.g., from 25 to 10^6 copies/reaction) [110].
- Limit of Detection (LOD) Determination: Perform replicate measurements (n≥20) of a sample with mutation copies near the expected LOD. The LOD is defined as the lowest concentration at which ≥95% of replicates are positive. The TEAM-PCR method achieved an LOD of 5 copies/reaction [110].
- Specificity Testing: Test the assay against a panel of samples known to be wild-type for the target mutation to confirm the absence of false-positive signals.
- Precision Assessment: Perform intra-assay (within-run) and inter-assay (between-run, between-day, between-operator) replicates at multiple concentrations (low, medium, high) to determine the coefficient of variation (CV).
The following workflow diagram illustrates the key stages of this validation process:
Figure 1: qPCR Assay Validation Workflow
Data Analysis and Interpretation
- Quantification: Use the standard curve to interpolate the copy number of the target mutation in unknown samples.
- Quality Control: Include positive controls (synthetic mutation), negative controls (wild-type DNA), and no-template controls (NTC) in every run. The reference gene (e.g., for input DNA quantification) must amplify consistently across samples.
- Result Reporting: For MRD monitoring, report the mutant allele frequency (MAF) as a percentage relative to the wild-type allele [109]. The CAP recommends clear reporting formats, including the removal of ambiguous terms like "(indeterminate)" in favor of definitive "Cannot be determined" answers when necessary [111].
Post-Analytical Phase: Data Management and Standardization
The high-throughput nature of qPCR biomarker screening generates vast amounts of data that require structured management.
- Metadata Registration: Adhere to standards like ISO/IEC 11179 for metadata registries to promote semantic interoperability and data reuse [112]. Document all data elements, including sample attributes, experimental conditions, and analysis parameters.
- Data Integration with Clinical Outcomes: In translational research, qPCR data (e.g., mutation levels) must be integrated with patient clinical data to validate the utility of the biomarker. This aligns with the "big data" approaches in cancer research, which leverage large-scale molecular and clinical datasets to advance discovery [113].
The clinical translation of high-throughput qPCR for cancer biomarkers is an iterative process that hinges on uncompromising standardization and quality control at every stage. By implementing the detailed protocols and validation frameworks outlined here—from pre-analytical sample handling to rigorous analytical validation and standardized data reporting—researchers can enhance the reliability, reproducibility, and clinical utility of their findings, thereby accelerating the development of non-invasive diagnostic and monitoring tools in oncology.
Conclusion
High-throughput qPCR stands as a powerful, accessible, and cost-effective pillar in the precision oncology toolkit, enabling the large-scale validation of cancer biomarkers essential for early detection and personalized treatment. Its strengths in throughput, sensitivity, and quantitative accuracy make it indispensable for profiling circulating nucleic acids and FFPE-derived samples. Future directions will be shaped by deeper integration with artificial intelligence for data interpretation, increased automation, and its evolving role alongside NGS in comprehensive multi-omics strategies. For researchers, overcoming challenges in standardization and miniaturization will be key to fully unlocking its potential in translating biomarker discoveries into routine clinical practice, ultimately improving patient outcomes in the global fight against cancer.
References
- 1. Global cancer burden growing, amidst mounting need for ... [https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services]
- 2. qPCR in Early Cancer Detection: Why It Still Leads the Pack [https://www.meridianbioscience.com/lifescience-blog/why-qpcr-still-leads-in-oncology/]
- 3. Advanced breast cancer diagnosis: Multiplex RT-qPCR for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10776909/]
- 4. Cancer biomarker discovery and validation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4511498/]
- 5. A high-throughput test enables specific detection of ... [https://www.nature.com/articles/s41467-023-39055-7]
- 6. Cancer Biomarkers - Emerging Trends and Clinical Implications for personalized treatment - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7616034/]
- 7. Bridging Discovery and Treatment: Cancer Biomarker [https://www.mdpi.com/2072-6694/17/22/3720]
- 8. Integrating AI and RNA biomarkers in cancer: advances in ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-025-02434-2]
- 9. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7283209/]
- 10. Blood-derived lncRNAs as biomarkers for cancer diagnosis [https://www.nature.com/articles/s41698-022-00283-7]
- 11. A high-throughput screening RT-qPCR assay for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9469018/]
- 12. Cancer Biomarkers: Reflection on Recent Progress, Emerging Innovations, and the Clinical Horizon - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468363/]
- 13. Size & Share Report, Molecular – 2034 Diagnostics Market 2025 [https://www.gminsights.com/industry-analysis/molecular-diagnostics-market-report]
- 14. Size to Hit USD 63.86 Billion By 2034 Molecular Diagnostics Market [https://www.precedenceresearch.com/molecular-diagnostics-market]
- 15. Industry Report Molecular -2035: Global... Diagnostics Market 2025 [https://www.businesswire.com/news/home/20250930129015/en/Molecular-Diagnostics-Market-Industry-Report-2025-2035-Global-Molecular-Diagnostics-Market-Set-to-Grow-to-USD-30.9-Billion-by-2035---ResearchAndMarkets.com]
- 16. Gene expression quantification for cancer biomarkers [https://www.takarabio.com/applications/cancer-research/cancer-biomarker-quantification?srsltid=AfmBOoqpm781e2yXvB6JjkyOu1xHub-ZCEyyazpME-d4UJerokv_QepX]
- 17. Introduction to Quantitative PCR (qPCR) Gene Expression ... [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/gene-expression-analysis-real-time-pcr-information/introduction-gene-expression.html]
- 18. A high-throughput screening RT-qPCR assay for ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.962220/full]
- 19. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOopGBnJYjqoOAjlTGL-K6ni_go_wODhcQ270w6eZsJ7xgw9jZlRA]
- 20. How To Interpret RT-qPCR Results [https://www.goldbio.com/blogs/articles/how-to-interpret-rt-qpcr-results?srsltid=AfmBOoqO6swH26ogLIenspMXiYaAWIrhm14PPXpb5ytYoDAnFpsOMtfm]
- 21. High-Throughput Screening for Biomarker Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4427806/]
- 22. Emerging biomarkers for early cancer detection and diagnosis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12359849/]
- 23. a roadmap for DNA methylation biomarkers in liquid biopsies [https://www.nature.com/articles/s41388-025-03624-5]
- 24. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOoqLpf8HBnY8ECMKYei8KkFLOxDiIpPnf1N2V0A4Re4AszkU_VYD]
- 25. pcr: an R package for quality assessment, analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5858653/]
- 26. qPCR in Early Cancer Detection: Why It Still Leads the Pack [https://www.meridianbioscience.com/cn/lifescience-blog/why-qpcr-still-leads-in-oncology/]
- 27. HTqPCR: high-throughput analysis and visualization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2788924/]
- 28. Microfluidics-enabled 96-well perfusion system for high- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7680692/]
- 29. Microfluidics-enabled 96-well perfusion system for high- ... [https://pubs.rsc.org/en/content/articlelanding/2020/lc/d0lc00615g]
- 30. Buying a qPCR Machine? Here's What You Should Know [https://biosistemika.com/blog/buying-new-qpcr-machine-2/]
- 31. High-Throughput Screening Assay Miniaturization [https://focus.gbo.com/blog/hts/the-power-of-small-high-throughput-screening-assay-miniaturization]
- 32. Cost saving with miniaturization and mosquito HV lease ... [https://www.sptlabtech.com/cost-saving-with-miniaturization-and-mosquito-hv-lease-scheme]
- 33. How to optimize qPCR in 9 steps? [https://westburg.eu/knowledge/pcr_qpcr/hate_optimizing_qpcr]
- 34. A Multiplex SNaPshot Assay as a Rapid Method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3157611/]
- 35. Optimized Multiplex Detection of 7 KRAS Mutations by ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0163070]
- 36. A rapid, multiplex digital PCR assay to detect gene variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10620117/]
- 37. Development and Optimization of a Multiplex Real-Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10948217/]
- 38. Systematic comparison of quantity and quality of RNA ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05890-5]
- 39. Extraction of Nucleic Acids from FFPE Samples [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rna-isolation/general-articles/extraction-of-nucleic-acids-from-ffpe-samples.html]
- 40. Liquid biopsy in cancer: current status, challenges and ... [https://www.nature.com/articles/s41392-024-02021-w]
- 41. Establishment of liquid biopsy procedure for the analysis ... [https://www.nature.com/articles/s41598-024-78497-x]
- 42. Comparative analysis of library preparation approaches for ... [https://www.nature.com/articles/s41598-025-12992-7]
- 43. Circular RNA-based liquid biopsy: a promising approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12635984/]
- 44. Enhancing Biomarker Detection in Cancer: A Comparative ... [https://pubmed.ncbi.nlm.nih.gov/39307310/]
- 45. High-Throughput Data Analysis for qPCR [https://www.neb.com/en-us/tools-and-resources/feature-articles/development-of-a-high-throughput-data-analysis-method-for-quantitative-real-time-pcr?srsltid=AfmBOoqIYq0FFYlTfl9JAMr74JKBpY_FGDm0re0Hufta8y5XINOe0ePt]
- 46. Biomarker Discovery and Validation: Statistical Considerations [https://pmc.ncbi.nlm.nih.gov/articles/PMC8012218/]
- 47. Statistical analysis of real-time PCR data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1395339/]
- 48. Evaluation of normalisation strategies for qPCR data ... [https://www.nature.com/articles/s41598-025-06657-8]
- 49. Data-driven normalization strategies for high-throughput ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2680405/]
- 50. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOoqi-S9T5ZO-UP5EgfIzUvYQUQSTapw2lHuShSdHewmbzrzvbeTR]
- 51. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 52. Quantitative PCR (qPCR) Data Analysis [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/gene-expression-analysis-real-time-pcr-information/precision-qpcr.html]
- 53. Quantitative PCR validation for research scientists: Part 1 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 54. A Detailed Guide to Quantitative RT-PCR (qRT-PCR) Protocols [https://www.clyte.tech/post/a-detailed-guide-to-quantitative-rt-pcr-qrt-pcr-protocols?srsltid=AfmBOoralpPY4sNXoujuzbfgWPgPLZuXzL3bd5cS2pcqlCbQWB8YUeIf]
- 55. Digital PCR: from early developments to its future application ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278255/]
- 56. Comparing the precision of two digital PCR applications for ... [https://www.nature.com/articles/s41598-025-13143-8]
- 57. MIQE Guidelines | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/why-choose-taqman-assays/publish-real-time-pcr-results.html]
- 58. Quantifying the Detection Sensitivity and Precision of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11634988/]
- 59. Emerging biomarkers for early cancer detection and diagnosis [https://eurjmedres.biomedcentral.com/articles/10.1186/s40001-025-03003-6]
- 60. Cancer in a Drop: Liquid biopsy insights from AACR 2025 [https://pmc.ncbi.nlm.nih.gov/articles/PMC12223554/]
- 61. Top 11 qPCR Papers Every Researcher Should Know [https://bitesizebio.com/62143/qpcr-papers/]
- 62. MIQE [https://en.wikipedia.org/wiki/MIQE]
- 63. Analyzing qPCR data: Better practices to facilitate rigor and ... [https://www.sciencedirect.com/science/article/pii/S2405580825004431]
- 64. Evaluation of PCR-enhancing approaches to reduce ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969724069250]
- 65. Systematic pan-cancer analysis of tumour purity [https://www.nature.com/articles/ncomms9971]
- 66. Live culture-based qPCR screening of Taq DNA ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1624735/full]
- 67. Putative biomarkers for predicting tumor sample purity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6933652/]
- 68. Use and Misuse of Cq in qPCR Data Analysis and Reporting [https://pmc.ncbi.nlm.nih.gov/articles/PMC8229287/]
- 69. High throughput, nanoliter quantitative PCR [https://www.sciencedirect.com/science/article/abs/pii/S1740674905000600]
- 70. Automation and miniaturization of high-throughput qPCR ... [https://www.sciencedirect.com/science/article/pii/S2472630324001237]
- 71. Miniaturization and optimization of 384-well compatible RNA ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0206194]
- 72. High-Throughput Data Analysis for qPCR [https://www.neb.com/en-us/tools-and-resources/feature-articles/development-of-a-high-throughput-data-analysis-method-for-quantitative-real-time-pcr?srsltid=AfmBOooCtAdLdckzKRCJYAkG7dbavOaxrLkX3ZlkR43UGjrB3-kQ0-cz]
- 73. Troubleshooting Non-Specific Amplification [https://bento.bio/resources/bento-lab-advice/troubleshooting-non-specific-amplification-with-bento-lab/]
- 74. How does HotStart PCR help minimize nonspecific ... [https://www.qiagen.com/us/resources/faq/2676]
- 75. Why hot start is so hot? [https://solisbiodyne.com/EN/why-hot-start-is-so-hot/]
- 76. How is Hot-Start Technology Beneficial For Your PCR [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/hot-start-technology-benefits-PCR.html]
- 77. An optimized protocol for stepwise optimization of real-time ... [https://www.nature.com/articles/s41438-021-00616-w]
- 78. Optimization of internal reference genes for qPCR in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8797320/]
- 79. Diagnostic value of genetic and epigenetic biomarker panels ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12098509/]
- 80. Optimization and Validation of a Folate Ligand-Targeted ... [https://pubmed.ncbi.nlm.nih.gov/40729092/]
- 81. A Cross-Comparison of High-Throughput Platforms for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9343840/]
- 82. Comparison of real-time PCR and nCounter NanoString ... [https://www.nature.com/articles/s41598-025-08036-9]
- 83. Optimization of the real-time PCR platform method using ... [https://www.nature.com/articles/s41598-025-16972-9]
- 84. The new gold rush in clinical diagnostics - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12584703/]
- 85. LightCycler® 480 System Technology [https://lifescience.roche.com/global/en/article-listing/article/lightcycler-480-system-technology.html]
- 86. LightCycler® 480 Detection Formats [https://lifescience.roche.com/global/en/article-listing/article/lightcycler-480-detection-formats.html]
- 87. QuantStudio 12K Flex Real-Time PCR System [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-instruments/quantstudio-systems/models/quantstudio-12-flex.html]
- 88. A Comparative Assessment of High-Throughput ... [https://www.mdpi.com/2076-3417/13/20/11229]
- 89. Benefits of NGS in Oncology [https://www.illumina.com/areas-of-interest/cancer/ngs-in-oncology.html]
- 90. Hybridization capture vs amplicon sequencing | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/targeted-sequencing-hybridization-capture-or-amplicon-sequencing/]
- 91. Hybridization Capture vs. PCR Amplification [https://www.cd-genomics.com/microbioseq/hybridization-capture-vs-pcr-amplification.html]
- 92. Quantitative comparison of HT-qPCR/16S rRNA ... [https://pubmed.ncbi.nlm.nih.gov/40972162/]
- 93. Comparison of Next-Generation Sequencing, Real-Time ... [https://www.mdpi.com/2076-2607/13/10/2344]
- 94. Evaluating sensitivity of NGS-based mutation detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12486888/]
- 95. Accurate quantification of cell-free Ceruloplasmin mRNA ... [https://www.nature.com/articles/s41598-025-99302-3]
- 96. Liquid biopsy for human cancer: cancer screening, monitoring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11130638/]
- 97. Assessing the Cost-Effectiveness of Next-Generation ... [https://www.sciencedirect.com/science/article/pii/S109830152402357X]
- 98. Recommendations for Method Development and ... [https://link.springer.com/article/10.1208/s12248-023-00880-9]
- 99. Colorectal cancer molecular profiling [https://pmc.ncbi.nlm.nih.gov/articles/PMC12477549/]
- 100. repurposing drugs to identify novel disruptors of 14-3 ... [https://www.nature.com/articles/s41419-025-08150-6]
- 101. Liquid Biopsy to Detect Minimal Residual Disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC8582541/]
- 102. Liquid biopsy: current technology and clinical applications [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-022-01351-y]
- 103. Modeling qRT-PCR dynamics with application to cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5756704/]
- 104. A Cross-Comparison of High-Throughput Platforms for ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.911613/full]
- 105. Clinical applications of cell-free DNA-based liquid biopsy ... [https://www.sciencedirect.com/science/article/pii/S1936523325002505]
- 106. Eleven Golden Rules of Quantitative RT-PCR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2518243/]
- 107. A comprehensive overview of minimal residual disease in ... [https://www.nature.com/articles/s41698-025-00984-9]
- 108. Liquid biopsy for monitoring minimal residual disease in ... [https://www.sciencedirect.com/science/article/pii/S2773160X24000242]
- 109. Cancer Research to Microbes: Harnessing the Power of ... [https://www.thermofisher.com/blog/behindthebench/harnessing-the-power-of-qpcr/]
- 110. Development and validation of an ultrasensitive qPCR ... [https://pubmed.ncbi.nlm.nih.gov/39812332/]
- 111. CAP Releases Q3 Cancer Protocol Updates with Expanded ... [https://newsroom.cap.org/latest-news/cap-releases-q3-cancer-protocol-updates-with-expanded-biomarker-reporting/s/0e8ba15e-9fad-4947-8e17-36489f973aab]
- 112. The ISO/IEC 11179 norm for metadata registries [https://www.sciencedirect.com/science/article/pii/S1532046412001827]
- 113. Big data in basic and translational cancer research - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9443637/]