Analytical Validation of qPCR Assays in Clinical Cancer Diagnostics: A Guide from Foundations to GxP Compliance
This article provides a comprehensive framework for the analytical validation of quantitative PCR (qPCR) assays in clinical cancer diagnostics.
Analytical Validation of qPCR Assays in Clinical Cancer Diagnostics: A Guide from Foundations to GxP Compliance
Abstract
This article provides a comprehensive framework for the analytical validation of quantitative PCR (qPCR) assays in clinical cancer diagnostics. Tailored for researchers, scientists, and drug development professionals, it covers the foundational principles of qPCR in oncology, detailed methodological setup for applications like liquid biopsies and minimal residual disease (MRD) monitoring, advanced troubleshooting and optimization techniques, and rigorous validation protocols for regulated environments. By synthesizing current best practices and comparing qPCR with emerging technologies like digital PCR, this guide aims to support the development of robust, reliable, and clinically actionable molecular assays that are essential for personalized cancer therapy.
The Unwavering Role of qPCR in Modern Clinical Oncology
Why qPCR Remains a Cornerstone for Targeted Cancer Mutation Detection
In the rapidly evolving landscape of molecular diagnostics, quantitative polymerase chain reaction (qPCR) maintains a fundamental position in targeted cancer mutation detection despite the emergence of sophisticated technologies like next-generation sequencing (NGS). Its persistence stems from a powerful combination of analytical sensitivity, operational efficiency, and economic practicality that remains unmatched for specific clinical applications. qPCR enables the detection of clinically actionable biomarkers at low concentrations while supporting molecular stratification for personalized therapy [1]. This technical guide objectively examines the performance characteristics of qPCR relative to alternative technologies, providing researchers and drug development professionals with experimental data and validation frameworks essential for implementing robust qPCR assays in cancer diagnostics.
The transformation of cancer diagnostics from late-stage diagnosis toward earlier detection and personalized treatment has intensified the need for technologies that can detect actionable biomarkers at low concentrations in limited sample material [1]. Within this context, qPCR continues to offer a compelling value proposition for targeted mutation detection, particularly in time-sensitive clinical scenarios and resource-constrained settings. The technology's robust performance characteristics, combined with ongoing innovations in chemistry and workflow integration, ensure its continued relevance in precision oncology.
Performance Comparison of Mutation Detection Technologies
The selection of an appropriate molecular detection platform requires careful consideration of analytical capabilities, operational parameters, and practical constraints. The table below provides a systematic comparison of qPCR against next-generation sequencing and digital PCR for key performance metrics relevant to cancer mutation detection.
Table 1: Comparative Analysis of Mutation Detection Technologies in Cancer Diagnostics
| Performance Parameter | qPCR | Next-Generation Sequencing (NGS) | Digital PCR (dPCR) |
|---|---|---|---|
| Analytical Sensitivity (VAF) | <0.1%–1% [1] | 1%–5% (standard); <1% (liquid biopsy) [2] | 0.08%–0.1% [3] [4] |
| Multiplexing Capability | Moderate (up to 11 genes) [5] | High (hundreds of genes) [6] | Limited (typically 3–5 targets) [4] |
| Turnaround Time | Hours (∼2–4 hours) [1] | Days (∼1–7 days) [3] [7] | Hours (∼3–6 hours) [4] |
| Cost Per Sample | $50–$200 [1] | $300–$3,000 [1] | $100–$300 (estimated) |
| Throughput | High (96/384-well formats) [1] | Variable (low to very high) [6] | Medium (limited by partitioning) [4] |
| Discovery Power | Limited to known variants [6] | High (hypothesis-free) [6] | Limited to known variants |
| Sample Input Requirements | Low (FFPE, cfDNA, fine needle aspirates) [1] | Moderate to high (depends on panel size) [5] | Low (comparable to qPCR) [4] |
| Quantification | Relative (Ct values) | Absolute (read counts) [2] | Absolute (molecular counting) [4] |
| Implementation Complexity | Low (standard equipment) [1] | High (specialized infrastructure) [7] | Medium (specialized equipment) |
qPCR demonstrates particular strength in clinical scenarios requiring rapid turnaround for a limited number of predefined targets, such as testing for EGFR mutations in non-small cell lung cancer or KRAS mutations in colorectal cancer [1] [3]. The technology provides a practical balance between sensitivity, speed, and cost, making it suitable for both centralized reference laboratories and hospital-based molecular pathology facilities [1] [7].
Key Experimental Applications in Cancer Research
Multiplexed qPCR for Comprehensive Lung Cancer Genotyping
Advanced qPCR systems now enable simultaneous detection of multiple clinically relevant mutations in a single reaction, maximizing data yield from minimal input material—a critical advantage in tissue-limited cases.
Experimental Protocol:
- Sample Preparation: DNA is extracted from various micro cell samples (MCSs) including puncture needle rinse samples and bronchoalveolar lavage fluid using the QIAamp DNA Micro Kit [5].
- qPCR Setup: Reactions are performed using the Lung Cancer 11-Gene Mutations Detection Kit on an Mx3000P PCR instrument [5].
- Thermal Cycling: Protocol-specific cycling conditions are applied with fluorescence detection at each cycle.
- Data Analysis: Threshold cycle (Ct) values are determined for each target, with positive calls made based on established cutoff values [5].
Performance Data: In a 2025 study analyzing 184 patients with suspected lung cancer, qPCR successfully detected mutations in 11 genes (including EGFR, KRAS, BRAF, ALK, ROS1) with 100% concordance between MCSs and paired tissue samples [5]. The methodology achieved a complete diagnostic turnaround time of only 24 hours, significantly shorter than the 5-7 days typically required for standard diagnostic pathways [5].
High-Sensitivity KRAS Mutation Detection Using LBDA Technology
The long blocker displacement amplification (LBDA) method represents an advanced qPCR-based approach for detecting low-frequency mutations with enhanced sensitivity.
Experimental Protocol:
- Primer/Blocker Design: A wild-type-specific nucleic acid blocker is designed to bind WT templates with higher affinity, suppressing their amplification while allowing mutant templates to be amplified [3].
- Reaction Setup: SYBR Green dye is incorporated to intercalate into accumulating double-stranded DNA products, generating a fluorescence signal proportional to mutant template abundance [3].
- Amplification Conditions: 50 cycles of: 98°C for 10s, 70°C for 30s, 72°C for 3min [3].
- Analysis: Fluorescence curves are analyzed to identify samples with significant amplification above background [3].
Performance Data: In colorectal cancer samples, this LBDA approach achieved a detection limit of 0.08% variant allele frequency with only 20 ng of synthetic DNA input [3]. When applied to 59 CRC tumor samples, the method identified KRAS mutations in 37.29% of cases, demonstrating 88% sensitivity and 100% specificity compared to NGS [3].
Table 2: Experimental Performance of qPCR-Based Methods in Cancer Detection
| Cancer Type | qPCR Method | Genes Detected | Sensitivity | Specificity | Reference |
|---|---|---|---|---|---|
| Lung Cancer | Multiplex qPCR (11-gene panel) | EGFR, KRAS, BRAF, ALK, ROS1, RET, MET, NTRK1-3, HER2 | 100% (vs. tissue) | 100% (vs. tissue) | [5] |
| Colorectal Cancer | LBDA qPCR | KRAS hotspots | 88% (vs. NGS) | 100% (vs. NGS) | [3] |
| Non-Small Cell Lung Cancer | Standard qPCR (cobas EGFR test) | EGFR exons 18-21 | 76.14% concordance with NGS | 76.14% concordance with NGS | [2] |
Analytical Validation Framework for qPCR Assays
Rigorous validation is essential for implementing reliable qPCR assays in clinical cancer research. The Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines provide a foundational framework for ensuring assay quality and reproducibility [8] [9].
Key Validation Parameters
- Inclusivity: Determines how well the qPCR detects all intended target variants. Validation should include up to 50 well-defined strains or variants reflecting the genetic diversity of the target [8].
- Exclusivity/Cross-reactivity: Assesses the assay's ability to exclude genetically similar non-targets. Both in silico analysis and experimental testing with near-neighbor species are required [8].
- Linear Dynamic Range: Established using a seven 10-fold dilution series of DNA standards analyzed in triplicate. The acceptable linearity (R²) value is typically ≥0.980, with primer efficiency between 90-110% [8].
- Limit of Detection (LOD): The lowest concentration at which the target can be reliably detected. Determined through serial dilution studies in relevant biological matrices [8] [9].
- Precision and Accuracy: Evaluated through replicate testing of samples across multiple runs, operators, and instruments [9].
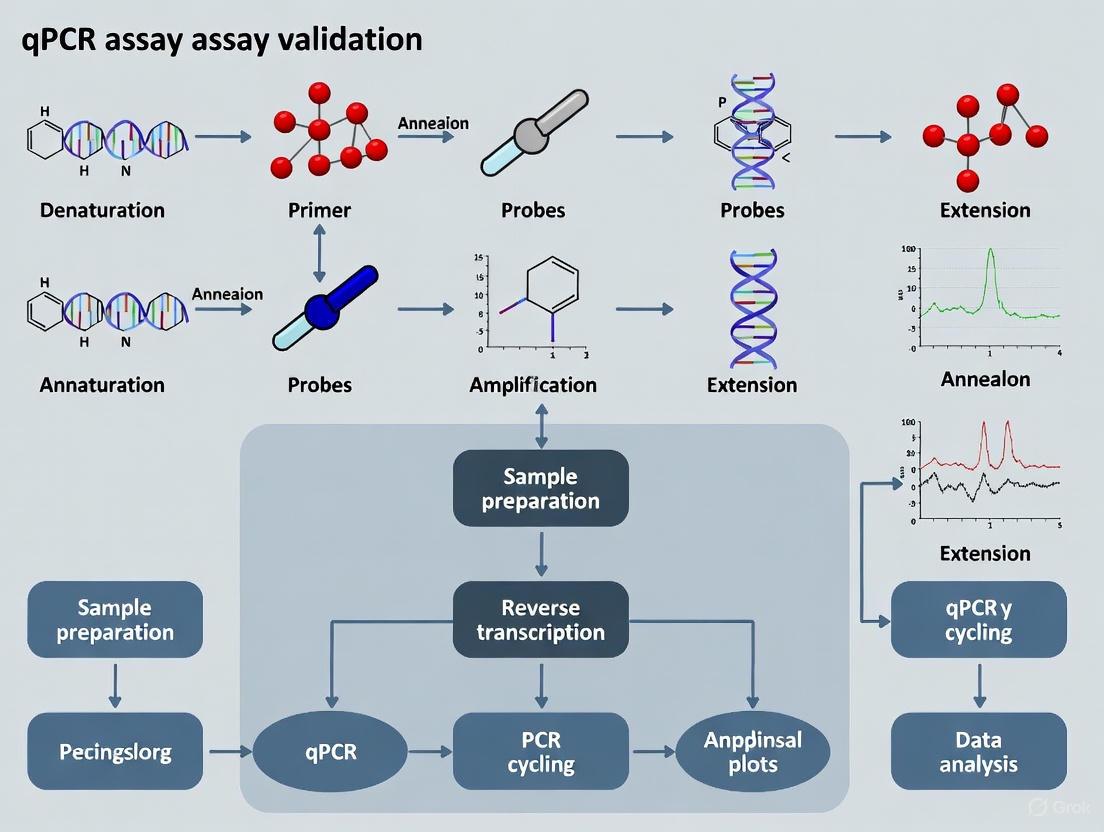
Figure 1: qPCR Assay Validation Workflow for Clinical Cancer Diagnostics
For laboratories implementing qPCR tests, whether as laboratory-developed tests (LDTs) or commercially available kits, verification of manufacturer's performance claims is essential [9]. This becomes particularly important for commercial assays, as performance may vary based on laboratory-specific factors including staff competency, equipment maintenance, and workflow systems [9].
qPCR Workflow Integration in Cancer Diagnostic Pathways
qPCR technology offers unique advantages in clinical workflows, particularly when integrated with traditional diagnostic methods to enhance overall efficiency and reliability.
Figure 2: qPCR Clinical Workflow for Cancer Mutation Detection
A 2025 study demonstrated the powerful synergy between traditional smear cytology (TSC) and qPCR testing for lung cancer diagnosis [5]. When used alone, TSC based on micro cell samples achieved a diagnostic yield of 78.9-93.5% across different sample types. However, when combined with qPCR genetic testing, the diagnostic yield increased to 81.6-98.1%, while maintaining a rapid 24-hour turnaround time [5]. This integrated approach exemplifies how qPCR complements established pathological methods to improve diagnostic accuracy without sacrificing speed.
Essential Research Reagent Solutions
Successful implementation of qPCR assays in cancer research requires carefully selected reagents and materials optimized for clinical samples. The following table details key components for robust qPCR-based mutation detection.
Table 3: Essential Research Reagents for qPCR-Based Cancer Mutation Detection
| Reagent Category | Specific Examples | Function & Features | Application Context |
|---|---|---|---|
| Master Mixes | Oncology-specific qPCR master mixes [1] | Inhibitor-resistant polymerases, optimized buffers for clinical matrices (plasma, FFPE, cfDNA) | High-sensitivity detection in challenging samples |
| Mutation Detection Kits | Lung Cancer 11-Gene Mutations Detection Kit [5] | Pre-optimized multiplex assays for simultaneous detection of key cancer drivers | Comprehensive genotyping with minimal sample input |
| Nucleic Acid Extraction | QIAamp DNA Micro Kit [5] | Efficient recovery from limited samples (fine needle aspirates, liquid biopsies) | Maximizing yield from low-input clinical specimens |
| Ambient-Stable Formulations | Lyophilized qPCR reagents [1] | Cold chain-independent transport and storage | Decentralized testing, resource-limited settings |
| Reference Standards | Biosynthetic DNA reference material [2] | Known variant allele frequencies for assay validation and quality control | Analytical validation, proficiency testing |
| Inhibition Resistance | Next-generation polymerases [1] | Tolerance to PCR inhibitors in clinical matrices (heparin, hemoglobin) | Reliable performance with direct clinical samples |
Modern qPCR reagent systems incorporate significant advancements including inhibitor-resistant enzymes, ambient-temperature stability, and enhanced multiplexing efficiency [1]. These innovations have substantially expanded the clinical utility of qPCR in oncology applications, enabling more reliable and efficient quantification of nucleic acids across diverse sample types and conditions.
qPCR maintains its cornerstone position in targeted cancer mutation detection through its unmatched combination of speed, cost-efficiency, and analytical robustness. The technology excels in clinical scenarios requiring rapid turnaround for a defined set of actionable mutations, particularly when sample material is limited or infrastructure constraints preclude more complex methodologies. While NGS offers superior discovery power for comprehensive genomic profiling, and digital PCR provides exceptional sensitivity for absolute quantification, qPCR remains the optimal solution for targeted mutation detection across a broad range of clinical and research settings [1] [6] [4].
For researchers and drug development professionals, qPCR represents a validated, accessible, and economically viable platform for precision oncology applications. Its continued evolution through advanced chemistries, improved multiplexing capabilities, and enhanced integration with complementary technologies ensures that qPCR will remain an indispensable tool in the cancer diagnostics arsenal for the foreseeable future.
Quantitative PCR (qPCR) remains a foundational technology in clinical oncology, enabling critical advancements in early cancer detection and personalized therapy selection. Its value is anchored in a unique combination of analytical sensitivity, rapid turnaround time, and cost-effectiveness, making it uniquely suited for informing therapeutic decision-making at scale, particularly in time-sensitive or resource-constrained settings [1]. Within the framework of analytical validation, qPCR assays provide robust, reproducible, and clinically actionable data. This guide objectively compares qPCR's performance against alternative methodologies like digital droplet PCR (ddPCR) and next-generation sequencing (NGS), providing supporting experimental data and detailed protocols to inform researchers and assay developers in the field of clinical cancer diagnostics.
Performance Comparison of qPCR, ddPCR, and NGS
The diagnostic performance of nucleic acid detection technologies varies significantly based on the application, target, and required sensitivity. The following tables summarize key comparative data.
Table 1: Comparative Diagnostic Performance in Detecting Circulating Tumor HPV DNA (ctHPVDNA) [10]
| Detection Platform | Sensitivity (Pooled) | Specificity (Pooled) | Key Strengths |
|---|---|---|---|
| Quantitative PCR (qPCR) | Lower than ddPCR and NGS (P<0.001) | Similar across all platforms | Cost-effective, rapid, widely established |
| Digital Droplet PCR (ddPCR) | Higher than qPCR, Lower than NGS (P=0.014) | Similar across all platforms | Absolute quantification, high sensitivity for low-frequency variants |
| Next-Generation Sequencing (NGS) | Greatest among the three platforms | Similar across all platforms | Multiplexing capability, discovery of novel variants |
Table 2: General Operational and Economic Comparison in Oncology Diagnostics [1]
| Parameter | qPCR | ddPCR | NGS |
|---|---|---|---|
| Approximate Cost per Test | $50 - $200 | ~$150 - $300 | $300 - $3,000 |
| Typical Turnaround Time | Hours | Several hours | Days |
| Variant Allele Frequency (VAF) Sensitivity | <0.1% | <0.1% | ~1-5% (varies by coverage) |
| Multiplexing Capability | Strong (4-6 plex) | Limited | Excellent (High-plex) |
| Infrastructure & Complexity | Low; widely accessible | Moderate | High; requires specialized infrastructure |
| Best Suited For | High-throughput, targeted screening; routine diagnostics | Ultra-sensitive quantification of known variants; liquid biopsies | Comprehensive genomic profiling; discovery |
Experimental Protocols and Methodologies
Protocol 1: Multiplex qPCR for Targeted Mutation Detection in NSCLC
This protocol is designed for the simultaneous detection of actionable mutations in genes like EGFR, KRAS, and BRAF from patient samples such as FFPE tissue or liquid biopsies [1].
- Nucleic Acid Extraction: Extract DNA from patient samples (e.g., FFPE tissue, plasma cfDNA) using a magnetic bead-based method. Quantify DNA using a spectrophotometer and dilute to a working concentration of 1-5 ng/μL.
- Reaction Setup: Prepare a 30 μL qPCR reaction mixture:
- 17 μL of qPCR master mix (containing inhibitor-resistant polymerase, dNTPs, and optimized buffer)
- 1 μL each of forward and reverse primers for multiple targets
- 1 μL of target-specific probes (e.g., TaqMan probes with different fluorophores)
- 10 μL of template DNA [11]
- Thermal Cycling: Run the reaction on a real-time PCR instrument with the following program:
- Initial Denaturation: 95°C for 10 minutes
- 40 Cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 1 minute [11]
- Data Analysis: Analyze amplification curves to determine Cq values. Use a standard curve for absolute quantification or ΔΔCq method for relative expression. Report clinically actionable mutations based on pre-validated Cq thresholds.
Protocol 2: Analytical Validation of a qPCR Assay for Residual Host Cell DNA
This method details the development and validation of a qPCR assay for quantifying residual Vero cell DNA in biological products, demonstrating the principles of analytical validation [11].
- Assay Design:
- Target Selection: Select highly repetitive, unique genomic sequences (e.g., a 172 bp tandem repeat or Alu repetitive elements) to maximize sensitivity.
- Primer/Probe Design: Design primers and probes targeting specific amplicons (e.g., 99 bp or 154 bp within the 172 bp sequence).
- Standard Curve and Linearity: Prepare a 10-fold serial dilution of the Vero genomic DNA standard, covering a range from 0.03 pg/μL to 30 pg/μL. Run the dilution series in triplicate to establish a standard curve. The assay must demonstrate a linear correlation with an R² value of >0.99 [11].
- Limit of Detection (LOD) and Quantification (LOQ):
- Specificity: Test the assay against genomic DNA from other common cell lines (e.g., CHO, HEK293) and bacterial strains to ensure no cross-reactivity [11].
- Precision and Accuracy: Assess intra- and inter-assay precision by calculating the relative standard deviation (RSD), which should be <20% across samples. Evaluate accuracy via spike-recovery experiments, with an ideal recovery rate of 85%-115% [11].
Visualizing the qPCR Workflow in Clinical Oncology
The following diagram outlines the standard workflow for applying qPCR in clinical cancer research, from sample collection to clinical decision-making.
Diagram Title: qPCR Clinical Workflow and Applications
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation of qPCR in clinical oncology relies on specialized reagents formulated to address the challenges of complex biological samples.
Table 3: Essential Reagents for Oncology qPCR Assay Development
| Reagent / Material | Function | Key Characteristics for Oncology Applications |
|---|---|---|
| Inhibitor-Resistant Master Mix | Provides enzymes, dNTPs, and optimized buffers for amplification. | Engineered to tolerate PCR inhibitors in clinical matrices (e.g., heparinized plasma, FFPE-derived nucleic acids) [1]. |
| Ambient-Stable Lyophilized Beads | Pre-formulated, room-temperature stable reaction pellets. | Reduces cold-chain costs, ideal for decentralized testing and OEM applications [1]. |
| Target-Specific Primers & Probes | Enables specific detection and quantification of mutant alleles. | Designed for high specificity and efficiency; crucial for multiplexed detection of actionable mutations [11]. |
| Quantified Genomic DNA Standards | Serves as a positive control and for generating standard curves. | Essential for assay validation, determining limits of detection (LOD), and ensuring quantitative accuracy [11]. |
| Internal Positive Control (IPC) | Distinguishes true negative results from PCR inhibition. | Added to each reaction to confirm that a negative result is due to the absence of the target, not assay failure. |
Advantages in Speed, Cost-Efficiency, and Scalability for Routine Diagnostics
Within the evolving landscape of clinical cancer diagnostics, the analytical validation of assays is paramount for ensuring reliable patient results. Quantitative PCR (qPCR) remains a foundational technology in this sphere, particularly when balanced against the capabilities of emerging methods like digital PCR (dPCR) and next-generation sequencing (NGS). This guide provides an objective comparison of these technologies, focusing on their performance in speed, cost, and scalability—critical factors for routine diagnostic workflows in research and drug development. By examining direct experimental data and validated protocols, this analysis aims to equip scientists with the evidence needed to make informed platform selections for clinical cancer research.
Technology Comparison: qPCR vs. dPCR vs. NGS
The selection of a nucleic acid detection platform involves trade-offs between analytical sensitivity, throughput, cost, and turnaround time. The table below summarizes the core performance characteristics of qPCR, dPCR, and NGS based on current literature and market data.
Table 1: Comparative Analysis of Nucleic Acid Detection Technologies in Clinical Diagnostics
| Feature | Quantitative PCR (qPCR) | Digital PCR (dPCR) | Next-Generation Sequencing (NGS) |
|---|---|---|---|
| Key Principle | Fluorescence-based real-time quantification relative to a standard curve [12] | Absolute nucleic acid quantification by end-point analysis of partitioned samples [12] | Massively parallel sequencing of clonally amplified DNA fragments [13] |
| Theoretical Sensitivity (Variant Allele Frequency) | ~1% (can be lower with optimized assays) [1] | <0.1% - 0.5% [12] [14] | ~1-2% (can be lower with deep sequencing) [13] |
| Absolute Quantification | No (requires standard curve) | Yes (via Poisson statistics) [12] | No (requires bioinformatic normalization) |
| Turnaround Time (Hands-on to result) | Several hours [13] [1] | Several hours [12] | Days to weeks [13] |
| Cost per Sample | $50 - $200 [1] | Higher than qPCR [12] | $300 - $3,000 [1] |
| Multiplexing Capability | High (with multiple probes/dyes) [1] [15] | Moderate (limited by fluorescence channels) | Very High (entire genomes/exomes) [13] |
| Best Suited For | High-throughput, rapid detection of known variants, routine screening [13] [1] | Detection of rare mutations, absolute quantification without standards, liquid biopsy monitoring [12] | Discovery of novel variants, comprehensive genomic profiling, complex mutation panels [13] |
| Infrastructure & Skill Demand | Widely accessible, standard molecular biology skills | Requires specialized instrumentation, moderate complexity | Demanding bioinformatics infrastructure and expertise [13] |
Experimental Data and Performance Benchmarks
Diagnostic Sensitivity in Liquid Biopsy Applications
Liquid biopsy, which involves analyzing circulating tumor DNA (ctDNA), presents a significant challenge for detection due to the low abundance of tumor DNA in a high background of wild-type DNA. A meta-analysis of circulating tumor HPV DNA (ctHPVDNA) detection across multiple cancer types provides a direct comparison of platform sensitivities.
Table 2: Comparative Sensitivity of ctHPVDNA Detection Platforms (Meta-Analysis Data) [16]
| Detection Platform | Relative Diagnostic Sensitivity | Specificity |
|---|---|---|
| Quantitative PCR (qPCR) | Baseline (Lowest) | Similar across platforms |
| Droplet Digital PCR (ddPCR) | Higher than qPCR (P < 0.001) | Similar across platforms |
| Next-Generation Sequencing (NGS) | Highest (P = 0.014 vs. ddPCR) | Similar across platforms |
This data demonstrates a clear tiered sensitivity, with NGS being the most sensitive, followed by dPCR, and then qPCR. However, this superior sensitivity comes at the cost of longer turnaround times and higher price points, making qPCR a robust choice for monitoring known, relatively abundant mutations.
Assay Performance in ESR1 Mutation Detection
A 2025 literature review of ESR1 mutation testing in advanced breast cancer provides a practical view of technology selection. The review identified 28 commercial assays for detecting ESR1 mutations in liquid biopsies, including four qPCR, four dPCR, and twelve NGS-based assays. The analysis concluded that while dPCR and NGS can offer high sensitivity, qPCR remains a clinically viable option with performance varying by the specific assay design and input DNA. The selection depends on the required analytical performance, desired turnaround times, and the available lab infrastructure and expertise [17]. This underscores that for many routine diagnostic contexts, qPCR provides a favorable balance of speed, cost, and sufficient accuracy.
Experimental Protocols for Analytical Validation
To ensure the reliability of qPCR assays in clinical research, the following key experimental protocols are employed for analytical validation.
Limit of Detection (LoD) and Variant Allele Frequency (VAF) Determination
- Objective: To determine the lowest concentration of a target mutant sequence that can be reliably detected by the qPCR assay.
- Methodology:
- Sample Preparation: Serially dilute synthetic mutant DNA targets into a background of wild-type genomic DNA. The dilutions should cover a range of Variant Allele Frequencies (e.g., from 5% down to 0.1%).
- qPCR Run: Amplify each dilution in a minimum of 20 replicates to establish a statistical confidence level (e.g., ≥95% detection rate).
- Data Analysis: The LoD is defined as the lowest VAF at which ≥95% of the replicates return a positive result. This validates the assay's sensitivity for detecting low-frequency variants, which is critical for liquid biopsy applications [1] [18].
Multiplexing Efficiency and Cross-Talk Validation
- Objective: To confirm that a single multiplex qPCR reaction can simultaneously and specifically detect multiple targets without interference.
- Methodology:
- Assay Design: Design primer-probe sets for multiple targets (e.g., mutations in EGFR, KRAS, and BRAF), each labeled with a spectrally distinct fluorescent dye.
- Specificity Testing: Run each primer-probe set individually in singleplex reactions and then combined in a multiplex reaction. Compare the amplification efficiency (measured by the Cq value) and the fluorescence signal in each channel between singleplex and multiplex setups.
- Analysis: A significant loss of sensitivity or an increase in background fluorescence (cross-talk) in the multiplex format indicates suboptimal assay conditions. Successful validation shows that the multiplex reaction performs with the same efficiency as the singleplex reactions, maximizing data output from minimal sample material [1] [15].
The following workflow diagram illustrates the decision-making process for selecting and validating a diagnostic technology based on clinical needs.
The Scientist's Toolkit: Key Research Reagent Solutions
The performance of a qPCR assay is heavily dependent on the quality and suitability of its core reagents. The following table details essential components for developing robust oncology diagnostics.
Table 3: Essential Reagents for qPCR Assay Development in Oncology Diagnostics
| Reagent / Material | Function / Key Characteristic | Application in Clinical Assays |
|---|---|---|
| High-Performance Master Mix | Optimized buffer and enzyme formulation for high sensitivity and specificity. | Core reagent for amplification; essential for detecting low-frequency variants in ctDNA [13] [1]. |
| Inhibitor-Resistant Polymerase | Engineered polymerase tolerant to PCR inhibitors found in clinical samples (e.g., from plasma, FFPE). | Ensures robust assay performance across diverse and challenging sample matrices [1]. |
| dUTP/UNG Contamination Control | Master mix containing dUTP and Uracil-N-Glycosylase (UNG) to prevent amplicon carryover. | Critical for high-throughput and reusable equipment settings to avoid false positives [13]. |
| Ambient-Stable (Lyo-Ready) Formulations | Reagents compatible with lyophilization for ambient-temperature storage and transport. | Enables decentralized testing, point-of-care use, and stable kit development for global distribution [13] [1]. |
| Multiplex Probe Systems | Sets of primers and probes labeled with different, non-overlapping fluorescent dyes. | Allows simultaneous detection of multiple mutations in a single reaction, conserving sample and increasing throughput [1] [15]. |
| Reference Templates | Synthetic DNA or well-characterized genomic DNA with known target mutations and wild-type sequences. | Serves as essential positive and negative controls for assay development, validation, and routine quality control. |
In the context of analytical validation for clinical cancer diagnostics, no single technology holds an absolute advantage; the choice is dictated by the specific clinical question and operational constraints. qPCR maintains its position as the optimal tool for high-speed, cost-effective, and scalable detection of known actionable mutations in routine diagnostics, especially where rapid therapeutic decisions are needed. dPCR provides superior sensitivity for absolute quantification of rare variants, while NGS offers unparalleled breadth for discovery and comprehensive profiling. A thorough understanding of the comparative data and validation protocols presented here empowers researchers and drug developers to strategically deploy qPCR, ensuring robust, reliable, and accessible diagnostic solutions.
The success of clinical cancer diagnostics and the development of targeted therapies hinge on the accurate molecular profiling of tumor material. In modern oncology, this analysis relies heavily on a variety of sample types, each with its own unique set of advantages and limitations. Formalin-fixed paraffin-embedded (FFPE) tissue has long been the gold standard, providing a stable, histologically-rich resource. More recently, the analysis of circulating cell-free DNA (cfDNA) from liquid biopsies has emerged as a powerful, minimally invasive tool for genomic profiling and monitoring therapy response [19] [20].
A significant challenge common to both sample types is the limited quantity and quality of nucleic acids available for testing. FFPE samples are notoriously compromised by chemical cross-linking and nucleic acid fragmentation, while cfDNA samples often contain very low concentrations of tumor-derived DNA (ctDNA) within a high background of wild-type DNA [20] [21]. This guide objectively compares the performance of qPCR assays across these challenging sample matrices, providing a framework for their analytical validation in clinical cancer research.
Sample Type Characteristics and Technical Hurdles
The pre-analytical variables and inherent properties of FFPE and liquid biopsy samples directly influence the choice of analytical platform and the design of the validation strategy. The table below summarizes the core challenges associated with each sample type.
Table 1: Core Characteristics and Challenges of Key Oncology Sample Types
| Sample Type | Core Characteristics | Primary Technical Hurdles |
|---|---|---|
| FFPE Tissue | - Chemically cross-linked and fragmented nucleic acids [21]- Long-term stability at room temperature- Allows for correlative histopathology | - Nucleic acid degradation impacts yield and quality [21]- PCR inhibition from residual formalin [1]- Challenging DNA/RNA co-extraction [21] |
| Liquid Biopsy (cfDNA) | - Minimally invasive, enables serial monitoring [20]- Represents tumor heterogeneity [22]- Short turnaround time [19] | - Extremely low input of circulating tumor DNA (ctDNA) [19]- High background of wild-type DNA [23]- Critical pre-analytical conditions (e.g., blood draw to plasma processing time) [22] |
| Low-Input Samples (e.g., Fine Needle Aspirates, Low-Volume Plasma) | - Minimal material, often precludes repeat testing- Essential for hard-to-biopsy cancers | - Risk of false negatives due to sampling error- Maximizing data output from minimal input is critical [1]- Assay sensitivity and robustness are paramount |
The following diagram illustrates the interconnected challenges and considerations when working with these sample types, from collection to analysis.
Performance Comparison: Experimental Data and Validation
Robust analytical validation is fundamental to generating reliable data from challenging samples. Key performance parameters must be rigorously established for each sample type and assay.
Analytical Sensitivity and Specificity
Sensitivity requirements are particularly stringent for liquid biopsy applications due to the low variant allele frequency (VAF) of ctDNA. For example, the Aspyre Lung assay, a multiplexed qPCR-based test, demonstrated a 95% limit of detection (LoD95) of 0.19% VAF for single nucleotide variants and indels in cfDNA, alongside 100% specificity for all targets [19]. This level of sensitivity is crucial for detecting minimal residual disease (MRD) and early resistance mutations.
Digital PCR (dPCR) platforms, which include droplet digital PCR (ddPCR) and BEAMing PCR, offer alternative approaches for ultra-sensitive detection. One study comparing BEAMing PCR to standard qPCR for EGFR mutation detection in NSCLC patients reported high concordance rates (98.8% for exon 19 and 95.5% for exon 21), confirming the reliability of sensitive methods for profiling ctDNA [23].
Table 2: Comparative Analytical Performance of PCR Assays Across Sample Types
| Assay / Technology | Reported Sensitivity (LoD) | Reported Specificity | Sample Type Validated On | Key Experimental Findings |
|---|---|---|---|---|
| Aspyre Lung qPCR (Multiplexed) | 0.19% VAF (SNV/Indels); 1 amplifiable copy (Fusions) [19] | 100% [19] | cfDNA, cfRNA, FFPE DNA/RNA [19] | Model trained on >13,500 contrived samples; enables single-workflow tissue and plasma testing [19]. |
| BEAMing dPCR (EGFR) | High sensitivity for mutant allele detection [23] | High specificity [23] | ctDNA from plasma [23] | 98.8% concordance with qPCR for exon 19 deletions; useful for mutation detection in background of normal DNA [23]. |
| Droplet Digital PCR (ddPCR) (EGFR) | High sensitivity [22] | Specificity validated with external controls [22] | cfDNA from plasma [22] | Pre-analytical conditions (shipping time, extraction) critically impact performance; in-house primers/probes can offer superior sensitivity [22]. |
| RT-qPCR (Reference Genes) | Varies with assay design | Varies with assay design | FFPE RNA, Cell line RNA [24] | ACTB, RPS23, RPS18, RPL13A genes are unstable in dormant cancer cells and are inappropriate references; validation of stable genes like B2M/YWHAZ is essential [24]. |
Nucleic Acid Yield and Quality
The quality of input material is a primary determinant of assay success. A comparative study of nucleic acid extraction kits from FFPE tissue highlights significant variability in performance. Omega Bio-tek's Mag-Bind FFPE DNA/RNA 96 Kit, for instance, demonstrated significantly higher DNA yield from FFPE lung and breast tumor samples compared to kits from two other manufacturers (Company T and Q). Furthermore, the average ΔCq value—a metric for FFPE DNA quality where a lower value indicates higher quality—was 3.10 for Omega Bio-tek versus 4.06 and 5.32 for competitors, indicating superior preservation of nucleic acid functionality [21].
For RNA from FFPE samples, the DV200 value (percentage of RNA fragments >200 nucleotides) is a key quality indicator. The same study found that the Omega Bio-tek kit yielded RNA with a DV200 of 70.97% for lung and 76.86% for colorectal FFPE tumors, outperforming other kits and exceeding the Illumina-recommended threshold of 20% for successful sequencing [21].
Essential Experimental Protocols
Standardized and optimized experimental protocols are critical for ensuring reproducibility and data reliability.
Pre-Analytical cfDNA Workflow for ddPCR
The CIRCAN study provides a detailed workflow for the detection of EGFR mutations in cfDNA using ddPCR [22]:
- Blood Collection and Processing: Blood is drawn into EDTA tubes. Plasma is separated via a two-step centrifugation protocol: first at 820 × g for 10 minutes, followed by a second centrifugation of the supernatant at 16,000 × g for 10 minutes to remove any remaining cellular debris.
- cfDNA Extraction: cfDNA is extracted from 1 mL of plasma using a commercially available DNA Micro Kit, following the manufacturer's instructions. The extracted DNA is eluted in a minimal volume.
- Droplet Digital PCR (ddPCR): The ddPCR reaction is set up using validated primer/probe sets for wild-type and mutant EGFR alleles (e.g., delEX19, L858R, T790M). The reaction mixture is partitioned into thousands of nanodroplets. Amplification is carried out on a thermal cycler, and droplets are read on a droplet reader to quantify the absolute number of mutant and wild-type DNA molecules.
Reference Gene Validation for RT-qPCR in Challenging Models
In novel experimental models, such as dormant cancer cells, classic reference genes can become unstable. A 2025 study established a protocol for validating reference genes in mTOR-suppressed cancer cells [24]:
- Model Generation: Treat cancer cell lines (e.g., A549, T98G) with a dual mTOR inhibitor (e.g., AZD8055) to induce a dormant phenotype.
- RNA Extraction and cDNA Synthesis: Extract total RNA from treated and control cells, and synthesize cDNA.
- Candidate Gene Screening: Perform RT-qPCR on a panel of 12 commonly used housekeeping genes (e.g., GAPDH, ACTB, B2M, YWHAZ).
- Stability Analysis: Use algorithms (e.g., geNorm, NormFinder) to rank the genes based on their expression stability. The study found that ACTB and ribosomal protein genes (RPS23, RPS18, RPL13A) were highly unstable under these conditions, while B2M and YWHAZ were among the most stable for A549 cells [24].
The Scientist's Toolkit: Key Research Reagent Solutions
Selecting the right reagents is fundamental to overcoming sample-specific challenges. The following table details key solutions mentioned in the literature.
Table 3: Essential Reagents and Kits for Challenging Sample Types
| Product / Solution | Primary Function | Key Features & Benefits | Supported Sample Types |
|---|---|---|---|
| Mag-Bind FFPE DNA/RNA 96 Kit (Omega Bio-tek) [21] | Co-extraction of DNA and RNA from FFPE samples. | - Uses non-toxic mineral oil for deparaffinization- Yields high-quality, functional nucleic acids with low ΔCq- Provides DNA-free RNA and RNA-free DNA in separate eluates | FFPE Tissues |
| Oncology-Focused qPCR Reagents (Meridian Bioscience) [1] | Master mixes for robust qPCR amplification. | - Engineered for inhibitor tolerance in clinical matrices (plasma, FFPE)- High sensitivity (<0.1% VAF detection)- Ambient-stable formulations for decentralized testing | cfDNA, FFPE-derived DNA, Low-input samples |
| Aspyre Lung Reagents (Biofidelity) [19] [1] | Targeted biomarker panel for NSCLC. | - Covers 114 genomic variants across 11 genes- Simultaneous DNA and RNA analysis from same workflow- Machine learning-powered data interpretation for high sensitivity | cfDNA, cfRNA, FFPE DNA/RNA |
| Droplet Digital PCR (ddPCR) Assays [22] | Absolute quantification of rare mutant alleles. | - High sensitivity and specificity for low-VAF variants- Digital counting of molecules enables precise quantification- Suitable for tumor-agnostic and tumor-informed approaches | ctDNA from Plasma |
The landscape of clinical oncology samples is inherently complex, dominated by FFPE tissues with compromised nucleic acids and liquid biopsies with ultra-low target concentrations. Success in this field requires a deep understanding of the specific challenges posed by each sample type. As the data shows, qPCR and dPCR technologies, when paired with optimized reagents and rigorous validation, are capable of meeting these demands, delivering the sensitivity, specificity, and robustness required for modern cancer diagnostics and drug development. By adhering to stringent validation guidelines and selecting tools fit for purpose, researchers can reliably generate meaningful data from even the most challenging samples, thereby accelerating the pace of discovery and precision medicine.
Building a Robust qPCR Assay: From Design to Data Interpretation
The precision of quantitative PCR (qPCR) in clinical cancer diagnostics hinges on the meticulous design of its core components: primers, probes, and the amplicon. For research aimed at predicting responses to neoadjuvant chemotherapy or detecting low-frequency cancer mutations, a robust assay design is not merely a preliminary step but the foundation of reliable, analytically valid results [1] [25] [26]. This guide objectively compares design strategies and presents supporting experimental data to inform the development of qPCR assays for cancer research.
Core Component Design and Comparison
The performance of a qPCR assay is directly governed by the biochemical properties of its primers and probes. Adherence to established design principles is crucial for achieving high specificity, sensitivity, and efficiency.
Primer Design Fundamentals
Primers are the critical determinants of assay specificity and efficiency. Optimal design requires balancing multiple biochemical parameters [27] [28].
- Length and Melting Temperature (Tm): Primers should be 18–22 base pairs long, with a Tm typically between 55–60°C. The forward and reverse primer Tm should be within 2°C of each other to ensure simultaneous annealing [27].
- GC Content and Sequence: Aim for a GC content of 35–65%, avoiding long stretches of a single nucleotide (homopolymers) or repeated G/C bases, especially at the 3' end, to prevent mis-priming [27].
- Specificity and Validation: Primer sequences must be checked for specificity using tools like NCBI BLAST to avoid amplification of non-target sequences, including pseudogenes. Furthermore, the optimal annealing temperature (Ta) must be determined empirically through a temperature gradient, as it is distinct from the in silico-calculated Tm and is vital for robustness [28].
Table 1: Key Design Parameters for qPCR Primers and Probes
| Parameter | Primers | TaqMan Probes |
|---|---|---|
| Optimal Length | 18–22 bp [27] | 20–30 bp [29] |
| Melting Temperature (Tm) | ~55–60°C [27] | 65–70°C; 8–10°C higher than primers [29] [30] |
| GC Content | 35–65% [27] | Avoid 5' G; more C than G [30] |
| Critical Checks | Specificity (BLAST), secondary structures (dimers, hairpins) [27] [28] | Avoid repetitive sequences, especially 4 consecutive Gs [29] |
Probe Selection and TaqMan Chemistry
TaqMan probes offer superior specificity compared to intercalating dye methods by providing an additional layer of sequence-specific detection [29].
- Working Principle: A TaqMan probe is a dual-labeled oligonucleotide with a 5' fluorophore and a 3' quencher. During PCR, when the probe binds to its target, the 5' to 3' exonuclease activity of the DNA polymerase cleaves the probe, separating the fluorophore from the quencher and generating a fluorescent signal [31] [29].
- Multiplexing Capability: TaqMan chemistry supports high-order multiplexing. Using different fluorophore-labeled probes, assays can detect up to five or six targets in a single reaction, which is invaluable for profiling multiple cancer biomarkers simultaneously from limited sample material [1] [31].
Amplicon Selection Criteria
The amplicon—the DNA region amplified by the primers—must be carefully selected to ensure efficient and specific detection.
- Length: For standard qPCR, ideal amplicon lengths are between 70–140 base pairs. This is short enough to ensure high amplification efficiency and is crucial when working with fragmented DNA from formalin-fixed paraffin-embedded (FFPE) tissue samples, a common source in cancer research [27] [26].
- Location: For mRNA quantification in RT-qPCR, primers should be designed to span an exon-exon junction. This ensures amplification of the spliced cDNA and not contaminating genomic DNA [27]. Assays must also be designed to avoid common single nucleotide polymorphisms (SNPs) within the binding sites [27].
Experimental Validation Protocols
A well-designed assay requires rigorous experimental validation to confirm its performance characteristics are fit for purpose in a clinical research setting.
Establishing Assay Efficiency and Dynamic Range
A core validation step is determining the amplification efficiency and dynamic range of the assay through a standard curve.
- Methodology: A serial dilution (e.g., 10-fold) of the target template (e.g., plasmid DNA or synthetic gBlock) with a known concentration is amplified. The Cq value is plotted against the logarithm of the starting quantity for each dilution [26].
- Data Interpretation: A slope between -3.1 and -3.6, a correlation coefficient (R²) greater than 0.99, and a calculated PCR efficiency between 90–110% are indicators of a highly efficient and precise assay [26]. Efficiencies outside this range suggest suboptimal primer/probe binding or issues with reaction components.
Assessing Analytical Sensitivity and Specificity
Sensitivity and specificity define an assay's ability to detect low amounts of the target and to distinguish it from related non-targets.
- Limit of Detection (LoD): The lowest concentration at which the target is detected in ≥95% of replicates. For example, a multiplex qPCR for duck viruses reported LoDs as low as 6.03 x 10¹ copies/μL for individual targets, demonstrating high sensitivity [32].
- Specificity Testing: The assay must be tested against a panel of non-target organisms or genetic variants to ensure no cross-reactivity. In the development of a 10-plex qPCR for bladder cancer, researchers confirmed primer specificity using Standard Nucleotide BLAST and wet-lab testing against non-target sequences [26].
Ensuring Reproducibility and Robustness
Reproducibility is assessed by testing the assay's performance across different operators, instruments, and days.
- Protocol: Run replicates of samples at high, medium, and low concentrations across multiple runs (inter-assay) and within the same run (intra-assay) [26].
- Acceptance Criteria: The results are typically expressed as the Coefficient of Variation (CV) of the Cq values. Both intra- and inter-assay CVs should generally be below 10%, confirming the assay is robust and generates consistent results [32] [26].
The following workflow diagram summarizes the key stages of qPCR assay design and validation.
Comparative Performance in Clinical Applications
qPCR holds a distinct position in the molecular diagnostics landscape, particularly when compared to broader profiling technologies like next-generation sequencing (NGS).
qPCR vs. Next-Generation Sequencing (NGS)
The choice between qPCR and NGS is driven by the clinical or research question, with each technology offering complementary strengths.
Table 2: Performance Comparison between qPCR and NGS in Cancer Diagnostics
| Characteristic | qPCR | Next-Generation Sequencing (NGS) |
|---|---|---|
| Best Application | Targeted mutation detection, rapid therapy selection [1] | Comprehensive genomic profiling, discovery [1] |
| Turnaround Time | Hours [1] | Days [1] |
| Cost per Sample | $50 – $200 [1] | $300 – $3,000 [1] |
| Multiplexing Scale | Moderate (e.g., 4-10 plex) [1] [32] | High (100s-1000s of targets) [1] |
| Analytical Sensitivity | High (can detect <0.1% VAF) [1] | Moderate (typically 1-5% VAF) [1] |
| Infrastructure Needs | Low, more accessible [1] | High, complex infrastructure [1] |
qPCR excels in time-sensitive, resource-conscious, and targeted applications. For example, in non-small cell lung cancer (NSCLC), multiplexed qPCR panels can simultaneously assess alterations in genes like EGFR, KRAS, and ALK, delivering results faster and using less input material than NGS [1]. This makes qPCR indispensable for large-scale screening and routine diagnostics where a defined set of actionable biomarkers is known.
The Critical Role of MIQE 2.0 Guidelines
The publication of the MIQE 2.0 guidelines is a critical response to widespread issues of poor transparency and reproducibility in qPCR-based research [25]. These guidelines provide a checklist of essential information that must be reported to ensure experimental rigor. Compliance is non-negotiable in clinical cancer research, as failures in proper sample handling, assay validation, and data normalization can lead to exaggerated sensitivity claims and overinterpreted results with real-world consequences for patient diagnosis and treatment [25] [28].
The Scientist's Toolkit: Research Reagent Solutions
Successful assay development relies on a suite of high-quality reagents and tools. The following table details key components for developing a robust qPCR assay for cancer diagnostics.
Table 3: Essential Reagents and Tools for qPCR Assay Development
| Item | Function / Key Feature | Example Application / Benefit |
|---|---|---|
| qPCR Master Mix | Contains polymerase, dNTPs, and optimized buffer. | Oncology-specific formulations are engineered for inhibitor resistance in clinical samples (e.g., plasma, FFPE) and high sensitivity for low-frequency variants [1]. |
| TaqMan Probes | Dual-labeled hydrolysis probes for specific target detection. | MGB probes enable shorter, more specific probes and higher-order multiplexing (up to 5-plex) for profiling multiple cancer biomarkers [31]. |
| Custom qPCR Primers | Sequence-specific oligonucleotides for target amplification. | HPLC-purified, desalted primers minimize synthesis impurities that increase background noise and decrease assay sensitivity and reproducibility [31] [28]. |
| RNA/DNA Extraction Kits | Purify nucleic acids from complex biological samples. | Kits designed for FFPE tissue or cell-free DNA are critical for obtaining high-quality input material from clinically relevant sample types [26]. |
| cDNA Synthesis Kits | Convert RNA to cDNA for RT-qPCR analysis. | Preamplification master mixes can be used to enable analysis of low-input and degraded RNA samples commonly encountered in cancer research [26]. |
| qPCR Assay Design Tool | In silico software for selecting optimal primers/probes. | Automated tools analyze sequence constraints to recommend primer/probe sets with appropriate Tm, GC%, and specificity [30]. |
The foundational elements of qPCR—primers, probes, and amplicons—must be designed with precision and validated with rigor. As shown, qPCR maintains a competitive edge over NGS for targeted, rapid, and cost-effective applications in clinical oncology, such as therapy selection and large-scale screening. By adhering to MIQE 2.0 guidelines and employing a disciplined approach to assay design and validation, researchers can ensure their qPCR data are robust, reproducible, and reliable, thereby solidifying the role of this powerful technology in the advancement of cancer diagnostics and personalized medicine.
In the field of clinical cancer diagnostics research, quantitative PCR (qPCR) remains a foundational tool for detecting oncogenes, monitoring minimal residual disease, and validating cancer biomarkers. Its high sensitivity, rapid turnaround, and cost-effectiveness make it uniquely suited for informing therapeutic decisions. A critical choice in developing a robust qPCR assay is the detection chemistry: the intercalating dye SYBR Green or the sequence-specific Hydrolysis Probes (often referred to as TaqMan probes). This guide provides an objective, data-driven comparison of these two chemistries to inform their application in validating cancer targets.
Mechanism of Detection: A Visual Guide
The core difference between the two chemistries lies in their mechanism for detecting amplified PCR products. The diagrams below illustrate the specific signaling pathways for each.
SYBR Green Detection Mechanism
Hydrolysis Probe Detection Mechanism
Direct Comparison: Performance and Practicality
The following table summarizes the key characteristics of each chemistry, supported by experimental data, to guide your selection.
| Feature | SYBR Green | Hydrolysis Probes (TaqMan) |
|---|---|---|
| Specificity | Lower; binds any dsDNA (e.g., primer-dimers, non-specific products). Requires post-run melt curve analysis for verification. [33] | Higher; requires binding of a sequence-specific probe, minimizing false positives from non-specific amplification. [33] [34] |
| Sensitivity (LOD) | Can be highly sensitive with optimization. One study achieved a detection limit of 3.16 TCID50/mL in infected tissues. [35] | Excellent sensitivity, consistently capable of detecting low-frequency variants below <0.1% variant allele frequency (VAF). Ideal for liquid biopsy and MRD. [1] |
| Cost | Relatively cost-benefit. No need for expensive labeled probes. [33] | More expensive due to the cost of fluorogenic probe synthesis and validation. [33] [36] |
| Experimental Workflow | Easier and faster setup. Requires only primer design. [33] [37] | More complex design. Requires optimization of primers and a probe with a higher Tm. [36] [37] |
| Multiplexing Potential | Not possible, as the dye binds to all double-stranded DNA products non-specifically. [38] | Excellent. Multiple targets can be detected in a single reaction by using probes labeled with different reporter dyes. [36] [1] |
| Best Suited For |
Quantitative Experimental Data
A direct comparative study on breast cancer tissues measuring adenosine receptor subtypes provides robust experimental data on the performance of both chemistries when optimized.
Table: Performance Comparison in Breast Cancer Gene Expression Analysis [33]
| Gene Target | SYBR Green (Normalized Expression) | Hydrolysis Probe (Normalized Expression) | PCR Efficiency | Correlation (P-value) |
|---|---|---|---|---|
| A1 Adenosine Receptor | 1.44 | 1.38 | >97% for both methods | Positive and Significant (P < 0.05) |
| A2A Adenosine Receptor | 2.38 | 2.43 | >97% for both methods | Positive and Significant (P < 0.05) |
| A2B Adenosine Receptor | 3.79 | 3.84 | >97% for both methods | Positive and Significant (P < 0.05) |
| A3 Adenosine Receptor | 3.55 | 3.58 | >97% for both methods | Positive and Significant (P < 0.05) |
Conclusion from Data: With the use of high-performance primers and proper optimization, SYBR Green can generate precise data comparable to the hydrolysis probe method. The correlation between the normalized expression data from both methods was positive and statistically significant for all genes tested. [33]
Detailed Experimental Protocols
Protocol 1: Hydrolysis Probe qPCR for miRNA (96-Well Plate)
This protocol, adaptable for cancer targets like microRNAs from liquid biopsies, highlights the specificity of probe-based chemistry. [34]
cDNA Synthesis (Reverse Transcription):
- Use 10 ng of total RNA extracted from serum or plasma in a 5 µL reaction volume.
- Prepare a master mix containing RT Buffer (1X), dNTPs (100 mM), RNase Inhibitor (20 U/µL), and reverse transcriptase (50 U/µL).
- Add 1 µL of a sequence-specific RT primer.
- Incubate on a thermal cycler: 16°C for 30 min, 42°C for 30 min, 85°C for 5 min (enzyme inactivation), and hold at 4°C.
Probe-based qPCR:
- Prepare a qPCR reagent mix containing a proprietary master mix, nuclease-free water, and a 20X probe-based assay mix (containing primers and probe).
- Combine 4.2 µL of this mix with 0.8 µL of cDNA in an optical 96-well plate.
- Seal the plate and centrifuge briefly.
- Run on a real-time PCR instrument with the following cycling conditions:
- Hold: 95°C for 20 sec (polymerase activation)
- 40-50 Cycles: 95°C for 1 sec (denaturation), 60°C for 30 sec (annealing/extension).
Protocol 2: SYBR Green qPCR for Gene Expression
This is a general protocol for SYBR Green-based detection, which requires a post-run melting curve analysis to verify specificity. [40]
Reaction Setup:
- Prepare a 50 µL reaction containing:
- 25 µL of 2X SYBR Green Master Mix (hot-start Taq polymerase, buffer, dNTPs, SYBR Green dye).
- 1 µL each of 10 µM forward and reverse primers (200 nM final concentration).
- Template DNA (e.g., up to 100 ng genomic DNA, or cDNA generated from up to 1 µg total RNA).
- Nuclease-free water to volume.
- Include a no-template control (NTC) to check for contamination.
- Prepare a 50 µL reaction containing:
Thermal Cycling and Melt Curve:
- UDG Incubation (Optional): 50°C for 2 minutes to prevent amplicon carryover.
- Polymerase Activation: 95°C for 10 minutes.
- Amplification (40 Cycles): 95°C for 15 sec, 60°C for 60 sec.
- Melting Curve Analysis: Program the instrument to gradually increase temperature from 60°C to 95°C (e.g., by 0.3°C/sec) while continuously monitoring fluorescence. A single sharp peak indicates a specific product; multiple peaks suggest primer-dimers or non-specific amplification.
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function & Application in Cancer Research |
|---|---|
| Inhibitor-Resistant Master Mix | Specially formulated polymerases and buffers to tolerate PCR inhibitors common in clinical samples like heparinized plasma, FFPE-derived nucleic acids, and cell-free DNA (cfDNA), ensuring reliable results. [1] |
| Ambient-Stable Master Mix | Lyophilized or stable liquid formulations that do not require a cold chain. Ideal for decentralized testing, resource-limited settings, or OEM diagnostic kit development. [1] |
| Pre-designed Assay Panels | Hydrolysis probe-based assays for multiplexed detection of clinically actionable mutations (e.g., in EGFR, KRAS, BRAF, ALK). Provide standardized, validated protocols for rapid deployment in labs. [1] |
| ROX Reference Dye | A passive dye included in the reaction to normalize for non-PCR-related fluctuations in fluorescence between wells, critical for accurate quantification in high-throughput settings. [40] |
| UDG (Uracil-DNA Glycosylase) | An enzyme included in many master mixes to prevent re-amplification of carryover PCR products between runs, a key quality control measure in clinical diagnostics. [40] |
The choice between SYBR Green and hydrolysis probes is not a matter of which is universally better, but which is optimal for your specific research context within clinical cancer diagnostics.
- Choose SYBR Green for initial, cost-effective screening, when working with non-standardized targets, or when melt curve analysis provides sufficient specificity for the assay.
- Choose Hydrolysis Probes when your objective requires the highest level of specificity in a complex biological matrix (e.g., plasma), when multiplexing several cancer targets, or when validating biomarkers with the utmost confidence for potential clinical application.
Ultimately, with careful optimization and validation, both chemistries are powerful tools for the analytical validation of qPCR assays in the fight against cancer.
Multiplexing Strategies for Simultaneous Multi-Gene Mutation Detection
Quantitative PCR (qPCR) remains a cornerstone technology in clinical cancer diagnostics, prized for its rapid turnaround time, cost-effectiveness, and high analytical sensitivity [1]. In the context of precision oncology, the ability to simultaneously interrogate multiple clinically actionable mutations from a single, often limited, patient sample is paramount. Multiplex qPCR strategies address this need by enabling the parallel detection of several DNA targets within one reaction, conserving precious sample material, reducing reagent costs, and streamlining laboratory workflow [41] [42]. This guide provides an objective comparison of contemporary multiplex qPCR approaches, evaluated against the rigorous demands of analytical validation for clinical research. The performance of these methods is critical for applications such as therapy selection, patient stratification, and tracking treatment resistance, where the accuracy and comprehensiveness of mutation profiling can directly impact patient management.
Comparative Analysis of Multiplex qPCR Strategies
The following table summarizes the key performance characteristics of three advanced multiplexing strategies, based on recent experimental studies.
Table 1: Performance Comparison of Multiplex qPCR Strategies for Mutation Detection
| Multiplexing Strategy | Theoretical Multiplexing Capacity | Demonstrated Sensitivity (Mutant Allele Fraction) | Key Advantages | Key Limitations / Challenges |
|---|---|---|---|---|
| Color Cycle Multiplex Amplification (CCMA) [43] | Up to 136 targets (with 4 colors) | Not explicitly quantified for low AF, but provides quantitative Ct data | Exceptional multiplexing capacity on standard instruments; quantitative output; suitable for syndromic panels. | Complexity in assay design (requires blocker oligonucleotides); newer approach with less extensive validation. |
| Multiplex Allele-Specific qPCR (with ARMS/Blockers) [41] [44] | Limited by available fluorescent channels (typically 4-6). | 0.05% - 0.5% [41]; ~5% for PIK3CA H1047R [44] | Well-established and optimized; very high sensitivity; robust on FFPE-derived DNA. | Lower multiplexing capacity per reaction tube; requires careful optimization to prevent primer interference. |
| Allele-Specific LNA qPCR [45] | Limited by available fluorescent channels. | Comparable to NGS; linear detection from 0% to 95% mutant [45] | Excellent specificity and sensitivity for single-nucleotide variants; LNA probes enhance allele discrimination. | Similar channel-limited multiplexing; cost of LNA-modified probes may be higher. |
Experimental Protocols and Workflows
Protocol: Multiplex Allele-Specific qPCR for PIK3CA Mutation Detection
This protocol, adapted from a study screening 515 colorectal cancer samples, is designed for high-sensitivity detection of hotspot mutations in genes like PIK3CA [41].
- Sample Preparation: DNA is extracted from formalin-fixed paraffin-embedded (FFPE) tissue sections using a dedicated kit (e.g., QIAamp DNA FFPE Tissue Kit). DNA quantity and quality are assessed, with fragmentation and chemical modifications expected [41].
- Assay Design:
- Primers: Allele-specific primers are designed with the 3' end complementary to the mutant sequence to preferentially amplify mutant alleles [44].
- Blockers: A wild-type blocking oligonucleotide (e.g., phosphate-modified to prevent extension) is included. It binds to the wild-type sequence, suppressing its amplification and enhancing the specificity for the mutant allele [44].
- Multiplexing: Multiple primer sets, each tagged with a unique fluorophore, are combined in a single master mix.
- Reaction Setup:
- Thermocycling: Conditions are optimized for the specific master mix. An example protocol is: 95°C for 10 min, followed by 50-60 cycles of 95°C for 15 sec and 60°C for 1 min, with fluorescence acquisition at the annealing/extension step [41].
- Data Analysis: The Cycle threshold (Ct) for each target is determined. A sample is considered positive for a mutation if the Ct value for the mutant-specific channel is significantly lower than that of a wild-type control, and/or falls within a validated positive range [44].
Protocol: Color Cycle Multiplex Amplification (CCMA)
CCMA is a novel strategy that uses temporal fluorescence patterns, rather than color alone, to dramatically increase multiplexing capacity [43].
- Core Principle: Each DNA target is programmed to induce a sequential, pre-determined pattern of fluorescence increases across different color channels over the course of the qPCR run. The identity of the target is determined by this unique fluorescence "permutation" [43].
- Implementation via Blocker Displacement Amplification (BDA):
- Blocker Design: For a given target, multiple amplicons are designed, each corresponding to a different fluorophore. Rationally designed oligonucleotide blockers are used to competitively bind to the template, programmably delaying the amplification of specific amplicons [43].
- Ct Delay: By adjusting the binding strength (thermodynamics) or concentration of these blockers, the Ct value for each color channel can be precisely delayed by several cycles relative to the first, unblocked amplicon. This creates the characteristic step-wise fluorescence pattern [43].
- Workflow: The workflow is similar to a standard multiplex qPCR from a sample preparation and run standpoint. However, the data analysis involves interpreting the order and Ct values of fluorescence emergence across all channels to identify the present pathogen or mutant target.
The following diagram illustrates the logical workflow for developing and validating a multiplex qPCR assay, from sample to clinical research application.
The PI3K-AKT-mTOR Signaling Pathway in Cancer
Mutations in genes like PIK3CA are frequently screened using multiplex qPCR. The PIK3CA gene encodes the catalytic subunit of PI3K, a key node in the PI3K-AKT-mTOR signaling pathway, which is crucial for cell survival, proliferation, and metabolism [41] [44]. This pathway is one of the most frequently altered in human cancers, making it a major focus for molecular diagnostics and targeted therapy.
The diagram below outlines the core components of this pathway, highlighting where common mutations occur and how targeted inhibitors intervene.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of a multiplex qPCR assay for clinical research relies on a suite of specialized reagents and materials. The following table details key components and their functions.
Table 2: Essential Research Reagent Solutions for Multiplex qPCR Assay Development
| Reagent / Material | Function and Importance in Assay Development |
|---|---|
| Specialized DNA Polymerase Master Mixes | Next-generation master mixes are engineered for inhibitor resistance (e.g., for FFPE, plasma), thermal stability for faster cycling, and enhanced multiplexing efficiency [1]. |
| Allele-Specific Primers | Primers designed with the variant base at the 3' end to preferentially initiate amplification from the mutant allele, forming the basis of allele discrimination [41] [44]. |
| Locked Nucleic Acid (LNA) Probes | Modified nucleic acid analogs incorporated into TaqMan probes to increase binding affinity (Tm) and greatly improve specificity for discriminating single-nucleotide variants [45]. |
| Wild-Type Blocker Oligonucleotides | Non-extendible oligonucleotides that bind to and suppress the amplification of the wild-type sequence, thereby dramatically improving the sensitivity for detecting low-abundance mutants [44]. |
| Fluorophore-Labeled Probes | TaqMan probes labeled with distinct fluorophores (e.g., FAM, HEX, Cy5, ROX) for each target, enabling simultaneous detection in a multiplex reaction [41] [43]. |
| Synthetic DNA Templates (gBlocks) | Defined, synthetic DNA fragments used as positive controls for assay validation, calibration curve generation, and determining limits of detection and quantification [43] [45]. |
| FFPE-Specific Nucleic Acid Extraction Kits | Optimized kits for recovering fragmented and cross-linked DNA from FFPE tissues, which is the most common source of material in clinical cancer diagnostics [41]. |
The choice of a multiplexing strategy for multi-gene mutation detection is a strategic decision that balances multiplexing capacity, sensitivity, specificity, and technical complexity. Traditional multiplex allele-specific qPCR remains a robust, highly sensitive, and well-validated choice for profiling a defined set of core hotspots. In contrast, emerging technologies like Color Cycle Multiplex Amplification offer a paradigm shift for applications requiring very high multiplexing without specialized instrumentation. For clinical research aimed at informing therapeutic decisions, the analytical validation of any chosen method—demonstrating precision, accuracy, sensitivity, and specificity against a reference standard—is non-negotiable. As the field advances, these qPCR-based multiplexing strategies will continue to be indispensable tools for enabling precision oncology in diverse laboratory settings.
The quantitative polymerase chain reaction (qPCR) and its counterpart for RNA analysis, reverse transcription qPCR (RT-qPCR), are foundational technologies in modern clinical cancer diagnostics research. Their power to precisely quantify nucleic acids enables critical applications from gene expression profiling of cancer biomarkers to validating oncogenic signatures. The reliability of any qPCR result, however, is entirely dependent on the integrity and execution of its core workflow: nucleic acid isolation, reverse transcription, and amplification. This guide provides an objective comparison of methodologies and critical reagents within this workflow, framing them within the essential context of analytical validation for robust and reproducible clinical cancer research.
Nucleic Acid Isolation: A Foundation of Data Quality
The initial step of nucleic acid isolation is arguably the most critical, as it determines the quality and quantity of the starting template. Variations in sample type and isolation methods can significantly impact downstream results, a point rigorously considered during assay development.
Sample Type Considerations
Research often relies on diverse sample types, each with unique challenges. Formalin-fixed paraffin-embedded (FFPE) tissues are a invaluable resource in oncology, but their fixation process can fragment RNA. Studies have demonstrated that successful qPCR with FFPE material requires a minimum RNA quality threshold (e.g., DV200 >15%) [46]. In contrast, fresh-frozen (FF) tissues and cell lines typically yield higher-quality RNA. Furthermore, the use of liquid biopsies, such as peripheral blood, is gaining traction for non-invasive diagnostics. One study successfully validated a five-gene pancreatic cancer signature from blood samples, confirming that with careful processing (e.g., RNA Integrity Number, RIN >7), reliable gene expression data can be obtained from these minimally invasive samples [47].
Comparison of Isolation Method Performance
The choice of isolation kit and protocol directly influences yield, purity, and the presence of enzymatic inhibitors. The following table summarizes key performance metrics for isolation strategies applicable to different sample types in cancer research.
Table 1: Comparison of Nucleic Acid Isolation Methods for Cancer Research Samples
| Sample Type | Recommended Method | Key Performance Metrics | Considerations for Clinical Validation |
|---|---|---|---|
| Cell Cultures | Direct lysis (e.g., Trizol) + column-based purification [48] | High yield and purity (A260/A280 ~1.8-2.0) from ~350μL Trizol [48] [49] | High throughput; suitable for screening; requires DNase treatment to eliminate gDNA [50]. |
| Fresh-Frozen Tissues | Column-based or magnetic bead purification | Yields high-quality RNA (RIN >8); optimal for sensitive gene expression assays | Considered the "gold standard" for analytical validation of new assays. |
| FFPE Tissues | Specialized FFPE RNA extraction kits | Requires DV200 >15%; stable performance across storage conditions [46] | High concordance with FF results can be achieved; fragmentation is a key variable to control [46]. |
| Peripheral Blood | Leucopak or whole blood RNA extraction (e.g., TRIzol LS) | Requires RIN >7 for reliable results in downstream assays [47] | Critical for liquid biopsy applications; sample processing time (<2h) is vital to prevent RNA degradation [47]. |
Reverse Transcription: Converting RNA to cDNA
The reverse transcription (RT) step converts RNA into more stable complementary DNA (cDNA), introducing another major source of technical variability. The choice between one-step and two-step RT-qPCR protocols is dictated by the research application.
- One-Step RT-qPCR: Combines the reverse transcription and PCR amplification in a single tube. This setup is optimal for high-throughput applications and minimizes pipetting steps, reducing the risk of contamination. It is often preferred in diagnostic settings for pathogen detection [51].
- Two-Step RT-qPCR: Physically separates the RT reaction from the PCR amplification. This approach is favored in gene expression studies in cancer research because it allows the generated cDNA to be used for multiple qPCR assays, enabling the validation of several gene targets from a single, consistent cDNA pool [48]. This provides greater flexibility and is more suited for assay development and validation.
A critical experimental protocol for the two-step method, as used in cancer cell research, is detailed below.
Table 2: Detailed Two-Step cDNA Synthesis Protocol [48]
| Step | Parameter | Specification |
|---|---|---|
| Input RNA | Amount | 1000 ng total RNA |
| Reaction Setup | Total Volume | 40 μL |
| Master Mix | SuperScript VILO MasterMix (8 μL) | |
| Water | DNAse/RNAse-free, to volume | |
| Thermocycling | Step 1: Primer Incubation | 25°C for 10 minutes |
| Step 2: Reverse Transcription | 42°C for 60 minutes | |
| Step 3: Enzyme Inactivation | 85°C for 5 minutes | |
| Step 4: Hold | 4°C indefinitely |
qPCR Amplification: Detection, Chemistry, and Assay Design
The amplification phase is where quantitative detection occurs. Key choices here involve detection chemistry and assay design, which directly influence the specificity, sensitivity, and cost of the experiment.
Detection Chemistries: Probe-Based vs. Intercalating Dyes
- TaqMan Probe-Based Assays: These assays (e.g., 5' nuclease assays) use a sequence-specific, dual-labeled probe with a 5' fluorophore and a 3' quencher. The polymerase degrades the probe during amplification, releasing the fluorophore and generating a fluorescent signal [50]. This technology offers superior specificity by ensuring that the detected signal originates only from the intended target, making it the preferred choice for clinical assay validation where distinguishing between homologous genes or splice variants is crucial [50].
- SYBR Green-Based Assays: This chemistry utilizes a dye that fluoresces brightly when intercalated into double-stranded DNA. While more cost-effective and easier to design, SYBR Green will bind to any dsDNA, including primer-dimers and non-specific products, which can lead to false-positive signals [49]. The requirement for post-amplification melt curve analysis to verify amplicon specificity is mandatory [49].
Table 3: Performance Comparison of qPCR Detection Chemistries
| Characteristic | TaqMan (Probe-Based) | SYBR Green (Dye-Based) |
|---|---|---|
| Specificity | High (sequence-specific probe) | Moderate (binds any dsDNA) |
| Background Signal | Low | Higher |
| Multiplexing Potential | High (multiple probe colors) | Low (single dye) |
| Assay Development Cost | Higher (requires probe) | Lower |
| Experimental Verification | Melt curve not required | Melt curve analysis essential [49] |
| Ideal Use Case | Clinically validated assays; multiplexing; splice variant detection [50] | Gene expression screening; assay optimization |
Primer and Probe Design Criteria
A well-designed assay is critical for accurate quantification. Adherence to established design parameters minimizes secondary structures and non-specific amplification.
- Primer Design: Primers should be 18-30 bases long with a Tm of ~60-62°C and GC content between 35-65% (ideally ~50%). Runs of more than four consecutive Gs should be avoided. To prevent amplification of genomic DNA, primers should be designed to span exon-exon junctions [50].
- Probe Design: For TaqMan assays, the probe should have a Tm 5-10°C higher than the primers and be limited to ~30 bases for optimal quenching. A guanine (G) base at the 5' end should be avoided as it can quench common dyes like FAM [50].
- Amplicon: The ideal amplicon size for probe-based assays is between 70-200 bp to ensure efficient amplification [50].
Analytical Validation in Cancer Research: A Case Study
Robust analytical validation ensures that a qPCR assay is reliable, reproducible, and fit for its intended purpose in clinical research. A prime example is the development of a multiplex qPCR assay to predict response to neoadjuvant chemotherapy (NAC) in muscle-invasive bladder cancer (MIBC) [46].
- Robustness Across Sample Types: The assay was validated to perform with high concordance between FFPE and fresh-frozen tissues, a critical feature for leveraging historical archives [46].
- Impact of Pre-Analytical Variables: The assay demonstrated stable performance across different RNA input levels (5–100 ng), and was minimally affected by tissue necrosis or different technicians, proving its reproducibility in a real-world diagnostic context [46].
- Performance Comparison of Commercial Kits: A study comparing five commercial SARS-CoV-2 RT-qPCR kits highlights the importance of independent validation. It found a wide heterogeneity in sensitivity (ranging from sub-optimal to 94%) and inter-test agreement, underscoring that not all kits perform equally even when targeting the same pathogen [52]. This principle directly translates to oncology, where different primer-probe sets for the same cancer biomarker can yield varying results.
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for the qPCR Workflow
| Item | Function | Example/Best Practice |
|---|---|---|
| RNA Extraction Kit | Purifies intact, protein/nuclease-free RNA from complex samples. | Zymo Research Direct-zol RNA Miniprep Kit; HiMedia HiPurAViral RNA kit [48] [52]. |
| Reverse Transcriptase | Synthesizes first-strand cDNA from RNA template. | SuperScript VILO MasterMix [48]. |
| qPCR Master Mix | Contains optimized buffer, dNTPs, polymerase, and fluorescence chemistry. | SYBR Green Master Mix; GoTaq Probe qPCR Master Mix [47] [52]. |
| Primers & Probes | Provide sequence-specific amplification and detection. | IDT PrimeTime qPCR Probe Assays; in-house designed primers [50] [47]. |
| Nuclease-Free Water | Serves as a reaction solvent without degrading nucleic acids. | Critical for preventing RNase and DNase contamination in all steps. |
| Controls | Ensures experimental validity and identifies contamination. | No-template control (NTC), no-RT control, positive amplification control [50]. |
Workflow and Logical Relationships
The following diagram illustrates the major decision points and their implications throughout the qPCR experimental workflow, highlighting how choices at each stage impact the final data quality and application suitability.
The qPCR workflow, from isolation to amplification, is a chain of interdependent steps where optimization and validation at each stage are non-negotiable for generating clinically actionable data in cancer research. The choice between sample types, isolation methods, one-step versus two-step protocols, and probe-based versus dye-based chemistries directly shapes the specificity, sensitivity, and reproducibility of the results. As evidenced by the development of validated assays for predicting chemotherapy response, a rigorous approach to analytical validation—assessing robustness across sample types, RNA inputs, and operators—is paramount. By understanding and objectively comparing the tools and methodologies outlined in this guide, researchers can design and execute qPCR experiments that reliably inform diagnostic and therapeutic development in oncology.
The Fundamentals of the Ct Value in qPCR
The Cycle threshold (Ct), also known as the quantification cycle (Cq) or crossing point (Cp), is a fundamental parameter in quantitative PCR (qPCR) data analysis. It represents the PCR cycle number at which the amplification curve of a reaction intersects a fluorescence threshold set within the exponential phase of amplification [53] [54]. This value is a relative measure of the concentration of the target nucleic acid in the reaction [55].
The relationship between Ct and target concentration is inverse and logarithmic. A lower Ct value indicates a higher starting concentration of the target sequence, as fewer amplification cycles are required for the fluorescent signal to cross the detection threshold. Conversely, a higher Ct value suggests a lower starting concentration [53] [54]. This correlation forms the basis for both qualitative detection and quantitative measurement in qPCR assays.
Mathematically, this relationship is described by the formula: ( Nc = N0 \times E^{Cq} ), where ( Nc ) is the number of amplicons at the Cq cycle, ( N0 ) is the initial number of target copies, and ( E ) is the PCR efficiency (fold-increase per cycle) [56]. The logarithmic form, ( Cq = \log(Nq) - \log(N0) / \log(E) ), clearly shows that the Cq value depends not only on the target concentration (( N0 )) but also on the PCR efficiency (E) and the level of the quantification threshold (( Nq )) [56].
Absolute vs. Relative Quantification Methods
In qPCR data analysis, two primary quantification methodologies are employed: absolute and relative quantification. The choice between them depends on the experimental goals.
Absolute Quantification
Absolute quantification determines the exact amount of a target sequence in a sample, expressed as a specific copy number or concentration. This method requires a standard curve generated from serial dilutions of a reference material with a known concentration [57] [55]. The standard curve plots the Ct values against the logarithm of the known concentrations. The concentration of an unknown sample is determined by comparing its Ct value to this standard curve [57].
This method is crucial for applications such as viral load measurement, determination of gene copy number, and microbiology [55]. The reference standards can be RNA transcripts, plasmid DNA, PCR fragments, or genomic DNA, and their copy number is calculated based on spectrophotometric measurements and known sequence length [57].
Relative Quantification
Relative quantification determines the change in the expression level of a target gene relative to a control sample and one or more reference genes. Instead of providing an exact copy number, it calculates the fold-difference in expression [57] [55]. This method requires a stable reference gene (e.g., a housekeeping gene) whose expression should not vary between experimental conditions [55].
The two most common calculation methods for relative quantification are:
- The Livak Method (2–ΔΔCT): This method assumes that the amplification efficiencies of the target and reference genes are approximately 100% and similar. It involves normalizing the target gene's Ct to the reference gene (ΔCt), and then normalizing this value to a control group (ΔΔCt) [58] [55].
- The Pfaffl Method: This method is more robust as it incorporates the actual amplification efficiencies of the target and reference genes, providing accurate results even when efficiencies are not ideal or differ between assays [58] [55].
The following workflow outlines the key decision points and steps for selecting and performing the appropriate quantification method:
Comparison of Quantification Methods
The table below summarizes the core differences, applications, and requirements for the two main quantification approaches.
| Feature | Absolute Quantification | Relative Quantification |
|---|---|---|
| Objective | Determine exact copy number or concentration of target [57] [55] | Determine fold-change in target levels relative to a control/reference [57] [55] |
| Primary Applications | Viral load, gene copy number, microbiome analysis [55] | Differential gene expression, developmental biology, diagnostic research [55] |
| Requires Standard Curve | Yes, with known-concentration standards [57] | No |
| Requires Reference Gene(s) | No | Yes, must be stably expressed [57] [55] |
| Key Assumption | Standard and target amplify with equal efficiency [57] | Reference gene expression is constant across samples [55] |
| Reported Results | Copy number, genome equivalents, concentration [57] | Fold-change, relative quantity [55] |
Essential Quality Controls and Validation Parameters
Robust interpretation of Ct values and reliable quantification depend on rigorous quality control measures. Adherence to these practices is critical for the analytical validation of qPCR assays, especially in clinical cancer diagnostics.
PCR Efficiency
PCR efficiency (E) is a critical parameter that must be assessed for every assay. It is a ratio of the number of amplified target molecules at the end of a cycle to the number present at the beginning [55]. Efficiency is calculated from a standard curve of serial dilutions using the formula: ( E = (10^{-1/slope} - 1) \times 100 ) [59]. The slope of the standard curve should be between -3.6 and -3.1, corresponding to an efficiency of 90%–110%, which is considered acceptable for reliable quantification [55] [59]. An efficiency of 100% means the target doubles perfectly every cycle, and a 10-fold dilution in template should result in a ~3.32 cycle difference in Ct [59].
Dynamic Range and Limit of Detection
The linear dynamic range is the range of template concentrations over which the fluorescent signal is directly proportional to the input concentration [8]. It is typically determined using a 6-8 order of magnitude dilution series, with an R² value of ≥ 0.980 considered acceptable for the standard curve [8]. The Limit of Detection (LOD) is the lowest concentration of the target that can be reliably detected, while the Limit of Quantification (LOQ) is the lowest concentration that can be reliably quantified with acceptable precision and accuracy [60] [8].
Specificity and Controls
Analytical specificity (or exclusivity) is the ability of an assay to distinguish the target from non-target sequences, such as closely related species or family members [60] [8]. This is typically confirmed in silico during assay design and validated experimentally by testing against a panel of non-targets. Inclusivity ensures the assay detects all intended target variants or strains [8]. Each run should include appropriate controls, such as no-template controls (NTC) to detect contamination, and positive controls to confirm assay functionality.
Experimental Protocols for Key Validations
Protocol for Determining PCR Efficiency and Dynamic Range
This protocol is essential for both absolute quantification and for validating an assay's performance before relative quantification studies [55].
- Standard Preparation: Prepare a minimum of five 10-fold serial dilutions of a known standard (e.g., plasmid DNA, PCR product, or synthetic oligonucleotide) [8] [55].
- qPCR Run: Amplify each dilution in triplicate using the same qPCR master mix and cycling conditions intended for sample analysis.
- Data Analysis:
- Record the average Ct value for each dilution.
- Plot the average Ct (y-axis) against the logarithm of the input concentration or dilution factor (x-axis).
- Perform linear regression to obtain the slope and R² value.
- Calculate the PCR efficiency using the formula: ( \text{Efficiency (%)} = (10^{-1/\text{slope}} - 1) \times 100 ) [55] [59].
- Acceptance Criteria: The assay is acceptable if the efficiency is between 90% and 110% and the R² is ≥ 0.980 [55] [59].
Protocol for Relative Quantification via the Pfaffl Method
This method is recommended when the amplification efficiencies of the target and reference genes are not equal or precisely 100% [58] [55].
- Sample Preparation: Run the target and reference genes for all test and control samples under validated conditions.
- Calculate Efficiency-Corrected ΔCt (wΔCT): For each sample, calculate the efficiency-weighted ΔCT value:
( w\Delta CT = \log2(E{target}) \times CT{target} - \log2(E{ref}) \times CT{ref} ) [58].
- Here, ( E{target} ) and ( E{ref} ) are the amplification efficiencies (e.g., 1.95 for 95% efficiency) of the target and reference genes, respectively.
- Calculate Fold Change (FC): Compute the fold change between the treatment (Tr) and control (Co) groups using the mean ( w\Delta CT ) values: ( FC = 2^{-(w\Delta \overline{CT}{Tr} - w\Delta \overline{CT}{Co})} ) [58].
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key reagents and materials required for developing and running a validated qPCR assay.
| Item | Function / Description | Key Considerations |
|---|---|---|
| Sequence-Specific Primers & Probes | To specifically amplify and detect the target sequence. | TaqMan probe-based assays offer superior specificity over intercalating dyes like SYBR Green, reducing false positives [59]. |
| qPCR Master Mix | Contains DNA polymerase, dNTPs, buffer, and salts. May include passive reference dyes (ROX). | Fluorescence can be affected by pH and salt concentration; use high-quality, consistent formulations [54]. |
| Reference Standard | For absolute quantification and efficiency determination. | Can be plasmid DNA, PCR fragments, or in vitro transcribed RNA. Copy number must be known [57]. |
| Reference Gene Assay | For relative quantification normalization. | Gene(s) must be stably expressed across all sample conditions (e.g., GAPDH, Actin). Validation is required [55]. |
| Nuclease-Free Water | Solvent for preparing reaction mixes. | Essential to prevent degradation of primers, probes, and templates. |
| qPCR Plates & Seals | Vessels for the reaction. | Must be optically clear for fluorescence detection and securely sealed to prevent evaporation. |
Advanced Analysis and Reporting for Rigor and Reproducibility
To ensure findings are robust and reproducible, especially in a clinical research context, several advanced practices are recommended.
- Statistical Analysis: For relative quantification, the efficiency-weighted ΔCt values (( w\Delta CT )) follow a normal distribution, allowing for the application of standard parametric statistical tests like t-tests or ANOVA for mean comparisons [58]. Analysis of Covariance (ANCOVA) is a flexible linear modeling approach that can offer greater statistical power and robustness compared to the 2−ΔΔCT method alone [61].
- Adherence to MIQE Guidelines: The Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines provide a checklist to ensure the reliability of results, promote inter-laboratory consistency, and increase experimental transparency [61] [8]. Reporting should include raw fluorescence data, exact primer/probe sequences, PCR efficiency, and evidence of reference gene stability.
- Data Sharing and FAIR Principles: Sharing raw qPCR fluorescence data along with detailed analysis scripts enhances transparency and allows other researchers to reproduce the analysis and findings [61]. Adhering to the FAIR (Findable, Accessible, Interoperable, Reproducible) principles is becoming a standard for rigorous scientific reporting.
Optimizing for Precision: A Step-by-Step Guide to Enhanced qPCR Performance
In the field of clinical cancer diagnostics research, the reliability of quantitative PCR (qPCR) data is paramount. The path from a discovery-oriented research use only (RUO) assay to a clinically validated tool requires meticulous optimization and validation. This process ensures that gene expression signatures, such as those predicting cancer prognosis or therapeutic response, are measured with the precision, accuracy, and robustness needed for clinical applications. A systematic approach to qPCR optimization is not merely a best practice but a fundamental requirement for generating data that can reliably inform diagnostic and therapeutic decisions. This guide outlines a comprehensive nine-step workflow, framed within the context of analytical validation, to achieve a perfectly optimized qPCR assay suitable for the rigorous demands of clinical cancer research.
A Systematic 9-Step Optimization Workflow
The following workflow provides a step-by-step guide for optimizing your qPCR assays, with a focus on meeting the performance criteria required for clinical research.
Step 1: Sample Preparation and Quality Control
The foundation of any reliable qPCR assay is high-quality input material. For RNA expression studies in cancer biomarkers, RNA samples should be of consistent quantity and quality to enable accurate comparison of gene expression levels [62]. The integrity of the purified nucleic acids must be verified using methods such as gel electrophoresis or a bioanalyzer [62]. This step is critical in a clinical context, as variations in sample quality are a major contributor to the lack of reproducibility in biomarker studies [60].
Step 2: Assay and Reagent Selection
Begin by using a high-quality, manufacturer-recommended qPCR reagent kit and be consistent with your choices [62]. The selection between one-step and two-step RT-qPCR protocols depends on the application. One-step RT-qPCR is advantageous for high-throughput processing of many samples with few targets, while two-step RT-qPCR allows for separate optimization of the reverse transcription and PCR steps, provides greater sensitivity for limited samples, and enables the creation of cDNA stocks for future analysis [63].
Step 3: Amplicon and Template Design
- Amplicon Length: Ideal template lengths are between 50 and 200 base pairs to ensure the reaction proceeds quickly and efficiently [62].
- Secondary Structure: The amplicon template should be checked for secondary structures that could hinder primer and probe binding [62] [64].
- Target Specificity: The target sequence must be unique to the genome of interest. For assays detecting residual host cell DNA in biologics, for example, this ensures the assay does not cross-react with other genetic material [11].
Step 4: Primer and Probe Design
Careful design of primers and probes is crucial for assay specificity and efficiency.
- Primer Guidelines:
- GC Content: Optimal GC content is between 40% and 60% [62].
- Melting Temperature (Tm): Ideal Tm is between 58°C and 65°C, with a maximum difference of 4°C between forward and reverse primers [62].
- 3' End: The last five nucleotides at the 3' end should contain no more than two Gs or Cs. Avoid three consecutive G or C bases (runs) [62].
- Length: Primers are typically designed to be at least 28 bp to reduce primer-dimer formation [62].
- Probe Guidelines:
Step 5: Thermal Cycler Protocol Optimization
Optimize the thermocycling protocol one parameter at a time [62]:
- Initial Denaturation/Activation: Typically 95°C for 30 seconds for short DNA templates, but duration may need adjustment for complex templates or to activate hot-start polymerases [62].
- Cycle Number: Standard is 40 cycles, but this can be reduced to 30 if the amplification curve reaches the plateau phase early [62].
- Denaturation: For short templates (<300 bp), 95°C for 5-15 seconds is often sufficient [62].
- Annealing & Extension: For a 2-step protocol, a combined annealing/extension step at ~60°C for 1 minute is a common starting point. The temperature can be optimized in 0.1°C increments [62]. Note that faster protocols with shortened cycle times may compromise sensitivity and increase variability compared to universal cycling conditions [65].
Step 6: Performance Parameter Validation
After optimization, key performance parameters must be validated to ensure the assay is fit for its clinical research purpose [60] [64]. The table below outlines these critical parameters.
Table 1: Key Performance Parameters for qPCR Assay Validation
| Parameter | Definition | Target Value/Range |
|---|---|---|
| Amplification Efficiency | The rate at of amplicon production per cycle. | 90–105% (Ideal: 100%) |
| Precision | The closeness of agreement between independent measurements. | Low variability (e.g., RSD <25% for low template concentrations) [11]. |
| Analytical Sensitivity (LOD) | The lowest concentration of the analyte that can be reliably detected. | Defined by the application; must be demonstrated [60] [66]. |
| Analytical Specificity | The ability to distinguish the target from non-target sequences. | No amplification in negative controls or non-target samples [60] [11]. |
| Linearity & Range | The interval of concentrations where results are directly proportional. | R² ≥ 0.985 for the standard curve [64]. |
| Robustness | The assay's tolerance to small, deliberate changes in protocol. | Maintains performance despite minor variations [64]. |
Step 7: Standard Curve and Data Analysis
The standard curve is the primary tool for assessing performance. Generate a dilution series of a template with known concentration and plot the Cq values against the log of the concentration [64]. An R² value of ≥0.985 indicates excellent linearity. Non-linearity at high concentrations suggests reaction saturation, while non-linearity at low concentrations may indicate the level is below the assay's sensitivity [64]. For data analysis, set the fluorescence threshold above the baseline and within the exponential phase of amplification, adjusting it for different dyes to ensure accurate Cq determination [64].
Step 8: Normalization Strategy
Employ a multi-faceted normalization approach to control for technical variability:
- Sample Normalization: Normalize purified nucleic acids through absorbance or fluorescence-based quantification [64].
- Assay Normalization: Use internal quality controls, such as reference genes, and external controls with known concentrations to assess PCR efficiency [64].
- Analysis Normalization: Include a passive reference dye (e.g., ROX) in the reaction mix to normalize for fluorescent signal intensity variations caused by pipetting inconsistencies or evaporation [64].
Step 9: System and Consumable Considerations
- qPCR Instrument: Newer systems using LEDs provide more uniform light distribution, which can improve detection and obviate the need for dyes like ROX for correction [62].
- Reaction Plates: Using white wells with ultra-clear caps can reduce light distortion from neighboring wells and increase signal reflection for optimal detection [62].
- Pipetting Accuracy: To minimize systemic variables, the sample should comprise about 40% of the total reaction volume, with the master mix making up the remaining 60%, allowing for more accurate pipetting in the mid-range of the pipette [64].
Experimental Protocols for Key Validation Experiments
Assay Linearity and Dynamic Range
This protocol determines the range of template concentrations over which the assay provides accurate and precise results.
- Methodology: Prepare a standard stock solution of the target nucleic acid (e.g., gDNA or cDNA) and serially dilute it across at least five orders of magnitude (e.g., from 10 ng/µL to 0.1 fg/µL) [11]. Run each dilution in triplicate on the qPCR platform.
- Data Analysis: Plot the mean Cq value for each standard against the log of its starting concentration. Perform linear regression analysis. The slope of the line is used to calculate amplification efficiency (E) using the formula: E = (10^(-1/slope) - 1) * 100%. The R² coefficient should be ≥0.985 [64].
Determination of Limit of Detection (LOD) and Limit of Quantification (LOQ)
This experiment establishes the lowest levels of analyte that can be detected and reliably quantified.
- Methodology: Prepare a minimum of six independent replicates of samples containing the target template at concentrations near the expected detection limit (e.g., 0.003 pg/reaction to 0.03 pg/reaction) [11]. Include no-template controls (NTCs) to confirm the absence of contamination.
- Data Analysis:
Analytical Specificity Testing
This protocol verifies that the assay detects only the intended target without cross-reacting with similar sequences.
- Methodology: Test the assay against a panel of nucleic acids from related but distinct organisms or cell lines. For a Vero cell DNA assay, for example, this would include testing against DNA from CHO, HEK293, and other common cell lines, as well as bacterial strains [11].
- Data Analysis: The assay should yield positive results only for the specific target (e.g., Vero DNA) and show no amplification (Cq > 40 or undetected) for all non-target samples in the panel [11].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for qPCR Optimization and Validation
| Item | Function & Importance |
|---|---|
| Nucleic Acid Isolation Kits | High-quality isolation is the first step to reliable results. Kits should be matched to the sample type (e.g., tissue, blood) to maximize yield and purity [62] [64]. |
| qPCR Master Mix | A high-quality, optimized master mix containing DNA polymerase, dNTPs, and buffer is essential for robust and efficient amplification. Consistency is key [62]. |
| Validated Primers & Probes | Primers and probes should be designed according to strict guidelines (e.g., Tm, GC content) and synthesized with high purity to ensure specificity and sensitivity [62] [64]. |
| Reference Genes | For gene expression normalization, reference genes must be validated for expression stability across the specific tissues and experimental conditions being studied [67]. |
| Nuclease-Free Water | The solvent for reaction preparation; must be free of nucleases to prevent degradation of primers, probes, and templates. |
| Standard Reference Material | A sample of known identity and concentration (e.g., genomic DNA National Standard) used to create standard curves for absolute quantification and assay validation [11]. |
Workflow Visualization
The following diagram illustrates the logical flow of the systematic 9-step optimization workflow, showing how each stage contributes to the final validated assay.
Systematic 9-Step qPCR Optimization Workflow
A methodical, nine-step approach to qPCR optimization is non-negotiable for generating data that meets the stringent requirements of clinical cancer diagnostics research. By rigorously addressing each stage—from initial sample quality control to final data normalization and performance validation—researchers can bridge the gap between basic research and clinically applicable assays. This systematic workflow ensures that qPCR results are not only statistically significant but also analytically valid, reproducible, and ultimately fit-for-purpose in the translation of molecular biomarkers into tools for patient diagnosis, prognosis, and prediction of therapeutic response.
Primer and Probe Design Refinements to Maximize Specificity and Sensitivity
In clinical cancer diagnostics research, the accuracy of quantitative PCR (qPCR) is paramount. The exquisite sensitivity and specificity required to detect low-frequency mutations, quantify gene expression biomarkers, or identify oncogenic pathogens hinge directly on the optimal design of primers and probes [28]. Despite the foundational nature of PCR, a significant number of published assays remain suboptimal, leading to reduced technical precision, false positives, or false negatives that can compromise diagnostic validity [28]. This guide objectively compares design strategies and their performance impacts, providing a framework for the analytical validation essential for robust clinical cancer research assays.
Foundational Principles of qPCR Oligonucleotide Design
Core Design Parameters for Primers
Successful qPCR assays are built upon primers that ensure specific and efficient amplification. Adherence to the following parameters is critical:
- Length and Melting Temperature (Tm): Primers should be 18-30 bases long, with an optimal Tm of 60–64°C. The Tms of the forward and reverse primer should not differ by more than 2°C to ensure both bind simultaneously and efficiently [68].
- GC Content: The GC content should be between 35% and 65%, with an ideal of 50%. Sequences should avoid regions of four or more consecutive G residues [68].
- Specificity and Secondary Structures: Primers must be free of strong secondary structures and self-complementarity. The ΔG value for any self-dimers, hairpins, and heterodimers should be weaker (more positive) than –9.0 kcal/mol [68]. It is crucial to use tools like NCBI BLAST to ensure primers are unique to the desired target sequence [68].
Advanced Design Considerations for Probes
Hydrolysis probes (e.g., TaqMan) are a gold standard in quantitative real-time PCR. Their design requires additional refinements:
- Location and Tm: The probe should be in close proximity to, but not overlapping, a primer-binding site. Its Tm should be 6–10°C higher than the Tm of the primers to ensure it binds before the primers and is fully hybridized during amplification [68] [69].
- Sequence and Quenchers: The 5' end of the probe should not be a guanine base, as it can quench the fluorophore. Double-quenched probes, which incorporate an internal quencher like ZEN or TAO, are recommended as they provide lower background and higher signal compared to single-quenched probes [68].
- Amplicon Characteristics: The amplicon itself should typically be 70–150 base pairs for optimal amplification efficiency [68]. When working with RNA targets, designing assays to span an exon-exon junction can reduce false positives from genomic DNA amplification [68].
Table 1: Key Design Parameters for qPCR Primers and Probes
| Component | Parameter | Optimal Range | Critical Considerations |
|---|---|---|---|
| Primers | Length | 18–30 bases | Based on Tm and binding efficiency. |
| Melting Temperature (Tm) | 60–64°C | Both primers should be within a 2°C Tm range. | |
| GC Content | 35–65% (50% ideal) | Avoid runs of 4 or more consecutive Gs. | |
| Specificity Check | BLAST analysis | Essential to confirm uniqueness to the target. | |
| Probes | Tm Relative to Primers | +6–10°C | Ensures probe is bound during primer extension. |
| Placement | Close to primer, no overlap | Prevents steric hindrance. | |
| 5' End | Avoid "G" | Prevents quenching of the fluorophore. | |
| Quencher Type | Double-quenched (e.g., ZEN) | Lowers background, increases signal-to-noise. | |
| Amplicon | Length | 70–150 bp | Allows for efficient amplification. |
| Location (for cDNA) | Span exon-exon junction | Reduces genomic DNA amplification. |
Comparative Analysis of Design Performance in Published Studies
Case Study 1: Specificity Failure in Leishmania Diagnostics
A 2025 study investigating visceral leishmaniasis diagnostics provides a stark example of the consequences of flawed probe design. Researchers evaluated the established LEISH-1/LEISH-2 primer pair with a TaqMan MGB probe and encountered unexpected amplification in all serologically negative dog and wild animal samples [70]. This critical specificity failure was primarily attributed to the probe. Subsequent in silico analysis revealed structural incompatibilities and low selectivity in the original probe sequence. To address this, the team designed a new oligonucleotide set, "GIO," which computational analyses showed had superior structural stability and specificity, highlighting the necessity for rigorous in silico and experimental validation before an assay is deployed in clinical or research settings [70].
Case Study 2: Sensitivity and Specificity Trade-offs in Equine Herpesvirus Detection
A direct comparison of three WOAH-recommended qPCR primer-probe sets for equid alphaherpesvirus 1 (EqAHV1) demonstrates the delicate balance between sensitivity and specificity [71].
- The High-Sensitivity, Low-Specificity Set (gB1P): This set, targeting the glycoprotein B (gB) gene, successfully detected low copy numbers (≤10 copies) of EqAHV1. However, it also erroneously amplified the closely related EqAHV4, rendering it unsuitable for diagnostic applications where distinguishing between these viruses is crucial [71].
- The Low-Sensitivity Set (gC1): The gC1 set, an ISO 17025-accredited method, failed to detect samples with ≤10 copies of EqAHV1. Its sensitivity could not be improved through optimization of primer and probe concentrations, suggesting a fundamental design limitation [71].
- The Optimal Performer (gB1H): The gB1H set, also targeting the gB gene, detected low copy numbers with high sensitivity and did not cross-react with EqAHV4. When tested on 120 clinical nasal swabs, it demonstrated slightly higher sensitivity than a legacy assay (gB1D), establishing it as the most reliable option [71].
This study underscores that primer and probe sequences must be evaluated not just for sheer sensitivity, but for their ability to uniquely identify the intended target within a complex biological background.
Table 2: Performance Comparison of Primer-Probe Sets from Clinical Evaluations
| Study / Target | Primer-Probe Set | Reported Sensitivity | Reported Specificity | Key Finding |
|---|---|---|---|---|
| Leishmania chagasi [70] | LEISH-1/LEISH-2 (with TaqMan MGB) | Not fully quantified | Failed (amplified negative controls) | Probe design flaws led to critical specificity failures. |
| New "GIO" Set | (Pending experimental validation) | High (in silico analysis) | Computational redesign showed superior structural stability and specificity. | |
| Equid Alphaherpesvirus 1 (EqAHV1) [71] | gB1P | Detected ≤10 copies | Low (cross-reacted with EqAHV4) | High sensitivity compromised by lack of specificity. |
| gC1 | Failed at ≤10 copies | 100% (no cross-reactivity) | Accredited method showed critically low sensitivity. | |
| gB1H | Detected ≤10 copies (high) | 100% (no cross-reactivity) | Optimal combination of high sensitivity and specificity. |
Experimental Protocols for Assay Validation
Protocol for In Silico Specificity and Structure Analysis
Before synthesizing oligonucleotides, comprehensive computational checks are mandatory.
- Sequence Alignment: Retrieve all relevant target and homologous sequences from curated databases (e.g., NCBI RefSeq). Use multiple sequence alignment software (e.g., MAFFT) to identify unique regions for primer and probe binding [70].
- Oligonucleotide Design: Use specialized tools (e.g., PrimerQuest, Eurofins GCG-based tools) that incorporate parameters for qPCR. Input the target sequence and set constraints for Tm, GC content, and amplicon length [68] [30].
- Specificity Check (BLAST): Perform a BLAST search for each primer and probe sequence against the entire genome of the organism(s) in the sample to ensure uniqueness [68] [28].
- Secondary Structure Analysis: Analyze all oligonucleotides for self-dimers, cross-dimers, and hairpins using tools like OligoAnalyzer. Reject designs with a ΔG value more negative than –9.0 kcal/mol [68].
- Folding and SNP Check: Use tools like UNAFold or RNAfold to predict secondary structures in the target amplicon that could impede amplification [68]. Verify that the probe binding site does not contain known single-nucleotide polymorphisms (SNPs) for conserved targets.
Protocol for Empirical Optimization of Annealing Temperature (Ta)
The optimal annealing temperature (Ta) must be determined experimentally, as it is dependent on the specific primers, probe, and master mix [28].
- Reaction Setup: Prepare a single positive control sample and a no-template control (NTC) for each condition.
- Temperature Gradient: Run the qPCR reaction using a thermal gradient across a range of temperatures (e.g., 55°C to 65°C) [68].
- Analysis: Identify the Ta that yields the lowest Cq (quantification cycle) for the positive control with no signal in the NTC. A robust assay will perform well over a broad temperature range, while an assay that works only in a narrow window is less robust [28].
Diagram 1: qPCR Assay Design and Validation Workflow. The pathway highlights critical, color-coded validation steps required for a clinically relevant assay.
A successful qPCR assay relies on high-quality reagents and bioinformatic tools.
Table 3: Research Reagent Solutions for qPCR Assay Development
| Item | Function / Description | Example Providers / Tools |
|---|---|---|
| Custom TaqMan Probes | Dual-labeled hydrolysis probes (fluorophore + quencher) for specific target detection. Available with MGB, QSY, or TAMRA quenchers for multiplexing. | Thermo Fisher Scientific [31] |
| qPCR Primers | Custom, desalted primers for specific target amplification. Available in dry or liquid format. | Thermo Fisher Scientific, IDT [68] [31] |
| qPCR Assay Design Tools | Bioinformatics tools for selecting optimal primer and probe sequences based on input parameters. | Eurofins Genomics, IDT SciTools (PrimerQuest, OligoAnalyzer) [68] [30] |
| In Silico Analysis Tools | Software for analyzing oligonucleotide properties, secondary structures, and specificity. | OligoAnalyzer Tool, UNAFold Tool, Primer-BLAST [68] |
| Master Mixes | Optimized buffered solutions containing DNA polymerase, dNTPs, and Mg2+ for robust qPCR amplification. | Various manufacturers (requires empirical testing) [28] |
The journey to a maximally specific and sensitive qPCR assay is systematic and iterative. It begins with meticulous in silico design adhering to established principles for primers and probes, and culminates in rigorous experimental validation. As demonstrated by comparative studies, failure at any step—from BLAST analysis to specificity testing against near-neighbor species—can render an assay unsuitable for the exacting demands of clinical cancer diagnostics research. By leveraging the available tools and validation protocols, researchers can ensure their qPCR data are both technically precise and biologically meaningful.
In the rigorous field of clinical cancer diagnostics research, the reliability of quantitative polymerase chain reaction (qPCR) assays is paramount. These assays depend fundamentally on the precise formulation of the PCR master mix, a pre-blended solution that ensures consistency, reduces setup errors, and enhances throughput. The analytical validation of diagnostic tests, particularly for detecting cancer biomarkers, requires robust and reproducible performance. At the heart of this performance are three critical components: Mg2+ ions, which act as an essential enzymatic cofactor; dNTPs, the fundamental building blocks of DNA; and the DNA polymerase, the engine of amplification. The selection and optimization of these components directly influence the sensitivity, specificity, and accuracy of qPCR assays, determining their suitability for clinical applications such as detecting minimal residual disease, profiling gene expression, or identifying somatic mutations. This guide provides an objective comparison of these core components, supported by experimental data, to inform their selection for validated clinical research assays.
The Core Components of a PCR Master Mix
A PCR master mix is a centralized mixture containing all the common reagents required for the DNA amplification process. Its use standardizes reactions, minimizes pipetting steps and contamination risk, and is indispensable for high-throughput applications commonly found in diagnostic laboratories [72] [73]. While master mixes can be prepared in-house, most clinical and research labs rely on commercial formulations, which are manufactured under stringent quality control to ensure lot-to-lot reproducibility, a critical factor for assay validation and regulatory compliance [9] [74].
The table below outlines the universal components of a standard master mix and their primary functions.
Table 1: Essential Components of a Standard PCR Master Mix
| Component | Function in the Reaction | Key Considerations for Clinical Diagnostics |
|---|---|---|
| DNA Polymerase | Enzyme that synthesizes new DNA strands by adding nucleotides to the growing primer chain. | Fidelity (accuracy), processivity (speed/continuity), and hot-start capability to minimize non-specific amplification at room temperature [75] [73]. |
| dNTPs (deoxynucleoside triphosphates) | The building blocks (dATP, dCTP, dGTP, dTTP) for new DNA synthesis. | High purity (e.g., 99%) is crucial to prevent premature termination of DNA chains; balanced concentrations are required for accurate base incorporation [72] [73]. |
| Mg2+ (Magnesium Ions) | An essential cofactor for DNA polymerase activity; it stabilizes the DNA duplex and the enzyme's active structure. | Concentration is critically optimized; it affects enzyme efficiency, primer annealing, and amplicon specificity. Often supplied as MgCl₂ or MgSO₄ [72] [76]. |
| Buffer System | Maintains the optimal pH and ionic conditions (e.g., KCl) for polymerase activity and stability. | Provides a stable chemical environment; often includes additives or stabilizers to enhance efficiency, especially with challenging samples like blood [72] [75]. |
Detailed Analysis of Key Components & Supporting Data
Magnesium Ions (Mg2+): The Crucial Cofactor
Magnesium ions are fundamental to the catalytic machinery of DNA polymerase. Structural and kinetic studies on the Klenow fragment of DNA polymerase I reveal that the active site employs two magnesium ions (metal A and metal B) coordinated by invariant aspartate residues (Asp705 and Asp882) [76]. Metal A facilitates the deprotonation of the primer's 3'-OH group, enabling nucleophilic attack on the incoming dNTP. Metal B stabilizes the structure of the dNTP and assists in the departure of the pyrophosphate leaving group [76]. The precise coordination of these ions is so critical that mutation of the aspartate ligands reduces polymerase activity to barely detectable levels [76].
Experimental Insight: Stopped-flow fluorescence assays demonstrate that the initial conformational changes in the polymerase-DNA-dNTP complex can occur at very low Mg2+ concentrations. However, higher concentrations are imperative for the final chemical step of covalent bond formation, consistent with the model that the second metal ion (metal A) may enter the active site after the initial fingers-closing conformational change [76]. This underscores that while minimal Mg2+ is needed for substrate binding, optimal concentrations are mandatory for efficient catalysis in diagnostic assays where every amplification cycle counts.
dNTPs: The Substrates and Building Blocks
Deoxynucleoside triphosphates provide both the energy and the nucleotides for the DNA synthesis reaction. The incorporation of a dNTP into the growing DNA strand involves a nucleophilic attack by the 3'-OH group of the primer on the α-phosphate of the dNTP, a reaction catalyzed by the Mg2+-bound polymerase active site. The subsequent release of pyrophosphate (PPi) provides the driving force for the reaction. The ability of DNA polymerases to discriminate against ribonucleotides (NTPs) is vital for maintaining genomic integrity. This specificity often arises from a steric gate mechanism, where a bulky amino acid side chain in the active site (e.g., a tyrosine) clashes with the 2'-OH group of a ribonucleotide, preventing its incorporation [77].
Table 2: Comparative Analysis of DNA Polymerase Formulations
| Polymerase / Master Mix | Key Features | Best-Suited Clinical Research Applications | Fidelity & Performance Notes |
|---|---|---|---|
| Standard Taq (e.g., ReadyMix) | Robust, cost-effective; available in hot-start formulations. | Routine amplification, high-throughput screening of known targets [73]. | Standard fidelity; suitable for well-optimized, non-multiplexed assays. |
| High-Fidelity (e.g., Q5, KOD) | Proofreading activity (3'→5' exonuclease) for error correction. | NGS library construction [75], detection of low-frequency somatic variants [73]. | ~50-100x higher accuracy than Taq; essential for sequencing and mutation detection. |
| Allele-Specific (e.g., PACE) | Patented chemistry for endpoint fluorescent genotyping. | SNP genotyping for pharmacogenetics, cancer risk alleles; uses unlabeled primers [72]. | High accuracy for SNP/Indel detection; cost-effective vs. probe-based assays. |
| Direct PCR (e.g., Q5 Blood) | Optimized to tolerate PCR inhibitors. | Genotyping directly from blood samples without DNA purification [75]. | Saves time and cost in sample processing; robust for crude samples. |
| One-Step RT-qPCR (e.g., TaqPath DuraPlex) | Integrates reverse transcription and qPCR in a single tube; benchtop-stable. | High-throughput viral/bacterial pathogen detection (relevant to oncology infections); gene expression from RNA [74] [78]. | Enables automation; stable at room temperature for >8 hours; high inhibitor tolerance. |
Optimizing Component Concentration: Experimental Evidence
The conventional use of master mixes at a 1x final concentration is not always a rigid requirement. Research validated during reagent-scarce periods, such as the COVID-19 pandemic, demonstrated that many master mixes could be diluted to a 0.5x or even 0.4x final concentration without a significant loss in qPCR efficiency or sensitivity for many assays [79]. In one study, amplification of a target (Assay D) with twelve different master mixes at reduced concentrations showed remarkably resilient Cq values compared to the standard 1x concentration [79].
Experimental Protocol: Master Mix Dilution Validation
- Preparation: Serially dilute a commercial 2x master mix with PCR-grade water to create working solutions at 1.0x, 0.8x, 0.7x, 0.5x, and 0.4x final concentrations.
- Assay Setup: Combine the diluted master mixes with a constant amount of template DNA (or RNA for RT-qPCR), primers, and probes. Include a no-template control (NTC) for each concentration to check for contamination.
- qPCR Run: Perform the qPCR run using the manufacturer's recommended thermal cycling protocol.
- Data Analysis: Calculate the amplification efficiency (E) from a standard dilution series using the formula: E = [10^(-1/slope)] - 1. The ideal efficiency is 100% (slope = -3.32). Compare the Cq values, efficiency, and fluorescence (ΔRn) across the different master mix concentrations to determine the acceptable dilution threshold before performance degradation (typically observed at 0.3x) [79].
This strategy can extend precious reagents and reduce costs, but it must be rigorously validated for each specific assay and sample type within the diagnostic laboratory before implementation [79] [9].
A Scientist's Toolkit: Key Reagents for Validation
The following list details essential materials and reagents required for the development and analytical validation of a qPCR assay in a clinical research context.
- Master Mix: The core reagent. Selection should be based on application (e.g., SYBR Green or probe-based for qPCR, high-fidelity for mutation detection) and validated for the specific instrument platform [74] [73].
- Primers & Probes: Sequence-specific oligonucleotides. Must be designed to target the biomarker of interest (e.g., a gene fusion, SNP, or expression marker) and optimized for concentration to maximize specificity and efficiency [9].
- Nuclease-Free Water: A critical component to avoid degradation of reagents and templates by environmental RNases and DNases.
- Positive Control Template: A well-characterized sample containing the target sequence at a known concentration. Essential for run-to-run monitoring of assay performance and for generating standard curves for quantification [9].
- Negative Control (NTC): A reaction mixture containing all components except the template DNA/RNA. Used to detect contamination or primer-dimer formation [9].
- Inhibition Control: A control sample spiked with a known amount of the target to identify the presence of PCR inhibitors in the sample matrix (e.g., from blood, stool) [9] [78].
Workflow for qPCR Assay Validation
The diagram below illustrates the critical stages of developing and validating a qPCR assay for clinical cancer research, from initial component selection to final data analysis.
In clinical cancer diagnostics research, the analytical validation of qPCR assays is paramount. Among the critical factors influencing assay performance—including sensitivity, specificity, and reproducibility—the optimization of thermocycling conditions stands out. The annealing temperature and cycle number are not mere procedural details; they are fundamental parameters that directly determine the efficacy of nucleic acid amplification and the accuracy of subsequent quantification [60]. Incorrect parameters can lead to false positives, false negatives, and unreliable data, ultimately compromising clinical decision-making. This guide objectively compares the performance of different optimization strategies and parameters, providing structured experimental data to inform robust assay development for clinical research.
Core Principles of Thermocycling Optimization
The annealing temperature (Tₐ) is the temperature at which primers bind to their complementary target sequences. An optimal Tₐ is high enough to promote specific primer binding and avoid mispriming, yet low enough to allow efficient hybridization. The cycle number determines the number of amplification cycles performed. While more cycles can increase the signal from low-abundance targets, excessive cycling can plateau the reaction, promote nonspecific amplification, and is not a substitute for poor reaction efficiency [80].
The goal of optimization is to find the condition that maximizes the amplification of the specific target while minimizing background noise. This process is a core component of the "fit-for-purpose" validation required for Clinical Research (CR) assays, which fill the gap between Research Use Only (RUO) and fully certified In Vitro Diagnostics (IVD) [60].
Experimental Methodologies for Optimization
Researchers employ several established methods to determine optimal thermocycling conditions. The methodologies below are foundational to the experimental data presented in this guide.
Annealing Temperature Gradient
A thermal cycler with a gradient functionality is used to test a range of annealing temperatures (e.g., 55°C to 65°C) across different wells of the same PCR plate within a single run [81] [80]. This allows for the direct comparison of amplification efficiency and specificity at different temperatures. The output, typically an amplification plot, is analyzed to identify the temperature that yields the lowest Cycle Threshold (Cₜ) value with a clean, sigmoidal curve, indicating high efficiency and specificity.
High-Resolution Melting (HRM) Analysis
Following amplification, HRM analysis can be used to verify amplicon specificity. The method involves slowly increasing the temperature and precisely monitoring the dissociation (melting) of double-stranded DNA. Each amplicon has a specific melting temperature (Tₘ) based on its GC content, length, and sequence [82] [83]. The presence of a single, sharp peak in the melting curve confirms a specific, homogeneous PCR product, whereas multiple or broad peaks indicate non-specific amplification or primer-dimer formation [83].
Digital PCR (dPCR) for Absolute Quantification
Droplet Digital PCR (ddPCR) partitions a sample into thousands of nanoliter-sized droplets, and PCR amplification occurs within each droplet. This allows for absolute quantification of the target nucleic acid without the need for a standard curve and is less susceptible to the effects of PCR inhibitors [80]. In optimization, ddPCR can be used to measure the absolute number of positive amplifications and mean fluorescence intensity, providing a robust metric to compare the performance of different primer-probe sets and annealing temperatures [80] [84].
Comparative Performance Data
The following tables synthesize experimental data from published studies to illustrate the impact of different optimization strategies.
Table 1: Impact of Annealing Temperature on Assay Performance in Pathogen Detection
| Target / Assay | Annealing Temp Range Tested | Optimal Temp Identified | Key Performance Outcome at Optimal Temp | Citation |
|---|---|---|---|---|
| Entamoeba histolytica qPCR | 59°C vs 62°C | 62°C | Higher amplification efficiency and specificity; reduced non-specific signal [80]. | [80] |
| SARS-CoV-2 Variant Melting Curve Assays | 60°C (fixed) | N/A | Melting curve analysis at 60°C enabled sensitive and specific identification of single-nucleotide polymorphisms (SNPs) with >99.4% agreement to sequencing [82]. | [82] |
| Plasmodium spp. HRM Analysis | 60°C (fixed) | N/A | HRM analysis post-amplification allowed clear differentiation of P. falciparum and P. vivax based on Tm differences, confirmed by sequencing [83]. | [83] |
Table 2: Optimization of Primer-Probe Sets and Cycle Number Using ddPCR
| Optimization Parameter | Method of Evaluation | Experimental Finding | Implication for Assay Development | Citation |
|---|---|---|---|---|
| Primer-Probe Set Efficiency | Absolute Positive Droplet (APD) count via ddPCR | From 20 candidate sets, only 2 maintained high efficiency at a stringent 62°C Tₐ. | High Tₐ is a key filter for selecting highly specific primer-probe sets [80]. | [80] |
| Cycle Threshold (Cₜ) Cut-off | Correlation of Cₜ values with APD from ddPCR | A logical, assay-specific cut-off Cₜ value of 36 was determined. Samples with Cₜ > 36 showed high false-positive rates. | ddPCR provides a logical strategy for setting a clinically relevant Cₜ cut-off to improve diagnostic accuracy [80]. | [80] |
| Assay Performance (Rain) | Droplet separation value and experience matrix | Annealing/extension temperature and oligonucleotide concentration were main parameters influencing clear separation of positive/negative droplets. | Objective metrics minimize "rain" (ambiguous signals) in ddPCR, crucial for precise GMO quantification and cancer biomarker analysis [84]. | [84] |
Optimization Workflow and Validation Context
The process of thermocycling optimization is systematic and should be integrated within the broader framework of analytical validation for clinical research. The diagram below outlines a standard workflow.
This optimization process is a critical component of the broader analytical validation of a Clinical Research (CR) qPCR assay, as shown in the following validation structure.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following reagents and materials are fundamental for the optimization experiments described in this guide.
Table 3: Key Reagents and Materials for qPCR Optimization
| Item | Function in Optimization | Application Example |
|---|---|---|
| Gradient Thermal Cycler | Enables simultaneous testing of multiple annealing temperatures in a single run, drastically speeding up optimization [81]. | Identifying the optimal Tₐ for a new cancer biomarker assay. |
| Hot-Start DNA Polymerase | Reduces non-specific amplification and primer-dimer formation by limiting polymerase activity until high temperatures are reached, improving assay specificity [80]. | Essential for all diagnostic qPCR assays to ensure clean amplification. |
| Fluorogenic Probes (e.g., TaqMan, EasyBeacon) | Provide sequence-specific detection and enable multiplexing. Probes like EasyBeacon allow for high-resolution melting curve analysis for SNP genotyping [82]. | Detecting specific mutations in cancer genes (e.g., KRAS, EGFR). |
| Synthetic DNA Standards (gBlocks) | Provide a consistent, quantifiable template for optimizing assay conditions and determining the limit of detection (LOD), independent of variable biological samples [85]. | Creating a standard curve for absolute quantification of a fusion transcript. |
| Digital PCR (ddPCR) System | Used for absolute quantification and to logically determine Cₜ cut-off values by correlating with absolute positive droplet counts, enhancing diagnostic accuracy [80] [84]. | Validating the performance of a qPCR assay for low-abundance circulating tumor DNA (ctDNA). |
The optimization of annealing temperature and cycle parameters is a non-negotiable step in developing a robust and clinically relevant qPCR assay. Data demonstrates that stringent annealing temperatures, logically determined cycle thresholds, and the use of advanced methods like HRM and ddPCR significantly enhance assay specificity and diagnostic accuracy. For clinical cancer diagnostics research, where results can influence patient management strategies, this rigorous, fit-for-purpose optimization and validation is not just best practice—it is a scientific and ethical imperative [60]. By adhering to structured optimization protocols and leveraging the appropriate technological toolkit, researchers can ensure their qPCR assays generate reliable, reproducible, and actionable data.
In clinical cancer diagnostics research, the analytical validation of quantitative PCR (qPCR) assays is paramount. The powerful ability of qPCR to quantify minimal residual disease (MRD), profile tumor biomarkers, and monitor therapeutic responses can be completely undermined by technical pitfalls such as PCR inhibition, high background noise, and primer-dimer formation [60] [86]. These issues directly compromise assay sensitivity and specificity, leading to false-negative results that could miss critical disease detection or false-positive findings that might prompt unnecessary treatments [60]. For researchers and drug development professionals, understanding and addressing these challenges is not merely about technical optimization—it is about ensuring that data generated from precious clinical samples is reliable, reproducible, and clinically actionable.
The transition of qPCR from a research-use-only (RUO) tool to a clinically validated assay requires rigorous validation guided by frameworks such as the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines and recent consensus from clinical consortia [87] [60] [8]. These guidelines emphasize that poor primer design combined with failure to optimize reaction conditions likely reduces technical precision and enables false positive or negative detection of amplification targets [87]. Within this context, we explore the specific pitfalls of inhibition, high background, and primer-dimer formation, providing comparative experimental data and validation protocols essential for robust assay development in cancer research.
Understanding the Pitfalls: Causes and Consequences
PCR Inhibition: The Hidden Assay Saboteur
PCR inhibition occurs when reaction components interfere with polymerase activity, reducing amplification efficiency. A significant and often overlooked source of inhibition stems from interactions between primers and genomic DNA present in the reaction, leading to exponential amplification of nonspecific amplicons that consume reagents [86]. This phenomenon is particularly problematic in sensitive applications like minimal residual disease (MRD) detection in lymphoid neoplasms, where inhibition can prevent reliable quantification below the 10^-4 level [86].
Inhibition is significantly influenced by primer characteristics. Studies demonstrate that primers with higher Tm values, higher G/C content, and particularly those terminating with G/C residues produce significantly more nonspecificity and inhibition than primers terminating with A/T [86]. The mechanistic basis for this lies in the stronger hydrogen bonding of G/C base pairs, which facilitates primer binding to off-target sequences in complex genomic DNA backgrounds. The consequences include reduced sensitivity, failed low-abundance target detection, and inaccurate quantification—all critical concerns in cancer biomarker studies where detecting rare mutation-bearing molecules directly impacts clinical interpretation.
High Background Noise: Compromising Signal Fidelity
High background fluorescence manifests as elevated baseline signals in qPCR amplification plots, reducing the signal-to-noise ratio and making accurate Cq determination challenging. This background noise stems from multiple sources, including nonspecific amplification, probe degradation, and the presence of contaminants that generate fluorescent signals independent of target amplification [88] [89].
In probe-based assays, high background frequently results from incomplete quenching of fluorophores, while in SYBR Green assays, it often indicates primer-dimer formation or nonspecific amplification [89] [68]. The consequences include reduced assay sensitivity, inaccurate quantification, and compromised linear dynamic range. For clinical cancer diagnostics, where precise quantification of gene expression changes or mutation load is essential, high background noise directly undermines data reliability and subsequent clinical interpretation.
Primer-Dimers: The Efficiency Competitors
Primer-dimer formation occurs when primers anneal to each other rather than to the target template, creating short, artifactual amplification products that compete with target amplification for reaction resources [90]. These structures are particularly problematic in SYBR Green assays where any double-stranded DNA product generates signal, but they can also impact probe-based assays by reducing overall amplification efficiency [90].
The formation of primer-dimers is favored by low annealing temperatures, excessive primer concentrations, and primers with complementary sequences, especially at their 3' ends [90] [68]. The consequences include reduced amplification efficiency, inaccurate quantification, and false-positive signals in no-template controls. In multiplex assays for cancer biomarker panels, primer-dimer formation becomes increasingly problematic as the number of primer pairs increases, potentially rendering portions of complex assays unusable for clinical decision-making.
Comparative Analysis: Experimental Data and Performance Metrics
Systematic Comparison of qPCR Pitfalls
Table 1: Comprehensive analysis of common qPCR pitfalls and their characteristics
| Pitfall | Primary Causes | Impact on Amplification | Effect on Quantification | Detection Methods |
|---|---|---|---|---|
| PCR Inhibition | Genomic DNA interactions [86], high primer Tm/G-C content [86], suboptimal polymerase concentration [86], PCR inhibitors in sample [88] | Reduced efficiency, delayed Cq, failed amplification at low targets [86] | Underestimation of target concentration, reduced sensitivity [86] | Standard curve analysis, inhibition index calculation [86], internal controls |
| High Background | Probe degradation [89], incomplete quenching [68], nonspecific amplification [90], contaminants [88] | Elevated baseline, reduced signal-to-noise ratio [89] | Inaccurate Cq determination, reduced linear dynamic range [89] | Baseline fluorescence analysis, no-template controls |
| Primer-Dimers | Complementary 3' ends [90] [68], low annealing temperatures [90], excessive primer concentration [90] | Early amplification in NTC, reduced target efficiency [90] | False positives, overestimation in SYBR Green [90] | Melt curve analysis (low Tm peak) [88], gel electrophoresis |
Quantitative Impact on Assay Performance
Table 2: Experimental data demonstrating performance impact of qPCR pitfalls
| Parameter | Optimal Performance | With Inhibition | With Primer-Dimers | Validation Threshold |
|---|---|---|---|---|
| Amplification Efficiency | 90–110% [8] [90] | Often <85% [86] | >110% or highly variable [90] | 90–110% [8] |
| Linear Dynamic Range | 6–8 orders of magnitude [8] | Reduced to 3–4 orders | Reduced range | R² ≥ 0.980 [8] |
| Limit of Detection (LOD) | Varies by assay; 10^-6 for HAT-PCR [86] | Failed at 10^-4 [86] | Compromised | Defined by CV <35% [60] |
| Inter-assay CV | <5% for Cq values [60] | >15% | Highly variable | <5% for Cq [60] |
| No-Template Control | No amplification [88] | Possible late amplification | Early amplification [88] | No amplification [88] |
Experimental data demonstrates that inhibition resulting from primer-genomic DNA interactions can reduce amplification efficiency by 13-21% depending on detection chemistry [90]. Similarly, suboptimal primer concentrations can cause efficiency deviations of similar magnitude, with elevated concentrations producing efficiencies >100% characterized by standard curve slopes less than -3.32 [90]. The HAT-PCR (High A/T PCR) approach, specifically designed to minimize these issues, enables reliable MRD detection down to 10^-6, a significant improvement over conventional qPCR that frequently fails below 10^-4 [86].
Methodologies for Detection and Validation
Experimental Protocols for Identifying Inhibition
Protocol 1: Inhibition Index Calculation This method quantifies the degree of inhibition caused by genomic DNA [86]:
- Perform qPCR with serial dilutions of target DNA both with and without added genomic DNA (500ng/reaction)
- Score results as positive or negative for amplification at each dilution
- Using limiting dilution analysis with Poisson statistics, calculate the mass of DNA containing one signal-producing sequence for wells with genomic DNA (m+) and without (m-)
- Calculate inhibition index as m-/m+
- An inhibition index significantly >1 indicates substantial inhibition requiring optimization
Protocol 2: Plasmid Test System for Primer Inhibition This approach tests individual primers for inhibition potential [86]:
- Construct a plasmid containing a single target sequence and quantify copies by digital PCR
- Set up amplification reactions with 3 copies of plasmid target per well
- Add test primer at 900 nmol/L concentration
- Run qPCR and judge inhibition to have occurred if amplification fails in all 3 replicates
- Compare with control reactions without test primer
Comprehensive Workflow for Troubleshooting qPCR Pitfalls
The following diagram illustrates a systematic approach to identifying and addressing common qPCR issues:
Primer and Probe Design Validation Protocol
Protocol 3: Comprehensive Primer/Probe Quality Control This protocol ensures optimal primer and probe characteristics [90] [68]:
- In Silico Analysis:
- Check specificity using NCBI BLAST against relevant genome
- Verify single amplicon as amplification product
- Screen for SNPs in binding regions using Ensembl genome browser
- Check for self-dimers, heterodimers, and hairpins using OligoAnalyzer Tool (ΔG > -9.0 kcal/mol)
Experimental Validation:
- Prepare seven 10-fold dilution series of DNA standard in triplicate
- Run qPCR and generate standard curve
- Calculate efficiency: E = [10^(-1/slope) - 1]
- Accept designs with efficiency 90-110% and R² ≥ 0.980
- Perform melt curve analysis for SYBR Green assays
Concentration Optimization:
- Test primer concentrations from 50-400 nM
- Identify concentration producing highest efficiency with minimal primer-dimer
- Ensure difference between primer Tms < 2-3°C
Research Reagent Solutions for Optimal qPCR Performance
Essential Reagents and Their Functions
Table 3: Key research reagents and solutions for mitigating qPCR pitfalls
| Reagent Category | Specific Examples | Function in Mitigating Pitfalls | Optimization Guidelines |
|---|---|---|---|
| Polymerase Systems | Hot-start Taq polymerases | Reduces primer-dimer formation by inhibiting activity until denaturation | Increase concentration to 2U/25μL to overcome inhibition [86] |
| Primer Design Tools | OligoAnalyzer, Primer-BLAST [68] | Identifies secondary structures and off-target binding | Ensure ΔG > -9.0 kcal/mol for dimers/hairpins [68] |
| Nuclease-Free Water | DEPC-treated water | Elimves RNase/DNase contamination preventing RNA degradation | Use in all reagent preparations |
| Probe Chemistry | Double-quenched probes (ZEN/TAO) [68] | Reduces background fluorescence vs single-quenched | 5' fluorophore, internal quencher, 3' blocker |
| DNase Treatment | RNase-free DNase I [68] | Removes genomic DNA preventing false positives | Treat RNA before reverse transcription |
| Inhibition Resistant Buffers | Custom Mg²⁺/K⁺ buffers | Optimizes cation concentrations for specificity | Adjust Mg²⁺ to 3-5mM based on assay [86] [90] |
| Standard Reference Materials | Certified reference DNA/RNA | Provides accurate quantification standards | Use in dilution series for standard curves |
The successful implementation of qPCR in clinical cancer research demands systematic approaches to overcome the technical pitfalls of inhibition, high background, and primer-dimer formation. Experimental evidence demonstrates that optimized primer design—focusing on Tm matching, appropriate GC content, and avoidance of G/C-rich 3' ends—combined with reaction condition optimization can significantly improve assay sensitivity and reliability [86] [90] [68]. The HAT-PCR approach, with its higher annealing temperatures and specialized primer design, represents one promising strategy for sensitive applications like MRD detection [86].
For researchers and drug development professionals, adhering to established validation frameworks such as the MIQE guidelines and recent clinical consensus recommendations provides a pathway to generating clinically actionable data [87] [60] [8]. The reagent solutions and methodologies presented here offer practical approaches for achieving the rigorous validation standards required in cancer diagnostics research. By implementing these systematic design and troubleshooting strategies, researchers can enhance the translational potential of their qPCR assays, ultimately contributing to improved cancer diagnostics and therapeutic monitoring.
Quantitative PCR (qPCR) stands as a fundamental technology in clinical cancer diagnostics research, where the accuracy of gene expression measurement directly impacts the identification of biomarkers and therapeutic targets. The reliability of these quantitative results is critically dependent on two primary quality assessment pillars: the calculation of amplification efficiency and the analysis of melting curves. Amplification efficiency, ideally approaching 100%, ensures that the initial template concentration is accurately reflected in the quantification cycle (Cq), while melting curve analysis verifies the specificity of the amplification product by detecting nonspecific products and primer-dimers. This guide objectively compares the performance of established and novel protocols for these quality assessments, providing supporting experimental data to underscore their application in validating qPCR assays for robust cancer research.
In the context of clinical cancer diagnostics, the analytical validation of qPCR assays is paramount. Inaccurate quantification of gene expression signatures, such as the 5-gene signature (LAMC2, TSPAN1, MYO1E, MYOF, and SULF1) for pancreatic cancer, can lead to misdiagnosis or incorrect assessment of therapeutic targets [91]. The quality of a qPCR result is not determined solely by the Cq value but by a suite of quality controls that assess the very foundations of the quantification process.
The first pillar, amplification efficiency, reflects the rate at which the PCR product is doubled during each cycle of the exponential phase. Deviations from an ideal 100% efficiency, caused by factors such as reaction inhibitors, poor primer design, or suboptimal reaction conditions, introduce significant bias in the calculated initial template concentration [92]. The second pillar, melting curve analysis, is a post-amplification assessment that determines the specificity of the reaction by characterizing the DNA melting behavior. It is an indispensable tool for verifying that a single, correct product has been amplified, free from artefacts such as primer-dimers or nonspecific products that can skew quantification [93] [94]. This guide provides a comparative analysis of methodologies for both processes, framing them within the essential workflow of analytical validation for cancer research.
Amplification Efficiency Calculations: Methods and Comparative Performance
Amplification efficiency (E) is a critical parameter that quantifies the performance of the qPCR reaction. The optimal value is E=2, representing 100% efficiency, where the product doubles every cycle. Acceptable efficiency typically falls within the range of 90–110% (E=1.9–2.1) [92]. Accurate determination of efficiency is non-negotiable for reliable gene expression ratios or fold-change calculations in diagnostic assays.
Experimental Protocols for Efficiency Determination
Standard Curve Method: A standard curve is generated using a serially diluted standard sample (e.g., recombinant plasmid or genomic DNA) with at least five different concentrations [92]. The Cq values are plotted against the logarithm of the initial template concentration. The slope of the resulting linear regression line is used to calculate the amplification efficiency using the formula: ( E = 10^{-1/slope} ) [92]. A slope of -3.32 corresponds to 100% efficiency. This method is widely used but can be compromised by inaccurate dilution of the standard and may not reflect the efficiency in unknown samples due to different sample matrices.
LinRegPCR Method: As implemented in the web-based LinRegPCR application, this method calculates the PCR efficiency from the exponential phase of each individual amplification curve, thereby accounting for sample-to-sample variation [95]. The analysis involves:
- Baseline Subtraction: An automated, user-independent baseline correction that does not rely on the noisy ground phase cycles.
- Identification of the Exponential Phase: The window of linearity is identified in the log(fluorescence) versus cycle number plot.
- Efficiency Calculation: The PCR efficiency is determined from the slope of the regression line through the exponential phase for each reaction. The mean efficiency of all reactions for the same assay is then used for further quantification, a method shown to yield highly reproducible results [95].
Comparative Performance Data
The following table summarizes the performance and characteristics of the two main methods for efficiency calculation.
Table 1: Comparison of qPCR Efficiency Calculation Methods
| Feature | Standard Curve Method | LinRegPCR Method |
|---|---|---|
| Principle | Uses external serial dilutions of a known standard [92] | Analyzes the exponential phase of individual sample amplification curves [95] |
| Reported Reproducibility | Can vary between machines and PCR runs [95] | Achieves the lowest variation and highest reproducibility in qPCR results [95] |
| Sample-Specific Efficiency | No, assumes efficiency is identical for standard and samples | Yes, calculates efficiency per reaction, accounting for sample-specific inhibitors [95] |
| Throughput | Lower, requires preparation of a separate dilution series | Higher, uses data from the main qPCR run without extra plates |
| Key Limitation | Inaccurate dilutions lead to erroneous efficiency values; does not reflect sample-specific effects [92] | Requires a well-defined exponential phase in the amplification curve for accurate calculation [95] |
Melting Curve Analysis: Protocols and Specificity Assessment
Melting curve analysis (MCA) is a powerful technique for verifying the specificity of qPCR amplicons, especially when using DNA-binding dyes like SYBR Green I. The principle is based on the gradual denaturation (melting) of double-stranded DNA as the temperature increases. The temperature at which 50% of the DNA is denatured is the melting temperature (Tm), a unique characteristic determined by the amplicon's length, GC content, and nucleotide sequence [93].
Experimental Protocol for Melting Curve Analysis
The standard workflow for MCA is as follows:
- Amplification: Complete the qPCR run.
- Melting: Immediately after amplification, run a melting protocol that slowly heats the reactions from a temperature below the expected Tm (e.g., 65°C) to a temperature above it (e.g., 95°C), while continuously monitoring the fluorescence [92].
- Data Processing: The raw fluorescence vs. temperature data is processed. The negative first derivative of the fluorescence (-dF/dT) is plotted against temperature to produce distinct peaks at the Tm of each DNA species present [95].
- Peak Calling and Interpretation: Peaks are identified, and their Tm is compared to the expected Tm of the specific target. A single, sharp peak at the expected Tm indicates a specific product. Multiple peaks indicate the presence of artefacts like primer-dimers (typically lower Tm) or nonspecific products (which may have a higher or lower Tm) [93] [92].
Advanced analysis tools like web-based LinRegPCR automate the steps of smoothing, normalization, peak calling, and assessment by comparing the observed Tm to the known Tm of the intended product [95].
Performance and Applications in Diagnostics
Melting curve analysis is crucial for diagnostic validation. Its performance can be characterized by its ability to:
- Identify Non-specific Amplification: A single, distinct peak between 80–90°C confirms robust specificity for a typical amplicon. A secondary peak below 80°C suggests primer-dimer formation, while a peak above 90°C may indicate genomic DNA contamination [92].
- Genotype and Detect Mutations: High-resolution melting (HRM) analysis can distinguish PCR products that differ by as little as a single base pair, enabling mutation screening and genotyping without the need for labeled probes [93].
- Correct for Artefact Bias: Critically, research has demonstrated that when the Tm of the correct product is known, the fluorescence contribution of the artefact peak can be quantified and used to correct the reported quantitative result, rather than forcing the researcher to discard the reaction [94]. This leads to more reliable and less biased quantitative data.
The following workflow diagram illustrates the logical process of integrating melting curve analysis for quality assessment and data correction.
The Scientist's Toolkit: Essential Reagents and Materials
The successful implementation of these quality assessment protocols relies on a set of key reagents and tools. The following table details essential solutions for a robust qPCR workflow in a clinical research setting.
Table 2: Key Research Reagent Solutions for qPCR Quality Assessment
| Reagent / Material | Function / Explanation | Application in Quality Control |
|---|---|---|
| Saturating DNA Dyes (e.g., LCGreen) | DNA-binding dyes that do not inhibit PCR at high concentrations and enable high-resolution melting (HRM) analysis [93]. | Essential for precise genotyping and mutation detection via HRM; provides superior data for melting curve analysis compared to standard SYBR Green I [93]. |
| Magnetic Bead Nucleic Acid Kits | Kits for automated or manual extraction of total nucleic acids from complex samples (e.g., blood, tissue) [96] [91]. | Critical for obtaining pure template, free of inhibitors that can adversely affect amplification efficiency. The quality of the input DNA directly impacts the validity of the efficiency calculation. |
| Validated Primers & Probes | Oligonucleotides designed for a specific target (e.g., a cancer biomarker gene) and validated for specificity and efficiency. | The cornerstone of assay specificity. Poorly designed primers are a primary source of non-specific amplification and primer-dimer formation, which are detected in melting curve analysis. |
| qPCR Master Mixes with UNG | Pre-mixed solutions containing enzymes, dNTPs, and buffers. Those containing Uracil-N-Glycosylase (UNG) prevent carryover contamination [96]. | Ensures reaction consistency for reliable efficiency measurements across samples and plates. UNG treatment maintains the integrity of results by degrading contaminants from previous amplifications. |
| Standard Reference Materials | Quantified standards (e.g., synthetic oligonucleotides, recombinant plasmids) of known concentration [11]. | Used to create standard curves for the absolute quantification of template and for independent validation of amplification efficiency. |
The rigorous analytical validation of qPCR assays for clinical cancer diagnostics is non-negotiable. This comparison guide demonstrates that a dual-focused approach, integrating both precise amplification efficiency calculations and specific melting curve analysis, is essential for generating reliable and interpretable data. While the standard curve method remains useful, the LinRegPCR approach offers superior reproducibility for efficiency determination. Similarly, melting curve analysis has evolved from a simple pass/fail check to a tool capable of quantitatively correcting for artefact amplification. By adopting these detailed quality assessment protocols and utilizing the essential research tools outlined, scientists and drug development professionals can ensure their qPCR data meets the high standards required for impactful cancer research and diagnostic development.
Ensuring Reliability: Validation Best Practices and Technology Positioning
In the field of clinical cancer diagnostics, the translation of research findings into reliable clinical applications hinges on rigorous analytical validation. For quantitative PCR (qPCR) assays, this process establishes the foundation of trustworthiness required for results that can inform patient management decisions, including diagnosis, prognosis, and prediction of treatment response [60]. The noticeable lack of technical standardization has been a significant obstacle in the clinical application of qPCR-based tests [60]. This guide objectively examines the core pillars of assay validation—sensitivity, specificity, and reproducibility—by comparing data and methodologies from recent cancer research studies. It provides a structured framework for researchers and drug development professionals to evaluate and implement robust qPCR assays in their own clinical research workflows.
Core Principles of qPCR Assay Validation
Defining the Validation Parameters
Analytical validation ensures that a qPCR test consistently performs according to its intended design and purpose. For clinical research, this process bridges the gap between basic Research Use Only (RUO) assays and fully certified In Vitro Diagnostic (IVD) products [60]. The key parameters are:
- Analytical Sensitivity: The lowest quantity of the analyte (e.g., a specific nucleic acid sequence) that can be reliably detected by the assay. This is often expressed as the Limit of Detection (LOD) [60] [64].
- Analytical Specificity: The ability of the assay to detect only the target analyte without cross-reacting with or being inhibited by other, non-target components in a sample [60].
- Reproducibility: The closeness of agreement between independent results obtained under varied conditions, such as different operators, instruments, or days. It encompasses both repeatability (same conditions) and intermediate precision (different conditions) [60] [64].
The required performance thresholds for these parameters are guided by the Context of Use (COU) and adhere to a Fit-for-Purpose (FFP) validation strategy. The COU is a formal statement describing the test's specific application, while FFP means the level of validation is sufficient to support that stated context [60].
Quantitative Comparison of Validation Parameters in Recent Cancer Studies
The following tables consolidate experimental data from recent studies to illustrate how sensitivity, specificity, and reproducibility are measured and achieved in practice.
Table 1: Experimental Data on Sensitivity and Specificity from Recent Cancer Studies
| Study & Cancer Type | Assay Description | Analytical Sensitivity (LOD) | Specificity & Cross-Reactivity Assessment |
|---|---|---|---|
| 10-Gene Panel (Bladder Cancer) [26] | Multiplex qPCR array (Nexus-Dx) | Reliable performance across RNA input levels of 5–100 ng. Required minimum RNA quality threshold (DV200 >15%). | Primer specificity confirmed via Primer-BLAST; amplification efficiencies between 90% and 110%. |
| 11-Gene Panel (Lung Cancer) [5] | qPCR on micro cell samples (MCSs) | Not explicitly stated in snippet. | 100% concordance in mutation detection between MCSs and paired tissue samples (n=38 for G1, n=108 for G2). |
| qPCR in Oncology (General) [1] | Multiplexed qPCR panels (e.g., for NSCLC) | Consistent detection of low-frequency variants at <0.1% Variant Allele Frequency (VAF). | Multiplexing capability allows specific detection of multiple mutations (e.g., EGFR, KRAS, BRAF) in a single reaction. |
Table 2: Experimental Data on Reproducibility and Robustness from Recent Cancer Studies
| Study & Cancer Type | Assay Robustness & Precision Evaluation | Key Outcome Measures |
|---|---|---|
| 10-Gene Panel (Bladder Cancer) [26] | Tested across FFPE and fresh-frozen tissues, different RNA qualities, and storage conditions (FFPE curls at ≤4°C for 2 weeks). Reproducibility confirmed across different technicians and time points. | High concordance between FFPE and fresh-frozen samples. Minimal impact from sample necrosis. Stable gene expression results. |
| 11-Gene Panel (Lung Cancer) [5] | Assay performed on diverse MCSs from needle rinses (EBUS-TBNA, PABL) and bronchoalveolar lavage fluid. | Rapid Turn-Around-Time (TAT of 24 hrs). Reliable results across different sample collection methods. |
Experimental Protocols for Core Parameter Validation
Protocol for Determining Analytical Sensitivity (Limit of Detection)
The following workflow is used to establish the minimum detectable concentration of an analyte with high confidence.
Detailed Methodology [64] [26]:
- Sample Preparation: A sample with a known, high concentration of the target analyte (e.g., a synthetic DNA template or characterized cell line RNA) is serially diluted in a matrix that mimics the clinical sample (e.g., negative plasma, FFPE-derived nucleic acids). A dilution series spanning several logs (e.g., from 100 ng to 1 pg) is typically created.
- Replicate Testing: Each dilution level is tested with a high number of replicates (e.g., n=8 to n=12) to account for stochastic variation, especially at very low concentrations.
- Data Analysis: The proportion of positive replicates (Cq value below a predefined threshold) is calculated for each dilution. The LOD is defined as the lowest concentration at which ≥95% of the replicates are positive [64]. For assays detecting very low copy numbers (less than ten copies per reaction), a consensus profile from a high number of technical replicates is recommended to minimize stochastic effects [64].
Protocol for Establishing Analytical Specificity
This protocol ensures the assay is specific for the intended target and is not affected by similar sequences or sample impurities.
Detailed Methodology [64] [26]:
- In Silico Analysis: Prior to testing, primer and probe sequences should be analyzed for specificity using tools like Standard Nucleotide BLAST (Primer-BLAST) to ensure they do not have significant homology with non-target sequences in relevant genomes [26].
- Wet-Lab Testing:
- Cross-Reactivity: Test the assay against nucleic acid samples that contain high concentrations of closely related sequences (e.g., homologous genes, common flora in the sample type, or other potential interferents).
- Inhibition Testing: Spike a known quantity of the target into different clinical sample matrices (e.g., plasma, FFPE extracts) and compare the Cq values to the target in a clean buffer. A significant delay in Cq (e.g., > 2 cycles) indicates the presence of inhibitors in the sample matrix. The use of inhibitor-resistant polymerases can mitigate this issue [1].
Protocol for Assessing Reproducibility
Reproducibility testing validates that the assay delivers consistent results under the expected variations of routine use.
Detailed Methodology [64] [26]:
- Experimental Design: Select a panel of samples that covers the assay's dynamic range (low, medium, and high analyte concentrations).
- Variable Conditions: Test these samples across multiple runs, on different days, using different operators, and if applicable, on different calibrated instruments.
- Statistical Analysis: Calculate the standard deviation (SD) and coefficient of variation (%CV) of the Cq values or reported concentrations for each sample across all these conditions. A low %CV indicates high reproducibility. Experimental tools like Plackett-Burman designs can be used to efficiently test the robustness of the assay by systematically altering multiple experimental parameters (e.g., incubation times, reagent volumes) simultaneously [64].
The Scientist's Toolkit: Essential Reagents and Materials
Successful assay validation depends on using high-quality, fit-for-purpose materials. The following table details key reagents and their critical functions.
Table 3: Essential Research Reagent Solutions for qPCR Assay Validation
| Reagent / Material | Critical Function in Validation | Key Considerations for Selection |
|---|---|---|
| Nucleic Acid Isolation Kits | To obtain pure, high-quality template from complex clinical matrices. | Match chemistry to sample type (e.g., FFPE, plasma). Assess yield and quality (e.g., DV200 value for FFPE RNA) [26]. |
| Inhibitor-Resistant Master Mix | To ensure robust amplification in the presence of PCR inhibitors from clinical samples. | Select master mixes engineered for tolerance to inhibitors in plasma, FFPE, etc. [1]. |
| Validated Primers & Probes | To ensure high specificity and efficiency for the intended target. | Confirm via BLAST; test for desired efficiency (90-110%) and specificity [64] [26]. Use multiple reference genes (e.g., TBP, ATP5E) for normalization [39] [26]. |
| Calibrators & Controls | To monitor assay performance, precision, and generate standard curves. | Include positive, negative, and quantitative controls in every run. Use controls with known concentrations to assess PCR efficiency [64]. |
| Ambient-Stable Kits | To reduce reliance on cold chain and enhance robustness for decentralized testing. | Lyophilized or ambient-stable reagents support scalability and deployment in varied settings [1]. |
Establishing the core validation parameters of sensitivity, specificity, and reproducibility is a non-negotiable prerequisite for employing qPCR assays in clinical cancer research. The data and protocols summarized here demonstrate that a fit-for-purpose approach, guided by the context of use, allows for the development of robust and reliable tests. As the field progresses, the integration of advanced reagents, meticulous experimental design, and thorough validation will continue to enhance the role of qPCR in enabling precision oncology, from early cancer detection and molecular stratification to monitoring treatment response and minimal residual disease.
For researchers in clinical cancer diagnostics, developing a qPCR assay is only half the challenge. The other, critical half is navigating the stringent Good Practices (GxP) regulations that ensure the reliability, traceability, and integrity of data supporting patient safety and product efficacy. This guide outlines the best practices for GxP compliance, framing them within the specific context of analytically validating qPCR assays for clinical cancer diagnostics research.
The GxP Framework in Life Sciences
GxP is a general abbreviation for the "Good Practice" quality guidelines and regulations mandated by global health authorities like the FDA and the European Medicines Agency (EMA) [97]. Its overarching goal is to build quality and safety into every step of the product lifecycle, from research and development to manufacturing and distribution, ensuring that products are safe, effective, and meet quality standards [98] [97].
For a researcher validating a qPCR assay, this translates to a framework of control and accountability. An error in pre-clinical data could lead to a flawed clinical trial design, potentially resulting in an unsafe drug being evaluated, thereby harming patients and compromising research integrity [97]. GxP provides the proactive framework designed to prevent such failures before they can happen.
The "x" in GxP encompasses several key disciplines, the most relevant for diagnostic research are:
- Good Laboratory Practice (GLP): Ensures the consistency, reliability, and traceability of non-clinical laboratory studies [99] [100].
- Good Clinical Practice (GCP): Governs the ethical and scientific integrity of clinical trials, ensuring patient safety and data reliability [99] [100].
- Good Manufacturing Practice (GMP): Ensures that products are consistently produced and controlled according to quality standards [98].
Core Principles of GxP: The Foundation for reliable data
At the heart of all GxP guidelines are core principles that create a culture of quality and accountability. For qPCR assay validation, these principles are not abstract concepts but daily practices.
The 5 Ps of GxP
A functional GxP system is built on five critical elements [99]:
- People: Personnel must be qualified and trained for their roles.
- Procedures: Processes must be clearly defined and documented.
- Products: Consistent quality of materials and products must be maintained.
- Premises & Equipment: Facilities and instruments must be suitable and validated.
- Processes: All operational processes must be reliable and validated.
ALCOA+ Principles for Data Integrity
In a regulated environment, the mantra is, "If it wasn't documented, it didn't happen." The ALCOA+ framework provides the global standard for ensuring all data—from a qPCR run output to a training record—is trustworthy [97].
| Principle | Description | Application in qPCR Validation |
|---|---|---|
| Attributable | It should be clear who created the data and when. | Unique login credentials for analysts running the qPCR instrument. |
| Legible | Data must be readable and permanent. | Secure, unalterable electronic records of amplification curves and Ct values. |
| Contemporaneous | Data should be recorded at the time of the activity. | Real-time entry of observations in an electronic lab notebook. |
| Original | The record is the source or a certified true copy. | Preserving the original qPCR data file, not a transcribed summary. |
| Accurate | Data must be correct, truthful, and free from errors. | No cherry-picking of qPCR data; all replicates must be reported. |
| Complete | All data must be present, including repeats. | Including all run data, even failed assays or out-of-spec results. |
| Consistent | Data should be in a chronological sequence. | Maintaining a validation study report that documents the workflow. |
| Enduring | Records must be maintained for the entire retention period. | Long-term, backed-up storage of all validation data. |
| Available | Records must be accessible for review and audit. | Ensuring data can be retrieved for regulatory inspection. |
Best Practices for GxP-Compliant qPCR Assay Validation
Transitioning from principle to practice requires a systematic approach. The following best practices, supported by experimental data, are essential for establishing a GxP-compliant qPCR assay.
Establish a Robust Training Program
All personnel must be thoroughly trained on the GxP principles and specific SOPs relevant to their roles before they begin their duties [97]. This includes scientists performing nucleic acid extraction, those operating qPCR instruments, and personnel responsible for data analysis. A capable Learning Management System (LMS) is critical for tracking this training, ensuring it is role-based, ongoing, and meticulously documented [101].
Validate All Computerized Systems
Any computer system used to create, modify, or store GxP data, including qPCR instrument software and data analysis platforms, must be formally validated [97]. This process, known as Computer System Validation (CSV), provides documented evidence that the system operates exactly as intended in a reliable and reproducible manner, ensuring it maintains data integrity and meets regulations like FDA 21 CFR Part 11 [102].
Implement Detailed Experimental Protocols
A GxP-compliant validation study requires a pre-defined, detailed protocol. The methodologies below, adapted from recent literature, illustrate the rigor required for key validation experiments.
Protocol 1: Determination of Limit of Detection (LOD) The LOD is the lowest concentration of an analyte that can be reliably detected. This protocol is adapted from the comprehensive evaluation of SARS-CoV-2 Ag-RDTs and Borrelia PCR assays [103] [104].
- Sample Preparation: Prepare a dilution series of the target analyte (e.g., a plasmid containing the target sequence or RNA from cultured cells) in a matrix that mimics the clinical sample (e.g., human DNA in buffer). A typical 10-fold serial dilution should span a range from below the expected LOD to well above it.
- qPCR Run: Analyze each dilution level with a minimum of 12-24 replicates across different days and by different analysts to determine a concentration at which 95% of the replicates test positive [103].
- Data Analysis: The LOD is determined as the lowest concentration where ≥95% of the replicates are positive. Statistical analysis, such as probit regression, can be used for precise calculation.
Protocol 2: Analytical Specificity Testing Specificity ensures the assay detects only the intended target and does not cross-react with other organisms or human DNA.
- Panel Creation: Assemble a panel of DNA/cDNA from closely related genetic species, other common pathogens, and human genomic DNA.
- Testing: Run the qPCR assay with each member of the panel as a template.
- Analysis: The assay is considered specific if no amplification occurs, or the Cycle threshold (Ct) values are significantly higher than those for the positive control, confirming no cross-reactivity [105] [104].
Protocol 3: Precision and Reproducibility Measurement Precision measures the agreement between independent test results under specified conditions.
- Experimental Design: Test multiple replicates (e.g., n=5) of at least two different analyte concentrations (low and high) within the same run (repeatability) and across different runs, days, and operators (intermediate precision).
- Statistical Analysis: Calculate the mean Ct value, standard deviation (SD), and coefficient of variation (%CV) for each set of replicates. A low %CV indicates high precision and robust assay performance [105].
Practice Good Documentation
Live by the ALCOA+ principles. All activities, from reagent preparation to instrument maintenance, must be documented accurately and at the time the work is performed. This includes maintaining batch records for reagents, calibration logs for equipment, and detailed notebooks for experimental procedures [97].
GxP-Compliant qPCR Assay Validation: An Experimental Data Perspective
The table below summarizes exemplary quantitative data from validation studies, demonstrating the level of detail and performance required for a GxP-compliant assay. These data, while from different fields, illustrate universal validation metrics.
Table 2: Comparative Analytical Performance of Molecular Assays
| Assay Description / Performance Metric | Limit of Detection (LOD) | Amplification Efficiency | Precision (Coefficient of Variation - CV) | Specificity (Cross-Reactivity) | Reference |
|---|---|---|---|---|---|
| Two-step qPCR for Carpione rhabdovirus (CAPRV2023) | 2 copies/μL | 104.7% | Intra-assay: 0.23-0.95%; Inter-assay: 0.28-1.95% | No cross-reactivity with other aquatic pathogens | [105] |
| One-step qPCR for Carpione rhabdovirus (CAPRV2023) | 15 copies/μL | 102.8% | CV = 0.81% | No cross-reactivity with other aquatic pathogens | [105] |
| Commercial RT-PCR for Borrelia burgdorferi | Variable by kit; 10-104 copies/5µL for most species | Not specified | Evaluated via delta Ct of replicates | 9 out of 11 kits cross-reacted with Relapsing Fever Borrelia | [104] |
| Multiplex MCDA-AuNPs-LFB for HBV/HCV | Comparable to qPCR | Not specified | Not specified | Undetectable cross-reactivity to non-target pathogens | [106] |
The Scientist's Toolkit: Essential Reagents & Materials
The quality of reagents directly impacts the validity of results. Sourcing materials from qualified suppliers is a GxP requirement.
Table 3: Essential Research Reagent Solutions for qPCR Assay Validation
| Item | Function in qPCR Validation | GxP-Compliance Consideration |
|---|---|---|
| Certified Reference Material | Provides a traceable standard for quantifying the target analyte and establishing the standard curve. | Must be obtained from a certified supplier with a valid Certificate of Analysis (CoA). |
| Master Mix Reagents | Contains enzymes, dNTPs, and buffer necessary for the PCR reaction. | Requires strict inventory control and stability testing to ensure consistent performance. |
| Primers & Probes | Specifically designed oligonucleotides for target amplification and detection. | Sequences must be documented, and each batch must be validated for performance. |
| Nuclease-Free Water | Used to prepare reagent mixes and dilutions, free of contaminants that could degrade reagents. | Treated as a critical reagent; quality must be assured by the manufacturer. |
| Positive & Negative Controls | Verify the assay is functioning correctly and that no contamination has occurred. | Controls must be well-characterized and included in every run. |
Visualizing the GxP-Compliant qPCR Workflow
The following diagram illustrates the integrated workflow of analytical validation and documentation in a GxP-compliant qPCR assay development process.
GxP qPCR Validation Workflow
For researchers in clinical cancer diagnostics, GxP compliance is not a bureaucratic obstacle but a fundamental component of scientific excellence. By integrating these best practices—from rigorous training and system validation to meticulous documentation following ALCOA+ principles—into the qPCR assay validation workflow, scientists can ensure their data is not only publishable but also audit-ready. This commitment to quality protects patient safety, builds trust with regulators, and ultimately turns a robust quality system from a cost of doing business into a strategic asset that accelerates the delivery of reliable cancer diagnostics.
In the field of clinical cancer diagnostics, the accurate detection and quantification of nucleic acids are foundational to personalized treatment strategies. Quantitative PCR (qPCR) has long been the gold standard for molecular analysis, providing robust, high-throughput detection of genetic alterations. The emergence of digital PCR (dPCR) represents a significant technological shift, offering alternative capabilities for absolute quantification. This guide provides an objective, data-driven comparison of these two pivotal technologies, focusing specifically on their performance in sensitivity and quantification within the context of analytical validation for clinical cancer research. The choice between these methods has profound implications for assay development, impacting everything from the detection of low-frequency mutations in liquid biopsies to the precise measurement of gene expression in tumor samples, ultimately guiding therapeutic decisions in oncology.
Fundamental Principles and Quantification Methods
The core difference between qPCR and dPCR lies not just in their workflows, but in their fundamental approach to quantification, which directly influences their application in sensitive diagnostic settings.
Quantitative PCR (qPCR)
qPCR, also known as real-time PCR, monitors the amplification of DNA in real-time during the exponential phase of the PCR reaction. The key output is the cycle threshold (Ct), the cycle number at which the fluorescent signal crosses a predefined threshold. This Ct value is inversely proportional to the initial amount of target nucleic acid. However, determining the actual concentration requires a standard curve generated from samples with known concentrations [107] [108]. This relative quantification can introduce variability, as it depends on the accuracy and stability of the reference standards. qPCR is celebrated for its high throughput, scalability, and cost-effectiveness, making it an indispensable tool for applications where these factors are primary, such as routine gene expression analysis and high-volume pathogen detection [1].
Digital PCR (dPCR)
dPCR takes a different approach by partitioning a single PCR reaction into thousands of individual reactions. The partitioning is achieved through water-in-oil droplets (droplet digital PCR, or ddPCR) or nanoscale wells on a chip (nanoplated-based dPCR) [109] [12]. Following end-point PCR amplification, each partition is analyzed as either positive (containing the target sequence) or negative. The absolute concentration of the target, in copies per microliter, is then calculated directly using Poisson statistics, without the need for a standard curve [108] [12]. This principle of "single-molecule counting" is what grants dPCR its key advantages: superior precision and accuracy for absolute quantification, and enhanced sensitivity for detecting rare genetic events [107].
Comparative Performance Data
Direct comparisons in recent studies highlight clear performance trade-offs between qPCR and dPCR, which are critical to consider for diagnostic assay validation.
Table 1: Comparative Analytical Performance of qPCR and dPCR
| Parameter | qPCR | Digital PCR | Experimental Context |
|---|---|---|---|
| Quantification Method | Relative (via standard curve) [108] | Absolute (via Poisson statistics) [108] [12] | Fundamental principle of operation. |
| Limit of Detection (LOD) | Moderate | Superior; Can detect single molecules [12] | LOD for dPCR platforms can be <0.5 copies/µL input [110]. |
| Precision & Accuracy | High for moderate to high abundance targets [1] | Superior, particularly for low abundance targets and absolute quantification [109] [110] | dPCR demonstrated superior accuracy for high viral loads of influenza A/B and SARS-CoV-2 [109]. |
| Tolerance to Inhibitors | Moderate; can be affected by sample matrix [107] | High; partitioning reduces inhibitor effect [107] [110] | Partitioning dilutes inhibitors, making dPCR more robust for complex samples like plasma or FFPE. |
| Multiplexing Capability | Strong; multiple targets in a single well [1] | Moderate; limited by available fluorescent channels [107] | qPCR is well-suited for targeted panels (e.g., in NSCLC [1]). |
| Throughput & Cost | High throughput, lower cost per sample [107] [1] | Lower throughput, higher cost per sample [109] [107] | qPCR is more suitable for large-scale screening [1]. |
Table 2: Platform-Specific dPCR Performance (from Direct Comparison Study)
| Platform | Partitioning Method | Limit of Detection (LOD) | Limit of Quantification (LOQ) | Key Finding |
|---|---|---|---|---|
| QX200 (Bio-Rad) | Droplet-based [110] | ~0.17 copies/µL input [110] | ~4.26 copies/µL input [110] | Precision significantly improved with optimal restriction enzyme (HaeIII vs. EcoRI) [110]. |
| QIAcuity One (QIAGEN) | Nanoplate-based [110] | ~0.39 copies/µL input [110] | ~1.35 copies/µL input [110] | Showed high precision across a wide concentration range with less variability from enzyme choice [110]. |
Experimental Protocols for Analytical Validation
Robust analytical validation is a prerequisite for deploying any PCR assay in a clinical cancer research setting. The following key experiments, drawn from recent validation studies, are essential for assessing performance.
Determining Limits of Detection and Quantification
The Limit of Detection (LOD) and Lower Limit of Quantification (LLOQ) define the sensitivity and dynamic range of an assay.
- Protocol Overview: A series of dilutions are prepared from a reference material of known concentration (e.g., synthetic oligonucleotides or cell line DNA). Each dilution is tested across multiple replicates and runs [111].
- LOD Determination: The LOD is defined as the lowest concentration at which the target is detected in ≥95% of replicates [111]. For dPCR, this can approach single-molecule levels, with studies reporting LODs below 0.5 copies/µL input [110].
- LLOQ Determination: The LLOQ is the lowest concentration that can be quantified with acceptable precision, typically defined by a coefficient of variation (CV) in copy number or Ct value. For example, the MPS2 urinary biomarker test established an LLOQ with a Crt standard deviation of ≤0.75 [111].
Assessing Assay Precision
Precision, or reproducibility, measures the agreement between repeated measurements of the same sample.
- Protocol Overview: A single sample is tested repeatedly under various conditions that might be encountered in a clinical lab [111].
- Tiers of Precision:
- Intra-run (Repeatability): Multiple replicates within a single run.
- Inter-run (Reproducibility): Replicates across different days and by different technicians.
- Inter-instrument: Replicates on different instruments of the same model.
- Acceptance Criteria: For qPCR-based tests like MPS2, precision is often deemed acceptable if the standard deviation of the Ct values is ≤0.5 across replicates [111]. dPCR typically demonstrates very low CVs for copy number concentration, especially at medium to high target levels [110].
Analyzing Linearity and Dynamic Range
This experiment confirms that the assay provides a linear response over a wide range of target concentrations.
- Protocol Overview: A sample is serially diluted (e.g., from 10^7 to 10^2 copies/reaction) and each dilution is tested [111].
- Data Analysis: The measured values (Ct for qPCR, copies/µL for dPCR) are plotted against the expected log concentration. The assay is considered linear if it demonstrates a high coefficient of determination (R² ≥ 0.975) and a PCR efficiency between 95% and 105% for qPCR [111]. dPCR maintains linearity across its dynamic range without efficiency calculations.
Application in Clinical Cancer Diagnostics
The choice between qPCR and dPCR is dictated by the specific clinical question and the nature of the biomarker.
- Detection of Rare Mutations and Liquid Biopsy: dPCR is the preferred tool for detecting rare somatic mutations (e.g., EGFR T790M in NSCLC) in cell-free DNA (cfDNA) due to its ability to detect variant alleles at frequencies as low as 0.1% or below [12]. This high sensitivity is crucial for non-invasive tumor monitoring and assessing emerging therapy resistance.
- Viral Load Monitoring and Pathogen Detection: In virology, dPCR demonstrates superior accuracy for high viral loads, as shown in a 2023-2024 study on respiratory viruses, making it valuable for quantifying oncolytic viruses or assessing latent infections in immunocompromised patients [109].
- High-Throughput Stratification and Routine Testing: For scenarios requiring rapid, cost-effective testing of well-characterized, higher-abundance biomarkers—such as screening for EGFR driver mutations or HPV status—multiplex qPCR panels offer an unbeatable combination of speed, throughput, and affordability [1].
- Copy Number Variation (CNV) Analysis: Both technologies can be used for CNV analysis. dPCR provides absolute copy number without a standard curve and has shown high precision in cross-platform evaluations for quantifying gene copies in eukaryotic cells [110].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of qPCR and dPCR assays in cancer research relies on specialized reagents and platforms.
Table 3: Key Reagents and Platforms for PCR-Based Cancer Diagnostics
| Item | Function | Example Use-Case / Note |
|---|---|---|
| qPCR Master Mixes | Provides optimized buffer, polymerase, dNTPs for efficient amplification. | Next-generation mixes are inhibitor-resistant and compatible with fast-cycling protocols, ideal for FFPE-derived DNA or cfDNA [1]. |
| dPCR Partitioning Reagents | Enables creation of stable droplets or loading of nanowells. | Includes droplet generation oil for ddPCR and specific nanoplate kits for systems like QIAcuity [109] [110]. |
| Validated Primer/Probe Assays | Target-specific oligonucleotides for detection. | Commercially available assays (e.g., for EGFR mutations) reduce development time and ensure reproducibility [1]. |
| Nucleic Acid Extraction Kits | Isulates high-quality DNA/RNA from complex clinical samples. | Kits specialized for sample type (e.g., urine, FFPE, plasma) are critical for success [112] [111]. |
| Synthetic Reference Standards | Acts as a positive control for assay development and validation. | Used to determine LOD, LLOQ, and linearity without biological variability [111]. |
| QX200 Droplet Digital PCR (Bio-Rad) | Droplet-based dPCR platform. | Used in comparative studies for its high sensitivity and precision, particularly after protocol optimization [110]. |
| QIAcuity (QIAGEN) | Nanoplate-based dPCR system. | Offers integrated partitioning and cycling; shown to have high throughput and reduced handling [109] [110]. |
| QuantStudio 12K Flex (Thermo Fisher) | High-throughput qPCR system. | Used with OpenArray technology for scalable testing, as in the MPS2 validation study [111]. |
The decision between qPCR and dPCR is not a matter of declaring one technology superior, but rather of matching the tool to the task at hand. qPCR remains the workhorse for high-throughput, cost-effective applications where relative quantification is sufficient and target abundance is not extremely low. Its established workflows, scalability, and strong multiplexing capabilities secure its role in routine cancer diagnostics. In contrast, dPCR excels in scenarios demanding ultimate sensitivity and absolute precision, such as liquid biopsy for rare mutation detection, validation of qPCR results, and absolute quantification without standard curves. As the field of clinical cancer research continues to evolve towards more sensitive and non-invasive diagnostics, dPCR is poised to play an increasingly critical role. However, its higher cost and lower throughput mean it will likely complement, rather than replace, the robust and economical qPCR in the well-equipped molecular diagnostic laboratory.
qPCR as an Orthogonal Validation Tool for Next-Generation Sequencing (NGS) Findings
In clinical cancer diagnostics, the imperative for accurate and reliable genomic data is paramount. Next-generation sequencing (NGS) delivers comprehensive profiling of tumor genomes but requires rigorous validation to ensure clinical-grade accuracy. Orthogonal validation, which verifies results using an independent methodological principle, is a cornerstone of this process [113]. Among available technologies, quantitative PCR (qPCR) has emerged as a powerful, targeted tool for confirming NGS-derived variants, offering a unique blend of speed, sensitivity, and cost-effectiveness that is well-suited to clinical workflows. This guide objectively compares the performance of qPCR and NGS, detailing how their synergistic application enhances the analytical validation of cancer diagnostics.
Understanding the fundamental operational differences between qPCR and NGS is critical for effectively deploying them in a complementary validation workflow. qPCR is a targeted amplification method that quantifies specific, pre-defined DNA or RNA sequences using primer probes, excelling in the rapid and sensitive detection of known variants [13] [6]. In contrast, NGS is a hypothesis-free approach that enables massively parallel sequencing of thousands to millions of DNA fragments simultaneously, providing the power to discover novel variants across the genome or exome without prior sequence knowledge [112] [6].
Table 1: Core Characteristics of qPCR and NGS
| Feature | qPCR | Next-Generation Sequencing (NGS) |
|---|---|---|
| Fundamental Principle | Target amplification with fluorescent probes | Massively parallel sequencing of DNA fragments |
| Discovery Power | Limited to known, pre-defined targets | High; detects known and novel variants [6] |
| Throughput | Low to medium; ideal for a limited number of targets | Very high; profiles hundreds to thousands of targets in one run [6] |
| Typical Turnaround Time | Several hours to 1 day | Several days to over a week [114] |
| Mutation Resolution | Excellent for single nucleotide variants (SNVs) and INDELs | Comprehensive; from single nucleotide variants to large chromosomal rearrangements [6] |
| Sensitivity (Variant Allele Frequency) | Can detect down to <1% with optimized assays [14] | Typically 1-5% for standard panels; can be lower with high depth [114] |
| Best-Suited Applications | Orthogonal validation, rapid screening, high-throughput clinical testing of known biomarkers | Comprehensive genomic profiling, discovery of novel variants, fusion detection [112] |
qPCR in Practice: Key Validation Studies and Performance Data
The application of qPCR for orthogonal confirmation is well-documented across various cancer genomic contexts. Its high specificity and sensitivity make it ideal for verifying critical biomarkers before clinical reporting.
A striking example of qPCR's performance is demonstrated in ovarian cancer detection. A 2025 study developed a qPCR algorithm using platelet-derived RNA, achieving a sensitivity of 94.1% and a specificity of 94.4% (AUC = 0.933) for detecting high-grade serous ovarian cancer. The 10-marker panel showed strong agreement with sequencing data (R² = 0.44-0.98), validating its use as a highly specific and cost-effective diagnostic tool [115].
In solid tumor profiling, a targeted NGS oncopanel for 61 cancer-associated genes was validated against known variants. The panel showed high sensitivity (98.23%) and near-perfect specificity (99.99%), performance metrics that are often confirmed initially using qPCR for key actionable mutations in genes like KRAS, EGFR, and PIK3CA [114]. This underscores qPCR's role in validating the most clinically relevant NGS findings.
Furthermore, machine learning models are now being developed to identify high-confidence single nucleotide variants (SNVs) from NGS data that may not require orthogonal confirmation. However, for low-confidence calls, complex variants, or when clinical actionability is high, qPCR remains a gold-standard confirmatory step to ensure >99.9% precision in variant reporting [116].
Table 2: Experimental Performance Data of qPCR in Diagnostic Applications
| Study Context | qPCR Assay Target/Type | Key Performance Metrics | Comparison to NGS |
|---|---|---|---|
| Ovarian Cancer Detection [115] | 10-gene panel from platelet RNA | Sensitivity: 94.1%Specificity: 94.4%AUC: 0.933 | Strong agreement with sequencing data (R² = 0.44-0.98) |
| Helicobacter pylori Detection [117] | IVD-certified kit (UreA gene) | Detected 16/40 samples (40.0%)Cq values: 17.51 - 32.21 | NGS detected 14/40 samples (35.0%); qPCR was slightly more sensitive |
| Limit of Detection (LOD) | Low-abundant mutation detection | Can detect SNPs with VAF as low as 0.1% - 0.5% [14] | Standard NGS panels typically have LOD of 2-5% VAF [114] |
Experimental Design: Orthogonal Validation Workflow
A robust orthogonal validation protocol ensures that NGS-identified variants are accurately confirmed by qPCR. The following workflow provides a detailed methodology for this critical process.
Stage 1: Variant Prioritization and Assay Design
The process begins by selecting NGS-derived variants that require confirmation. Priority should be given to: a) Clinically actionable mutations (e.g., in KRAS, EGFR, BRAF) that will directly influence treatment decisions; b) Low-confidence calls where NGS quality metrics (e.g., low mapping quality, strand bias, low allele frequency) indicate potential false positives; and c) Variants with allele frequencies near the assay's limit of detection [116] [114].
For qPCR assay design, two primary chemistries are available. TaqMan Probe-Based Chemistry uses a sequence-specific fluorescent probe for superior specificity and is ideal for discriminating single-nucleotide polymorphisms. SYBR Green Chemistry is more cost-effective and uses a dye that binds double-stranded DNA, but requires careful optimization to ensure specificity via melt-curve analysis. Assays must be designed to be highly specific, with primers and probes checked for potential secondary structures or off-target binding. The use of a digital PCR (dPCR) system can be considered for absolute quantification without a standard curve, offering the highest precision for low-VAF variant confirmation [14].
Stage 2: Wet-Lab Procedures and Data Acquisition
- Nucleic Acid Extraction: Use the same original DNA or RNA sample that was subjected to NGS to avoid sample heterogeneity. For FFPE tissues, kits like the AllPrep DNA/RNA FFPE Kit (Qiagen) are recommended to handle fragmented and cross-linked nucleic acids [112]. Quantify input DNA using a fluorometer (e.g., Qubit, Thermo Fisher Scientific) for accuracy, and use ≥ 50 ng of DNA as input to ensure robust detection, as lower inputs can lead to dropouts [114].
- qPCR Run Setup: Plate samples in at least three technical replicates to account for pipetting error and ensure statistical robustness. The run must include a standard curve (for SYBR Green), a no-template control (NTC) to check for contamination, and known positive and negative controls to verify assay performance. The positive control should be a sample with a known, low VAF of the target variant to challenge the assay's sensitivity.
- Data Acquisition: Perform the run on a calibrated real-time PCR instrument. The primary data output is the quantification cycle (Cq) for each replicate. Analyze the melt curve for SYBR Green assays to confirm a single, specific amplification product. Exclude any replicate with abnormal amplification or Cq values that are outliers from the mean.
Stage 3: Data Analysis and Concordance Assessment
Calculate the mean Cq value for the test sample and controls. For TaqMan genotyping assays, the scatter plot will clearly cluster samples as wild-type, heterozygous, or mutant. To correlate Cq with VAF from NGS, a standard curve can be generated using serially diluted positive control material.
A result is considered concordant when the qPCR genotyping call (e.g., "mutant detected") matches the NGS call. For low-VAF variants, a detection Cq value that is significantly earlier than the negative control and within the expected range of the standard curve confirms the NGS finding. The threshold for concordance should be established during assay validation; for clinical applications, the positive percent agreement (sensitivity) and negative percent agreement (specificity) between NGS and qPCR should exceed 99% for high-confidence SNVs [116].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of an orthogonal validation workflow depends on critical reagents and instruments.
Table 3: Essential Reagents and Tools for Orthogonal Validation
| Item | Function/Role | Example Products & Specifications |
|---|---|---|
| Nucleic Acid Extraction Kits | Isolate high-quality DNA/RNA from diverse clinical samples (FFPE, blood, frozen tissue). | Qiagen AllPrep DNA/RNA FFPE Kit [112]; Promega Maxwell RSC kits for automated extraction. |
| qPCR Master Mixes | Provide optimized buffers, enzymes, and dNTPs for efficient and specific amplification. | Meridian Bioscience Lyo-Ready and dUTP master mixes; ambient-temperature stable formulations [13]. |
| TaqMan Assays | Enable highly specific detection and quantification of known SNVs/INDELs. | Thermo Fisher Scientific TaqMan SNP Genotyping Assays; Custom-designed assays for novel variants. |
| Digital PCR Systems | Provide absolute quantification of variant allele frequency without a standard curve; highest sensitivity. | Bio-Rad QX200 Droplet Digital PCR; Thermo Fisher QuantStudio Absolute Q Digital PCR. |
| NGS Library Prep Kits | Prepare sequencing libraries for the initial comprehensive genomic profiling. | Sophia Genetics NGS library kits; Illumina TruSeq stranded mRNA kit [112] [114]. |
| Fluorometers & qPCR Instruments | Accurately quantify nucleic acid input and perform the real-time PCR reaction. | Thermo Fisher Qubit Fluorometer; QuantStudio 5 Real-Time PCR System [112]. |
The integration of qPCR as an orthogonal tool for NGS validation represents a robust and efficient paradigm in clinical cancer genomics. While NGS offers unparalleled discovery power, qPCR provides the speed, cost-effectiveness, and analytical specificity required to confidently verify critical genomic findings for patient care [13] [6]. This complementary relationship enhances the overall precision of diagnostic workflows, ensuring that treatment decisions are based on highly reliable data.
Emerging technologies like CRISPR/Cas-based diagnostics and machine learning triage of NGS variants are shaping the future of diagnostic validation [116] [14]. However, the fundamental principle of orthogonal confirmation remains unchanged. By leveraging the distinct strengths of both qPCR and NGS, clinical and research laboratories can optimize their validation strategies, streamline reporting, and ultimately advance the field of precision oncology.
The translation of quantitative PCR (qPCR)-based tests from research discoveries to clinical applications has been hampered by a significant lack of technical standardization, creating a substantial obstacle for their use in clinical cancer diagnostics [60]. Despite thousands of biomarker studies published to date, few have successfully transitioned to clinical practice, primarily due to irreproducible findings across laboratories [60]. This case study examines the framework for multi-laboratory validation (MLV) of clinical qPCR assays, focusing on the specific requirements for analytical validation in cancer research contexts.
The distinction between research-use-only (RUO) assays and properly validated clinical research (CR) assays is substantial. CR assays fill the critical gap between RUO and fully certified in vitro diagnostics (IVD), undergoing more thorough validation without yet reaching the status of a certified IVD assay [60]. For clinical cancer diagnostics, where qPCR is increasingly used for early detection, molecular stratification, and personalized therapy, proper validation is not optional—it is fundamental to ensuring reliable patient management decisions involving diagnosis, prognosis, prediction, and therapeutic monitoring [60] [1].
Analytical Validation Framework for Clinical qPCR Assays
Core Performance Parameters
The validation of a qPCR assay for clinical research requires rigorous assessment of multiple analytical performance parameters, each serving a distinct purpose in establishing assay reliability [60] [9] [8]. These parameters collectively ensure that the assay generates accurate, reproducible, and clinically meaningful results across multiple laboratory environments.
Table 1: Essential Analytical Validation Parameters for Clinical qPCR Assays
| Parameter | Definition | Acceptance Criteria | Clinical Significance |
|---|---|---|---|
| Analytical Sensitivity (LOD) | The lowest concentration of analyte that can be reliably detected [60] | Concentration with ≥95% detection rate [9] | Determines ability to detect low-abundance targets (e.g., rare mutations, minimal residual disease) |
| Analytical Specificity | Ability to distinguish target from nontarget analytes [60] | No amplification of non-target sequences [8] | Ensures accurate target detection in complex biological samples |
| Inclusivity | How well the qPCR detects all intended target strains/isolates [8] | Detection of all genetic variants intended to be captured | Critical for detecting diverse molecular subtypes in heterogeneous cancers |
| Exclusivity (Cross-reactivity) | How well the qPCR excludes genetically similar non-targets [8] | No detection of closely related non-target sequences | Prevents false positives from genetically similar but clinically distinct markers |
| Linear Dynamic Range | Range of template concentrations where signal is directly proportional to input [8] | R² ≥ 0.980; efficiency 90%-110% [8] | Enables accurate quantification across clinically relevant concentration ranges |
| Precision | Closeness of agreement between repeated measurements [60] | CV < 10-15% for replicate measurements | Ensures consistent results across repeated testing and multiple operators |
| Trueness (Accuracy) | Closeness of measured values to true values [60] | <0.5 log difference from reference standard | Confirms quantitative accuracy for clinical decision-making |
Sample Preparation and Quality Control Considerations
Proper sample acquisition, processing, storage, and RNA purification represent critical preanalytical factors that must be standardized across participating laboratories [60]. The validation plan must establish a quality assurance framework before testing begins, including consideration of external quality assessment reagents and protocols for resolving discrepant results [9]. For rare biomarkers or emerging targets where quality assurance panels may be unavailable, laboratories may need to collaborate with providers to produce suitable reference materials [9].
Multi-Laboratory Validation Study Design
Experimental Protocol for Interlaboratory Validation
A robust MLV study follows a standardized protocol that ensures comparable results across participating facilities while accounting for real-world variability in equipment, reagents, and technical staff [118] [119].
Participating Laboratory Selection: Laboratories should be selected based on their demonstrated experience with qPCR methodologies and clinical sample handling. A study validating Salmonella detection in frozen fish involved 14 participating laboratories [118], while a separate interlaboratory study for water quality testing engaged 16 laboratories [119]. Each laboratory is typically assigned a unique identifier to maintain anonymity during data analysis.
Sample Preparation and Distribution: The central coordinating facility prepares identical sets of blind-coded test samples representing a range of target concentrations, including negative controls. For the Salmonella validation study, each laboratory received 24 blind-coded frozen fish test portions for parallel testing with both the qPCR method and the reference culture method [118]. Sample panels should include:
- Positive samples across the assay's dynamic range
- Negative samples without the target
- Cross-reactivity challenges with genetically similar non-targets
- Inhibitor-containing samples to assess robustness
Reference Material Implementation: The use of standardized reference materials, such as the NIST SRM 2917—a linearized double-stranded plasmid DNA construct containing multiple qPCR target sequences—significantly improves interlaboratory consistency [119]. Each participating laboratory uses the same reference material to generate calibration models, with dilution series typically consisting of five to six 10-fold dilution concentrations with at least three replicate measurements per dilution level [119].
Data Collection and Analysis: All laboratories follow identical protocols for nucleic acid extraction, qPCR setup, amplification, and data analysis. The threshold cycle (Ct) values, amplification efficiency, and quantitative results are compiled centrally for statistical analysis. Performance metrics including false positives, false negatives, limit of detection, and precision are calculated for each laboratory and compared across sites [118] [119].
Statistical Analysis and Acceptance Criteria
The statistical framework for MLV studies focuses on establishing equivalence between the validated qPCR method and reference methods, as well as consistency across participating laboratories.
For the Salmonella detection validation, researchers calculated the fractional positive rate (required to be 25%-75% by FDA guidelines), compared negative and positive deviation rates against acceptability limits defined by ISO 16140-2:2016, and determined the relative level of detection (RLOD) to compare detection capabilities between methods [118]. An RLOD of approximately 1 indicates equivalent performance between the new qPCR method and the reference method [118].
In the NIST SRM 2917 interlaboratory study, researchers employed a Bayesian Markov Chain Monte Carlo approach to characterize within- and between-lab variability [119]. This advanced statistical method helps establish appropriate data acceptance metrics for future implementations. The study found that 99.5% of single-instrument run calibration models exhibited efficiency values between 90% and 110%—the expert-recommended range [119].
Comparative Performance Data: qPCR vs. Alternative Platforms
Analytical Sensitivity Across Detection Platforms
The diagnostic performance of qPCR must be understood in the context of alternative nucleic acid detection technologies, particularly for clinical applications in oncology.
Table 2: Comparative Analytical Performance of Nucleic Acid Detection Platforms
| Platform | Reported Sensitivity for ctDNA Detection | Key Advantages | Limitations | Ideal Clinical Context |
|---|---|---|---|---|
| qPCR | Varies by assay; suitable for variants >1% VAF [1] | Rapid turnaround (hours), cost-effective ($50-$200/test), high-throughput capability [1] | Lower sensitivity than advanced platforms for very rare targets [16] | High-abundance mutation detection, time-sensitive treatment decisions |
| ddPCR | Higher than qPCR (P < 0.001) [16] | Absolute quantification without standards, exceptional sensitivity for rare variants [16] | Higher cost per sample, limited multiplexing capability | Low variant allele frequency detection, minimal residual disease monitoring |
| NGS | Highest sensitivity (P = 0.014 vs. ddPCR) [16] | Comprehensive profiling, discovery capability, highest multiplexing [16] | Highest cost ($300-$3,000/test), longest turnaround (days), complex data analysis [1] | Unbiased mutation discovery, comprehensive genomic profiling |
A meta-analysis of circulating tumor HPV DNA detection across multiple cancer types demonstrated a clear hierarchy in analytical sensitivity, with NGS showing the greatest sensitivity, followed by ddPCR, and then qPCR (ddPCR > qPCR, P < 0.001; NGS > ddPCR, P = 0.014) [16]. This performance stratification must be balanced against practical considerations including cost, turnaround time, and infrastructure requirements when selecting the appropriate platform for specific clinical applications.
Practical Implementation Considerations
Beyond pure analytical performance, qPCR offers distinct practical advantages that contribute to its enduring position in clinical cancer diagnostics:
- Turnaround Time: qPCR delivers clinically actionable results within hours compared to days for NGS platforms, making it particularly valuable for time-sensitive therapeutic decisions [1]
- Infrastructure Requirements: qPCR requires less complex infrastructure than NGS and is more amenable to automation and implementation in resource-limited settings [1] [120]
- Multiplexing Capability: Advanced qPCR master mixes and probe systems enable detection of multiple mutations in a single reaction, crucial for cancers with complex mutational profiles [1]
- Regulatory Compliance: qPCR assays are generally easier to validate and implement within existing regulatory frameworks compared to more complex NGS workflows [1]
Implementation Framework and Reagent Solutions
Research Reagent Solutions for Oncology qPCR Assays
Successful multi-laboratory validation requires careful selection and standardization of reagents across participating sites. The following table outlines essential reagent components and their functions in clinical qPCR assays.
Table 3: Essential Research Reagent Solutions for Clinical qPCR Validation
| Reagent Component | Function | Key Characteristics for Clinical Assays |
|---|---|---|
| Hot-Start DNA Polymerase | Catalyzes DNA synthesis during PCR | Engineered to remain inactive at room temperature, preventing non-specific amplification and improving specificity [1] |
| Reverse Transcriptase | Converts RNA to cDNA for gene expression analysis | High efficiency with complex RNA secondary structures, minimal RNase H activity [121] |
| qPCR Master Mix | Provides optimized buffer conditions and detection chemistry | Contains inhibitors-resistant polymerses, optimized for clinical matrices (plasma, FFPE), compatible with multiplex detection [1] |
| Fluorescent Probes (TaqMan) | Sequence-specific detection of amplified targets | Enable multiplexing through different fluorophores, high quenching efficiency, minimal background fluorescence [122] |
| Reference DNA/RNA | Calibration and standardization across runs | Certified concentration, high purity, stability across freeze-thaw cycles [119] |
| Inhibition Resistance Additives | Counteract PCR inhibitors in clinical samples | Engineered buffers that tolerate inhibitors common in plasma, whole blood, and FFPE-derived nucleic acids [1] |
Workflow Optimization for Multi-Laboratory Consistency
Standardization of the entire qPCR workflow is essential for reproducible results across multiple laboratories. The following diagram illustrates the critical stages in multi-laboratory validation of clinical qPCR assays.
Multi-Lab qPCR Validation Workflow
Multi-laboratory validation represents a critical step in the translation of qPCR assays from research tools to clinically applicable tests for cancer diagnostics. Through standardized protocols, appropriate statistical analysis, and careful attention to preanalytical and analytical variables, qPCR assays can achieve the reproducibility required for meaningful clinical interpretation. While emerging technologies like ddPCR and NGS offer advantages for specific applications, qPCR maintains a vital role in clinical cancer diagnostics due to its unique combination of speed, cost-effectiveness, and scalability [1] [16]. The implementation of rigorous multi-laboratory validation frameworks ensures that qPCR continues to provide reliable, actionable genomic information for cancer researchers and clinicians worldwide.
Conclusion
The analytical validation of qPCR assays is a critical, multi-faceted process that underpins their reliability in clinical cancer diagnostics. From foundational design to rigorous troubleshooting and final validation, each step ensures that these assays deliver the speed, sensitivity, and specificity required for time-sensitive therapeutic decisions. While qPCR offers an unparalleled combination of cost-effectiveness, scalability, and robust performance for targeted mutation detection, its role is evolving alongside technologies like digital PCR. The future of clinical oncology will likely see qPCR maintaining its position as a workhorse for routine screening and MRD monitoring, while increasingly being integrated into complementary diagnostic workflows with dPCR and NGS. Future efforts should focus on standardizing validation protocols across laboratories and further optimizing assays for emerging biomarkers and complex sample matrices to expand access to precision medicine.
References
- 1. qPCR in Early Cancer Detection: Why It Still Leads the Pack [https://www.meridianbioscience.com/lifescience-blog/why-qpcr-still-leads-in-oncology/]
- 2. The Advantage of Targeted Next-Generation Sequencing ... [https://www.mdpi.com/1422-0067/25/14/7908]
- 3. Analysis of Colorectal Cancer Gene Mutations and Application ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12110167/]
- 4. Digital PCR outperforms quantitative real-time PCR for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288185/]
- 5. Innovative diagnostic approaches for lung cancer [https://www.nature.com/articles/s41598-025-13743-4]
- 6. NGS vs qPCR [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/advantages/ngs-vs-qpcr.html]
- 7. Emerging Molecular Technology in Cancer Testing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11536271/]
- 8. Quantitative PCR validation for research scientists: Part 1 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 9. Validating Real-Time Polymerase Chain Reaction (PCR) Assays [https://pmc.ncbi.nlm.nih.gov/articles/PMC7834634/]
- 10. Comparing the Diagnostic Performance of Quantitative PCR ... [https://pubmed.ncbi.nlm.nih.gov/38103593/]
- 11. Development and validation of a qPCR assay for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12536431/]
- 12. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 13. The Growing Demand for Personalized Diagnostics [https://www.meridianbioscience.com/lifescience-blog/the-growing-demand-for-personalized-diagnostics-navigating-the-benefits-and-limits-of-qpcr-vs-ngs/]
- 14. Building the Future of Clinical Diagnostics: An Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11854988/]
- 15. Multiplex PCR Assay Market CAGR, Segments and ... [https://www.towardshealthcare.com/insights/multiplex-pcr-assay-market-sizing]
- 16. Comparing the Diagnostic Performance of Quantitative ... [https://www.sciencedirect.com/science/article/pii/S1525157823002921]
- 17. literature review and comparison of assays for ESR1 ... [https://pubmed.ncbi.nlm.nih.gov/41186491/]
- 18. Liquid biopsy in cancer: current status, challenges and ... [https://www.nature.com/articles/s41392-024-02021-w]
- 19. Development of a Machine Learning Model for Aspyre ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12366736/]
- 20. Integrating Circulating Tumor DNA into Clinical Management ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12346868/]
- 21. Comparison Analysis of Yield, Quality and Next Generation ... [https://omegabiotek.com/application-notes/comparison-analysis-of-yield-quality-and-next-generation-sequencing-from-ffpe-samples-using-three-total-nucleic-acid-isolation-kits/]
- 22. Evaluation of pre-analytical conditions and comparison ... [https://www.oncotarget.com/article/21256/text/]
- 23. Comparative analysis of EGFR mutations in circulating ... [https://www.nature.com/articles/s41598-025-85160-6]
- 24. Selection and validation of reference genes for RT-qPCR ... [https://www.nature.com/articles/s41598-025-02951-7]
- 25. MIQE 2.0 and the Urgent Need to Rethink qPCR Standards [https://pmc.ncbi.nlm.nih.gov/articles/PMC12154065/]
- 26. Development and analytical validation of a multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12454996/]
- 27. How to Design Primers for PCR Experiments [https://www.zymoresearch.com/blogs/blog/how-to-design-primers-for-pcr-experiments?srsltid=AfmBOopYkFSrwwqutHxj9M4EkYj6k6QYJSivwv1D0H1C-8SIz-Cgq-CD]
- 28. qPCR primer design revisited - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC5702850/]
- 29. Precision and Accuracy with TaqMan Probes [https://synbio-tech.com/taqman-probe]
- 30. qPCR Primer & Probe Design Tool [https://eurofinsgenomics.eu/en/ecom/tools/qpcr-assay-design/]
- 31. TaqMan Probes and qPCR Primers [https://www.thermofisher.com/us/en/home/life-science/oligonucleotides-primers-probes-genes/applied-biosystems-custom-primers-probes.html]
- 32. Taqman-probe-based multiplex real-time RT-QPCR for ... [https://www.sciencedirect.com/science/article/pii/S0032579125007515]
- 33. Comparison of SYBR Green and TaqMan methods in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3988599/]
- 34. Probe-based Real-time PCR Approaches for Quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4541571/]
- 35. Development and Validation of a SYBR Green Real Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8069436/]
- 36. Overview: Types of PCR Probes [https://www.goldbio.com/blogs/articles/overview-types-pcr-probes?srsltid=AfmBOoroJ9Dd-YooPZvxrFXAXh4iVGHYsltPRXlq-UALrtTv3SBcpmsz]
- 37. qPCR: Plexor vs Hydrolysis Probes [https://bitesizebio.com/2473/qpcr-plexor-vs-hydrolysis-probes/]
- 38. What's the Right Master Mix for Your qPCR Experiment? ... [https://www.thermofisher.com/blog/behindthebench/whats-the-right-master-mix-for-your-qpcr-experiment-lets-ask-taqman/]
- 39. Cancer Research to Microbes: Harnessing the Power of ... [https://www.thermofisher.com/blog/behindthebench/harnessing-the-power-of-qpcr/]
- 40. SYBR GreenER qPCR SuperMix Universal [https://www.thermofisher.com/us/en/home/references/protocols/nucleic-acid-amplification-and-expression-profiling/pcr-protocol/sybr-greener-qpcr-supermix-universal.html]
- 41. Development of a multiplex allele-specific qPCR approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9708691/]
- 42. PCR (qPCR, dPCR, Singleplex, Multiplex) Markets ... [https://www.businesswire.com/news/home/20250616322572/en/PCR-qPCR-dPCR-Singleplex-Multiplex-Markets-Forecasts-2025-2030---Revolutionizing-Diagnostics-with-Cutting-Edge-Precision-Fueling-Opportunities---ResearchAndMarkets.com]
- 43. Advancing quantitative PCR with color cycle multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417387/]
- 44. A simple and robust real-time qPCR method for the ... [https://www.nature.com/articles/s41598-018-22473-9]
- 45. High throughput AS LNA qPCR method for the detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11007389/]
- 46. Development and analytical validation of a multiplex ... [https://www.sciencedirect.com/science/article/pii/S1936523325002591]
- 47. Integrating traditional machine learning with qPCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11754184/]
- 48. QPCR Protocol [https://www.protocols.io/view/qpcr-protocol-dxq57my6.html]
- 49. Universal SYBR Green qPCR Protocol [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/qpcr/sybr-green-qpcr?srsltid=AfmBOopk_5gCesjIQ6oqkn2Dxep9IiCPGBIimhd3Fd7kpAJSe6nlKmsj]
- 50. qPCR Steps: Creating a Successful Experiment | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/successful-qpcr/]
- 51. RT-qPCR Testing and Performance Metrics in the COVID- ... [https://www.mdpi.com/1422-0067/25/17/9326]
- 52. Comparative analysis of the diagnostic performance of five ... [https://www.nature.com/articles/s41598-021-00852-z]
- 53. Understanding Ct Values in Real-Time PCR [https://www.thermofisher.com/blog/behindthebench/understanding-ct-values/]
- 54. What is a Ct Value and does it differ from qPCR Cq Values? [https://bitesizebio.com/24581/what-is-a-ct-value/]
- 55. How To Interpret RT-qPCR Results [https://www.goldbio.com/blogs/articles/how-to-interpret-rt-qpcr-results?srsltid=AfmBOop69HTUL4DZ646w0yIDtwZFApFpDSAHKmACdy8USyifdV0nEUjJ]
- 56. Use and Misuse of Cq in qPCR Data Analysis and Reporting [https://pmc.ncbi.nlm.nih.gov/articles/PMC8229287/]
- 57. Absolute Quantification qPCR | Relative ... [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/pcr-quantification/pcr-quantification]
- 58. rtpcr: a package for statistical analysis and graphical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530193/]
- 59. qPCR and qRT-PCR analysis: Regulatory points to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7786041/]
- 60. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 61. Analyzing qPCR data: Better practices to facilitate rigor and ... [https://www.sciencedirect.com/science/article/pii/S2405580825004431]
- 62. How to optimize qPCR in 9 steps? [https://westburg.eu/knowledge/pcr_qpcr/hate_optimizing_qpcr]
- 63. One-step vs. two-step RT-qPCR—tips for choosing the right ... [https://www.takarabio.com/about/bioview-blog/tips-and-troubleshooting/one-step-vs-two-step-rt-qpcr?srsltid=AfmBOoqNq3XgrLAJo0mFpAgqvaw5clhMTXzVkpI2JlBxEI3pSZ_-9FrI]
- 64. How to build qPCR assays like a pro [https://www.news-medical.net/whitepaper/20240617/How-to-build-qPCR-assays-like-a-pro.aspx]
- 65. Faster quantitative real-time PCR protocols may lose ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1297710/]
- 66. Multi-laboratory validation of a modified real-time PCR ... [https://www.sciencedirect.com/science/article/pii/S0740002025000073]
- 67. Evaluation and validation of reference genes for RT-qPCR ... [https://www.nature.com/articles/s41598-025-22650-7]
- 68. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 69. How to Choose a PCR Probe [https://www.goldbio.com/blogs/articles/how-to-choose-pcr-probe?srsltid=AfmBOorAxm8ehVoiEdh7tq5_GgM8_qGHhRRvKUCwsaJZhbRCo3Z-1LKw]
- 70. Specificity of Primers and Probes for Molecular Diagnosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566895/]
- 71. Comparative analysis of 3 qPCR primer–probe sets for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12504209/]
- 72. What is a PCR Master Mix? Components & Functions ... [https://3crbio.com/blog/what-is-a-pcr-master-mix/]
- 73. PCR Master Mix [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/pcr-master-mix?srsltid=AfmBOoqD4Zt9TpjptDl3uTmKJ7WlLn5fQKawmkG3kzGoHII2C1FxJouD]
- 74. TaqPath qPCR and RT-qPCR Master Mixes [https://www.thermofisher.com/vi/en/home/clinical/diagnostic-development/molecular-diagnostic-test-development/consumables-reagents-clinical-reagents/taqpath-qpcr-master-mixes.html]
- 75. FAQ: What are the differences between the numerous Q5 ® ... [https://www.neb.com/en-us/faqs/2023/07/24/what-are-the-differences-between-the-numerous-q5-polymerase-products-available?srsltid=AfmBOooSk6jsMcsoltEPXuqxZ70fZT1GwY2GbOZvEvQ9VjtHtd2HMUb4]
- 76. Distinct Roles of the Active-site Mg2+ Ligands, Asp882 and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3030377/]
- 77. Crystal structures of phage NrS-1 N300-dNTPs-Mg ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X19310605]
- 78. New Master Mix Helps Increase Productivity and ... [https://www.labmanager.com/new-master-mix-helps-increase-productivity-and-performance-31776]
- 79. Maximising the Use of Scarce qPCR Master Mixes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9368830/]
- 80. Optimization of TaqMan-based quantitative PCR diagnosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12165414/]
- 81. Thermal Cyclers vs. qPCR Machines: What's the Difference? [https://www.excedr.com/blog/thermal-cyclers-vs-qpcr-machines-whats-the-difference]
- 82. Validation and advantages of using novel RT-qPCR ... [https://www.nature.com/articles/s41598-022-17339-0]
- 83. Optimization of the real-time PCR platform method using ... [https://www.nature.com/articles/s41598-025-16972-9]
- 84. Optimization of digital droplet polymerase chain reaction ... [https://www.sciencedirect.com/science/article/pii/S2214753515300127]
- 85. Optimization and validation of a consolidated Set ... [https://www.sciencedirect.com/science/article/pii/S2215016125004443]
- 86. Inhibition of the PCR by genomic DNA - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10120932/]
- 87. qPCR primer design revisited [https://www.sciencedirect.com/science/article/pii/S221475351730181X]
- 88. qPCR Troubleshooting Guide [https://azurebiosystems.com/qpcr-resources/troubleshooting/]
- 89. Troubleshooting qPCR: Interpreting Amplification Curves [https://blog.biosearchtech.com/thebiosearchtechblog/bid/63622/qpcr-troubleshooting]
- 90. Troubleshooting fine‐tuning procedures for qPCR system ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6647679/]
- 91. Integrating traditional machine learning with qPCR ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1539120/full]
- 92. Understanding Fluorescent Quantitative PCR (qPCR) Curves [https://www.cd-genomics.com/resource-understanding-fluorescent-quantitative-pcr-curves.html]
- 93. Melting Curve Analysis - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/melting-curve-analysis]
- 94. Removal of artifact bias from qPCR results using DNA ... [https://pubmed.ncbi.nlm.nih.gov/31682470/]
- 95. Web-based LinRegPCR: application for the visualization and ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04306-1]
- 96. Establishment and validation of a simple and accurate ... [https://www.nature.com/articles/s41598-025-92803-1]
- 97. Complete Guide to GxP: Compliance, QMS & Best Practices [https://www.qualityfwd.com/blog/gxp-compliance/]
- 98. Understanding GxP Compliance: A Guide to Good ... [https://www.labmanager.com/understanding-gxp-compliance-a-guide-to-good-practices-in-regulated-industries-33604]
- 99. GxP Compliance: A Comprehensive Guide to 2025 [https://www.compliancequest.com/what-is-gxp-compliance-with-fda-regulations/]
- 100. GxP Compliance for Regulated Industries [https://reesscientific.com/blog/gxp-compliance-overview-ensuring-quality-across-regulated-industries]
- 101. GxP Compliance Training: 3 Pillars for Effective ... [https://www.gyrus.com/blogs/gxp-compliance-training/]
- 102. GxP Managed Services: A 2025 Analysis for Life Sciences [https://intuitionlabs.ai/articles/gxp-managed-services-landscape-2025]
- 103. Comparison of the analytical and clinical sensitivities of 34 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12502672/]
- 104. Analytical comparison of 11 CE-IVD marked real-time PCR ... [https://link.springer.com/article/10.1007/s10096-025-05214-5]
- 105. One-step and two-step qPCR assays for CAPRV2023 [https://www.frontiersin.org/journals/veterinary-science/articles/10.3389/fvets.2025.1620997/full]
- 106. The new gold rush in clinical diagnostics: from standard ... [https://www.sciencedirect.com/org/science/article/pii/S2165049725017615]
- 107. Quantitative or digital PCR? A comparative analysis for ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993624001584]
- 108. Digital PCR (dPCR) vs Real-Time PCR (qPCR) [https://firalismolecularprecision.com/2025/04/09/dpcr-qpcr-differences/]
- 109. Comparative Performance of Digital PCR and Real-Time RT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12474457/]
- 110. Comparing the precision of two digital PCR applications for ... [https://www.nature.com/articles/s41598-025-13143-8]
- 111. Analytical Validation of MyProstateScore 2.0 [https://www.mdpi.com/2075-4418/15/7/923]
- 112. Clinical and analytical validation of a combined RNA ... [https://www.nature.com/articles/s43856-025-00934-3]
- 113. Antibody Validation Essentials: Orthogonal Strategy - CST Blog [https://blog.cellsignal.com/hallmarks-of-validation-orthogonal-strategy]
- 114. A targeted next-generation sequencing panel for ... [https://www.nature.com/articles/s41598-025-08039-6]
- 115. Innovative qPCR Algorithm Using Platelet-Derived RNA for ... [https://pubmed.ncbi.nlm.nih.gov/40227838/]
- 116. Determination of high-confidence germline genetic variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12326712/]
- 117. Comparison of Next-Generation Sequencing, Real-Time ... [https://www.mdpi.com/2076-2607/13/10/2344]
- 118. Multi-laboratory validation study of a real-time PCR method ... [https://pubmed.ncbi.nlm.nih.gov/40484534/]
- 119. Interlaboratory performance and quantitative PCR data ... [https://www.sciencedirect.com/science/article/abs/pii/S0043135422011071]
- 120. qPCR Instruments Market Size, Trends & Forecast 2025- ... [https://www.futuremarketinsights.com/reports/qpcr-instruments-market]
- 121. qPCR Enzymes and Kit 2025 Trends and Forecasts 2033 [https://www.archivemarketresearch.com/reports/qpcr-enzymes-and-kit-309200]
- 122. Real-Time PCR | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr.html]