AI-Powered Virtual Screening: Computational Protocols Revolutionizing Anticancer Drug Discovery
This comprehensive review explores the transformative role of computational virtual screening protocols in accelerating anticancer drug discovery.
AI-Powered Virtual Screening: Computational Protocols Revolutionizing Anticancer Drug Discovery
Abstract
This comprehensive review explores the transformative role of computational virtual screening protocols in accelerating anticancer drug discovery. It examines foundational concepts, from target identification to drug repurposing, and details state-of-the-art methodologies including molecular docking, molecular dynamics simulations, and AI-accelerated platforms. The article provides practical insights for troubleshooting common challenges and presents rigorous validation frameworks through case studies across various cancer targets. By synthesizing recent advances and real-world applications, this work serves as an essential resource for researchers and drug development professionals seeking to leverage computational approaches for developing more effective and targeted cancer therapies.
Virtual Screening Foundations: From Basic Concepts to Anticancer Applications
Virtual screening (VS) is a computational technique that automatically searches through libraries of molecules to identify structures most likely to bind effectively to a therapeutic target, such as a protein receptor or enzyme implicated in cancer progression [1]. In the pharmaceutical industry, VS has demonstrated efficacy as a strategy for effectively identifying bioactive molecules, presenting the potential to drastically speed up the drug discovery phase, which is often hindered by huge costs and high failure rates [1]. In oncology, this approach is particularly valuable for identifying novel anti-tumor agents and targeted therapies [2].
The two primary computational strategies in virtual screening are ligand-based screening and structure-based screening [1].
Ligand-Based Screening: This approach is used when the 3D structure of the target protein is unknown but there is information about known active ligands. It involves creating a pharmacophore model—a set of structural features essential for biological activity—from a collection of active ligands, or performing 2D chemical similarity analyses to find new molecules that resemble known actives [1]. This method is computationally efficient, often requiring only a single CPU to screen thousands of compounds rapidly [1].
Structure-Based Screening (Molecular Docking): This method is employed when the three-dimensional structure of the target protein is available. It involves computationally "docking" candidate small molecules into the binding site of the target protein and scoring their complementary to predict binding affinity [3] [1] [4]. This approach is more computationally intensive and typically relies on a parallel computing infrastructure to manage large datasets and run multiple comparisons simultaneously [1].
AI-Accelerated Virtual Screening Platforms
The advent of artificial intelligence (AI) and machine learning (ML) has transformed virtual screening, enabling the efficient exploration of ultra-large chemical libraries containing billions of compounds [3] [5]. AI-driven platforms can slash development timelines and boost success rates by learning complex patterns from large datasets of chemical compounds and biological targets [5].
State-of-the-Art Platforms and Performance
Recent advances have led to the development of highly accurate, open-source platforms. One such platform, RosettaVS, incorporates an improved physics-based force field (RosettaGenFF-VS) and uses active learning to triage the most promising compounds for expensive docking calculations [3]. This platform has demonstrated state-of-the-art performance on standard benchmarks.
Table 1: Performance Benchmarking of RosettaGenFF-VS on CASF-2016 Dataset
| Benchmark Metric | Performance of RosettaGenFF-VS | Comparison to Second-Best Method |
|---|---|---|
| Docking Power (Pose Prediction) | Top-performing method for distinguishing native binding poses from decoys [3] | Superior performance across a broad range of ligand RMSDs [3] |
| Screening Power (Top 1% Enrichment Factor) | EF~1%~ = 16.72 [3] | Outperformed second-best method (EF~1%~ = 11.9) by a significant margin [3] |
| Success Rate (Identifying Best Binder) | Excelled at placing the best binder within the top 1%, 5%, and 10% of ranked molecules [3] | Surpassed all other methods in the benchmark [3] |
In practical applications, this AI-accelerated platform successfully screened multi-billion compound libraries against two unrelated oncology-relevant targets: the ubiquitin ligase KLHDC2 and the human voltage-gated sodium channel NaV1.7. The screening process was completed in less than seven days, identifying hit compounds with single-digit micromolar binding affinities [3].
Workflow of an AI-Accelerated Screening Platform
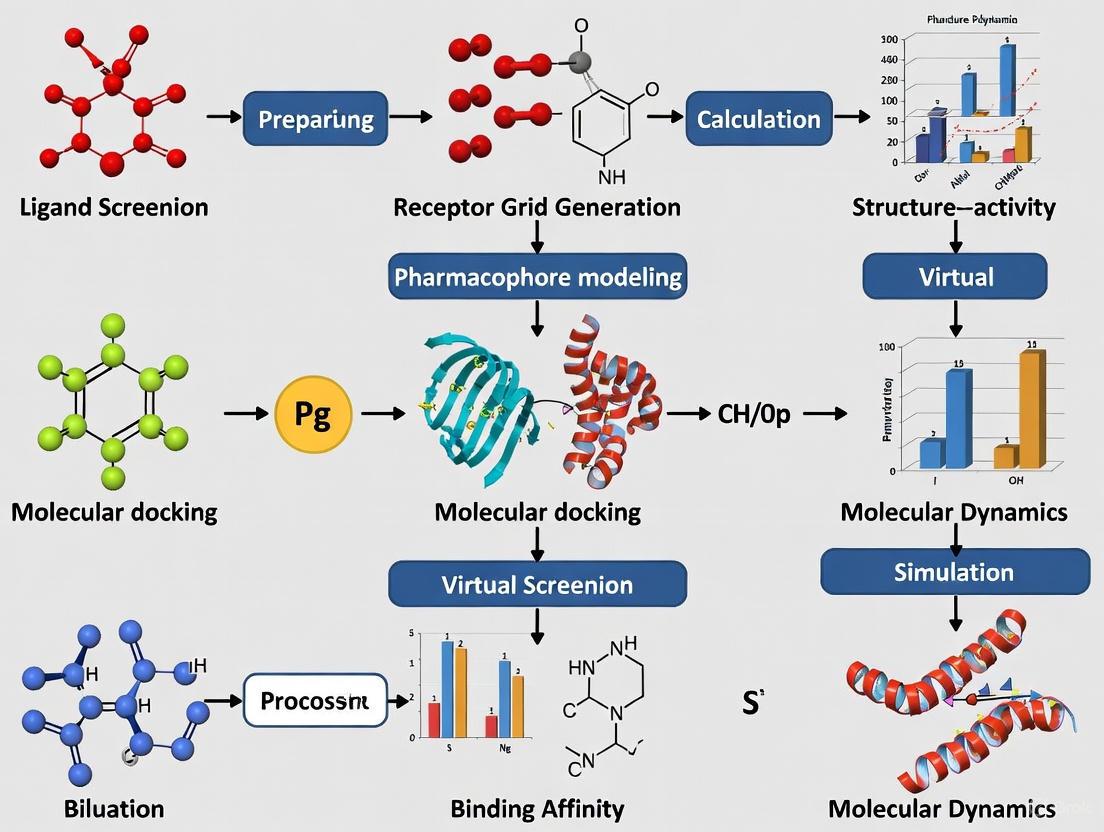
AI-powered platforms integrate multiple components into a cohesive and iterative workflow. The following diagram illustrates the key stages of this process, from initial library preparation to final experimental validation.
Key Experimental Protocols
This section provides detailed methodologies for implementing structure-based and AI-enhanced virtual screening campaigns in an oncology context.
Protocol for Structure-Based Virtual Screening with RosettaVS
This protocol is adapted from a successful screening campaign against the oncology target KLHDC2, which yielded a 14% hit rate with micromolar affinities [3].
Objective: To identify novel, high-affinity small-molecule binders to a defined binding site on an oncology target protein from an ultra-large chemical library.
Required Reagents and Resources:
- Target Structure: A high-resolution 3D structure of the protein target (e.g., from X-ray crystallography or cryo-EM) in PDB format [3] [4].
- Compound Library: A file (e.g., in SDF or SMILES format) of small molecules to screen. Publicly available libraries include ZINC and PubChem [4].
- Computational Infrastructure: A high-performance computing (HPC) cluster. The referenced study used a cluster with 3000 CPUs and one GPU per target [3].
- Software: The OpenVS platform incorporating RosettaVS [3].
Step-by-Step Procedure:
- Target Preparation:
- Obtain the protein structure and prepare it using molecular modeling software (e.g., Maestro, MOE).
- Add hydrogen atoms, assign partial charges, and define protonation states of key residues at physiological pH.
- Define the binding site coordinates based on known ligand interactions or structural data.
Ligand Library Preparation:
- Download and curate the compound library. Apply filters for drug-like properties (e.g., Lipinski's Rule of Five) and remove pan-assay interference compounds (PAINS) [4].
- Generate biologically relevant, low-energy 3D conformers for each molecule in the library.
Hierarchical Docking with RosettaVS:
- Stage 1 - VSX (Virtual Screening Express) Mode: Perform rapid, initial docking of the entire library. In this mode, the receptor is treated as rigid to maximize speed [3].
- Stage 2 - VSH (Virtual Screening High-precision) Mode: Take the top-ranked hits from the VSX stage (e.g., top 1-5%) and re-dock them using the high-precision mode. This mode allows for full receptor side-chain flexibility and limited backbone movement, which is critical for accurately modeling induced fit upon ligand binding [3].
Scoring and Hit Selection:
- Score the final docking poses from the VSH stage using the RosettaGenFF-VS scoring function, which combines enthalpy (∆H) calculations with a new model estimating entropy changes (∆S) upon binding [3].
- Rank the compounds based on their predicted binding affinity.
- Visually inspect the top 100-500 ranked compounds to assess binding pose rationality and key interactions.
Experimental Validation:
- Procure the top-ranked virtual hit compounds from commercial suppliers or synthesize them.
- Validate binding using biochemical assays (e.g., fluorescence polarization, surface plasmon resonance) to determine half-maximal inhibitory concentration (IC~50~) or dissociation constant (K~d~) [3].
- Confirm functional activity in cell-based assays relevant to the oncology target (e.g., cell proliferation, apoptosis) [2].
Protocol for AI-Guided Hit Identification
This protocol leverages machine learning to enhance the efficiency of virtual screening [3] [5].
Objective: To employ a target-specific neural network, trained concurrently with docking calculations, to prioritize compounds for docking and improve hit rates.
Required Reagents and Resources:
- All resources from Protocol 3.1.
- An active learning framework integrated into the virtual screening platform (e.g., OpenVS) [3].
Step-by-Step Procedure:
- Initial Sampling and Model Training:
- Start by docking a small, random subset (e.g., 0.1%) of the ultra-large library.
- Use the docking scores and molecular descriptors from this subset as training data to initialize a target-specific machine learning model (e.g., a deep neural network).
Iterative Active Learning Cycle:
- Predict: Use the trained ML model to predict the docking scores for the entire undocked portion of the library.
- Select: Prioritize and select a new batch of compounds for docking that the model predicts will have high affinity.
- Update: Dock the newly selected batch of compounds and add their results (true docking scores) to the training set.
- Retrain: Update the ML model with the expanded training data.
- Repeat this cycle until a predefined stopping criterion is met (e.g., number of compounds docked or convergence of hit discovery).
Final Hit Selection and Validation:
- After the active learning cycle is complete, the top-ranked compounds from the final model and their docking poses are analyzed.
- Proceed with experimental validation as described in Step 5 of Protocol 3.1.
Successful implementation of virtual screening requires a collection of specialized computational and experimental resources.
Table 2: Key Research Reagent Solutions for Virtual Screening
| Resource Category | Specific Examples | Function and Application in Virtual Screening |
|---|---|---|
| Public Compound Databases | ZINC [4], PubChem [4] | Provide libraries of commercially available, synthesizable small molecules for screening. |
| Bioactivity Databases | ChEMBL [4], BindingDB [4] | Contain experimental bioactivity data for model training and validation. |
| Protein Structure Repository | Protein Data Bank (PDB) [4] | Source of 3D structural data for target preparation in structure-based screening. |
| Docking & VS Software | RosettaVS (OpenVS) [3], Glide [4], AutoDock Vina [3] | Core programs for predicting protein-ligand complex structures and binding affinities. |
| AI/ML Platforms | Aurigene.AI [6], BenevolentAI [5], Owkin [7] | Offer predictive and generative AI models for target identification, compound scoring, and lead optimization. |
| Computing Infrastructure | High-Performance Computing (HPC) Clusters [3] [1], NVIDIA GPUs [3] [6] | Provide the necessary parallel processing power for large-scale docking and AI model training. |
The discovery and development of novel anticancer agents represent a complex, risky, and costly endeavor, traditionally requiring over 15 years and exceeding $1.8 billion USD per approved drug [8]. Within this landscape, computational methodologies have become crucial components of drug discovery programs, significantly accelerating the identification and optimization of potential therapeutic compounds [8]. Computer-Aided Drug Discovery and Design (CADDD) harnesses various sources of information and computational techniques to facilitate the development of new drugs that modulate therapeutically relevant protein targets in cancer [8]. These approaches have evolved from serendipitous discovery to rational design, enabling researchers to make in silico improvements before resource-intensive laboratory experimentation [8].
Computational drug design approaches are broadly classified into two families: ligand-based and structure-based methods [8]. Ligand-based methods utilize existing knowledge of active compounds against a target to predict new chemical entities with similar behavior, while structure-based methods rely on three-dimensional structural information of the target to determine whether new compounds are likely to bind and interact effectively [8]. The integration of both approaches has become common in virtual screening, enhancing strengths while mitigating the limitations of each method individually [8]. This document provides detailed application notes and experimental protocols for key computational methodologies within the context of virtual screening for anticancer drug discovery.
Key Terminology and Computational Concepts
Core Methodologies
Molecular Docking predicts the preferred orientation of a small molecule (ligand) when bound to its target binding site, enabling the prediction of binding affinity and molecular interactions [9] [10]. This technique is fundamental for structure-based virtual screening, allowing researchers to prioritize compounds with the highest predicted binding energies for further investigation.
Quantitative Structure-Activity Relationship (QSAR) modeling correlates the structural properties of compounds with their biological activity through statistical methods [11]. These models enable the prediction of biological activity for novel compounds based on their structural descriptors, guiding the optimization of lead compounds in anticancer development.
ADMET profiling predicts the Absorption, Distribution, Metabolism, Excretion, and Toxicity properties of candidate molecules [12] [10]. These computational assessments are critical early in the discovery process to eliminate compounds with unfavorable pharmacokinetic or safety profiles, reducing late-stage attrition.
Molecular Dynamics (MD) Simulation analyzes the physical movements of atoms and molecules over time, providing insights into the stability and dynamics of protein-ligand complexes under biologically relevant conditions [9] [13]. These simulations validate docking results and assess the temporal stability of binding interactions.
Pharmacophore Modeling identifies the essential structural features responsible for a compound's biological activity [12]. This approach schematically illustrates the critical components of molecular recognition, enabling the identification of novel compounds that share these key features regardless of their overall chemical structure.
Integrated Workflows in Anticancer Discovery
Modern computational drug discovery employs integrated workflows that combine multiple methodologies. For example, a typical structure-based virtual screening workflow might include: structure-based pharmacophore modeling, virtual screening of compound libraries, molecular docking of top hits, ADMET profiling, and final validation through molecular dynamics simulations [9] [10] [14]. Such integrated approaches have successfully identified potential inhibitors for various cancer targets, including PD-L1, VEGFR-2, c-Met, MCL1, and XIAP [9] [10] [13].
Table 1: Performance Metrics of Computational Methods in Anticancer Discovery
| Method | Reported Enrichment | Library Size Screened | Success Rate | Key Applications in Cancer |
|---|---|---|---|---|
| Pharmacophore Modeling | Early enrichment factor (EF1%) = 10.0 [14] | 52,765 - 407,270 compounds [9] [13] | AUC: 0.98 [14] | XIAP, MCL1, VEGFR-2/c-Met inhibitors [10] [13] [14] |
| Molecular Docking | Binding affinity improvements from -6.8 kcal/mol to -11.2 kcal/mol [14] | 1.28 million compounds [10] | 18 hit compounds from 1.28 million [10] | PD-L1, VEGFR-2/c-Met dual inhibitors [9] [10] |
| QSAR Modeling | IC50 prediction below median value [13] | 407,270 compounds [13] | Sub-nanomolar potency achievement [13] | MCL1 inhibitor optimization [13] |
| MD Simulations | Stable conformation maintenance at 100 ns [9] [10] | 2-4 final candidates [10] [14] | Binding free energy validation [10] | PD-L1, VEGFR-2/c-Met, XIAP complex stability [9] [10] [14] |
| AI-Enhanced Screening | >50-fold hit enrichment vs traditional methods [15] | 26,000+ virtual analogs [15] | 4,500-fold potency improvement [15] | MAGL inhibitor optimization [15] |
Table 2: ADMET Profiling Parameters for Anticancer Candidate Selection
| Parameter | Optimal Range | Computational Tools | Impact on Candidate Selection |
|---|---|---|---|
| Aqueous Solubility | Level 3 (reference value) [10] | SwissADME [15] | Ensures adequate bioavailability |
| Blood-Brain Barrier Penetration | Level 3 (reference value) [10] | ADMET predictors [10] | Minimizes CNS-related side effects |
| Cytochrome P450 2D6 Inhibition | Non-inhibitor preferred [10] | PreADMET [14] | Reduces drug-drug interaction potential |
| Hepatotoxicity | Non-toxic preferred [10] | PreADMET [14] | Prevents liver damage |
| Human Intestinal Absorption | Level 0 (good absorption) [10] | SwissADME [15] | Ensures oral bioavailability |
| Plasma Protein Binding | Moderate to high [14] | PreADMET [14] | Influences drug distribution and half-life |
Experimental Protocols for Key Methodologies
Protocol 1: Structure-Based Pharmacophore Modeling
Application Context: Identification of natural PD-L1 inhibitors from marine natural products [9].
Principle: Structure-based pharmacophore modeling defines the essential steric and electronic features necessary for molecular recognition at a drug target's binding site [12] [9].
Procedure:
- Protein Preparation: Obtain the 3D crystal structure of the target protein (e.g., PD-L1, PDB ID: 6R3K) from the RCSB Protein Data Bank. Remove water molecules, add hydrogen atoms, complete missing amino acid residues, and minimize energy using force fields (e.g., CHARMM) [9] [10].
- Pharmacophore Generation: Using molecular interaction data between the protein and a known co-crystallized ligand, generate pharmacophore features including:
- Model Validation: Validate the pharmacophore model using a decoy set containing known active compounds and inactive molecules. Calculate the Enrichment Factor (EF) and Area Under the Curve (AUC) of the Receiver Operating Characteristic (ROC) curve. A reliable model typically has AUC >0.7 and EF >2 [10] [14].
- Virtual Screening: Employ the validated pharmacophore model to screen large compound databases (e.g., ZINC, COCONUT, ChemDiv) to identify hits containing the essential features [9] [13] [14].
Protocol 2: Molecular Docking and Binding Affinity Assessment
Application Context: Identification of VEGFR-2 and c-Met dual inhibitors [10].
Principle: Molecular docking predicts the preferred orientation and binding affinity of small molecules within a target's binding site through scoring functions [9] [10].
Procedure:
- Ligand Preparation: Obtain 3D structures of compounds from databases. Prepare ligands by removing salts, adding hydrogen atoms, generating possible tautomers and protonation states, and minimizing energy [10].
- Binding Site Definition: Define the protein's binding site coordinates based on the known location from co-crystallized ligands or computational prediction methods [10] [14].
- Docking Execution: Perform docking simulations using software such as AutoDock or molecular operating environment. Apply appropriate search algorithms and scoring functions [15] [9].
- Pose Analysis and Scoring: Analyze the predicted binding poses and interactions (hydrogen bonds, hydrophobic interactions, π-π stacking). Rank compounds based on docking scores and evaluate binding free energies using methods like MM-GBSA or MM-PBSA [10] [13].
Protocol 3: ADMET Profiling
Application Context: Early-stage filtering of potential MCL1 inhibitors [13].
Principle: ADMET prediction evaluates the pharmacokinetic and safety profiles of compounds using computational models [12] [10].
Procedure:
- Compound Filtering: Apply Lipinski's Rule of Five and Veber's rules to filter compounds for drug-likeness [10].
- ADMET Prediction: Calculate key ADMET descriptors using tools such as PreADMET or SwissADME:
- Hit Selection: Prioritize compounds with favorable ADMET properties based on predefined cut-off values (e.g., solubility level >3, absorption level 0) [10].
Protocol 4: Molecular Dynamics Simulation
Application Context: Validation of XIAP inhibitor binding stability [14].
Principle: MD simulations assess the stability and dynamics of protein-ligand complexes under biologically relevant conditions over time [9] [14].
Procedure:
- System Setup: Place the protein-ligand complex in a solvation box with explicit water molecules. Add counterions to neutralize the system [9] [14].
- Energy Minimization: Perform energy minimization to remove steric clashes using steepest descent or conjugate gradient algorithms [14].
- Equilibration: Equilibrate the system in NVT (constant Number, Volume, Temperature) and NPT (constant Number, Pressure, Temperature) ensembles to achieve stable temperature and pressure [9].
- Production Run: Conduct production MD simulations for sufficient duration (typically 100-200 ns) using software like GROMACS or AMBER. Apply periodic boundary conditions and maintain constant temperature and pressure [9] [10] [14].
- Trajectory Analysis: Analyze root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and hydrogen bonding patterns to evaluate complex stability [9] [14].
Workflow Visualization of Computational Protocols
Diagram 1: Integrated Computational Workflow for Anticancer Drug Discovery. This workflow illustrates the sequential integration of computational methods from target identification to lead candidate selection, highlighting the screening and optimization phases.
Research Reagent Solutions
Table 3: Essential Computational Tools and Databases for Virtual Screening
| Resource Category | Specific Tools/Databases | Key Functionality | Application in Protocol |
|---|---|---|---|
| Protein Structure Databases | RCSB Protein Data Bank (http://www.rcsb.org/) [8] [10] | Provides 3D structural data of biological macromolecules | Source for target protein structures (e.g., XIAP PDB: 5OQW) [14] |
| Compound Libraries | ZINC Database, COCONUT, ChemDiv [10] [13] [14] | Curated collections of commercially available compounds for virtual screening | Source of natural products and synthetic compounds for screening [9] [13] |
| Pharmacophore Modeling | Discovery Studio, LigandScout [10] [14] | Generation and validation of structure-based and ligand-based pharmacophore models | Essential for Protocol 1: Structure-based pharmacophore modeling [14] |
| Molecular Docking | AutoDock, SwissDock, Molecular Operating Environment [15] [9] | Prediction of ligand binding modes and binding affinities | Core component of Protocol 2: Molecular docking assessment [15] [9] |
| ADMET Prediction | PreADMET, SwissADME, pkCSM [10] [14] | Prediction of absorption, distribution, metabolism, excretion, and toxicity properties | Required for Protocol 3: ADMET profiling [10] [14] |
| Molecular Dynamics | GROMACS, AMBER, CHARMM [9] [10] [14] | Simulation of molecular systems over time to analyze stability and dynamics | Implementation of Protocol 4: MD simulation validation [9] [14] |
| Validation Data Sources | DUD-E (Database of Useful Decoys: Enhanced) [10] [14] | Provides decoy compounds for validation of virtual screening methods | Used for pharmacophore model validation in Protocol 1 [14] |
The integration of computational protocols outlined in this document has transformed the landscape of anticancer drug discovery. Through structured workflows combining pharmacophore modeling, molecular docking, ADMET profiling, and molecular dynamics simulations, researchers can efficiently identify and optimize promising therapeutic candidates with higher precision and reduced resource expenditure. These methodologies have demonstrated significant success across various cancer targets, including PD-L1, VEGFR-2, c-Met, MCL1, and XIAP, leading to novel inhibitors with validated binding stability and favorable drug-like properties [9] [10] [13]. As these computational approaches continue to evolve with advancements in artificial intelligence and machine learning, their predictive power and efficiency in anticancer drug discovery are expected to further increase, potentially reducing both timelines and attrition rates in the development of novel cancer therapeutics [15] [11].
The Role of Target Identification and Validation in Cancer Therapeutics
The discovery and development of effective cancer therapeutics are fundamentally reliant on the precise identification and validation of molecular targets. A drug target is a biological molecule, typically a protein, that plays a pivotal role in a disease pathway and whose modulation by a therapeutic agent is expected to yield a clinical benefit [16] [17]. In oncology, the landscape of target discovery has been revolutionized by rapid and affordable tumor profiling, which has led to an explosion of genomic data and facilitated the development of targeted therapies against specific oncogenic lesions [18]. However, the inherent complexity of cancer, characterized by different gene mutations and omics profiles across cancer types, demands a rigorous and multi-faceted approach to distinguish true therapeutic targets from mere biological noise [19] [17]. This document outlines standardized protocols and application notes for target identification and validation, framed within a modern computational paradigm for anticancer drug discovery.
Target Identification: Approaches and Protocols
Target identification is the initial critical step focused on discovering and prioritizing "druggable" biological molecules involved in cancer pathophysiology. An ideal target possesses several key properties: a pivotal role in the disease, confined expression to specific locations, the existence of a 3D model for druggability assessment, suitability for high-throughput screening, and a favorable predicted toxicity profile upon modulation [16]. The following protocols describe core identification strategies.
Protocol: Multi-Omics Data Analysis for Target Discovery
Principle: Integrative analysis of transcriptomics and proteomics data from cancer cell lines and patient tissues to identify genes and proteins significantly overexpressed or dysregulated in specific cancer types [19].
Materials:
- Cancer Cell Line Encyclopedia (CCLE): A publicly available database containing RNA-Seq transcriptomics data for over 1000 cancer cell lines [19].
- Proteomics Datasets: Tandem mass tag (TMT)-based quantitative proteomics data from cancer cell lines (e.g., from 375 cell lines as described in Nusinow et al.) [19].
- Bioinformatics Software: Tools for statistical analysis (e.g., R/Bioconductor), pathway enrichment analysis (e.g., GSEA, DAVID).
Procedure:
- Data Acquisition: Download RNA-Seq and proteomics data for 16 or more common cancer types (e.g., AML, breast cancer, NSCLC) from repositories such as the CCLE.
- Differential Expression Analysis: For each cancer type, identify significant transcripts and proteins by comparing their expression levels against all other cancer types. Apply false discovery rate (FDR) corrections to adjust for multiple hypothesis testing.
- Biotype Analysis: Classify significant transcripts into biotypes (e.g., protein-coding, lincRNA, pseudogene) to prioritize protein-coding targets.
- Pathway Enrichment Analysis: Input lists of significant transcripts and proteins into pathway analysis tools to identify biological pathways (e.g., olfactory transduction, GPCR signaling, mRNA processing) that are characteristic of each cancer type.
- Target Prioritization: Prioritize molecular targets that are:
- Statistically significant at both the transcript and protein levels.
- Enriched in cancer-specific pathways with known therapeutic relevance.
- Linked to patient survival or treatment response in clinical datasets.
Protocol: Functional Genomics Screening
Principle: Use RNA interference (RNAi) or CRISPR-Cas9 screens to systematically knock down or knock out genes in cancer cells to identify those essential for cell survival or proliferation (synthetic lethality) [18] [17].
Materials:
- shRNA or CRISPR Libraries: Genome-wide or pathway-focused libraries.
- Cancer Cell Lines: Representative of the cancer type of interest.
- Next-Generation Sequencing (NGS) Platform: For quantifying guide RNA abundance.
Procedure:
- Library Transduction: Transduce a population of cancer cells with the shRNA or CRISPR library at a low multiplicity of infection (MOI) to ensure single integration events.
- Negative Selection: Culture the transduced cells for multiple population doublings. Cells carrying guides targeting essential genes will be depleted from the population.
- Genomic DNA Extraction and Sequencing: Extract genomic DNA from cells at the beginning (T0) and end (Tfinal) of the experiment. Amplify the integrated shRNA or guide RNA sequences and sequence them using NGS.
- Hit Identification: Calculate the depletion of individual guides between T0 and Tfinal using specialized bioinformatics pipelines (e.g., MAGeCK). Genes targeted by significantly depleted guides are considered essential for cancer cell fitness and are candidate therapeutic targets.
Computational Approaches in Identification
Computer-aided drug design (CADD) has emerged as a powerful technology to make drug discovery quicker, cheaper, and more efficient [20] [21]. Ligand-based virtual screening uses known active compounds to search large chemical databases for structurally similar molecules. Conversely, structure-based virtual screening uses the 3D structure of a target protein to computationally "dock" millions of small molecules and predict their binding affinity and pose [22] [21]. Machine learning models are now being employed to further accelerate this process by predicting docking scores without explicitly performing costly docking calculations, thereby enabling the virtual screening of ultra-large libraries [22].
Table 1: Summary of Target Identification Approaches
| Approach | Core Principle | Key Outputs | Considerations |
|---|---|---|---|
| Multi-Omics Analysis [19] | Integrative analysis of transcriptomics and proteomics data from cancer cell lines and tissues. | Lists of significant transcripts/proteins; enriched cancer-specific pathways. | Requires robust statistical correction; validation is essential. |
| Functional Genomics [18] | Systematic gene knockdown/knockout to identify genes essential for cancer cell survival. | Ranked list of candidate essential genes (synthetic lethal interactions). | Can have off-target effects; in vivo validation is often needed. |
| Computational Virtual Screening [22] [21] | Using computer simulations to identify hit molecules that bind to a defined target. | Predicted high-affinity ligands for a target protein. | Highly dependent on the quality of the target protein structure. |
Target Validation: Confirming Therapeutic Relevance
Once a candidate target is identified, it must be rigorously validated to confirm its functional role in the disease and that its modulation provides a therapeutic effect. Validation is a critical step to justify the substantial investment in subsequent drug discovery campaigns [17] [23].
Protocol: In vivo Target Validation Using Inducible RNAi
Principle: Combine inducible RNAi technology with genetically engineered mouse models (GEMMs) to assess the impact of target inhibition on tumor growth and to probe potential toxicities in a physiologically relevant in vivo context [18].
Materials:
- Inducible shRNA System: Vectors allowing doxycycline-dependent expression of shRNAs.
- Genetically Engineered Mouse Models (GEMMs): Models of specific cancers (e.g., lymphoma, melanoma).
- Imaging and Histology Equipment: For monitoring tumor growth and analyzing tissue samples.
Procedure:
- Model Generation: Generate GEMMs in which the expression of a specific shRNA targeting the candidate gene can be induced systemically or in a tissue-specific manner upon administration of doxycycline.
- Tumor Monitoring: Induce shRNA expression in tumor-bearing mice and monitor tumor progression and regression using caliper measurements or in vivo imaging.
- Toxicity Assessment: Closely monitor mice for signs of toxicity in normal tissues. Perform histopathological analysis on key organs post-study.
- Target Engagement Analysis: Validate that the shRNA-mediated knockdown leads to a significant reduction in the target protein levels within the tumors.
Protocol: Digital Pathology-Based Biomarker Analysis
Principle: Utilize multiplexed immunohistochemistry (IHC) or immunofluorescence (IF) coupled with whole-slide imaging and artificial intelligence (AI)-based analysis to quantitatively validate target expression and its spatial relationship within the tumor microenvironment (TME) [24] [25].
Materials:
- Tissue Microarrays (TMAs): Containing patient tumor samples and normal tissue controls.
- Multiplex Staining Kits: Such as Tyramide Signal Amplification (TSA)-based kits for simultaneous detection of multiple proteins.
- Whole Slide Scanner: For high-resolution digitization of stained slides.
- Digital Pathology Software: Open-source (e.g., QuPath, ImageJ) or commercial (e.g., HALO).
Procedure:
- Multiplex Staining: Perform TSA-based multiplex IHC/IF on TMAs. A typical panel may include antibodies against the target protein (e.g., TROP2), tumor markers (PanCK), immune cell markers (CD8), and checkpoint inhibitors (PD-L1) [25].
- Slide Digitization: Scan the stained slides using a whole-slide scanner to create high-resolution digital images.
- Algorithmic Analysis: Use digital pathology software to train an AI algorithm to identify and quantify different cell types and the expression levels (membrane, cytoplasmic, nuclear) of the target protein.
- Spatial and Clinical Correlation: Correlate the quantitative target expression data (e.g., nuclear TROP2) with patient clinical outcomes such as progression-free survival (PFS) or response to immunotherapy to establish clinical relevance [25].
Table 2: Key Metrics for Target Validation and Qualification [23]
| Validation Component | Assessment Metrics (in ascending order of priority) |
|---|---|
| Target Validation (Human Data) | Tissue expression profile → Genetic association in humans (e.g., GWAS) → Clinical experience with target modulation (e.g., known drugs) |
| Target Qualification (Preclinical Data) | Phenotypic data from genetically engineered models → Evidence of target engagement and pathway modulation → Demonstrated efficacy in translational disease models |
Integration with Computational Drug Discovery
The identified and validated targets seamlessly feed into the computational pipeline for drug discovery. A highly validated target with a known or homology-modeled 3D structure becomes the foundation for structure-based drug design.
Workflow: From Validated Target to Lead Compound
The diagram below illustrates the integrated computational workflow, from a validated target to a optimized lead compound ready for experimental testing.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table details key reagents and platforms essential for conducting the experiments described in these protocols.
Table 3: Research Reagent Solutions for Target Identification & Validation
| Category / Reagent | Specific Example(s) | Function in Research |
|---|---|---|
| Omics Platforms | RNA-Seq (CCLE), TMT-based Proteomics [19], Single-cell & Spatial Transcriptomics [25] | Generate comprehensive molecular profiles of cancers for target discovery. |
| Functional Genomics Tools | shRNA Libraries, CRISPR-Cas9 Systems [18] [17] | Perform genome-wide loss-of-function screens to identify essential genes. |
| Preclinical Models | Cancer Cell Line Encyclopedia (CCLE) [19], Genetically Engineered Mouse Models (GEMMs) [18] | Provide in vitro and in vivo systems for target validation and efficacy testing. |
| Digital Pathology | Tyramide Signal Amplification (TSA) Kits [24], Whole Slide Scanners, HALO/QuPath Software [24] [25] | Enable multiplexed protein detection and quantitative, spatially resolved biomarker analysis. |
| Computational Tools | Molecular Docking Software, XGBoost, Attention-based LSTM Networks [22] | Accelerate virtual screening and predict protein-ligand interactions. |
| Assay Development | Biochemical & Cellular Assays, High-Throughput Screening (HTS) [16] | Test and validate target engagement and functional activity of small molecules. |
| nor-4 | nor-4, CAS:163180-50-5, MF:C14H18N4O4, MW:306.32 g/mol | Chemical Reagent |
| UK-2A | UK-2A | UK-2A is a natural product-derived Qi site inhibitor fungicide for agricultural research. This product is For Research Use Only (RUO). Not for human or veterinary use. |
Target identification and validation form the critical, non-negotiable foundation of modern cancer drug discovery. The convergence of multi-omics, functional genomics, and advanced computational methods has created a powerful, integrated pipeline. This pipeline enables researchers to move from genomic data to a validated, "druggable" target with higher confidence and efficiency. By adhering to the rigorous protocols outlined herein—from multi-omics analysis and in vivo validation to AI-powered digital pathology and computational screening—researchers can de-risk the drug development process and prioritize the most promising targets for intervention. This structured approach is essential for translating the wealth of cancer genomic data into safe and effective therapeutics for patients.
Drug Repurposing Strategies for Oncology Applications
Drug repurposing represents a paradigm shift in oncology drug development, seeking to identify new therapeutic uses for existing drugs already approved for other conditions. This strategy significantly accelerates therapeutic development while reducing costs and risks associated with novel drug discovery [26] [27]. The established safety profiles, known pharmacokinetics, and existing clinical experience with these compounds enable researchers to bypass early-phase development stages, focusing resources directly on efficacy validation in oncological contexts [27] [28].
Computational approaches have revolutionized drug repurposing by enabling systematic, high-throughput screening of existing drug libraries against cancer-specific targets. The integration of bioinformatics, artificial intelligence (AI), and molecular modeling has transformed the field, allowing researchers to predict drug-target interactions with increasing accuracy and identify promising repurposing candidates from thousands of existing compounds [29] [20]. The global drug repurposing market, valued at US$29.4 billion in 2024 and projected to reach US$37.3 billion by 2030, reflects the growing importance of these approaches, with oncology representing the largest therapeutic segment [30].
Promising Repurposed Drug Candidates in Oncology
The Repurposing Drugs in Oncology (ReDO) database has identified 268 non-cancer drugs with published evidence of anticancer activity, demonstrating the substantial potential of this approach [27]. Table 1 summarizes the evidence levels and characteristics of these repurposing candidates.
Table 1: Evidence Profile for 268 Drugs in the ReDO Database
| Characteristic | Number of Drugs | Percentage |
|---|---|---|
| Included in WHO Essential Medicines List | 87 | 32% |
| Off-patent | 226 | 84% |
| Supported by in vitro evidence | 264 | 99% |
| Supported by in vivo evidence | 247 | 92% |
| Supported by human data (case reports, observational studies, or clinical trials) | 194 | 72% |
| Tested in clinical trials | 178 | 66% |
| Meeting all favorable criteria (WHO EML + off-patent + human data) | 67 | 25% |
Source: Adapted from ReDO_DB summary statistics [27]
These repurposing candidates originate from diverse therapeutic areas, with cardiovascular, nervous system, and alimentary tract medications being the most common sources, as shown in Table 2.
Table 2: Therapeutic Origins of Repurposing Candidates by ATC Classification
| Anatomical Therapeutic Chemical (ATC) Category | Number of Drugs |
|---|---|
| Cardiovascular System | 56 |
| Nervous System | 49 |
| Alimentary Tract and Metabolism | 39 |
| Musculo-Skeletal System | 31 |
| Antiinfectives for Systemic Use | 26 |
| Dermatologicals | 23 |
| Genito Urinary System and Sex Hormones | 23 |
| Sensory Organs | 22 |
| Antiparasitic Products, Insecticides and Repellents | 20 |
Source: Adapted from ReDO_DB analysis [27]
Clinically Evaluated Repurposing Candidates
Randomized controlled trials (RCTs) provide the highest quality evidence for repurposed drugs in oncology. Recent RCTs have evaluated several promising candidates:
Metformin: Originally an antidiabetic medication, metformin has been studied in various cancers including prostate, lung, and pancreatic malignancies. Its mechanisms involve activation of AMP-activated protein kinase (AMPK), inhibition of mTOR signaling, and reduction of insulin-like growth factor levels [28].
Propranolol: This beta-blocker, used for cardiovascular conditions, has demonstrated potential in multiple myeloma and, when combined with etodolac, in breast cancer. Proposed mechanisms include inhibition of β-adrenergic signaling pathways that influence tumor growth and metastasis [28].
Mebendazole: An antiparasitic agent showing promise in colorectal cancer through tubulin polymerization inhibition and interference with glucose uptake in cancer cells [28].
Sulconazole: Originally an antifungal, sulconazole inhibits PD-1 expression in immune and cancer cells by blocking NF-κB and calcium signaling, representing an immunomodulatory approach [26].
Olaparib: While already approved for BRCA-mutant cancers, olaparib has shown potential for repurposing in lung cancer, demonstrating improved progression-free survival as monotherapy compared to combination regimens [26].
Computational Framework for Drug Repurposing
Virtual Screening Methodologies
Virtual screening (VS) comprises computational techniques to identify structures most likely to bind to drug targets from large libraries of small molecules [31]. The two primary approaches are structure-based and ligand-based methods, which can be integrated in hybrid frameworks for enhanced accuracy [31] [29].
Diagram 1: Computational virtual screening methodologies for drug repurposing
Integrated Repurposing Workflow
A systematic computational repurposing workflow combines multiple data sources and validation steps to identify high-probability drug-target matches for oncology applications.
Diagram 2: Integrated computational repurposing workflow for oncology
Experimental Protocols
Structure-Based Virtual Screening Protocol
Objective: Identify potential inhibitors for protein kinase CK2α, a crucial cancer target, through structure-based virtual screening.
Materials and Reagents:
- Molecular Target: Zea mays CK2α crystal structure (PDB ID: 4RLK)
- Compound Library: 5,000 small molecules for initial screening
- Software Tools: AutoDock Vina, AutoDock, Desmond Molecular Dynamics
- Computing Infrastructure: Linux cluster with batch queue processor
Procedure:
Target Preparation:
- Obtain 3D structure of CK2α from Protein Data Bank (4RLK)
- Remove water molecules and add polar hydrogen atoms
- Define binding site coordinates based on known active site residues
Molecular Docking:
- Perform initial screening of 5,000 compounds using AutoDock Vina
- Select top 50 compounds based on docking scores for refined analysis
- Execute refined docking with AutoDock using more precise parameters
- Analyze binding poses and interaction patterns (hydrogen bonds, π-stacking, hydrophobic interactions)
Molecular Dynamics Simulations:
- Run 100 ns simulations using Desmond with OPLS-2005 force field
- Analyze Root Mean Square Deviation (RMSD) for complex stability
- Calculate Root Mean Square Fluctuation (RMSF) for residue flexibility
- Monitor hydrogen bonding patterns throughout simulation trajectory
Hit Identification:
- Prioritize compounds demonstrating stable binding throughout MD simulation
- Select candidates with consistent protein-ligand interactions for experimental validation [32]
Computational Repurposing Database Screening Protocol
Objective: Systematically identify off-target repurposing opportunities using validated computational databases.
Materials and Reagents:
- Database Resources: Probe Miner (PM), Broad Institute Drug Repurposing Hub, TOPOGRAPH
- Validation Set: FDA-approved therapies with known biomarkers
- Analysis Tools: Custom scripts for data integration and scoring
Procedure:
Database Curation:
- Compile FDA-approved oncology drugs from Table of Pharmacogenomic Biomarkers in Drug Labeling
- Manually curate drug-target combinations to ensure accuracy
- Filter for small molecule inhibitors (n=67) for repurposing analysis
Platform Validation:
- Use Probe Miner quantitative score (0-1 scale) to assess compound selectivity
- Apply inclusion threshold of PM quantitative score ≥0.25
- Calculate sensitivity and specificity for identifying known gene-targetable drugs
- Validate approach against known FDA-approved drug-biomarker combinations
Tumor Genomic Analysis:
- Sequence tumors using Illumina TruSight Oncology 500 or FoundationOne CDx panels
- Analyze 523 genes for single nucleotide variations and insertions/deletions
- Call amplifications with limit of detection of 2.2x fold change
- Determine microsatellite instability status and tumor mutational burden
Variant Classification:
- Review NGS reports for genomic variants of significance
- Classify variants as gain-of-function (GOF) or loss-of-function (LOF)
- Annotate pathogenic mutations with orthogonal functional validation
- Classify variants of unknown significance using FATHMM pathogenicity scoring
Repurposing Event Identification:
- Assess GOF mutations for targeting compounds across PM, Broad Institute DRH, and TOPOGRAPH
- Prioritize compounds not indexed in TOPOGRAPH and excluding FDA-approved biomarkers
- Validate predictions through experimental assays [33]
AI-Driven Drug Repurposing Protocol
Objective: Leverage artificial intelligence and machine learning to identify novel drug-disease relationships for oncology repurposing.
Materials and Reagents:
- AI Platform: Predictive Oncology's PEDAL platform (92% accuracy in tumor response prediction)
- Biobank Resources: 150,000+ tumor samples across 130+ cancer types
- Drug Library: ~150 FDA-approved drugs with known clinical indications
- Computing Infrastructure: AI servers with GPU acceleration
Procedure:
Data Integration:
- Construct knowledge graphs integrating drug-target-disease relationships
- Apply large language models (LLMs) to analyze biomedical literature
- Incorporate real-world medical data from electronic health records
- Integrate multi-omics data (transcriptomics, toxicogenomics, functional genomics)
Model Training:
- Train machine learning models on known drug response data
- Validate models using cross-validation techniques
- Optimize parameters to achieve >90% prediction accuracy
- Establish confidence thresholds for prediction reliability
Candidate Identification:
- Screen tumor samples against FDA-approved drug library
- Rank opportunities based on AI-predicted efficacy scores
- Prioritize candidates for diseases with unmet therapeutic needs
- Apply ensemble methods combining multiple AI approaches
Experimental Validation:
- Test top predictions using patient-derived tumor models
- Generate dose-response curves for prioritized drug-cancer pairs
- Validate mechanisms of action through pathway analysis
- Confirm selectivity and safety profiles [34]
Table 3: Computational Tools and Databases for Drug Repurposing
| Resource Name | Type | Primary Function | Application in Oncology Repurposing |
|---|---|---|---|
| Probe Miner (PM) | Database | Indexes >1.8M compounds against 2,220 human targets with quantitative scoring | Identifies potent and selective compounds for specific proteins; validated for FDA-approved drug prediction |
| Broad Institute Drug Repurposing Hub | Database | Curated collection of repurposing candidates and their targets | Provides well-annotated compound-target relationships for hypothesis generation |
| TOPOGRAPH | Database | Maps drug-target interactions and polypharmacology | Filters out non-specific interactions to improve repurposing candidate quality |
| AutoDock Vina | Software | Molecular docking and virtual screening | Performs initial high-throughput screening of compound libraries against cancer targets |
| Desmond | Software | Molecular dynamics simulations | Assesses binding stability and conformational changes in protein-ligand complexes |
| TruSight Oncology 500 | Sequencing Panel | Analyzes 523 genes for variants, fusions, and splice variants | Comprehensive genomic profiling to identify targetable alterations in tumors |
| FoundationOne CDx | Sequencing Panel | Analyzes 324 genes with TMB and MSI assessment | FDA-approved comprehensive genomic profiling for therapy selection |
| PEDAL Platform | AI Tool | Predicts tumor response to drugs with 92% accuracy | AI-driven drug response prediction using extensive tumor biobank data |
Table 4: Key Chemical and Biological Reagents
| Reagent | Specifications | Experimental Role | Considerations for Repurposing Studies |
|---|---|---|---|
| FDA-Approved Drug Library | ~150 compounds with diverse indications | Screening against tumor models | Prioritize off-patent compounds with favorable safety profiles |
| Patient-Derived Tumor Cells | 150,000+ samples across 130 cancer types | Ex vivo drug response testing | Maintain biological relevance and tumor heterogeneity |
| CK2α Protein | Zea mays crystal structure (PDB: 4RLK) | Structure-based screening target | Representative kinase model for cancer signaling pathways |
| Molecular Dynamics Force Field | OPLS-2005 parameters | Simulation of protein-ligand interactions | Balance between computational efficiency and physical accuracy |
| NGS Library Prep Kits | TSO-500 or FoundationOne CDx | Tumor genomic profiling | Ensure coverage of clinically actionable cancer genes |
Signaling Pathways in Drug Repurposing for Oncology
The efficacy of repurposed drugs in oncology often derives from their action on critical cancer signaling pathways. Understanding these mechanisms is essential for rational repurposing strategy design.
Diagram 3: Signaling pathways and mechanisms of action for repurposed drugs in oncology
Computational drug repurposing represents a transformative approach in oncology, offering accelerated pathways to new cancer therapies by leveraging existing pharmacological agents. The integration of structure-based virtual screening, AI-driven prediction platforms, and systematic database mining has created a robust framework for identifying high-probability repurposing candidates.
The promising clinical results from randomized controlled trials of drugs like metformin, propranolol, and mebendazole validate this computational approach [28]. Furthermore, the establishment of large-scale collaborations between organizations like Predictive Oncology and Every Cure demonstrates the growing recognition of computational repurposing as a viable strategy for addressing unmet needs in oncology [34].
As computational methods continue to evolve, particularly through advances in artificial intelligence and machine learning, the precision and efficiency of drug repurposing will further improve. The availability of extensive tumor biobanks, comprehensive genomic databases, and validated screening platforms creates an unprecedented opportunity to systematically explore the vast landscape of existing drugs for new anticancer applications. This approach promises to deliver safe, effective, and affordable cancer therapies in significantly reduced timeframes, ultimately benefiting patients through expanded treatment options and improved outcomes.
The identification of novel anticancer agents relies heavily on the screening of diverse chemical libraries to find compounds that can modulate specific biological targets. Publicly available chemical libraries and databases provide an indispensable resource for virtual screening (VS), a computational approach that dramatically reduces the time and financial costs associated with early drug discovery [35]. These libraries vary significantly in size, content, structural diversity, and design methodology, making the selection of appropriate screening collections crucial for successful hit identification [36]. Within the context of anticancer research, specifically targeting oncogenic drivers like the V600E-BRAF kinase—a key therapeutic target in melanoma, colorectal cancer, and thyroid cancer—the strategic use of these libraries enables researchers to efficiently identify potent inhibitors with superior pharmacokinetic properties [35].
The construction of virtual chemical libraries can be achieved through various methods, including using known reaction schemas with available reagents, functional group-based approaches, de novo design, molecular graph decoration, and morphing/transformation techniques [37]. This protocol outlines the key publicly available libraries, provides methodologies for their utilization in virtual screening workflows, and demonstrates their application through a case study on V600E-BRAF inhibitor identification.
Key Public Chemical Libraries and Databases
Major Public Compound Databases
Table 1: Major Public Compound Databases for Anticancer Virtual Screening
| Database Name | Key Characteristics | Size | Special Features | Relevance to Anticancer Research |
|---|---|---|---|---|
| PubChem | NCBI's repository of chemical molecules and their activities | 72+ million compounds (as exemplified by a specific anticancer set [35]) | Links to bioactivity data, screening assays, and toxicity information | Source of anticancer compounds with known biological activities [35] |
| ZINC15 | Curated collection of commercially available compounds for virtual screening | Over 100 million compounds (as of 2015) [36] | Includes 37 vendors offering >100,000 compounds each [36] | Foundation for building targeted screening libraries against cancer targets |
| ChEMBL | Manually curated database of bioactive drug-like molecules | Not specified in sources | Contains drug-like molecules with binding, functional ADMET data | Reference for similarity searches in anticancer library design [38] |
| Traditional Chinese Medicine Compound Database (TCMCD) | Natural products from Chinese medicinal herbs | 57,809 molecules [36] | High structural complexity and unique scaffolds [36] | Source of natural compounds with potential anticancer activity |
Specialized Anticancer Libraries
Table 2: Specialized Anticancer Screening Libraries
| Library Name | Composition | Design Methodology | Key Features | Cancer Targets/Models |
|---|---|---|---|---|
| Life Chemicals Anticancer Library | 9,100 drug-like molecules [38] | 2D similarity search against ChEMBL and BindingDB with 80% similarity cut-off [38] | PAINS/reactive groups filtered; Ro5 compliance indicated [38] | 12,000 reference anticancer agents; various cancer cell lines and targets [38] |
| Life Chemicals Docking Set | 4,500 structurally diverse molecules [38] | Molecular docking against cancer-focused targets [38] | Focus on synthetically feasible compounds | MRP1, TNF targets [38] |
| CCSMD Database | Combinatorial library built from smart reaction modules [39] | Virtual synthesis through amide reactions [39] | High actual hit rate (76.92%) in validation [39] | Discovered CDK6 inhibitors with IC50 values ~1.3 μM [39] |
| Natural Product Libraries (e.g., Anticancer Bioscience) | 17,636 crude extracts, 1,211 fractions, 2,452 pure compounds [40] | Collection from Traditional Chinese Medicine herbs and plants [40] | Structural diversity unavailable in synthetic libraries [40] | Targets difficult to drug with synthetic compounds [40] |
DNA-Encoded Chemical Libraries (DELs)
DNA-encoded chemical libraries (DELs) represent a powerful alternative approach for hit identification, combining aspects of combinatorial chemistry with biological selection methods. These libraries consist of organic molecules covalently coupled to distinctive DNA fragments that serve as amplifiable barcodes, enabling the screening of millions to billions of compounds in a single test tube [41]. DELs can be categorized as either single pharmacophore libraries (one DNA fragment coupled to a chemical building block) or dual pharmacophore libraries (pairs of chemical building blocks attached to complementary DNA strands) [41]. The screening process involves incubating the DEL with an immobilized target protein, washing away non-binders, and identifying binding molecules through PCR amplification and high-throughput sequencing of the DNA barcodes [41].
Experimental Protocols for Virtual Screening
Protocol 1: Structure-Based Virtual Screening Using Molecular Docking
Application: Identification of potential V600E-BRAF kinase inhibitors [35]
Materials and Reagents:
- Target Structure: Crystal structure of V600E-BRAF (PDB code: 3OG7) [35]
- Compound Library: 72 anticancer compounds from PubChem database [35]
- Software Tools: Molecular docking software (e.g., Molegro Virtual Docker), structure visualization tool (e.g., Discovery Studio) [35]
- Reference Ligand: Vemurafenib (FDA-approved V600E-BRAF inhibitor) [35]
Procedure:
- Target Preparation:
Ligand Preparation:
Docking Validation:
Docking Simulation:
Interaction Analysis:
Diagram 1: Workflow for structure-based virtual screening of V600E-BRAF inhibitors
Protocol 2: Library Enumeration and Design Using Pre-validated Reactions
Application: Construction of virtual combinatorial libraries for anticancer screening [37]
Materials and Reagents:
- Chemical Reaction Schemas: Pre-validated or reported reactions (e.g., amide bond formation) [37] [39]
- Building Blocks: Commercially available chemical reagents [37]
- Software Tools: Open-source chemoinformatics tools (e.g., DataWarrior, KNIME) [37]
- Molecular Representation: SMILES, SMARTS, or InChI notations [37]
Procedure:
- Reaction Selection:
Building Block Selection:
Library Enumeration:
Library Characterization:
Library Filtering:
Diagram 2: Chemical library enumeration workflow using pre-validated reactions
Protocol 3: Analysis of Scaffold Diversity in Compound Libraries
Application: Comparative assessment of purchasable screening libraries for anticancer virtual screening [36]
Materials and Reagents:
- Compound Libraries: Standardized subsets of commercial libraries (e.g., Mcule, Enamine, ChemDiv) [36]
- Software Tools: Pipeline Pilot, Molecular Operating Environment (MOE), Tree Maps software [36]
- Analysis Methods: Murcko framework analysis, Scaffold Tree generation, RECAP fragmentation [36]
Procedure:
- Library Standardization:
Fragment Generation:
Diversity Assessment:
Visualization:
Library Selection:
Table 3: Essential Research Reagent Solutions for Anticancer Virtual Screening
| Resource Category | Specific Tools/Resources | Function | Application in Anticancer Research |
|---|---|---|---|
| Compound Databases | PubChem, ZINC15, ChEMBL | Source of screening compounds and bioactivity data | Identification of compounds with known activity against cancer targets [35] |
| Cheminformatics Tools | DataWarrior, KNIME, Pipeline Pilot | Library enumeration, property calculation, and filtering | Design of targeted libraries against specific oncogenic targets [37] |
| Molecular Modeling Software | Molegro Virtual Docker, Spartan, MOE | Structure-based design, docking, and quantum calculations | Docking against cancer targets like V600E-BRAF kinase [35] |
| ADMET Prediction Platforms | SwissADME, pkCSM | Prediction of drug-likeness and pharmacokinetic properties | Optimization of anticancer candidates for favorable properties [35] |
| Specialized Screening Libraries | Life Chemicals Anticancer Library, Natural Product Libraries | Focused sets for specific target classes | Screening against cancer cell lines and specific oncogenic targets [38] [40] |
Case Study: Identification of V600E-BRAF Kinase Inhibitors
The V600E-BRAF mutation, present in 60% of melanomas and 10-70% of other cancers, represents a critical therapeutic target in oncology [35]. Despite the availability of FDA-approved inhibitors like dabrafenib and vemurafenib, resistance frequently emerges after 5-8 months of treatment, necessitating the discovery of novel chemotypes [35]. A recent study demonstrated the successful application of computational protocols to identify new V600E-BRAF inhibitors from a set of 72 anticancer compounds in the PubChem database [35].
Methodology Overview: Researchers employed an integrated in silico approach combining:
- Molecular docking simulation against V600E-BRAF crystal structure (PDB: 3OG7)
- Pharmacokinetic evaluation using SwissADME and pkCSM
- Density functional theory (DFT) computations for electronic structure analysis [35]
Results: The screening identified five top-ranked molecules (compounds 12, 15, 30, 31, and 35) with excellent docking scores (MolDock score ≥90 kcal molâ»Â¹, Rerank score ≥60 kcal molâ»Â¹) that formed hydrogen bonds and hydrophobic interactions with essential residues in the V600E-BRAF binding site [35]. DFT calculations revealed favorable frontier molecular orbital characteristics and reactivity parameters, while drug-likeness predictions indicated superior pharmacokinetic properties compared to existing inhibitors [35].
Significance: This case study demonstrates how publicly available chemical libraries, when screened with robust computational protocols, can yield promising hit compounds for further development as anticancer agents. The identified compounds showed potential as candidates for overcoming resistance to current V600E-BRAF inhibitors, highlighting the value of virtual screening in addressing challenging problems in oncology drug discovery [35].
Publicly available chemical libraries and databases provide an essential foundation for computational approaches to anticancer drug discovery. The strategic selection and application of these resources, combined with robust virtual screening protocols, can significantly accelerate the identification of novel therapeutic agents for oncology targets. As library enumeration methods continue to advance and new screening technologies like DNA-encoded libraries mature, the opportunities for discovering innovative cancer therapies through computational means will continue to expand. The protocols and resources outlined in this application note provide researchers with a comprehensive toolkit for leveraging these powerful approaches in their anticancer drug discovery efforts.
Computational Methodologies: AI, Docking, and Dynamics in Practice
Structure-Based Virtual Screening (SBVS), often used interchangeably with molecular docking, has become an indispensable tool in modern drug discovery pipelines [42] [43]. This computational approach predicts the preferred orientation and binding affinity of a small molecule (ligand) when bound to a macromolecular target, typically a protein [44]. In the context of anticancer drug discovery, SBVS provides a rapid and cost-effective method to identify novel chemical entities from vast virtual libraries, significantly accelerating the hit identification phase [21]. The process fundamentally involves two core components: a search algorithm that explores possible ligand conformations and orientations within the target's binding site, and a scoring function that estimates the binding strength of each generated pose [45] [43]. The integration of these components into robust protocols allows researchers to prioritize a manageable number of compounds for experimental validation, making the drug discovery process more rational and efficient [46].
Key Components of a Docking Protocol
A successful SBVS campaign relies on the careful setup and execution of several interconnected steps. The diagram below illustrates the typical workflow.
Molecular Preparation
The initial and critical phase involves preparing the structures of both the target and the ligands.
- Target Preparation: The 3D structure of the protein target is most often obtained from the Protein Data Bank (PDB) [45] [47]. If an experimental structure is unavailable, computationally modeled structures (e.g., from AlphaFold) can be used [47]. Preparation typically involves:
- Adding missing hydrogen atoms and assigning protonation states to amino acid residues using tools like
PropKaorH++[45]. - Removing water molecules and original co-crystallized ligands, unless they are integral to binding.
- Adding partial atomic charges and optimizing the structure via energy minimization [47].
- Adding missing hydrogen atoms and assigning protonation states to amino acid residues using tools like
- Ligand Preparation: Small molecule structures are sourced from databases like ZINC, PubChem, or ChEMBL [45] [47]. Ligand preparation includes:
Binding Site Definition
Docking accuracy is greatly improved when the search is focused on a specific region of the protein. If the binding site is unknown (e.g., from a co-crystallized ligand), cavity detection programs like DoGSiteScorer, CASTp, or DeepSite can predict potential binding pockets [45] [47]. Performing a "blind docking" over the entire protein surface is computationally expensive and often less accurate [43].
Docking Execution: Search Algorithms and Scoring Functions
The core of SBVS involves generating and evaluating ligand poses.
- Search Algorithms: These algorithms explore the translational, rotational, and conformational degrees of freedom of the ligand within the defined binding site. They are broadly classified as follows [44] [45] [43]:
- Systematic Search: Explores torsional angles incrementally (e.g.,
FlexXuses incremental construction). - Stochastic Search: Uses random changes to find low-energy poses (e.g.,
AutoDock VinaandGOLDuse Monte Carlo and Genetic Algorithms, respectively). - Shape-Matching: Quickly fits the ligand to a defined cavity based on geometric and chemical complementarity [44].
- Systematic Search: Explores torsional angles incrementally (e.g.,
- Scoring Functions: These functions rank the generated poses by estimating the binding affinity. They fall into three main categories [48] [43]:
- Force Field-Based: Calculate energy terms based on molecular mechanics.
- Empirical: Use weighted sums of heuristic interaction terms.
- Knowledge-Based: Derive potentials from statistical analyses of known protein-ligand complexes in the PDB.
Table 1: Popular Molecular Docking Software and Their Key Characteristics
| Software | Search Algorithm | Scoring Function Type | License | Reference |
|---|---|---|---|---|
| AutoDock Vina | Iterated Local Search | Empirical / Knowledge-Based | Free (Apache) | [45] |
| GLIDE | Systematic + Optimization | Empirical | Commercial | [42] [45] |
| GOLD | Genetic Algorithm | Physics-based, Empirical, Knowledge-based | Commercial | [45] [3] |
| DOCK | Anchor-and-grow incremental construction | Physics-based | Academic | [42] [45] |
| RosettaVS | Genetic Algorithm | Physics-based (RosettaGenFF-VS) | Free (Rosetta) | [3] |
Advanced SBVS Protocols and Considerations
Accounting for Flexibility
A significant challenge in docking is treating molecular flexibility. While most protocols treat the receptor as rigid, this can limit accuracy. Advanced protocols incorporate flexibility through various methods [43] [47]:
- Side-chain Flexibility: Allowing side-chains in the binding site to rotate during docking.
- Ensemble Docking: Docking against multiple receptor conformations (e.g., from NMR or molecular dynamics simulations) to account for backbone movement [43].
- Induced-Fit Docking: More advanced (and computationally intensive) methods that explicitly model the conformational changes in the receptor upon ligand binding.
Target-Specific and Machine Learning Scoring
Generic scoring functions may not be optimal for all targets. The development of Target-Specific Scoring Functions (TSSFs) can significantly improve virtual screening performance [48]. Furthermore, machine learning and deep learning models are increasingly being integrated into docking pipelines. These models, such as DeepScore, can be trained on specific target data to better distinguish true binders from non-binders, potentially reducing false-positive rates [48] [45] [3].
Consensus Methods
To improve the reliability of hit selection, consensus scoring—using multiple scoring functions to rank compounds—is a widely adopted strategy. A compound that ranks highly across several different scoring functions is more likely to be a true active [48] [45].
Performance Evaluation and Experimental Validation
Evaluating Docking Performance
Before launching a prospective SBVS campaign, it is crucial to validate the chosen protocol. This is typically done using benchmarking datasets like the Directory of Useful Decoys: Enhanced (DUD-E) [48] [3]. Key metrics include:
- Enrichment Factor (EF): Measures the concentration of true active compounds found in a top fraction of the ranked library compared to a random selection [49] [3].
- Area Under the Curve (AUC) of ROC: Represents the overall ability of the method to discriminate actives from inactives [49].
- Early Recognition Metrics: Metrics like BEDROC and RIE are designed to specifically reward methods that rank actives very early in the list, which is critical for practical VS [49].
Table 2: Common Metrics for Evaluating Virtual Screening Performance
| Metric | Description | Interpretation | Utility in VS |
|---|---|---|---|
| AUC-ROC | Area Under the Receiver Operating Characteristic Curve | Overall ability to rank actives above inactives. Value of 0.5 is random; 1.0 is perfect. | Measures global performance but may not reflect early enrichment. |
| Enrichment Factor (EF) | Fraction of actives found in a top percentage (e.g., 1%) of the screened library vs. random. | An EF of 10 in the top 1% means a 10-fold enrichment over random. | Directly measures early enrichment, which is highly relevant for VS. |
| BEDROC / RIE | Exponentially weights the rank of actives to emphasize early recognition. | A single metric that focuses on early ranks. More sensitive to early performance than AUC. | Specifically designed to address the "early recognition" problem in VS. |
Experimental Validation
The ultimate test of any SBVS protocol is experimental validation. Promising computational hits must be procured or synthesized and tested in biochemical or cell-based assays [42]. A comprehensive validation cascade includes:
- In vitro binding/activity assays (e.g., measuring ICâ‚…â‚€, Káµ¢) to confirm potency [42].
- Cellular assays to demonstrate activity in a more physiologically relevant context [42] [21].
- Co-crystallization of the hit compound with the target protein, which provides the highest level of validation by confirming the predicted binding mode [42] [3].
Successful case studies, such as the discovery of hits against the ubiquitin ligase KLHDC2 and the sodium channel Naáµ¥1.7 using the RosettaVS protocol, underscore the power of well-validated SBVS approaches. In these studies, high-resolution X-ray crystallography confirmed the predicted docking poses, demonstrating remarkable agreement between computation and experiment [3].
Table 3: Key Resources for Structure-Based Virtual Screening
| Resource Category | Examples | Primary Function |
|---|---|---|
| Protein Structure Databases | Protein Data Bank (PDB), AlphaFold Database | Source for 3D atomic coordinates of target proteins, either experimentally determined or computationally predicted. |
| Small Molecule Databases | ZINC, PubChem, ChEMBL, DrugBank | Provide 2D or 3D structures of commercially available or known bioactive compounds for virtual screening libraries. |
| Structure Preparation Tools | UCSF Chimera, AutoDockTools, Open Babel, Schrodinger Maestro | Prepare protein and ligand structures for docking (add H atoms, assign charges, optimize hydrogen bonding). |
| Docking Software | AutoDock Vina, GLIDE, GOLD, DOCK, RosettaVS | Core platforms that perform the conformational sampling (posing) and scoring of ligands. |
| Binding Site Prediction | DoGSiteScorer, CASTp, DeepSite, COACH | Predict potential binding pockets on a protein surface when the active site is unknown. |
| Benchmarking Sets | DUD-E, CASF-2016 | Standardized datasets used to validate and benchmark the performance of docking protocols and scoring functions. |
In the landscape of anticancer drug discovery, ligand-based computational approaches provide powerful tools for identifying and optimizing novel therapeutic candidates when the structural information of the biological target is limited or unavailable. These methods rely on the principle that molecules with similar structural or physicochemical properties often exhibit similar biological activities. Quantitative Structure-Activity Relationship (QSAR) modeling and pharmacophore modeling represent two cornerstone methodologies in this domain, enabling researchers to distill critical features responsible for anticancer activity from known active compounds [11] [50]. Within the broader context of computational protocols for virtual screening, these ligand-based strategies offer a cost-effective and efficient solution for prioritizing compounds with a high likelihood of efficacy from extensive chemical libraries, thereby accelerating the early stages of anticancer drug development [51] [52].
Theoretical Background
Quantitative Structure-Activity Relationship (QSAR)
QSAR is a computational methodology that correlates quantitative descriptions of molecular structure with a specific biological activity. The fundamental hypothesis is that a direct, quantifiable relationship exists between a compound's molecular properties and its biological response [11]. Once established, this mathematical model can predict the activity of new, untested compounds.
- Molecular Descriptors: These are numerical representations of a molecule's physicochemical properties. They can range from simple atomic counts to complex electronic or topological indices.
- Biological Activity Data: Typically expressed as IC50, Ki, or EC50 values, this data represents the potency of a series of compounds against a specific anticancer target or cell line.
- Statistical Modeling: Algorithms such as Partial Least Squares (PLS) or multiple linear regression are used to derive the mathematical relationship between the descriptors and the biological activity [11].
Pharmacophore Modeling
A pharmacophore is defined as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [53]. In simpler terms, it is an abstract representation of the essential molecular features a compound must possess to bind to a target.
- Pharmacophoric Features: Common features include hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), hydrophobic regions (H), and positive or negative ionizable groups [53] [54].
- Ligand-Based Pharmacophore Generation: This approach constructs the pharmacophore model based on a set of known active ligands. It identifies the common spatial arrangement of features shared among these diverse but active molecules, which is presumed to be responsible for their biological activity [53] [55].
Application Note 1: Ligand-Based 3D-QSAR Pharmacophore Modeling for Topoisomerase I Inhibitors
Background
DNA Topoisomerase I (Top1) is a well-validated anticancer target. While natural products like Camptothecin (CPT) and its derivatives are known Top1 poisons, they suffer from limitations such as instability and toxicity [55]. This application note details a protocol for discovering novel Top1 inhibitors using a 3D-QSAR pharmacophore model, virtual screening, and molecular docking.
Experimental Protocol
Step 1: Compound Selection and Dataset Preparation
- A diverse set of 62 CPT derivatives with known experimental IC50 values against the A549 lung cancer cell line was compiled from literature [55].
- The dataset was divided into a training set (29 compounds) and a test set (33 compounds). The training set included molecules spanning a wide activity range (IC50 from 0.003 μM to 11.4 μM) and diverse chemical substitution patterns to ensure a robust model.
Step 2: Conformational Analysis and Pharmacophore Generation
- The 3D structures of the training set compounds were generated and energy-minimized using a force field like CHARMM [55].
- The HypoGen algorithm in Discovery Studio was employed to generate 3D-QSAR pharmacophore models [55]. The algorithm creates models that correlate the spatial arrangement of features with the experimental activity.
- The top model, Hypo1, was selected based on its high correlation coefficient and low root mean square deviation.
Step 3: Pharmacophore Model Validation
- The model's predictive ability was assessed using the test set molecules. A strong correlation (r = 0.87) between the experimental and estimated activities was reported, indicating good predictive power [55].
- The model was used as a 3D query for virtual screening.
Step 4: Virtual Screening of Chemical Databases
- The validated Hypo1 model was used to screen over 1 million drug-like molecules from the ZINC database [55].
- The screened hits were subjected to a multi-tiered filtration process:
- Lipinski's Rule of Five: To ensure drug-likeness and favorable ADME properties.
- SMART Filtration: To remove compounds with undesirable or reactive functional groups.
- Activity Filtration: Retention of compounds with an estimated activity better than 1.0 μM.
Step 5: Molecular Docking and Toxicity Assessment
- The filtered hits were docked into the active site of the Top1-DNA cleavage complex (PDB ID: 1T8I) to study binding modes and interactions.
- The top six molecules from docking were evaluated for toxicity using the TOPKAT program, yielding three final "hit" molecules (e.g., ZINC68997780, ZINC15018994) with potential as novel Top1 inhibitors [55].
The workflow for this protocol is summarized in the diagram below.
Key Research Reagents and Computational Tools
Table 1: Essential research reagents and software used in the Top1 inhibitor discovery protocol.
| Item Name | Type | Function/Description | Source |
|---|---|---|---|
| CPT Derivatives | Chemical Dataset | 62 molecules with known Top1 inhibitory activity (IC50). Used as training/test sets. | Literature [55] |
| ZINC Database | Chemical Library | A public database containing over 1 million commercially available "drug-like" compounds for virtual screening. | https://zinc.docking.org [55] |
| Discovery Studio | Software Suite | Integrated platform for molecular modeling, pharmacophore generation (HypoGen), and virtual screening. | Commercial Software [55] |
| CHARMM Force Field | Computational Parameter Set | A set of mathematical parameters for calculating molecular energies and forces during geometry optimization. | Academic/Commercial [55] |
| TOPKAT | Software Module | Predictive tool for assessing potential toxicity of small molecules based on their structure. | Commercial Software [55] |
Application Note 2: Developing an Ensemble Pharmacophore for EGFR Inhibitors
Experimental Protocol
Step 1: Ligand Selection and Alignment
- A set of known EGFR kinase inhibitors with diverse chemical scaffolds was selected. The inhibitors were obtained from protein-ligand complexes in the PDB (e.g., 5HG8, 5UG8) [53].
- The 3D structures of the ligands were extracted and aligned based on the maximum common scaffold to ensure a consistent frame of reference for feature comparison [53].
Step 2: Pharmacophore Feature Extraction
- For each aligned ligand, key pharmacophoric features (HBD, HBA, and Hydrophobic) were identified and their 3D coordinates recorded [53].
Step 3: Clustering to Generate Ensemble Pharmacophore
- The coordinates of each feature type (all HBDs, all HBAs, all Hydrophobic) were clustered independently using the k-means clustering algorithm [53].
- The k-means algorithm partitions the data into 'k' clusters, where each feature point belongs to the cluster with the nearest mean. This helps identify the most representative locations for each feature type across all ligands.
- The central point (centroid) of the most relevant cluster for each feature type was selected to form the final ensemble pharmacophore model.
Step 4: Virtual Screening
- The ensemble pharmacophore model was used as a 3D query to screen a database of compounds.
- Molecules that matched all the essential features of the ensemble model were identified as potential hits for further experimental testing.
The workflow for generating an ensemble pharmacophore is visualized below.
Key Parameters and Considerations
Table 2: Key parameters and their impact on ligand-based pharmacophore modeling.
| Parameter | Description | Impact on Model Quality |
|---|---|---|
| Conformational Sampling | The method used to generate representative 3D conformations of each ligand. | More thorough sampling improves the chance of finding the bioactive conformation but is computationally expensive. For virtual screening, faster protocols can be sufficient [56]. |
| Chemical Diversity of Input Ligands | The degree of structural variation among the known active compounds used to build the model. | High diversity leads to a more general and robust model that can identify novel scaffolds (scaffold hopping) [54]. |
| Number of Pharmacophore Features | The count of features (e.g., HBD, HBA) included in the final model. | Too few features can lead to promiscuous hits; too many can make the model too restrictive and miss valid actives. |
| Alignment Method | The technique used to superimpose the input ligands (e.g., common scaffold, flexible alignment). | The choice of alignment directly defines the spatial arrangement of features and is critical for model accuracy [53]. |
Comparative Analysis and Protocol Selection
Choosing the appropriate ligand-based method depends on the available data and the project's goals. The following table compares the two featured approaches.
Table 3: Comparison between 3D-QSAR Pharmacophore and Ensemble Pharmacophore approaches.
| Aspect | 3D-QSAR Pharmacophore (HypoGen) | Ensemble Pharmacophore |
|---|---|---|
| Primary Requirement | A set of ligands with quantitative biological activity data (IC50/Ki). | A set of known active ligands, activity data beneficial but not strictly required. |
| Key Output | A predictive model that estimates biological activity of new compounds. | A consensus set of features representing the common interaction pattern. |
| Major Strength | Directly links structural features to potency; useful for lead optimization. | Excellent for scaffold hopping and identifying structurally diverse hits. |
| Best Suited For | Projects where understanding the structural determinants of potency is critical. | Projects focused on finding novel chemotypes from large libraries when activity data is scarce. |
| Computational Cost | Higher, due to the iterative algorithm and need for conformational analysis of a diverse set. | Lower to moderate, depending on the number of ligands and the complexity of alignment. |
Ligand-based approaches, namely QSAR and pharmacophore modeling, are indispensable tools in the modern computational toolkit for anticancer drug discovery. As demonstrated in the application notes, these protocols can systematically translate the information encoded in known active compounds into predictive models and actionable queries for virtual screening. The integration of these methods with other computational techniques, such as molecular docking and toxicity prediction, creates a powerful, multi-tiered filter that efficiently transitions from vast chemical libraries to a manageable number of high-priority experimental candidates. Future advancements in this field are likely to be driven by the integration of machine learning and artificial intelligence with traditional methods, further enhancing the precision and speed of discovering novel anticancer agents from the ever-expanding chemical space [11] [51].
AI and Machine Learning in Accelerated Virtual Screening
The field of anticancer drug discovery is in the midst of a transformative shift, driven by the integration of artificial intelligence (AI) and machine learning (ML). Virtual screening (VS), a computational technique used to search libraries of small molecules to identify those most likely to bind to a drug target, has become a cornerstone of this evolution [31]. By leveraging AI, researchers can now screen billions of compounds in a matter of days, dramatically accelerating the identification of hit molecules and optimizing lead compounds for a fraction of the traditional cost and time [57] [58]. This protocol details the application of advanced AI-accelerated virtual screening methodologies within the specific context of anticancer drug discovery, providing a structured framework for researchers to efficiently identify novel therapeutic candidates.
Core AI and ML Methodologies in Virtual Screening
The application of AI in virtual screening can be broadly categorized into ligand-based and structure-based approaches, with hybrid methods and advanced ML techniques enhancing the capabilities of both.
Ligand-Based Virtual Screening
Ligand-based methods are employed when the 3D structure of the target protein is unknown but information about active ligands is available.
- Pharmacophore Modeling: This technique involves defining an ensemble of steric and electronic features necessary for optimal supramolecular interactions with a biological target. When creating a model, it is preferred to use multiple, structurally diverse rigid molecules to ensure different features that occur during binding are captured [31].
- Shape-Based Screening: Approaches like Rapid Overlay of Chemical Structures (ROCS) use Gaussian functions to define molecular volumes and are considered an industry standard for identifying molecules with similar three-dimensional shapes to known actives, implying a similar mode of action [31].
- Quantitative Structure-Activity Relationship (QSAR): QSAR models are predictive models built using information from a set of known active and inactive compounds. They help prioritize compounds for lead discovery by correlating chemical structure with biological activity [31].
Structure-Based Virtual Screening
Structure-based methods rely on the known three-dimensional structure of the target protein.
- Molecular Docking with AI Acceleration: This is the most used structure-based technique, involving the prediction of a ligand's preferred orientation (pose) within a target's binding site and the estimation of its binding affinity (scoring). AI has been instrumental in enhancing the speed and accuracy of these simulations. For instance, the RosettaVS protocol incorporates a modified docking approach with two modes: a Virtual Screening Express (VSX) mode for rapid initial screening and a Virtual Screening High-precision (VSH) mode that includes full receptor flexibility for final ranking of top hits [57].
- AI-Driven Affinity Prediction: Beyond docking, advanced AI models are used to predict key binding affinity constants, such as the half-maximal inhibitory concentration (IC50), which is crucial for understanding drug-target interaction strength even without the 3D structures of all drug targets [59].
Hybrid and Machine Learning Approaches
Hybrid methods that leverage both structural and ligand similarity are being developed to overcome the limitations of traditional approaches [31].
- Machine Learning Algorithms: Supervised ML techniques are widely used to build predictive models from training datasets of known active and inactive compounds. These include:
- Support Vector Machines (SVM)
- Random Forest
- k-Nearest Neighbors (k-NN)
- Neural Networks These models calculate the probability that a compound is active and then rank each compound based on this probability, significantly enriching the screening library with promising candidates [31].
- Active Learning: Modern platforms, such as the open-source OpenVS platform, use active learning techniques. These systems simultaneously train a target-specific neural network during docking computations to intelligently select the most promising compounds for more expensive, high-fidelity docking calculations, making the screening of billion-compound libraries feasible [57].
Table 1: Key Machine Learning Algorithms and Their Applications in Anticancer Virtual Screening
| Algorithm | Primary Function | Advantages in Anticancer VS |
|---|---|---|
| Random Forest | Classification & Regression | Handles high-dimensional data; robust against overfitting [31]. |
| Support Vector Machines (SVM) | Classification | Effective in high-dimensional spaces; versatile with different kernels [60]. |
| Graph Neural Networks (GNN) | Link Prediction & Classification | Integrates data from multiple sources (e.g., for synthetic lethality prediction) [59]. |
| Heterogeneous Graph Convolutional Networks | Drug-Target Interaction (DTI) Prediction | Predicts DTIs without requiring 3D target structures [59]. |
Experimental Protocols
This section provides a detailed, step-by-step guide for conducting an AI-accelerated virtual screening campaign for anticancer drug discovery.
Protocol 1: AI-Accelerated Structure-Based Virtual Screening
Aim: To identify novel hit compounds against a defined anticancer target (e.g., a kinase or ubiquitin ligase) from an ultra-large chemical library. Background: This protocol is based on the OpenVS platform and RosettaVS method, which have been proven to discover hit compounds with single-digit micromolar binding affinity in less than seven days [57].
Materials and Reagents:
- Target Protein Structure: A high-resolution 3D structure of the target protein (e.g., from X-ray crystallography, NMR, or a highly accurate predicted model from AlphaFold2 [58]).
- Compound Library: A database of small molecules in an appropriate format (e.g., SDF, SMILES). Examples include ZINC, PubChem, or in-house libraries [60].
- Computational Infrastructure: A high-performance computing (HPC) cluster with multiple CPUs (e.g., 3000+ CPUs) and GPUs (e.g., RTX2080 or higher) [57].
- Software: OpenVS platform (integrated with RosettaVS), or commercial equivalents.
Procedure:
- Target Preparation:
- Obtain the 3D structure of the target protein (PDB format).
- Preprocess the structure: add hydrogen atoms, assign partial charges, and optimize side-chain conformations for residues in the binding pocket.
- Define the binding site coordinates based on known ligand interactions or structural analysis.
Ligand Library Preparation:
- Curate the initial compound library (e.g., multi-billion molecules).
- Apply pre-filtering to enhance drug-likeness: use rules like Lipinski's Rule of Five to remove compounds with poor pharmacokinetic properties [61].
- Prepare ligand structures: generate 3D conformations, assign correct tautomeric and protonation states at physiological pH.
AI-Accelerated Docking and Active Learning:
- Initial Phase (VSX Mode): Initiate the OpenVS platform. The active learning algorithm will begin by docking a diverse subset of the library using the fast VSX mode, which allows for rapid initial screening.
- Model Training: Simultaneously, a target-specific neural network is trained on the fly using the docking results to learn the features of compounds that score well.
- Iterative Screening: The trained model predicts and prioritizes the next batch of compounds most likely to be active, which are then fed back into the docking process. This iterative loop continues until the chemical space is sufficiently explored.
High-Precision Docking (VSH Mode):
- The top-ranking hits (e.g., 1-2% of the initial library) from the VSX screening are subjected to a more computationally intensive docking run using the VSH mode.
- This mode incorporates full receptor side-chain flexibility and limited backbone movement, which is critical for accurately modeling induced fit upon ligand binding [57].
Post-Processing and Hit Selection:
- Rescoring and Ranking: Analyze the VSH results. Use the RosettaGenFF-VS scoring function, which combines enthalpy (ΔH) and entropy (ΔS) changes upon binding, to generate a final ranked list of compounds [57].
- Cluster Analysis: Cluster the top-scoring compounds by chemical similarity to ensure structural diversity among the selected hits.
- Visual Inspection: Manually inspect the predicted binding poses of the top-ranked, structurally diverse hits to confirm plausible binding modes and key interactions.
The following workflow diagram illustrates this multi-stage protocol:
Protocol 2: Ligand-Based Virtual Screening with Metabolomics and ML for MoA Prediction
Aim: To predict the Mode of Action (MoA) of novel anti-proliferative drug candidates and identify new hits based on metabolic profiling. Background: This protocol is particularly useful for anticancer drug discovery when the protein target is ambiguous, but phenotypic screening data is available. It leverages the fact that drugs with similar MoAs induce distinct metabolic changes in cancer cells [62].
Materials and Reagents:
- Cell Lines: Relevant cancer cell lines (e.g., PC-3 prostate cancer cells).
- Reference Compound Set: A panel of drugs with known MoAs affecting various cancer metabolism pathways.
- Analytical Instrumentation: Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS) for metabolomic profiling.
- Software: Machine learning environment (e.g., Python with scikit-learn, TensorFlow).
Procedure:
- Metabolomic Profiling of Reference Compounds:
- Treat cancer cells with a set of reference drugs with known MoAs.
- Using LC-MS/MS, profile the low molecular weight intermediates of central carbon and energy metabolism (CCEM) to generate metabolic fingerprints for each reference drug [62].
Machine Learning Model Training:
- Use the metabolic profiles from step 1 as the feature set (input X) and the known MoAs as labels (output Y).
- Train a multi-class classification ML model (e.g., Random Forest or Support Vector Machine) to recognize the unique metabolic patterns associated with each MoA.
Profiling and Prediction for Novel Candidates:
- Treat the same cancer cells with novel, uncharacterized drug candidates.
- Generate their metabolic profiles using the same LC-MS/MS protocol.
- Input the new metabolic data into the trained ML model to predict the most likely MoA for each candidate.
Similarity-Based Virtual Screening:
- Use the metabolic profile of a known active compound (the "query") as a benchmark.
- Screen a database of metabolic profiles from novel compounds to find those with similar patterns.
- Prioritize novel compounds whose metabolic profiles are most similar to the query, as they are predicted to share a similar MoA [62].
Table 2: Key Research Reagents and Computational Tools for AI-Driven VS
| Category/Item | Specific Examples | Function in Protocol |
|---|---|---|
| AI Screening Platforms | OpenVS, NVIDIA NIM Blueprint | Provides integrated, scalable environments for running AI-accelerated VS workflows [57] [58]. |
| Docking & Scoring Software | RosettaVS, Autodock Vina, Schrödinger Glide, DiffDock NIM | Predicts ligand binding pose and affinity to the target protein [57] [58]. |
| Generative AI Models | MolMIM NIM | Generates novel molecules with optimized properties (e.g., solubility, low toxicity) rather than screening existing libraries [58]. |
| Compound Databases | PubChem, ZINC, ChEMBL, DrugBank | Provides large collections of small molecules for screening [60]. |
| Target Structure Sources | Protein Data Bank (PDB), AlphaFold2 NIM | Provides 3D structural data of the biological target for structure-based screening [58]. |
Performance and Validation
The success of a virtual screen is ultimately defined by its ability to identify molecules with novel chemical structures that bind to the target, rather than just a high number of hits [31].
Retrospective vs. Prospective Validation
- Retrospective Benchmarking: This involves measuring a method's ability to retrieve known active molecules from a library spiked with decoys. Standard benchmarks like DUD (Directory of Useful Decoys) and CASF (Comparative Assessment of Scoring Functions) are used. For example, RosettaGenFF-VS achieved a top 1% enrichment factor (EF1%) of 16.72 on the CASF-2016 benchmark, significantly outperforming other methods [57].
- Prospective Validation: This is the conclusive proof of a method's utility, where the computationally identified hits are subjected to experimental testing (e.g., IC50 measurements). A study screening two unrelated targets (KLHDC2 and NaV1.7) reported hit rates of 14% and 44%, respectively, with all hits exhibiting single-digit µM affinity. The predicted binding pose for a KLHDC2 ligand was later validated by a high-resolution X-ray crystal structure [57].
Performance Metrics
The following metrics are crucial for evaluating virtual screening performance:
Table 3: Key Metrics for Evaluating Virtual Screening Performance
| Metric | Description | Interpretation |
|---|---|---|
| Enrichment Factor (EF) | Measures the concentration of active compounds found in a top fraction (e.g., 1%) of the screened library compared to a random selection. | A higher EF indicates better early recognition of true positives. |
| Area Under the Curve (AUC) | The area under the Receiver Operating Characteristic (ROC) curve, which plots the true positive rate against the false positive rate. | An AUC of 1.0 represents a perfect screen; 0.5 represents random selection. |
| Hit Rate | The percentage of tested virtual hits that are confirmed to be active in experimental assays. | The primary metric for success in a prospective screen. |
| Binding Affinity (IC50/Ki) | The experimental measure of a compound's potency in inhibiting the target. | Validates the predictive power of the scoring function. |
Application in Anticancer Drug Discovery
The protocols outlined herein are uniquely positioned to address critical challenges in oncology drug development. AI-driven virtual screening can help tackle undruggable targets, tumor heterogeneity, and drug resistance [59]. For instance, the integration of multi-omics data (genomics, epigenomics, proteomics) through AI models allows for a more holistic approach to target identification and compound efficacy prediction [59] [60]. A specific application involves predicting synthetic lethality—a promising approach for discovering anticancer drug targets that selectively kill cancer cells while sparing healthy ones—using graph neural networks that incorporate knowledge graphs (e.g., KG4SL model) [59]. The ability to screen billions of molecules rapidly also opens the door to repurposing existing drugs for new anticancer indications, as ML models can find unexpected connections between drugs and targets based on shared patterns in large-scale biological data [60].
Molecular Dynamics Simulations for Binding Stability Analysis
Molecular dynamics (MD) simulations have become an indispensable computational tool in modern anticancer drug discovery, providing critical insights into binding stability that bridge the gap between static structural information and dynamic biological function [8]. Within virtual screening pipelines, MD simulations serve as a powerful validation step that assesses the temporal stability of protein-ligand complexes identified through molecular docking [63] [64]. This analytical approach enables researchers to filter out false positives and prioritize the most promising drug candidates by evaluating how potential therapeutics interact with cancer-related targets at an atomic level over time [11]. The implementation of MD simulations has proven particularly valuable in the exploration of natural products as anticancer agents, where it helps elucidate binding mechanisms and stability for complex plant-derived compounds [11] [64]. As computational resources have advanced, MD simulations have evolved from supplementary analyses to central components in rational drug design protocols, offering unprecedented insights into molecular recognition events that underlie successful cancer treatments [51] [8].
Quantitative Data from MD Simulations in Anticancer Research
MD simulations generate substantial quantitative data that researchers analyze to evaluate binding stability. The tables below summarize key parameters and their significance in assessing anticancer drug-target interactions.
Table 1: Key Quantitative Parameters from MD Simulations for Binding Stability Assessment
| Parameter | Interpretation | Typical Value Range | Research Application |
|---|---|---|---|
| RMSD (Root Mean Square Deviation) | Measures structural stability of protein-ligand complex | < 2-3 Ã… indicates stability [64] | Tracking conformational changes during simulation |
| RMSF (Root Mean Square Fluctuation) | Quantifies residual flexibility | High values indicate flexible regions | Identifying mobile domains affecting binding |
| Radius of Gyration | Assesses protein compactness | Consistent values suggest structural maintenance | Evaluating overall protein folding stability |
| Binding Free Energy (MM-GBSA/PBSA) | Predicts binding affinity | More negative values indicate stronger binding [65] [63] | Ranking candidate compounds by affinity |
| Hydrogen Bonds | Measures specific interactions | Consistent H-bonds suggest stable binding [66] | Evaluating interaction quality and persistence |
Table 2: Representative Binding Stability Data from Recent Anticancer Studies
| Study Focus | Simulation Duration (ns) | Key Findings | Binding Free Energy (kcal/mol) |
|---|---|---|---|
| Lignans as MDM2-p53 Inhibitors [63] | 100 | Stable complexes with minimal RMSD fluctuation | -7.24 to -7.53 [63] |
| Pinocembrin Derivatives Targeting MMP9 [65] | Not specified | CID-25149104 and CID-42607886 showed most stable binding | Below -70 (MM-GBSA) [65] |
| Ficus carica Compounds [64] | Not specified | β-bourbonene demonstrated stable binding with multiple targets | Calculated via MM-PBSA/GBSA [64] |
Experimental Protocols for Binding Stability Analysis
System Preparation and Equilibration
The initial step involves preparing the protein-ligand complex for simulation. Researchers typically retrieve protein structures from the Protein Data Bank, while ligand structures are optimized using molecular modeling software [64]. For a recent study on lignans as MDM2-p53 interaction inhibitors, researchers prepared the MDM2 crystal structure (bound to Nutlin-3a) by removing water molecules and adding polar hydrogen atoms [63]. The system is then solvated in a water box with appropriate dimensions to accommodate the complex, followed by the addition of ions to neutralize the system charge [67]. Energy minimization is performed using steepest descent algorithms to remove steric clashes, followed by stepwise equilibration under NVT (constant Number of particles, Volume, and Temperature) and NPT (constant Number of particles, Pressure, and Temperature) ensembles to stabilize temperature and pressure [67].
Production MD Simulation and Analysis
Following equilibration, production MD simulations are conducted using software such as AMBER or GROMACS [68] [64]. In the investigation of Ficus carica bioactive compounds, researchers used AMBER16 available through the LARMD platform to simulate protein-ligand complexes [64]. Simulations typically run for time scales ranging from 100 nanoseconds to several microseconds, depending on system size and computational resources [63]. During this phase, trajectories are saved at regular intervals for subsequent analysis. For the MDM2-p53 inhibitor study, researchers conducted 100 ns simulations and analyzed trajectories for RMSD, radius of gyration, and hydrogen bonding patterns to evaluate complex stability [63]. Additional analyses include calculating binding free energies using MM-PBSA/GBSA methods and performing per-residue energy decomposition to identify key interacting residues [68] [64].
Table 3: Essential Computational Tools for MD Simulations in Anticancer Drug Discovery
| Tool/Resource | Specific Examples | Application in Workflow |
|---|---|---|
| Molecular Dynamics Software | AMBER [68] [64], GROMACS [67] | Running production MD simulations |
| Visualization & Analysis | VMD, PyMOL, MDTraj | Trajectory analysis and visualization |
| Binding Energy Calculations | MM-PBSA, MM-GBSA [65] [64] | Quantifying binding affinities |
| Force Fields | AMBER force fields, CHARMM, GROMOS | Defining atomic interactions and parameters |
| System Preparation Tools | PDB2GMX, tleap | Preparing protein structures for simulation |
| Specialized Analysis | Principal Component Analysis (PCA), Dynamic Cross-Correlation Matrix (DCCM) [65] | Identifying essential dynamics and correlated motions |
Workflow Visualization
MD Simulation Workflow
Analysis Framework
ADMET Prediction for Early-Stage Toxicity Assessment
Within anticancer drug discovery, the high attrition rates of candidate molecules due to unforeseen toxicity and unfavorable pharmacokinetics remain a significant challenge [69]. The evaluation of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties is thus critical from the earliest stages of research. Traditional experimental ADMET profiling is often resource-intensive and low-throughput, creating a bottleneck in the development pipeline [70]. The integration of in silico ADMET prediction tools offers a powerful strategy to mitigate this risk, enabling researchers to prioritize compounds with a higher probability of clinical success by flagging potential toxicity issues and suboptimal pharmacokinetic profiles before significant resources are invested [15] [71]. This Application Note details standardized computational protocols for ADMET prediction, framed within the context of virtual screening for anticancer drug discovery.
Key ADMET Properties and Prediction Tools
Early-stage in silico toxicity assessment focuses on a core set of properties that are major contributors to compound failure. The following table summarizes the key endpoints, their significance in toxicity assessment, and common benchmarks for evaluation.
Table 1: Key ADMET Properties for Early-Stage Toxicity Assessment in Anticancer Drug Discovery
| ADMET Property | Significance in Toxicity Assessment | Common Prediction Models/Benchmarks |
|---|---|---|
| Hepatotoxicity | Predicts drug-induced liver injury (DILI), a major cause of drug withdrawal [70]. | DILIrank dataset; models trained on ~475 compounds annotated for hepatotoxic potential [70]. |
| hERG Inhibition | Identifies compounds with potential for cardiotoxicity via blockade of the hERG potassium channel [70]. | Binary classification models based on a 10 µM inhibition threshold; hERG Central database with >300,000 records [70]. |
| Ames Mutagenicity | Assesses genotoxic potential through bacterial reverse mutation assay prediction [72] [73]. | In silico models using random forest algorithms on public toxicity databases [73]. |
| CYP450 Inhibition | Predicts drug-drug interactions by identifying compounds that inhibit key metabolic enzymes (e.g., CYP3A4, CYP2D6) [72] [74]. | Classification models predicting inhibition for major CYP isoforms [73] [74]. |
| Human Oral Bioavailability | Estimates the fraction of an orally administered dose that reaches systemic circulation, critical for dosing efficacy [73] [69]. | Quantitative and classification models using molecular descriptors and fingerprints [73]. |
To support the prediction of these properties, a suite of software tools and databases has been developed. The selection of an appropriate tool depends on the specific project needs, considering whether open-source or commercial solutions are required.
Table 2: Research Reagent Solutions for ADMET Prediction
| Tool Name | Type | Key Function(s) | Relevance to Protocol |
|---|---|---|---|
| ADMET Predictor [72] | Commercial Software Platform | Predicts over 175 properties including solubility, metabolic stability, DILI, Ames mutagenicity, and integrated PBPK simulations. | Flagship platform for comprehensive ADMET profiling; includes "ADMET Risk" score for compound prioritization. |
| FP-ADMET [73] | Open-Source Software | Repository of fingerprint-based predictive models for over 50 ADMET endpoints using Random Forest algorithm. | Provides open-access models for key toxicity endpoints; useful for organizations with limited commercial software access. |
| SwissADME [73] [74] | Free Web Tool | Evaluates pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of small molecules. | Quick assessment of drug-likeness and key pharmacokinetic parameters during initial virtual screening. |
| admetSAR [73] [74] | Free Web Tool / Platform | Provides models for ADMET properties for both drug discovery and environmental risk assessment. | Useful for wide-scope toxicity screening and predictions based on molecular fingerprints. |
| Tox21 [70] | Public Database | Qualitative toxicity data for 8,249 compounds across 12 biological targets related to nuclear receptor and stress response pathways. | Benchmark dataset for training and validating predictive toxicity models. |
Integrated Computational Protocol for ADMET Profiling in Anticancer Discovery
The following workflow integrates in silico ADMET profiling into a typical virtual screening pipeline for anticancer lead optimization, as demonstrated in studies on EGFR inhibitors and caged xanthone derivatives [75] [76]. The diagram below outlines the key stages from initial compound library generation to the final selection of optimized leads.
Protocol Steps
Pharmacophore-Based Virtual Screening:
- Objective: To rapidly reduce a large virtual compound library to a manageable number of hits based on essential structural features for biological activity.
- Methodology: Using a matrix compound with known anticancer activity (e.g., Erlotinib for EGFR inhibitors [75]), define a pharmacophore model. Screen databases like DrugBank against this model to select compounds that match the critical pharmacophoric features.
- Output: A focused library of 20-30 candidate molecules for further analysis [75].
Preliminary ADMET Filtering:
- Objective: To remove compounds with obvious drug-likeness violations or high structural alerts for toxicity.
- Methodology: Subject the focused library to screening using tools like SwissADME [74]. Apply Lipinski's Rule of 5 (or its "soft" thresholds as in ADMET Risk [72]) to filter for oral bioavailability. Assess other simple parameters like synthetic accessibility [76].
- Output: A refined list of compounds that pass initial drug-likeness criteria.
Multi-Parameter ADMET Profiling:
- Objective: To perform a deep and quantitative assessment of toxicity and pharmacokinetic risks.
- Methodology: Use robust platforms like ADMET Predictor [72] or FP-ADMET [73] to predict critical endpoints detailed in Table 1. This includes:
- Toxicity: Hepatotoxicity (DILI), hERG inhibition, Ames mutagenicity, carcinogenicity.
- Metabolism: CYP450 inhibition profiles.
- Pharmacokinetics: Plasma protein binding (Fup), intrinsic clearance, fraction absorbed.
- Output: A comprehensive ADMET risk score for each compound, enabling direct comparison and prioritization [72] [76].
Molecular Docking and Binding Affinity Assessment:
- Objective: To validate the binding mode and affinity of top-ranked ADMET candidates to the specific anticancer target (e.g., EGFR, Mpro).
- Methodology: Perform molecular docking studies using software like AutoDock or GOLD [75] [76]. Analyze hydrogen bonding interactions, hydrophobic contacts, and binding energies with the target's active site residues. This step confirms that promising ADMET profiles are coupled with maintained efficacy.
- Output: A ranked list of compounds based on binding affinity and interaction quality.
Advanced Simulations for Validation:
- Objective: To dynamically assess the stability of the protein-ligand complex and validate reactivity insights.
- Methodology:
- Molecular Dynamics (MD) Simulation: Run 100 ns MD simulations to evaluate the conformational stability, binding free energy, and interaction persistence of the lead complex in a solvated environment [75] [71].
- Density Functional Theory (DFT): Perform DFT calculations to understand the electronic properties (e.g., HOMO-LUMO energies) and chemical reactivity of the lead molecule [75].
- Output: Validated, stable lead candidates with a low predicted ADMET risk and high potential for in vitro and in vivo success.
Case Study: Application in Anti-EGFR Drug Discovery
A recent study on EGFR tyrosine kinase inhibitors for cancers like non-small cell lung cancer exemplifies this protocol [75]. Researchers began with virtual screening of the DrugBank database using a pharmacophore model, identifying 23 initial hits. These compounds underwent rigorous ADMET prediction, which prioritized the molecule DB03365. Docking studies confirmed its strong binding to the EGFR active site via multiple hydrogen bonds. Subsequent DFT analysis revealed high reactivity based on its HOMO-LUMO band gap. Finally, a 100 ns MD simulation demonstrated that DB03365 formed stable interactions with key residues in the EGFR protein, outperforming the reference compound Erlotinib in these in silico assays [75]. This integrated approach showcases how computational protocols can de-risk the early discovery process and identify promising lead molecules for experimental validation.
The discovery of novel tubulin inhibitors represents a cornerstone of anticancer drug development. Microtubules, fundamental components of the eukaryotic cytoskeleton, are critically involved in cell division, morphology, and intracellular transport. Their dynamics are a clinically validated target for cancer chemotherapy, as disrupting microtubule function halts mitosis and induces apoptosis in rapidly dividing cancer cells [77] [78] [79]. However, the clinical utility of existing microtubule-targeting agents (MTAs) is often limited by drug resistance, toxicity, and narrow therapeutic windows [80] [79]. This case study details a successful drug discovery campaign that leveraged modern virtual screening protocols to identify a novel, potent tubulin inhibitor, providing a template for computational approaches in anticancer research.
Background: Tubulin as a Therapeutic Target
Microtubule Structure and Dynamics
Microtubules are hollow, cylindrical filaments composed of α- and β-tubulin heterodimers. These heterodimers assemble in a head-to-tail fashion to form protofilaments, which then associate laterally to form the mature microtubule. The structure has an inherent polarity, with a slow-growing minus end (α-tubulin exposed) and a fast-growing plus end (β-tubulin exposed) [80] [78]. Microtubule function is governed by dynamic instability—stochastic phases of growth (polymerization) and shrinkage (depolymerization) driven by the hydrolysis of GTP bound to β-tubulin [80] [78] [79]. This precise dynamics is essential for proper mitotic spindle formation and chromosome segregation during cell division.
Binding Sites for Inhibitors
Tubulin possesses several distinct binding sites for inhibitory compounds. The most therapeutically exploited are:
- The Paclitaxel site: Binds microtubule-stabilizing agents like taxanes, located on the inner surface of the microtubule.
- The Vinca alkaloid site: Binds microtubule-destabilizing agents like vinblastine, located at the plus ends.
- The Colchicine site: Binds microtubule-destabilizing agents, situated at the interface between the α- and β-tubulin monomers [80] [79].
Inhibitors targeting the colchicine site, such as the compound discovered in this case study, are of particular interest because they are often less susceptible to efflux pump-mediated drug resistance, a common problem with taxanes [80].
Computational Discovery Protocol
The following section outlines the step-by-step computational protocol used to identify the novel tubulin inhibitor, designated as compound 89.
Virtual Screening Workflow
The overall process integrated both structure-based and ligand-based virtual screening techniques to efficiently navigate a vast chemical library.
Step-by-Step Protocol
Step 1: Library Preparation
- Objective: Prepare a diverse chemical library for screening.
- Procedure:
- Obtain the Specs library, containing 200,340 commercially available compounds [77].
- Prepare all compound structures using a molecular modeling suite (e.g., Schrodinger's Maestro or OpenEye toolkits). This includes:
- Generating realistic 3D conformations.
- Assigning correct protonation states at physiological pH (7.4) using tools like
Epik. - Performing energy minimization to refine molecular geometries.
- Key Reagents:
- Specs Library: A large collection of small molecules for hit discovery.
Step 2: Structure-Based Virtual Screening
- Objective: Identify compounds that favorably bind to the target tubulin sites.
- Procedure:
- Retrieve high-resolution crystal structures of tubulin in complex with colchicine or paclitaxel (e.g., from the Protein Data Bank, PDB IDs: 1SA0, 1JFF).
- Prepare the protein structure by adding hydrogen atoms, assigning bond orders, and optimizing side-chain orientations for unresolved residues.
- Define the binding site for docking using a grid box centered on the known ligand (e.g., colchicine) in the structure.
- Perform molecular docking of the prepared library against the defined binding sites using software such as Glide or AutoDock Vina.
- Rank compounds based on docking scores and visual inspection of binding poses, focusing on key interactions like hydrogen bonding and hydrophobic contacts [77] [31].
Step 3: Ligand-Based Virtual Screening
- Objective: Complement structure-based methods by identifying compounds similar to known active tubulin inhibitors.
- Procedure:
- Curate a set of known tubulin inhibitors (e.g., colchicine, combretastatin A-4) to serve as reference queries.
- Perform similarity searching using molecular fingerprints (e.g., ECFP4 fingerprints) and the Tanimoto coefficient as a similarity metric.
- Alternatively, employ pharmacophore modeling to define the essential steric and electronic features necessary for tubulin inhibition [31].
- Screen the library against these pharmacophore models or for shape similarity using tools like ROCS (Rapid Overlay of Chemical Structures) [31].
Step 4: Hit Selection and Triaging
- Objective: Generate a manageable list of high-priority candidates for experimental testing.
- Procedure:
- Consolidate results from structure-based and ligand-based screening.
- Apply drug-like filters (e.g., Lipinski's Rule of Five) to prioritize compounds with favorable physicochemical properties.
- Cluster compounds by scaffold to ensure structural diversity among hits.
- Select the top 93 candidates for purchase and experimental validation [77].
Table 1: Key Parameters for the Virtual Screening Protocol
| Step | Software/Tool | Key Parameters | Output |
|---|---|---|---|
| Library Prep | LigPrep, MOE, RDKit | pH = 7.4 ± 0.5, Force Field: OPLS4/MMFF94 | Prepared 3D molecular library |
| Molecular Docking | Glide (SP or XP mode), AutoDock Vina | Grid Box: ~20ų, Pose Sampling: Flexible | Docking scores & binding poses |
| Similarity Search | RDKit, Canvas | Fingerprint: ECFP4, Metric: Tanimoto | Similarity scores (Tanimoto > 0.7) |
| Pharmacophore | Phase, MOE | Features: H-bond Donor/Acceptor, Hydrophobic | Pharmacophore matches |
| Hit Selection | In-house scripts | Rules: Lipinski's Rule of 5, Diversity | 93 prioritized candidates |
Experimental Validation & Characterization
The 93 virtual screening hits underwent rigorous experimental characterization to confirm tubulin targeting and antitumor efficacy. The lead compound, a nicotinic acid derivative designated compound 89, emerged from this process [77].
Biochemical and Cellular Assays
Protocol 1: Tubulin Polymerization Assay
- Objective: To determine if compound 89 directly inhibits tubulin polymerization in vitro.
- Materials:
- Purified bovine or porcine brain tubulin (>97% pure).
- GTP solution in polymerization buffer (80 mM PIPES pH 6.9, 2 mM MgClâ‚‚, 0.5 mM EGTA).
- Test compound (compound 89) and controls (e.g., colchicine for inhibition, paclitaxel for promotion).
- UV-Vis spectrophotometer with temperature-controlled cuvette holder.
- Procedure:
- Prepare a solution of tubulin (3 mg/mL) in ice-cold polymerization buffer.
- Add GTP to a final concentration of 1 mM and the test compound (at various concentrations, e.g., 1-10 µM).
- Transfer the mixture to a pre-chilled cuvette in the spectrophotometer.
- Initiate polymerization by rapidly raising the temperature to 37°C.
- Monitor the increase in turbidity (a proxy for polymer mass) by measuring the absorbance at 340 nm every minute for 60 minutes.
- Expected Outcome: Compound 89 demonstrated significant inhibition of tubulin polymerization, comparable to the colchicine control, confirming its direct action on tubulin [77].
Protocol 2: Cell Viability Assay (MTT/XTT)
- Objective: To evaluate the antiproliferative activity of compound 89 against a panel of human cancer cell lines.
- Materials:
- Cancer cell lines (e.g., HeLa, MCF-7, A549).
- Cell culture media and reagents.
- Test compound serially diluted in DMSO.
- MTT or XTT reagent.
- Multiplate reader.
- Procedure:
- Seed cells in 96-well plates at a density of 5,000 cells/well and incubate for 24 hours.
- Treat cells with a range of compound 89 concentrations for 72 hours.
- Add MTT reagent and incubate for 4 hours to allow formazan crystal formation.
- Solubilize crystals with DMSO or SDS solution.
- Measure the absorbance at 570 nm. Calculate the % cell viability and the half-maximal inhibitory concentration (ICâ‚…â‚€).
- Expected Outcome: Compound 89 exhibited potent anti-tumor efficacy in vitro with low ICâ‚…â‚€ values across multiple cancer cell lines [77].
Target Engagement and Mechanism of Action
Protocol 3: Competitive Binding Assay
- Objective: To confirm that compound 89 binds specifically to the colchicine site on tubulin.
- Materials:
- Purified tubulin.
- Fluorescent colchicine derivative (e.g., DAPI-colchicine).
- Test compound (compound 89).
- Fluorescence plate reader.
- Procedure:
- Incubate tubulin with the fluorescent colchicine probe in the presence of increasing concentrations of compound 89.
- Measure the fluorescence intensity (excitation/emission specific to the probe).
- Plot fluorescence intensity vs. compound concentration to determine the ICâ‚…â‚€ for displacement.
- Expected Outcome: An EBI competitive binding assay confirmed that compound 89 competitively displaces a known colchicine-site binder, validating its binding to the colchicine site [77].
Molecular docking studies further suggested that compound 89 binds selectively to the colchicine site, forming key interactions with tubulin residues that explain its high affinity [77].
The following diagram illustrates the multifaceted mechanism of action of compound 89, culminating in apoptosis.
In Vivo Efficacy and Safety
- Objective: Evaluate the antitumor efficacy and toxicity of compound 89 in animal models.
- Model: Mice bearing human tumor xenografts or patient-derived organoids.
- Dosing: Compound 89 administered at therapeutic doses (specific dose and route, e.g., oral or intraperitoneal, determined from prior pharmacokinetic studies).
- Endpoints: Tumor volume measurement, body weight change, and histological analysis of key organs for toxicity.
- Outcome: The study demonstrated significant anti-tumor efficacy in vivo with robust activity in patient-derived organoids. Crucially, no observable toxicity was noted at therapeutic doses, indicating a favorable safety profile [77].
Table 2: Summary of Key Experimental Findings for Compound 89
| Assay Type | Model System | Key Result | Significance |
|---|---|---|---|
| Tubulin Polymerization | Purified tubulin in vitro | Inhibited polymerization | Confirmed direct target engagement |
| Binding Assay | EBI competitive assay | Bound to colchicine site | Defined binding site, suggests mechanism |
| Cellular Viability | Human cancer cell lines | Low ICâ‚…â‚€ values | Demonstrated potent anti-proliferative effect |
| In Vivo Efficacy | Mouse xenograft models | Significant tumor growth inhibition | Confirmed efficacy in complex biological system |
| In Vivo Safety | Mice (therapeutic doses) No observable toxicity | Indicated a wide therapeutic window | |
| Organoid Model | Patient-derived organoids | Robust antitumor activity | Predicted clinical relevance and translation potential |
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and their applications in tubulin inhibitor discovery, as utilized in this case study and the broader field.
Table 3: Essential Research Reagents for Tubulin Inhibitor Discovery
| Reagent / Material | Function / Application | Example/Catalog Source |
|---|---|---|
| Purified Tubulin Protein | In vitro biochemical assays (polymerization, binding). | Porcine brain tubulin (Cytoskeleton, Inc.) |
| Colchicine, Paclitaxel | Reference controls for inhibition and stabilization. | Sigma-Aldrich |
| Fluorescent Colchicine Probes | Competitive binding assays to determine binding site. | DAPI-colchicine analogues |
| Cancer Cell Line Panel | In vitro cytotoxicity and mechanism studies. | e.g., HeLa, MCF-7, A549 (ATCC) |
| Patient-Derived Organoids | High-fidelity ex vivo tumor models for efficacy testing. | Internally generated or biobanks |
| Virtual Screening Compound Library | Source of chemical starting points for computational screening. | Specs Library, ZINC Database |
| Molecular Docking Software | Structure-based prediction of ligand binding. | Glide, AutoDock Vina, GOLD |
| Pharmacophore Modeling Software | Ligand-based design and screening. | Schrodinger Phase, MOE |
| Animal Xenograft Models | In vivo evaluation of antitumor efficacy and toxicity. | Mouse models (e.g., nude mice) |
| Pdnhv | Pdnhv, CAS:251362-87-5, MF:C47H68O11, MW:809 g/mol | Chemical Reagent |
| ApCp | ApCp Polysaccharide |
This case study exemplifies a modern, integrated pipeline for successful anticancer drug discovery. The strategic application of virtual screening enabled the efficient identification of a novel chemical scaffold—a nicotinic acid derivative—from a library of over 200,000 compounds. The subsequent rigorous target validation confirmed compound 89 as a potent tubulin inhibitor that binds the colchicine site, inhibits polymerization, and modulates the PI3K/Akt pathway. Its compelling efficacy in both cell-based and more complex in vivo and patient-derived organoid models, coupled with an absence of observed toxicity, underscores its potential as a candidate for advancing next-generation microtubule-targeted chemotherapies [77]. This end-to-end protocol, from in silico screening to in vivo validation, provides a robust framework for researchers aiming to accelerate the discovery of targeted cancer therapeutics.
The p21-activated kinase 2 (PAK2) is a serine/threonine kinase that plays a critical role in regulating cellular signaling pathways, cytoskeletal organization, cell motility, survival, and proliferation [81] [82]. As a member of the Group I PAK family, PAK2 serves as a crucial effector linking Rho GTPases to cytoskeleton reorganization and nuclear signaling, and its dysregulation has been implicated in various cancers and cardiovascular diseases [81] [82] [83]. Despite its promise as a drug target, the development of novel PAK2 inhibitors has proven challenging due to the labor-intensive nature and high costs of traditional drug discovery approaches [81].
Drug repurposing has emerged as a strategic alternative, offering to accelerate the identification of therapeutic agents by screening existing FDA-approved compounds against new biological targets [81]. This approach leverages existing safety and pharmacokinetic data, potentially reducing development timelines and costs. Within anticancer research, computational protocols for virtual screening have become indispensable tools for efficiently exploring large chemical spaces and identifying promising drug candidates [84] [85] [20]. This case study details the application of a systematic, structure-based drug repurposing strategy to identify FDA-approved drugs as potential PAK2 inhibitors, providing a comprehensive protocol within the broader context of computational methods for anticancer drug discovery.
Background and Significance
PAK2 Biology and Therapeutic Relevance
PAK2 is encoded by the PAK2 gene located on chromosome 3q29 in humans and shares structural similarities with other Group I PAKs, containing a p21-binding domain (PBD) and an auto-inhibitory domain (AID) that maintains the kinase in an inactive conformation [82]. Unlike other PAK family members, PAK2 can be activated through proteolytic cleavage by caspases during apoptosis, suggesting a role in regulating apoptotic events [82]. The kinase functions as a downstream effector of Rac or Cdc42 and participates in diverse cellular processes through phosphorylation of various substrates, including merlin, c-Jun, Caspase-7, Paxillin, and STAT5 [82] [83].
In cancer, PAK2 signaling modulates critical oncogenic pathways. Research has demonstrated that PAK2 activity maintains the c-MYC transcriptional program and, in specific mutational contexts such as FLT3-ITD and KIT D816V mutated cells, promotes STAT5 nuclear translocation and transcription of the anti-apoptotic protein BCL-XL [83]. Its involvement in these key survival and proliferation pathways positions PAK2 as an attractive therapeutic target for anticancer drug development.
Computational Drug Repurposing in Oncology
Traditional drug discovery is a time-consuming and expensive process, often requiring over 10 years and investments exceeding $1 billion to bring a new drug to market [20]. Computational drug discovery technologies have dramatically impacted cancer therapy development by providing efficient methods for lead compound identification and optimization [20]. Virtual screening, particularly structure-based molecular docking, has become a routine computational method in computer-aided drug design (CADD), enabling researchers to identify potentially highly active compounds from large ligand databases by evaluating binding affinities between receptors and ligands [85].
The recent explosion of chemical libraries beyond a billion molecules has necessitated more efficient virtual screening approaches [84]. Methods like Deep Docking (DD) enable up to 100-fold acceleration of structure-based virtual screening by docking only a subset of a chemical library iteratively synchronized with ligand-based prediction of remaining docking scores [84]. These advancements make computational repurposing of existing drug libraries particularly feasible and efficient for target-focused discovery campaigns.
Materials and Methods
Research Reagent Solutions
The following table details key reagents, software tools, and data resources essential for implementing the PAK2 inhibitor repurposing protocol.
Table 1: Essential Research Reagents and Computational Tools for PAK2 Virtual Screening
| Category | Specific Resource | Function/Application |
|---|---|---|
| Chemical Libraries | FDA-Approved Compound Library (3,648 compounds) [81] | Source of repurposing candidates with known safety profiles |
| Structural Data | PAK2 Protein Structure (PDB ID not specified in search results) | Target structure for molecular docking studies |
| Docking Software | AutoDock Vina [85] | Molecular docking to predict ligand-receptor binding |
| Molecular Dynamics | Desmond [32] or similar MD software | Simulation of protein-ligand complex stability (300 ns) |
| Visualization/Analysis | RDKit [84], Open Babel [84] | Cheminformatics analysis and molecule manipulation |
| Validation Tools | Molecular Dynamics Simulation (300 ns) [81] | Assessment of binding stability and interactions |
Computational Workflow Protocol
The integrated protocol for PAK2 inhibitor identification combines structure-based virtual screening with molecular dynamics validation, as detailed below.
Target Preparation
The three-dimensional structure of PAK2 was obtained from the Protein Data Bank. The protein structure was prepared by adding hydrogen atoms, assigning partial charges, and removing water molecules and co-crystallized ligands not directly involved in the active site [85]. The binding site was defined based on known catalytic residues and literature evidence of the PAK2 active site [81].
Compound Library Curation
A library of 3,648 FDA-approved compounds was compiled and prepared for virtual screening [81]. Ligand preparation included generating 3D structures, optimizing geometry, enumerating possible tautomers and stereoisomers, and assigning appropriate protonation states at physiological pH [84] [85]. The prepared compounds were stored in a searchable database format for efficient screening.
Virtual Screening Protocol
Structure-based virtual screening was performed using a molecular docking approach with the following detailed steps:
- Initial Docking: The entire prepared compound library was docked into the PAK2 active site using AutoDock Vina or similar docking software [81] [85]. Docking parameters included an exhaustiveness value of 8-16 to ensure adequate sampling of conformational space.
- Pose Analysis: For each compound, multiple binding poses were generated and scored based on predicted binding affinity (in kcal/mol). The top-scoring poses for each compound were retained for further analysis.
- Interaction Analysis: The binding modes of top-hit compounds were visually inspected using molecular visualization software. Specific attention was paid to hydrogen bonding interactions with key PAK2 residues, hydrophobic contacts, and other intermolecular interactions contributing to binding stability and specificity [81].
- Selectivity Assessment: Comparative docking against other PAK isoforms (PAK1 and PAK3) was performed to evaluate selectivity and preferential targeting of PAK2 [81].
The following diagram illustrates the complete virtual screening workflow:
Molecular Dynamics Validation
To validate the stability of predicted protein-ligand complexes and confirm binding modes observed in docking studies, molecular dynamics (MD) simulations were conducted [81] [32]. The protocol included:
- System Preparation: The top protein-ligand complexes were solvated in explicit water molecules in a triclinic box with periodic boundary conditions. Ions were added to neutralize system charge.
- Simulation Parameters: Simulations were performed for 300 ns using the OPLS-2005 force field in Desmond [81] [32]. Temperature was maintained at 300 K using a Nosé-Hoover thermostat, and pressure at 1 bar using a Martyna-Tobias-Klein barostat.
- Trajectory Analysis: The stability of complexes was assessed by calculating Root Mean Square Deviation (RMSD) of protein backbone and ligand atoms, Root Mean Square Fluctuation (RMSF) of protein residues, and hydrogen bonding patterns throughout the simulation trajectory [81] [32].
Results and Discussion
Identification of Top Hit Compounds
The systematic virtual screening of 3,648 FDA-approved compounds against PAK2 identified two top-hit candidates: Midostaurin and Bagrosin [81]. Both compounds demonstrated high predicted binding affinity and specificity to the PAK2 active site. Interaction analysis from molecular docking revealed that both compounds formed stable hydrogen bonds with key PAK2 residues, suggesting a potential inhibitory mechanism [81].
Table 2: Virtual Screening Results for Top PAK2 Hit Compounds
| Compound Name | Predicted Binding Affinity | Key Interactions | Selectivity Profile | Therapeutic Class |
|---|---|---|---|---|
| Midostaurin | High binding affinity to PAK2 active site [81] | Stable hydrogen bonds with key PAK2 residues [81] | Preferentially targets PAK2 over PAK1 and PAK3 [81] | Kinase inhibitor (FDA-approved for AML) |
| Bagrosin | High binding affinity to PAK2 active site [81] | Stable hydrogen bonds with key PAK2 residues [81] | Preferentially targets PAK2 over PAK1 and PAK3 [81] | Not specified in search results |
Validation of Binding Stability
Molecular dynamics simulations conducted for 300 ns demonstrated good thermodynamic properties for the stable binding of both Midostaurin and Bagrosin to PAK2 [81]. The RMSD values for both protein and ligands stabilized during the simulations, indicating complex stability. Hydrogen bond analysis confirmed the persistence of key interactions observed in the docking studies. The performance of both identified compounds was comparable to the control inhibitor IPA-3 in terms of binding stability [81].
PAK2 Signaling and Inhibitor Mechanism
PAK2 occupies a central position in multiple oncogenic signaling pathways. The diagram below illustrates key pathways regulated by PAK2 and the potential mechanism by which identified inhibitors disrupt these signaling cascades:
As illustrated, PAK2 inhibition potentially disrupts multiple downstream oncogenic processes: (1) reduction of c-MYC transcriptional activity and expression of ribosomal proteins; (2) inhibition of STAT5 phosphorylation at Tyr699, particularly relevant in FLT3-ITD mutated cells; and (3) subsequent downregulation of anti-apoptotic BCL-XL expression [83]. These multifaceted effects on critical cancer survival pathways underscore the therapeutic potential of effective PAK2 inhibitors.
Advantages of the Repurposing Approach
The identification of Midostaurin as a PAK2 inhibitor is particularly noteworthy as it is already FDA-approved for acute myeloid leukemia (AML), suggesting potential for rapid clinical translation for PAK2-dependent cancers [81]. The repurposing approach offers significant advantages over de novo drug discovery, including:
- Accelerated Timeline: By leveraging existing compounds with known safety profiles, the development timeline from target identification to clinical trials can be significantly reduced [81] [20].
- Reduced Costs: Utilizing compounds that have already passed Phase I safety trials eliminates substantial early-development costs [81].
- Established Manufacturing: Production processes for FDA-approved drugs are already optimized and scalable [81].
- Known Pharmacokinetics: Existing human pharmacokinetic data informs optimal dosing strategies for new indications [81].
This case study demonstrates the successful application of computational protocols for identifying repurposed PAK2 inhibitors through systematic virtual screening. The integration of molecular docking with molecular dynamics validation provides a robust framework for evaluating compound-target interactions in silico. The identification of Midostaurin and Bagrosin as potential PAK2 inhibitors highlights the value of drug repurposing strategies in anticancer drug discovery.
While the computational results are promising, the study represents only the initial phase of inhibitor development. Future work should focus on experimental validation of PAK2 inhibition by Midostaurin and Bagrosin using biochemical and cellular assays [81]. Additionally, structure-activity relationship studies could guide the optimization of these compounds for enhanced potency and selectivity against PAK2.
The integration of machine learning approaches, such as those successfully implemented for predicting response to PAK inhibitors in AML [83], could further refine patient selection and enable personalized therapeutic applications. As computational methods continue to advance, particularly with AI-enabled screening platforms like Deep Docking [84], the efficiency and scope of drug repurposing efforts will expand, accelerating the discovery of novel therapeutic applications for existing drugs.
Optimization Strategies: Overcoming Computational Challenges in Screening
Addressing Receptor Flexibility in Docking Simulations
The accurate prediction of how a small molecule ligand binds to its macromolecular target is a cornerstone of structure-based drug design, particularly in anticancer drug discovery. Conventional docking simulations often treat the protein receptor as a rigid body, a simplification that severely limits their predictive accuracy for many flexible targets. Induced fit effects, where the binding site conformation changes upon ligand binding, are a common phenomenon in biological systems [86]. For kinases—a prevalent class of anticancer targets—this flexibility is a defining characteristic, as they often switch between active and inactive states, a transition that can be exploited for designing selective inhibitors [87] [88].
Addressing receptor flexibility is therefore not merely an incremental improvement but a fundamental necessity for improving the success rate of virtual screening campaigns in oncology. This protocol outlines practical strategies and detailed methodologies for incorporating receptor flexibility into docking simulations, framed within the context of discovering new anticancer therapeutics.
Key Methodological Approaches for Incorporating Receptor Flexibility
Several computational strategies have been developed to manage receptor flexibility, each with distinct advantages, computational costs, and ideal use cases.
Table 1: Core Methodologies for Managing Receptor Flexibility in Docking
| Method | Key Principle | Advantages | Limitations | Representative Software |
|---|---|---|---|---|
| Ensemble Docking | Docking against a collection of discrete receptor conformations [89] [88]. | Captures large-scale backbone motions; computationally efficient after ensemble generation. | Quality depends on the diversity and relevance of the conformational ensemble. | AutoDock Suite [90], MedusaDock [91] |
| Flexible Sidechains | Specifying key binding site sidechains as flexible during the docking search [92]. | Models local induced fit at the binding site; more affordable than full flexibility. | Limited to sidechain motions; cannot model backbone shifts. | AutoDock4 [90] [92] |
| Full Backbone & Sidechain Flexibility | Modeling both backbone and sidechain movements during docking. | Most comprehensive flexibility model. | Extremely computationally intensive; challenging conformational search. | MedusaDock (with backbone ensemble) [91], FlexScreen [86] |
| Interactive Docking with Flexibility | User-guided docking in virtual reality with real-time flexibility modeling. | Leverages human intuition and expertise; immediate feedback. | Requires specialized VR hardware; not suited for high-throughput screening. | DockIT [93] |
The following workflow diagram provides a strategic decision pathway for selecting the most appropriate method based on the characteristics of the drug target and the project's goals.
Experimental Protocols
This section provides detailed, step-by-step protocols for implementing two of the most widely used flexibility methods in anticancer virtual screening.
Protocol 1: Ensemble Docking with Multiple Receptor Conformations
Ensemble docking involves screening ligands against a pre-generated set of receptor structures to account for backbone and large-scale sidechain movements [89]. This method is particularly effective for kinase targets like CDK2 or VEGFR2, which exhibit distinct active and inactive states [89].
Workflow Overview:
- Ensemble Generation: Collect multiple receptor conformations from experimental structures or molecular dynamics (MD) simulations.
- Structure Preparation: Prepare each conformation for docking.
- Parallel Docking: Dock the ligand library against each receptor conformation.
- Pose Integration & Ranking: Combine results and select the top poses.
Detailed Methodology:
- Construct the Conformational Ensemble:
- Source 1: Experimental Structures. Retrieve multiple PDB structures of the target protein, ideally in different conformational states (e.g., apo, holo, with different ligands). Select representative conformations using structural clustering based on backbone root-mean-square deviation (RMSD) of the binding site residues [89].
- Source 2: Molecular Dynamics (MD). Perform an explicit-solvent MD simulation of the apo receptor (e.g., 50-100 ns). Extract snapshots from the trajectory and cluster them to obtain a representative set of conformations that sample the protein's intrinsic dynamics [89] [88].
Prepare Receptor and Ligand Coordinates:
- For each receptor conformation, use a tool like AutoDockTools (ADT) to add polar hydrogen atoms and assign Gasteiger charges [90] [92]. Save the prepared structures in PDBQT format.
- Prepare the ligand library by generating 3D structures, assigning charges, and defining rotatable bonds. Convert all ligands to PDBQT format.
Perform Parallel Docking:
- Using a docking program like AutoDock Vina [90], dock the entire ligand library against each receptor conformation in the ensemble. Use a consistent grid box size and center for all runs to ensure the same binding site is targeted.
- The Vina command for a single conformation would be:
Integrate and Rank Results:
- Collect all docking poses and their scores from every run.
- Use a statistical approach, such as Naïve Bayesian classification, to integrate scores across the different receptor conformations. This method weights the contribution of each conformation based on its ability to discriminate known binders from non-binders, yielding a consensus ranking that often outperforms docking against any single rigid structure [89].
- Alternatively, a simple approach is to take the best score for each ligand across all ensembles.
Protocol 2: Docking with Selective Sidechain Flexibility
This method is ideal when the binding site is known and flexibility is primarily confined to a few key sidechains, such as those forming the "hinge region" in kinases or gating residues. This is implemented in AutoDock4 [92].
Workflow Overview:
- Identify Flexible Residues: Select sidechains expected to undergo motion upon ligand binding.
- Define Receptor Flexibility: Specify selected residues and their torsional degrees of freedom.
- Run Flexible Docking: Perform a docking search that optimizes both ligand pose and sidechain conformations.
- Analyze Results: Cluster and analyze the generated poses and sidechain rotamers.
Detailed Methodology:
- Identify Flexible Residues:
- Analyze MD simulations or a series of crystal structures to identify sidechains in the binding site that display high conformational variability.
- In the absence of such data, residues with long, polar sidechains (e.g., Arginine, Lysine, Glutamate) that line the binding pocket are common candidates.
Prepare the Receptor with Flexible Residues:
- Load the receptor structure into AutoDockTools.
- Use the "Flexible Residues" menu to select the target sidechains. ADT will automatically separate these residues from the rigid backbone and assign them torsional degrees of freedom.
- The output will be two files: a rigid receptor file (
.pdbqt) and a flexible residues file (.pdbqt).
Run the Docking Simulation:
- Configure the docking parameters in ADT, including the grid box size and location to encompass the binding site and the flexible residues.
- Use the Lamarckian Genetic Algorithm (LGA) in AutoDock4, which is efficient at searching the combined conformational space of the ligand and the flexible sidechains [92].
- A typical docking run will generate 50-100 poses, which should be clustered using a 2.0 Ã… RMSD tolerance to identify consensus binding modes.
Validation:
- Cross-docking validation: Test the protocol's ability to reproduce a known crystallographic pose of a ligand, especially one that induces a conformational change in the selected sidechains. A successful prediction will have a low ligand RMSD to the crystal structure and correctly recapitulate the sidechain orientations.
The Scientist's Toolkit: Essential Research Reagents & Software
Table 2: Key Software and Resources for Flexible Docking
| Tool / Resource | Type | Primary Function in Protocol | Application Notes |
|---|---|---|---|
| AutoDockTools [90] [92] | Graphical Interface | Prepares receptor/ligand PDBQT files; defines flexible residues and docking grid. | Essential for setup and analysis; automates batch processing for virtual screening. |
| AutoDock Vina [90] | Docking Program | Fast, turnkey docking for ensemble docking protocols. | Optimized for speed; uses a simple scoring function. Ideal for initial screening steps. |
| AutoDock4 [92] | Docking Program | Docking with selective receptor flexibility (flexible sidechains). | Platform for advanced methods; empirical free energy force field. |
| GROMACS [93] | MD Simulation Package | Generates conformational ensembles from MD trajectories. | Provides physically realistic receptor dynamics; computationally intensive. |
| Raccoon2 [90] | Virtual Screening GUI | Streamlines virtual screening workflow management, job distribution, and result analysis. | Manages large ligand libraries and multiple receptor targets efficiently. |
| RosettaVS [3] | Docking & VS Platform | High-precision docking and screening with receptor flexibility. | Open-source; combines physics-based scoring with active learning for ultra-large libraries. |
| DockIT [93] | Interactive VR Tool | Allows researchers to manipulate and dock ligands in real-time within a flexible receptor. | Useful for educational purposes and intuitive lead optimization; not for high-throughput. |
| IMR-1 | IMR-1, MF:C15H15NO5S2, MW:353.4 g/mol | Chemical Reagent | Bench Chemicals |
| CSC-6 | CSC-6, MF:C18H12F3NO2S2, MW:395.4 g/mol | Chemical Reagent | Bench Chemicals |
Performance Benchmarking and Quantitative Data
The integration of receptor flexibility consistently improves docking performance. The following table summarizes key quantitative evidence from benchmark studies.
Table 3: Performance Benchmarking of Flexible Docking Methods
| Method / Approach | Test System / Benchmark | Key Performance Metric | Result | Reference |
|---|---|---|---|---|
| MedusaDock with Backbone Ensemble | CSAR2011 Benchmark (35 diverse complexes) | Success Rate (Pose Prediction <2.5 Ã… RMSD) | 80% (28/35 cases) | [91] |
| Ensemble Docking (Naïve Bayesian) | Kinase Targets (ALK, CDK2, VEGFR2) | Virtual Screening Enrichment | Outperformed docking to any single rigid structure | [89] |
| AutoDock4 Flexible Sidechains | 87 HIV Protease Complexes (Cross-docking) | Redocking Accuracy | Improved accuracy when flexible sidechains (e.g., ARG8) were modeled. | [92] |
| RosettaVS (with flexibility) | CASF-2016 & DUD Benchmarks | Top 1% Enrichment Factor (EF1%) & Pose Prediction | EF1% = 16.72, outperforming other physics-based methods. | [3] |
The integration of receptor flexibility is a critical advancement that elevates computational docking from a simplistic modeling exercise to a more physiologically accurate tool in structure-based anticancer drug discovery. As demonstrated by benchmark studies, methods like ensemble docking and flexible sidechain modeling significantly improve the rate of successful pose prediction and the enrichment of true hits in virtual screening [91] [89].
The field continues to evolve with emerging trends such as the incorporation of AI and active learning to make flexible docking of ultra-large libraries feasible [3], and the use of interactive VR tools to leverage expert intuition in designing ligands for flexible targets [93]. By adopting the protocols outlined in this document, researchers can systematically address the challenge of protein flexibility, thereby increasing the likelihood of discovering novel and effective anticancer agents.
The advent of ultra-large chemical libraries, often referred to as "chemical spaces," represents a paradigm shift in early-stage anticancer drug discovery. These libraries contain billions to trillions of readily available, synthetically accessible compounds, offering unprecedented opportunities for identifying novel therapeutic agents [94] [95]. However, this expansion introduces significant computational scalability challenges, particularly when performing structure-based virtual screening with full receptor and ligand flexibility. Conventional virtual high-throughput screening (vHTS) methods become prohibitively expensive when applied to libraries of this magnitude [94]. This application note details specialized protocols and solutions designed to overcome these scalability barriers, enabling efficient exploration of ultra-large chemical spaces within the context of anticancer drug discovery research.
Ultra-large chemical spaces are constructed combinatorially from lists of available substrates and validated chemical reactions, rather than being fully enumerated. This approach generates astronomical numbers of virtual compounds while ensuring synthetic accessibility [95]. The table below summarizes key commercially available chemical spaces relevant to anticancer drug discovery.
Table 1: Key Commercial Ultra-Large Chemical Spaces for Anticancer Drug Discovery
| Space Name | Size (No. of Compounds) | Vendor/Partner | Key Traits | Accessibility |
|---|---|---|---|---|
| xREAL [95] | 4.4 trillion | Enamine Ltd. | Exclusive access via infiniSee; >80% synthesis success rate | Make-on-demand |
| eXplore [95] | 5 trillion | eMolecules | Drug- & lead-like compounds; "Do-it-yourself" or CRO synthesis | Make-on-demand |
| REAL Space [94] [95] | 82.97 billion | Enamine Ltd. | Drug-like properties; 172 in-house reactions | Make-on-demand |
| GalaXi [95] | 25.8 billion | WuXi LabNetwork | Rich in sp³ motifs; diverse scaffolds | Make-on-demand |
| Freedom Space [95] | 142 billion | Chemspace | ML-based filtering; >80% synthesis success rate | Make-on-demand |
| Synple Space [95] | 1 trillion | Synple Chem | Cartridge-based automated synthesis | Make-on-demand |
Core Computational Methodology: The Evolutionary Algorithm Solution
Algorithm Selection and Rationale
To address the computational intractability of exhaustive flexible docking on billion-compound libraries, we implemented RosettaEvolutionaryLigand (REvoLd), an evolutionary algorithm specifically designed for ultra-large combinatorial chemical spaces [94]. Unlike traditional vHTS that docks all library members, REvoLd treats the chemical space as a fitness landscape and evolves populations of molecules toward improved binding affinity against a specific cancer target. This meta-heuristic approach requires only a few thousand docking calculations to identify promising compounds, offering an efficiency improvement of 869 to 1622-fold over random screening [94].
REvoLd Experimental Protocol
Table 2: Key REvoLd Hyperparameters Optimized for Ultra-Large Library Screening [94]
| Parameter | Optimized Value | Functional Role | Impact on Screening |
|---|---|---|---|
| Population Size | 200 individuals | Maintains genetic diversity | Prevents premature convergence |
| Generations | 30 | Optimization duration | Balances exploration vs. exploitation |
| Selection Rate | 25% (50 individuals) | Determines who reproduces | Controls selection pressure |
| Crossover Rate | Increased | Combines promising solutions | Enhances structural recombination |
| Mutation Rate | Multiple steps introduced | Introduces novel variations | Promotes exploration of chemical space |
Detailed Protocol Workflow:
Initialization: Generate a random starting population of 200 molecules from the combinatorial library, ensuring they are synthetically accessible based on available building blocks and reaction rules [94].
Docking and Fitness Evaluation: Perform flexible protein-ligand docking using RosettaLigand against the specific anticancer target (e.g., kinase, protease). Use the resulting binding energy as the fitness score for each molecule [94].
Selection: Apply tournament selection to identify the top 50 molecules (25% of population) based on docking scores for reproduction [94].
Reproduction with Crossover and Mutation:
- Crossover: Recombine pairs of high-fitness molecules by exchanging molecular fragments at compatible sites to create offspring [94].
- Fragment Replacement Mutation: Switch single fragments in promising molecules with low-similarity alternatives from the building block library [94].
- Reaction Switching Mutation: Change the core reaction scheme of a molecule while searching for similar fragments compatible with the new reaction [94].
Second Optimization Round: Implement an additional crossover and mutation cycle excluding the very fittest molecules to allow less optimal ligands to contribute valuable genetic material [94].
Generational Advancement: Combine parents and offspring, select the best 200 individuals for the next generation, and repeat the process for 30 generations [94].
Hit Identification and Validation: After multiple independent runs (recommended: 20 runs), select top-ranking compounds for experimental validation in anticancer assays [94].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Ultra-Large Library Screening
| Reagent/Resource | Function/Purpose | Example Sources/Identifiers |
|---|---|---|
| Chemical Building Blocks | Core substrates for combinatorial library construction | Enamine, WuXi, Ambinter building block collections [95] |
| Validated Reaction Rules | SMARTS patterns defining chemically feasible compound generation | eXplore Cookbook; 185+ curated reactions in GalaXi [95] |
| Target Protein Structures | High-resolution structures for structure-based docking | RCSB PDB; cancer-related targets (kinases, etc.) [50] |
| Docking Software | Flexible protein-ligand docking platform | RosettaLigand within Rosetta software suite [94] |
| Resource Identifiers | Unique identification of key biological resources | Antibody Registry; Addgene (plasmids); Resource Identification Portal [96] |
| VE607 | VE607|SARS-CoV-2 Inhibitor|For Research Use | VE607 is a small molecule inhibitor that blocks SARS-CoV-2 viral entry by stabilizing the Spike RBD. This product is for Research Use Only. |
| SAV13 | SAV13, MF:C19H13Cl2FN2O4, MW:423.2 g/mol | Chemical Reagent |
Workflow and Pathway Visualizations
REvoLd Screening Workflow
Chemical Space Structure
Implementation Considerations for Anticancer Research
Successful implementation of these protocols requires careful consideration of several factors specific to anticancer drug discovery:
- Target Selection: Prioritize cancer targets with well-characterized active sites and available high-resolution structures to maximize docking reliability [50].
- Chemical Space Selection: Diversify screening across multiple chemical spaces (e.g., REAL, GalaXi, eXplore) as their building blocks and reaction knowledge differ significantly, increasing the probability of identifying novel scaffolds [95].
- Protocol Adaptation: The evolutionary parameters (population size, mutation rates) may require optimization for specific target classes, particularly for challenging anticancer targets like protein-protein interactions [94].
- Experimental Validation: Always plan for synthesis and biological testing of top-ranking virtual hits to confirm anticancer activity, as in silico predictions require experimental validation [94] [50].
These scalability solutions enable research teams to leverage ultra-large chemical libraries effectively, transforming them from computational burdens into valuable resources for discovering novel anticancer therapeutics.
Balancing Computational Speed vs. Accuracy in Screening Workflows
In the field of anticancer drug discovery, virtual screening has become an indispensable computational technique for rapidly identifying promising candidate molecules that can interact with specific cancer-related biological targets. The core challenge in this process lies in balancing the computational speed of the screening workflow against the accuracy of its predictions. This speed-accuracy tradeoff (SAT) is a fundamental phenomenon documented across computational and decision-making systems, where adjustments to prioritize one factor inevitably impact the other [97]. For research teams working against the clock to discover new oncology therapeutics, strategically managing this tradeoff can significantly impact project timelines and resource allocation.
The underlying mechanism of SAT can be conceptually understood through the threshold hypothesis, which postulates that SAT results from adjustments to the decision threshold [97]. In practical terms for virtual screening, this means that when computational speed is prioritized, the decision threshold for identifying a "hit" is lowered, enabling faster screening based on less accumulated evidence. Conversely, when accuracy is prioritized, this threshold is raised, requiring more evidence accumulation at the expense of increased computational time and resources [97]. Understanding and strategically manipulating this balance is crucial for designing efficient screening pipelines in anticancer drug discovery.
Quantitative Landscape of Speed-Accuracy Tradeoffs
The relationship between computational speed and accuracy manifests differently across various screening approaches and parameters. The following tables summarize key quantitative relationships observed in computational screening workflows.
Table 1: Impact of Screening Parameters on Speed-Accuracy Balance
| Screening Parameter | Impact on Speed | Impact on Accuracy | Typical Use Case |
|---|---|---|---|
| High-Throughput Docking | Very Fast | Low to Moderate | Initial screening of large libraries (>1 million compounds) |
| Standard Precision Docking | Moderate | Moderate | Intermediate screening of focused libraries (100,000-1 million compounds) |
| High Precision Docking | Slow | High | Final evaluation of top candidates (<100,000 compounds) |
| Coarse-Grained Scoring | Fast | Lower | Rapid filtering and clustering |
| Multi-Parameter Scoring | Slower | Higher | Prioritization for experimental validation |
| Limited Conformational Sampling | Faster | Reduced | Initial binding pose estimation |
| Extended Conformational Sampling | Slower | Improved | Refined binding affinity predictions |
Table 2: Comparative Performance of Screening Architectures
| Screening Architecture | Relative Speed | Relative Accuracy | Computational Demand |
|---|---|---|---|
| Ligand-Based Similarity | Fastest | Low to Moderate | Low |
| Pharmacophore Screening | Fast | Moderate | Low to Moderate |
| Rigid Receptor Docking | Moderate | Moderate | Moderate |
| Flexible Side-Chain Docking | Slow | High | High |
| Full Flexible Docking | Slowest | Highest | Very High |
Protocol for Automated Virtual Screening Pipeline
This protocol outlines a standardized workflow for structure-based virtual screening, balancing speed and accuracy through a multi-stage approach [98].
Stage 1: Library Generation and Preparation
Objective: Create a structured, screening-ready compound library with appropriate molecular diversity.
Materials and Reagents:
- Compound databases (e.g., ZINC, ChEMBL, in-house collections)
- Chemical structure standardization tools
- Molecular descriptor calculation software
- High-performance computing cluster or cloud resources
Methodology:
- Library Curation
- Source compounds from commercial, public, or proprietary databases
- Apply drug-like filters (e.g., Lipinski's Rule of Five for oral availability)
- Remove compounds with undesirable chemical properties or reactive functional groups
- Standardize chemical representations (tautomer, ionization state)
Library Enumeration
- Generate physiologically relevant tautomers and protonation states
- Create 3D conformers using rule-based or knowledge-based methods
- Optimize geometries using molecular mechanics force fields
- Output in standardized formats (SDF, MOL2) for docking
Library Profiling
- Calculate molecular descriptors (MW, logP, TPSA, hydrogen bond donors/acceptors)
- Perform diversity analysis to ensure chemical space coverage
- Cluster compounds to enable representative subset screening if needed
Stage 2: Receptor and Grid Preparation
Objective: Prepare the target protein structure and define the binding site for docking calculations.
Materials and Reagents:
- Protein Data Bank (PDB) structures or homology models
- Molecular visualization software (PyMOL, Chimera)
- Protein preparation tools (Schrödinger Protein Preparation Wizard, MOE)
- Grid generation software (AutoDock Tools, UCSF DOCK)
Methodology:
- Protein Structure Preparation
- Select high-resolution crystal structure (preferably <2.5 Ã…) of target protein
- Add missing side chains and loop regions using modeling tools
- Assign appropriate protonation states for residues in binding site
- Optimize hydrogen bonding network
- Perform energy minimization to relieve steric clashes
Binding Site Definition
- Identify binding site from co-crystallized ligand or literature data
- Define grid box dimensions to encompass entire binding site
- Set grid spacing (0.2-0.5 Ã…) balancing precision and computational cost
- Include key water molecules if evidence supports their structural role
Grid Parameter Optimization
- Validate grid parameters with known binders and decoys
- Adjust box size to minimize unnecessary computation while ensuring coverage
- Save grid parameter file for docking simulations
Stage 3: Multi-Level Docking and Evaluation
Objective: Execute a tiered docking approach to efficiently identify high-affinity binders.
Materials and Reagents:
- Docking software (AutoDock Vina, Glide, GOLD)
- Scoring functions (empirical, force-field based, knowledge-based)
- High-performance computing resources with CPU/GPU capabilities
- Result analysis and visualization tools
Methodology:
- High-Throughput Docking (Speed-Optimized)
- Use fast docking algorithms with simplified scoring
- Employ rigid receptor and flexible ligand approximations
- Set moderate conformational sampling parameters
- Execute parallel docking jobs on computing cluster
- Apply initial cutoff based on docking score to reduce candidate pool
Standard Precision Docking (Balanced Approach)
- Use more sophisticated scoring functions
- Include limited receptor flexibility if computationally feasible
- Increase conformational sampling for top candidates
- Re-dock compounds passing initial cutoff with higher precision
- Apply more stringent scoring cutoff for candidate selection
High Precision Docking (Accuracy-Optimized)
- Use most accurate docking protocols available
- Include explicit water molecules in binding site if appropriate
- Employ molecular mechanics-based refinement for top candidates
- Perform binding pose clustering and analysis
- Generate final ranked list of candidates for experimental validation
Stage 4: Result Ranking and Analysis
Objective: Systematically analyze and prioritize docking hits for further investigation.
Materials and Reagents:
- Data analysis frameworks (Python/R scripts, KNIME, Pipeline Pilot)
- Visualization tools (LibMols, Spotfire, Tableau)
- Structural analysis software (Schrödinger, MOE, PyMOL)
Methodology:
- Multi-Parameter Ranking
- Combine docking scores with additional filters (ADMET properties, synthetic accessibility)
- Apply consensus scoring from multiple scoring functions if available
- Rank compounds by integrated score incorporating multiple parameters
Binding Mode Analysis
- Visually inspect top scoring poses for key interactions
- Verify formation of crucial hydrogen bonds, hydrophobic contacts, etc.
- Check for reasonable binding geometries and intermolecular complementarity
- Identify potential false positives from strained conformations
Hit List Finalization
- Select diverse chemotypes to mitigate scaffold-specific risks
- Apply final filters based on project-specific criteria
- Generate report with structures, scores, and interaction diagrams
- Transfer top candidates to experimental validation pipeline
Workflow Visualization
Diagram 1: Tiered virtual screening workflow with progressive focus on accuracy.
Diagram 2: Decision points for speed versus accuracy optimization.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Computational Tools for Virtual Screening Workflows
| Tool Category | Specific Solutions | Primary Function | SAT Consideration |
|---|---|---|---|
| Compound Libraries | ZINC, ChEMBL, DrugBank | Source of screening compounds | Larger libraries increase accuracy potential but reduce speed |
| Docking Software | AutoDock Vina, Glide, GOLD | Molecular docking simulations | Precision settings directly control speed-accuracy balance |
| Scoring Functions | Empirical, Force-Field, Knowledge-Based | Ranking binding affinity | Multiple functions increase accuracy at computational cost |
| MD Simulation | GROMACS, AMBER, NAMD | Molecular dynamics analysis | Provides high accuracy but extremely computationally demanding |
| Scripting Frameworks | Python, R, KNIME | Workflow automation | Customization allows precise SAT optimization |
| Visualization Tools | PyMOL, Chimera, Schrodinger | Structural analysis | Essential for manual verification of automated results |
| HPC Infrastructure | CPU Clusters, GPU Accelerators | Computational resources | More resources enable higher accuracy within practical timeframes |
| IQ-1 | IQ-1, MF:C21H22N4O2, MW:362.4 g/mol | Chemical Reagent | Bench Chemicals |
| DSTMS | DSTMS, CAS:945036-56-6, MF:C25H30N2O3S, MW:438.6 g/mol | Chemical Reagent | Bench Chemicals |
The effective balancing of computational speed and accuracy in anticancer drug discovery screening workflows requires thoughtful consideration of the specific research context. For initial stages of discovery where broad chemical space exploration is valuable, speed-optimized approaches enable efficient triaging of large compound libraries. As the focus narrows to lead optimization, accuracy-optimized protocols become essential for reliable prediction of binding affinities and interactions.
Implementation of the tiered screening protocol outlined in this document allows research teams to dynamically adjust the speed-accuracy tradeoff according to project needs. By employing a multi-stage approach that progresses from high-throughput methods to high-precision validation, researchers can maximize the efficiency of computational resources while maintaining scientific rigor in the identification of promising anticancer compounds.
Validating Force Fields and Scoring Functions for Cancer Targets
The accurate prediction of how potential drug molecules interact with cancer-related protein targets is a cornerstone of modern computational drug discovery. This process relies heavily on two fundamental components: force fields that describe the physical forces between atoms, and scoring functions that predict binding affinity [99] [100]. The validation of these computational tools against experimentally characterized cancer targets ensures their predictive reliability for identifying and optimizing novel anticancer agents. This protocol details the methodologies for rigorously validating force fields and scoring functions, specifically within the context of virtual screening campaigns for anticancer drug discovery. The procedures are designed to be integrated into a broader computational workflow, contributing to the development of more effective and targeted cancer therapies.
Theoretical Background
Force Fields in Molecular Simulations
In the context of chemistry and molecular modeling, a force field is a computational model that describes the potential energy of a system of atoms and molecules [100]. The basic functional form for molecular systems is typically decomposed into bonded and non-bonded interactions:
E_total = E_bonded + E_nonbonded
Where:
E_bonded = E_bond + E_angle + E_dihedralE_nonbonded = E_electrostatic + E_van der Waals[100]
Force fields are categorized based on their granularity: all-atom force fields provide parameters for every atom, including hydrogen; united-atom potentials treat hydrogen and carbon atoms in methyl and methylene groups as single interaction centers; and coarse-grained potentials sacrifice chemical details for computational efficiency in simulating large macromolecules [100]. The parameters for these energy functions are derived from laboratory experiments, quantum mechanical calculations, or both, and are stored in force field databases such as openKim, TraPPE, and MolMod [100].
Scoring Functions in Molecular Docking
Scoring functions are algorithms used to predict the binding affinity of a ligand to a protein target, which is crucial for ranking compounds in virtual screening [101] [102]. They are broadly classified into three categories:
- Force-Field Based Scoring Functions: Utilize energy terms from molecular mechanics force fields, often combined with implicit solvation models like Poisson-Boltzmann/Surface Area (PB/SA) or Generalized-Born/Surface Area (GB/SA) to estimate binding free energies [102].
- Empirical Scoring Functions: Decompose the binding free energy into weighted empirical terms (e.g., hydrogen bonding, hydrophobic effects), with coefficients derived from linear regression against experimental binding affinity data [102] [103].
- Knowledge-Based Scoring Functions: Derived by converting the statistical frequencies of interatomic pairs observed in structural databases of protein-ligand complexes into pseudo-energy potentials using inverse Boltzmann relationships or more advanced iterative methods [102].
Relevance to Cancer Drug Discovery
Computational methods have a significant impact on anticancer drug design by reducing the time and cost associated with traditional drug development [99]. The validation of these tools is particularly critical for cancer targets, many of which, such as protein kinases, transcription factors, and RAS family members, have been historically classified as "undruggable" due to the lack of well-defined active sites [99] [104]. Successful examples of targeted cancer therapies, such as imatinib (Bcr-Abl inhibitor) and trastuzumab (HER2 inhibitor), underscore the importance of precise molecular recognition, which begins with accurate computational predictions [104].
Experimental Protocols
Validation of Scoring Functions using the CSAR Benchmark
Principle: This protocol uses a standardized, high-quality benchmark to assess the accuracy of a scoring function in predicting binding affinities and poses. The Community Structure-Activity Resource (CSAR) benchmark is a curated set of diverse protein-ligand complexes with reliable experimental binding constants (Kd) and high-resolution crystal structures [102].
Table 1: Key Components of the CSAR Benchmark for Validation
| Component | Description | Significance in Validation |
|---|---|---|
| Complex Diversity | 345 protein-ligand complexes | Tests general applicability across different target classes. |
| Data Quality | Experimentally determined Kd values and high-resolution X-ray structures. | Minimizes introduction of experimental errors into validation. |
| Ligand Properties | Drug-like, non-covalently bound molecules. | Ensures relevance to real-world drug discovery. |
Procedure:
- Data Preparation: Download the CSAR benchmark set (http://www.csardock.org/). Prepare the structures by removing water molecules and adding hydrogen atoms. Ensure all atomic assignments are consistent with electron density maps.
- Binding Affinity Prediction: For each complex in the benchmark, use the scoring function to calculate a score. For knowledge-based functions like ITScore, the total energy is calculated by summing all protein-ligand atomic pairs:
E_ITScore = Σ u_ij(r)[102]. - Pose Prediction (Optional): Re-dock the native ligand into the protein binding site. Assess the scoring function's ability to identify the native crystal structure pose as the lowest-energy conformation.
- Correlation Analysis: Calculate the Pearson correlation coefficient (R²) between the predicted scores and the experimental log(Kd) values across all 345 complexes. A higher R² indicates better predictive power. For reference, the knowledge-based scoring function ITScore 2.0 achieved an R² of 0.54 on this benchmark [102].
- Comparative Analysis: Compare the performance of your scoring function against established benchmarks like the VDW scoring function, force field scoring (e.g., from DOCK), and other empirical functions [102].
Validation of Force Fields using Free Energy Calculations
Principle: This protocol validates a force field by assessing its accuracy in predicting absolute binding free energies (ΔG) using advanced sampling methods, such as the MovableType (MT) algorithm, which offers a balance between computational speed and accuracy [105].
Table 2: Research Reagent Solutions for Free Energy Validation
| Reagent / Resource | Function / Description | Application in Protocol |
|---|---|---|
| MovableType Software | A software package using numerical integration to estimate atomic partition functions and molecular free energy. | Core engine for performing free energy calculations [105]. |
| CASF-2016 Benchmark | Industry-standard set containing 57 protein targets and 285 ligands. | Validates robustness across a broad range of protein classes [105]. |
| PDBBind Database | A curated database of protein-ligand complexes with binding affinity data. | Provides structures and experimental Kd/IC50 values for validation [105]. |
| Protein Data Bank (PDB) | Repository for 3D structural data of proteins and nucleic acids. | Source of high-resolution input structures for the calculations [105]. |
Procedure:
- System Setup: Obtain high-resolution X-ray structures of protein-ligand complexes from the PDBBind database or the CASF-2016 benchmark. Prepare the structures by adding hydrogen atoms and assigning protonation states relevant to physiological pH.
- Parameter Assignment: Assign force field parameters (e.g., AMBER, CHARMM) to the protein and ligands. For the MT method, this includes defining atom types and partial charges.
- MovableType Calculation: Execute the MT free energy calculation. The method approximates the molecular partition function as a product of atomic partition functions. It samples atom-pair Boltzmann factors within a defined range of motion from the initial coordinates to generate an energy-state ensemble [105].
- Result Analysis: The output is a predicted binding free energy (ΔGpred) in kcal/mol. Compare this value to the experimental binding free energy, derived from the experimental Kd using the relationship: ΔGexp = -RT ln(Kd).
- Performance Metrics: Calculate the root-mean-square error (RMSE), Pearson correlation (R²), and mean unsigned error (MUE) between the predicted and experimental ΔG values across the entire test set (e.g., the 285 complexes in CASF-2016). A lower RMSE and higher R² indicate a more accurate force field.
The following diagram illustrates the logical workflow and key decision points for the validation protocols described above:
Data Analysis and Interpretation
Quantitative Performance Metrics
The performance of force fields and scoring functions should be evaluated using standardized quantitative metrics. The following table summarizes the expected performance ranges based on published validation studies:
Table 3: Expected Performance Metrics from Established Benchmarks
| Computational Tool | Benchmark Used | Key Performance Metric | Reported Value |
|---|---|---|---|
| ITScore 2.0\n(Knowledge-Based Scoring Function) | CSAR (345 complexes) | Pearson Correlation (R²) for binding affinity | 0.54 [102] |
| MovableType (MT)\n(Free Energy Method) | CASF-2016 (285 complexes) | RMSE for binding free energy (ΔG) | Comparable to or better than other methods (exact value not reported) [105] |
| Drug Sensitivity Score (DSS3)\n(Cell-based Scoring) | Primary AML patient cells | Accuracy in clustering drugs by Mechanism of Action (MoA) | Systematically improved vs. IC50 or Activity Area (p < 0.0005) [106] |
Application to Cancer Target Workflow
Validated computational tools must be integrated into a practical workflow for anticancer drug discovery. The flow chart below outlines this process, from initial target selection to the final experimental validation of computational predictions.
Troubleshooting
- Poor Correlation with Experimental Affinities: If the R² value from the CSAR benchmark is low, verify the preparation of the protein-ligand complexes. Check for correct protonation states and consider using a different class of scoring function (e.g., empirical instead of knowledge-based) or re-parameterizing the force field for specific chemical motifs common in your cancer targets [102] [103].
- Inability to Reproduce Native Poses: If the scoring function fails to identify the crystal ligand pose as the lowest-energy state, investigate the handling of solvent effects and explicit hydrogen bonding. Methods that incorporate explicit hydration or improved hydrogen bond potentials can significantly improve pose prediction accuracy [107] [102].
- High Computational Cost: For free energy methods, if the MT protocol is too slow for your screening pipeline, reduce the number of energy states sampled or explore the use of rigid-receptor approximations, which are faster and have been shown to maintain robust predictability for many systems [105].
The rigorous validation of force fields and scoring functions against standardized benchmarks is a critical prerequisite for their successful application in anticancer drug discovery. The protocols outlined here for benchmarking binding affinity prediction (using CSAR) and free energy estimation (using MovableType) provide a robust framework for assessing computational tools. By integrating these validated methods into a structured virtual screening workflow, researchers can enhance the predictive accuracy of their computational models, thereby increasing the likelihood of identifying novel, effective, and selective anticancer therapeutics.
Multi-Stage Hybrid Screening Approaches for Efficiency
Within the broader context of computational protocols for anticancer drug discovery, Multi-Stage Hybrid Virtual Screening (VS) represents a powerful strategy for efficiently identifying novel therapeutic candidates from ultra-large chemical libraries. Conventional single-stage VS methods often struggle to balance computational expense with comprehensive coverage, particularly as publicly accessible compound libraries now contain hundreds of millions to billions of synthesizable molecules [108] [51]. The hybrid approach addresses this challenge by integrating multiple computational techniques—typically combining fast ligand-based filtering with more computationally intensive structure-based methods—in a sequential workflow that progressively enriches for promising candidates while rapidly eliminating unsuitable compounds [109] [8]. This protocol is particularly valuable in anticancer drug discovery, where researchers must identify potent cytotoxic payloads with specific target interactions and favorable drug-like properties from extraordinarily large chemical spaces [108] [11].
Quantitative Outcomes of Multi-Stage Screening
The efficiency gains achieved through multi-stage hybrid screening are demonstrated by the progressive enrichment of compound libraries at each filtration stage. The following table summarizes the results from a large-scale case study targeting microtubule inhibitors:
Table 1: Library Reduction Through Sequential Screening Stages
| Screening Stage | Compounds Remaining | Reduction Rate | Key Criteria Applied |
|---|---|---|---|
| Initial Compound Library | ~900 million | - | Collected from ZINC12, ChEMBL, PubChem, QM9 [108] |
| Drug-like Property Filter | 90 million | 90% | Lipinski's Rule of Five [109] |
| Fragment-based Similarity Screening | 150,000 (threshold 0.4) to 12,915 (threshold 0.6) | 99.8%+ | Tanimoto similarity >0.4-0.6 to approved anticancer drugs [108] [109] |
| Molecular Docking | 1,000 | 93-99% | Docking score with β-tubulin [108] |
| ADMET & Synthetic Validation | 5-20 | 95-99% | Absorption, distribution, metabolism, excretion, toxicity & synthetic accessibility [108] [109] |
This sequential refinement demonstrates how multi-stage screening can efficiently distill billions of initial compounds down to a manageable number of high-priority candidates for experimental validation, achieving a final enrichment factor exceeding 99.999% [108] [109].
Experimental Protocol for Multi-Stage Hybrid Screening
Stage 1: Library Preparation and Drug-Like Property Filtering
Purpose: To assemble a comprehensive compound library and remove molecules with poor drug-like properties.
Methodology:
- Compound Collection: Download structures from multiple databases including ZINC12, ChEMBL, PubChem, and QM9 to create an initial library of approximately 900 million molecules [108] [109].
- Data Standardization: Standardize molecular structures, normalize representations, and remove duplicates using tools such as RDKit [109].
- Drug-like Filtering: Apply Lipinski's Rule of Five (RO5) criteria:
- Output: A refined library of drug-like compounds (approximately 90 million molecules) for subsequent screening stages.
Stage 2: Ligand-Based Similarity Screening
Purpose: To identify compounds structurally similar to known active molecules.
Methodology:
- Reference Set Preparation: Compile a set of 220-312 approved small-molecule anticancer drugs from sources such as the Anticancer Fund database [109].
- Fragment Generation: Deconstruct reference drugs into molecular fragments to capture essential pharmacophoric features [108].
- Similarity Calculation: Compute structural similarity between database compounds and reference active compounds using the Tanimoto coefficient and fingerprint-based methods (e.g., RDKit) [109].
- Threshold Application: Retain compounds exceeding similarity thresholds (typically 0.4-0.6) to reference actives [108] [109].
- Output: A subset of 12,915 to 150,000 compounds with structural similarities to known anticancer drugs.
Stage 3: Structure-Based Virtual Screening
Purpose: To evaluate filtered compounds based on predicted binding affinity to the biological target.
Methodology:
- Target Preparation: Obtain a three-dimensional structure of the target protein (e.g., β-tubulin for microtubule inhibitors) from the Protein Data Bank. Prepare the structure by adding hydrogen atoms, assigning partial charges, and defining binding site coordinates [108] [110].
- Molecular Docking: Perform docking simulations using software such as AutoDock Vina to predict binding poses and scores for each compound [108] [110].
- Pose Analysis: Examine predicted binding modes to ensure meaningful molecular interactions.
- Ranking and Selection: Rank compounds based on docking scores and select the top 1,000 candidates for further evaluation [108].
Stage 4: ADMET Prediction and Synthetic Accessibility
Purpose: To prioritize compounds with favorable pharmacological profiles and feasible synthesis pathways.
Methodology:
- ADMET Profiling: Predict key absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties using tools such as ADMET modeling approaches [108] [11].
- Synthetic Accessibility Assessment: Evaluate the feasibility of chemical synthesis using retrosynthetic analysis tools or scoring functions [108].
- Final Selection: Apply stringent filters to select the 5-20 most promising candidates for experimental validation [108] [109].
Workflow Visualization
Diagram 1: Multi-Stage Hybrid Screening Workflow. This diagram illustrates the sequential filtration process, showing the dramatic reduction in compound numbers at each stage while progressively applying more computationally intensive methods.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Computational Tools and Databases for Multi-Stage Screening
| Category | Tool/Database | Specific Function | Application in Protocol |
|---|---|---|---|
| Compound Databases | ZINC12, ChEMBL, PubChem | Source of screening compounds | Provide initial molecular libraries (~900M compounds) [108] [109] |
| Cheminformatics | RDKit | Chemical informatics and fingerprinting | Calculate Tanimoto similarity, generate fragments [109] |
| Molecular Docking | AutoDock Vina | Protein-ligand docking and scoring | Predict binding affinity to target (e.g., β-tubulin) [108] [110] |
| Structure Analysis | Pharmit | Pharmacophore modeling and screening | Create 3D pharmacophore queries from receptor-ligand structures [110] |
| Dynamics Validation | GROMACS | Molecular dynamics simulations | Confirm structural stability over 100-200 ns simulations [108] [110] |
| ADMET Prediction | ADMET Modeling Tools | Property prediction | Evaluate drug-like properties and toxicity profiles [108] [11] |
Case Study: Selective PARP-1 Inhibitor Identification
A recent study demonstrates the application of this multi-stage approach to identify selective PARP-1 inhibitors [110]. The researchers began with a library of nearly 450,000 phthalimide-containing compounds and applied this optimized workflow:
Stage 1: Generated a 3D pharmacophore model based on essential interactions of a known selective PARP-1 inhibitor (compound IV) using the Pharmit web server, resulting in 165 compounds that matched the pharmacophore features [110].
Stage 2: Performed molecular docking of the 165 compounds into the active sites of both PARP-1 and PARP-2 using AutoDock Vina, identifying 5 compounds with better docking scores than the reference compound and potential selectivity for PARP-1 over PARP-2 [110].
Stage 3: Conducted molecular dynamics simulations over 200 ns using GROMACS software to confirm the structural stability and binding modes of the top candidate (MWGS-1), demonstrating its higher affinity and selectivity for PARP-1 compared to PARP-2 [110].
This case study exemplifies how the multi-stage approach successfully identified a selective inhibitor while minimizing false positives that can occur with single-method screening approaches.
Pathway Visualization of Computational Screening Strategy
Diagram 2: Hybrid Screening Strategy Integration. This diagram shows how multi-stage approaches combine the complementary strengths of different computational methods to achieve both efficiency and accuracy in anticancer drug discovery.
Multi-stage hybrid virtual screening represents a sophisticated computational protocol that dramatically enhances the efficiency of anticancer drug discovery. By strategically combining ligand-based and structure-based methods in a sequential workflow, researchers can effectively navigate ultra-large chemical spaces exceeding hundreds of millions of compounds to identify promising therapeutic candidates. The documented success of this approach in discovering microtubule inhibitors and selective PARP-1 inhibitors validates its utility in the broader context of computational protocols for anticancer drug discovery [108] [110]. As chemical libraries continue to expand and computational methods evolve, these multi-stage hybrid approaches will become increasingly essential for leveraging the full potential of virtual screening in oncology drug development.
Active Learning and AI-Triaged Screening Methods
The discovery of anticancer therapeutics is increasingly leveraging artificial intelligence (AI) to navigate the immense scale of available chemical space. Traditional high-throughput screening (HTS), while instrumental in identifying active compounds, is often hampered by high costs, low success rates, and extensive resource requirements [111]. AI-triaged screening, particularly methods incorporating active learning, represents a transformative approach. These methods use machine learning models to iteratively select the most promising compounds for evaluation, dramatically reducing the number of molecules that require expensive experimental or computational testing [112] [113]. Within anticancer research, this enables the rapid identification of novel chemotypes targeting key enzymes upregulated in cancers, such as AKR1C3 in prostate and breast cancers, and Src kinase across various human cancers [114] [115].
The core principle of active learning is its iterative, closed-loop workflow. A surrogate machine learning model is initially trained on a small subset of data. It then prioritizes compounds from a large library for the next round of evaluation (e.g., docking or biochemical assays). The results from this evaluation are used to retrain and refine the model, which then selects the next batch of candidates. This cycle significantly improves sample efficiency, allowing researchers to identify a majority of top-hit candidates after screening only a tiny fraction of an ultra-large library [112] [113].
Quantitative Performance of AI-Screening Methods
The integration of AI into virtual screening has yielded substantial quantitative improvements in efficiency and accuracy compared to traditional methods. The following tables summarize key performance metrics from recent studies and platforms.
Table 1: Benchmark Performance of AI-Triaged Screening in Virtual Screening
| Method / Platform | Key Innovation | Library Size | Performance Highlight | Reference |
|---|---|---|---|---|
| Pretrained Transformer/GNN | Bayesian Optimization & Active Learning | 99.5 million compounds | Identified 58.97% of top-50,000 hits after screening only 0.6% of the library (8% improvement over previous baseline) | [112] [113] |
| OpenVS (RosettaVS) | Physics-based forcefield (RosettaGenFF-VS) with active learning | Multi-billion compounds | Achieved an enrichment factor (EF1%) of 16.72 on CASF-2016, outperforming the second-best method (EF1%=11.9) | [57] |
| AMLSF | Active learning for negative molecular selection | DUD-E dataset | Significantly increased the number of active molecules in the top 1000 ranked compounds, reducing the false positive rate | [116] |
| AtomNet (AIMS Program) | Deep learning for structure-based design | >15 quadrillion synthesizable compounds | Identified drug-like hits for 296 academic targets; 21 targets confirmed via dose-response validation | [114] |
Table 2: Experimental Validation from Selected Anticancer Discovery Campaigns
| Target | Cancer Relevance | AI Screening Method | Experimental Outcome | Reference |
|---|---|---|---|---|
| AKR1C3 | Upregulated in prostate, breast, and other cancers | AtomNet (via AIMS Awards) | Identified a novel 7-hydroxycoumarin scaffold inhibitor; binding mode validated by X-ray crystallography | [115] |
| Src Kinase | Key enzyme in multiple human cancers | AtomNet | Successful identification of drug-like hits, contributing to a larger study with a 14% hit rate for some targets | [114] |
| KLHDC2 | Human ubiquitin ligase | OpenVS (RosettaVS) | Discovered 7 hits (14% hit rate) with single-digit µM affinity; pose prediction validated by X-ray crystallography | [57] |
| NaV1.7 | Human voltage-gated sodium channel | OpenVS (RosettaVS) | Discovered 4 hits (44% hit rate) with single-digit µM binding affinity | [57] |
Application Notes: Protocol for an AI-Triaged Anticancer Virtual Screening Campaign
This protocol outlines a typical workflow for identifying novel inhibitors against a cancer target (e.g., AKR1C3 or Src kinase) using an AI-triaged active learning approach. The process integrates computational AI screening with experimental validation to form a closed loop.
The diagram below illustrates the iterative cycle of AI-triaged screening.
Protocol Steps
Step 1: Target Selection and Library Preparation
- Target Definition: Select a well-defined anticancer target (e.g., a kinase or enzyme like AKR1C3) with a known or predicted binding site [57] [115].
- Compound Library Curation: Assemble an ultra-large library of commercially available or synthesizable compounds. Libraries can range from millions to billions of molecules, such as the Mcule library or others containing over 15 quadrillion compounds [114] [115].
Step 2: Initial Sampling and Surrogate Model Training
- Initial Random Sampling: Perform computational docking (e.g., using RosettaVS express mode or Autodock Vina) or a small-scale biochemical assay on a randomly selected subset of the library (e.g., 0.1-0.5%) [57] [116]. This creates the initial training data.
- Model Selection and Training: Train a surrogate machine learning model on the initial data. Best performance is achieved with pretrained models such as:
- Pretrained Transformer-based Language Models: Treat compounds as SMILES strings [112] [113].
- Graph Neural Networks (GNNs): Model molecules as graphs of atoms and bonds to capture structural information [112] [113].
- Multimodal Models (e.g., UMME): Integrate multiple data types like molecular graphs, protein sequences, and assay information for enhanced accuracy [117].
Step 3: Active Learning Cycle
- Prediction and Prioritization: The trained surrogate model predicts the binding affinity or activity for all remaining compounds in the large library.
- Candidate Selection: An active learning algorithm (e.g., Bayesian optimization) selects the next batch of compounds (e.g., the top 1,000 highest-scoring molecules) for the next round of evaluation. This step focuses on the most promising regions of chemical space [112] [113] [116].
- Iterative Refinement: The newly tested compounds are added to the training set, and the surrogate model is retrained. This iterative loop (Steps 3-5 in the diagram) continues until a predefined stopping criterion is met, such as identifying a sufficient number of hit candidates or exhausting resources.
Step 4: Experimental Validation and Hit Confirmation
- In-depth Computational Evaluation: Top-ranked compounds from the final cycle undergo more rigorous docking (e.g., RosettaVS high-precision mode with full receptor flexibility) and molecular dynamics simulations to assess binding stability [57].
- Biochemical and Cellular Assays: Selected compounds are synthesized or acquired and tested in dose-response experiments (e.g., IC50 determination) against the target enzyme in cancer-relevant cell lines [114] [115].
- Structural Validation: For the most promising hits, determine a high-resolution co-crystal structure (e.g., via X-ray crystallography at a synchrotron beamline) to confirm the predicted binding pose and guide further optimization, as demonstrated with the AKR1C3 inhibitor [115].
The Scientist's Toolkit: Key Research Reagents and Computational Solutions
Implementing an AI-triaged screening protocol requires a combination of computational tools and experimental reagents.
Table 3: Essential Reagents and Computational Tools for AI-Triaged Screening
| Item Name | Function/Description | Application in Protocol |
|---|---|---|
| Ultra-Large Compound Library | Collections of billions of synthesizable small molecules (e.g., ZINC, Mcule). | Provides the chemical space for the AI model to explore and select from. |
| Pretrained AI Model (e.g., Transformer, GNN) | A machine learning model pre-trained on vast chemical data for property prediction. | Serves as the initial surrogate model to boost sample efficiency and accelerate the active learning process [112] [113]. |
| Docking Software (e.g., RosettaVS, Autodock Vina) | Programs that predict how a small molecule binds to a protein target. | Used for the initial random sampling and high-precision evaluation of top hits [57]. |
| Active Learning Framework | Computational script/package for Bayesian optimization or other query strategies. | Automates the iterative process of selecting the most informative compounds for the next round of testing [112] [116]. |
| Target Protein & Assay Reagents | Purified protein, buffers, substrates, and cellular lines for the cancer target. | Essential for experimental validation of AI-prioritized hits via biochemical and cellular assays [114] [115]. |
| Crystallization & X-ray Diffraction Platform | Resources for protein-ligand co-crystallization and structural determination. | Provides atomic-level validation of the binding mode of confirmed hit compounds, closing the discovery loop [115]. |
Advanced Applications: Multimodal AI and Combination Therapies
Beyond single-target screening, AI-triaged methods are expanding into more complex areas of anticancer drug discovery.
- Multimodal AI for Drug Combinations: Models like MADRIGAL integrate structural, pathway, cell viability, and transcriptomic data to predict the clinical outcomes of drug combinations from preclinical data. This is crucial for designing effective combination therapies for complex diseases like acute myeloid leukemia, while anticipating potential adverse drug interactions [118].
- Reducing False Positives with Curated Negative Data: The AMLSF method highlights the importance of high-quality negative data (inactive molecules) for training machine learning scoring functions. By actively learning and iteratively improving the selection of negative examples, this method significantly reduces the false positive rate in docking-based virtual screening, saving valuable experimental resources [116].
- Generative AI for de novo Design: AI is evolving from simply screening existing libraries to generating novel molecular structures. Pipelines that integrate generative models with docking and molecular dynamics simulations enable the de novo design of compounds against specific cancer targets, such as mutant IDH1 [117].
Validation Frameworks: From Computational Predictions to Experimental Confirmation
Benchmarking Virtual Screening Performance Metrics
Within anticancer drug discovery, the efficient identification of novel therapeutic compounds is paramount. Structure-based virtual screening (SBVS) serves as a cornerstone computational technique for this task, enabling researchers to rapidly prioritize potential drug candidates from libraries containing billions of molecules by predicting their binding to a protein target of interest, such as an enzyme critical for cancer cell survival [31] [61]. The practical utility of any virtual screening (VS) campaign, however, depends entirely on the computational models' ability to truly enrich active molecules over inactive ones. Consequently, rigorous benchmarking using appropriate performance metrics is not merely an academic exercise but a fundamental prerequisite for successful lead identification. This protocol details the key metrics and methodologies for assessing VS performance, specifically contextualized for research in anticancer drug discovery. A paradigm shift is currently underway, moving from traditional global accuracy metrics toward those emphasizing early enrichment, which is precisely aligned with the practical constraints of experimental follow-up in a laboratory setting [119].
Performance Metrics for Virtual Screening
The evaluation of virtual screening models requires metrics that reflect the real-world goal of identifying the maximum number of true active compounds within a limited selection chosen for experimental testing. The following metrics are essential for comprehensive benchmarking.
Key Metrics and Their Interpretation
- Positive Predictive Value (PPV): Also known as precision, PPV is increasingly recognized as a critical metric for modern VS campaigns. It measures the proportion of predicted active compounds that are truly active (PPV = True Positives / (True Positives + False Positives)) [119]. In a practical VS scenario, where a researcher can only test a small batch of compounds—for instance, the 128 that fit on a single 1536-well plate—a model with high PPV ensures that this limited experimental resource is not wasted on false positives. This directly translates to a higher hit rate. Recent studies demonstrate that models optimized for PPV, even when trained on imbalanced datasets, can achieve hit rates at least 30% higher than those trained using traditional balanced datasets [119].
- Enrichment Factor (EF): The EF quantifies how much better a model is at identifying active compounds compared to random selection at a specific early threshold of the screened library [120] [3]. It is defined as the fraction of actives found in the top X% of the ranked list divided by the fraction of actives expected in a random X% of the library. The top 1% (EF1%) is a commonly used benchmark. For example, state-of-the-art methods like RosettaGenFF-VS have achieved an EF1% of 16.72 on standard benchmarks, significantly outperforming other methods [3]. A higher EF indicates better early enrichment.
- Bayes Enrichment Factor (EFB): A recent innovation addresses a fundamental limitation of the traditional EF, whose maximum value is capped by the ratio of inactives to actives in the benchmark set. The EFB is calculated as the fraction of actives scoring above a threshold divided by the fraction of random molecules (not presumed inactives) scoring above the same threshold [121]. This metric is particularly valuable for estimating model performance on ultra-large libraries where true inactive-to-active ratios are immense, as it does not rely on carefully curated decoy sets and allows for enrichment estimation at very low selection fractions [121].
- Area Under the Receiver Operating Characteristic Curve (AUC-ROC): The AUC-ROC measures the overall ability of a model to discriminate between active and inactive compounds across all possible classification thresholds [120] [3]. While useful for a global assessment, a high AUC-ROC does not guarantee good performance specifically among the very top-ranked compounds, which is the primary focus of VS [119].
- Balanced Accuracy (BA): Balanced accuracy is the average of sensitivity and specificity. While historically used for model assessment, its relevance is now being questioned for VS tasks. Optimizing for BA often leads to models that perform well across the entire dataset but may fail to concentrate the true actives in the very top ranks, which is detrimental to practical screening efficiency [119].
Table 1: Summary of Key Virtual Screening Performance Metrics
| Metric | Formula (Simplified) | Key Interpretation | Advantage | Limitation |
|---|---|---|---|---|
| Positive Predictive Value (PPV) | TP / (TP + FP) | Hit rate; proportion of selected compounds that are truly active. | Directly measures practical experimental efficiency. | Sensitive to the number of compounds selected. |
| Enrichment Factor (EF) | (TPX% / NX%) / (Total Actives / Total Compounds) | Measures early enrichment in the top X% of rankings. | Intuitive; standard for retrospective benchmarking. | Maximum value is limited by the dataset's active/inactive ratio [121]. |
| Bayes Enrichment Factor (EFB) | (Fraction of Actives above Threshold) / (Fraction of Random Compounds above Threshold) | Estimates true enrichment using random compounds instead of decoys. | Unaffected by dataset composition; suitable for ultra-large libraries [121]. | A newer metric, not yet universally adopted. |
| AUC-ROC | Area under the ROC curve | Overall discriminative power across all thresholds. | Provides a global performance assessment. | Does not specifically focus on early enrichment. |
| Balanced Accuracy (BA) | (Sensitivity + Specificity) / 2 | Overall accuracy in classifying actives and inactives. | Useful for balanced classification tasks. | Not optimal for prioritizing top-ranked hits in VS [119]. |
The Metric Selection Workflow
The choice of metric should be guided by the specific goal of the virtual screening campaign. The following diagram illustrates the decision process for selecting the most appropriate primary metric.
Experimental Protocols for Benchmarking
This section provides a detailed, step-by-step protocol for conducting a rigorous virtual screening benchmark, suitable for assessing performance against anticancer drug targets.
Protocol: Benchmarking a VS Pipeline on a Known Target
1. Objective: To evaluate the accuracy and enrichment performance of a virtual screening workflow using a dataset with known active and decoy molecules for a specific protein target (e.g., an oncogenic kinase).
2. Materials and Data Preparation
- Protein Target Preparation:
- Obtain a high-resolution 3D structure of the target protein from the Protein Data Bank (PDB). For anticancer research, relevant targets may include kinases (e.g., BCR-ABL), apoptosis regulators, or epigenetic enzymes.
- Prepare the protein structure using standard software (e.g., OpenEye's Make Receptor [120] or Schrodinger's Protein Preparation Wizard). This involves adding hydrogen atoms, assigning protonation states, optimizing hydrogen bonding networks, and removing water molecules and co-crystallized ligands not critical for binding.
- Benchmark Dataset Curation:
- Actives: Collect a set of known bioactive molecules for the target from public databases such as ChEMBL [119] or BindingDB [122]. Ensure chemical diversity to avoid bias.
- Decoys: Generate a set of decoy molecules that are physically similar but chemically distinct from the actives to avoid artificial enrichment. Use a validated protocol like DEKOIS 2.0 [120], which creates property-matched decoys. A typical active-to-decoy ratio is 1:30 to 1:100 [120].
3. Virtual Screening Execution
- Molecular Docking:
- Select one or more docking programs for evaluation (e.g., AutoDock Vina, PLANTS, FRED, Glide) [120] [122].
- Dock every molecule (actives and decoys) from the benchmark dataset into the defined binding site of the prepared protein structure. Use a consistent configuration for all runs.
- Retain multiple poses per molecule but record the best (top-scoring) pose and its score for the initial ranking.
- Re-scoring with Machine Learning (Optional but Recommended):
- To improve performance, re-score the docking poses using a pretrained Machine Learning Scoring Function (ML SF) such as CNN-Score or RF-Score-VS v2 [120] [3].
- This step can significantly enhance the differentiation between actives and decoys. For instance, re-scoring with CNN-Score has been shown to improve EF1% from worse-than-random to over 28 for specific targets [120].
4. Performance Assessment and Analysis
- Ranking and Metric Calculation:
- Rank all compounds from the benchmark set based on their docking or ML SF score.
- Calculate the key metrics described in Section 2.1: PPV at various batch sizes (e.g., top 50, 100, 500), EF1%, EFB, and AUC-ROC.
- Visualization with pROC-Chemotype Plots:
- Go beyond single-number metrics by generating pROC-Chemotype plots [120]. These plots analyze the structural diversity (chemotypes) of the actives retrieved at early enrichment, ensuring that the model is not just retrieving a single, highly scored chemical series but a variety of promising scaffolds. This is crucial for lead discovery in anticancer research.
Table 2: The Scientist's Toolkit: Essential Reagents and Resources for VS Benchmarking
| Category | Item / Resource | Description / Function |
|---|---|---|
| Computational Tools | AutoDock Vina [120] [3] | A widely used, open-source molecular docking program. |
| FRED & PLANTS [120] | Alternative docking programs often used in benchmarking studies. | |
| CNN-Score / RF-Score-VS [120] | Pretrained Machine Learning Scoring Functions for pose re-scoring. | |
| RosettaVS [3] | A state-of-the-art physics-based VS method that models receptor flexibility. | |
| Data Resources | Protein Data Bank (PDB) [120] | Primary repository for 3D structural data of proteins and nucleic acids. |
| ChEMBL / PubChem [119] | Public databases of bioactive molecules with drug-like properties. | |
| DEKOIS 2.0 [120] | A database for benchmarking docking and VS, providing ready-made datasets with actives and decoys. | |
| ZINC [122] | A free database of commercially available compounds for virtual screening. | |
| Benchmarking Sets | DUD-E / LIT-PCBA | Benchmarking sets designed for VS validation, containing actives and confirmed inactives or decoys. |
| CASF-2016 [3] | A standard benchmark for scoring function evaluation. | |
| BayesBind [121] | A recently introduced benchmark designed to prevent data leakage when testing ML models. |
Advanced Applications and Case Studies in Anticancer Discovery
The following case studies illustrate the application of these benchmarking principles in a context relevant to drug discovery.
Case Study 1: Benchmarking Against a Resistant Cancer Target
A rigorous benchmarking study was performed on both the wild-type (WT) and a drug-resistant quadruple-mutant (Q) variant of Plasmodium falciparum Dihydrofolate Reductase (PfDHFR), a model system with parallels to drug resistance in cancer [120]. The protocol involved:
- Targets: WT and Q PfDHFR crystal structures (PDB: 6A2M and 6KP2).
- Methods: Three docking tools (AutoDock Vina, PLANTS, FRED) were evaluated, with and without re-scoring by ML SFs (CNN-Score, RF-Score-VS v2).
- Results: The optimal performance was achieved through hybrid workflows. For the WT, PLANTS with CNN re-scoring yielded an EF1% of 28. For the resistant Q variant, FRED with CNN re-scoring was most effective, achieving a remarkable EF1% of 31 [120]. This demonstrates that the optimal VS pipeline can be target-dependent, especially when dealing with mutations, and that ML re-scoring can dramatically improve performance.
Case Study 2: AI-Accelerated Screening for Oncology Targets
The development of the OpenVS platform showcases the application of advanced VS to high-value targets using an AI-accelerated, active learning approach [3].
- Targets: The platform was successfully used to discover hits for two unrelated targets: a ubiquitin ligase (KLHDC2) and the human voltage-gated sodium channel (NaV1.7).
- Method: The protocol used a two-stage docking process with RosettaVS: a fast VSX mode for initial filtering, followed by a high-precision VSH mode for final ranking. Active learning was employed to efficiently triage billions of compounds.
- Results: The campaign resulted in a 14% hit rate for KLHDC2 and a 44% hit rate for NaV1.7, with all hits showing single-digit micromolar affinity. The entire screening process for each target was completed in less than seven days [3]. This highlights the power of modern, metrics-driven VS platforms to rapidly identify high-quality leads from ultra-large chemical spaces, a capability directly transferable to the search for novel oncology therapeutics.
Benchmarking virtual screening performance is a critical step in building confidence for prospective drug discovery campaigns, particularly in the challenging field of anticancer research. This application note has underscored the necessity of moving beyond traditional metrics like balanced accuracy and toward early enrichment metrics such as PPV and EF1%, which better reflect real-world experimental constraints. The provided protocols and case studies offer a framework for researchers to rigorously evaluate their VS pipelines. By adopting these best practices—including the use of robust benchmark sets, hybrid docking/ML-scoring workflows, and advanced metrics like the Bayes Enrichment Factor—discovery scientists can significantly improve the odds of successfully identifying novel, potent chemical starting points for the development of next-generation cancer therapeutics.
Comparative Analysis of Docking Software and Scoring Functions
Molecular docking is an indispensable tool in modern computational drug discovery, providing critical insights into how small molecule ligands interact with biomolecular targets at an atomic level [123] [124]. The core of docking protocols relies on scoring functions—computational methods that approximate the binding affinity between a ligand and its protein target by calculating their interaction energy [125]. These functions enable researchers to predict binding poses and identify potential drug candidates through virtual screening of compound libraries containing thousands to billions of molecules [126] [127].
The predictive performance of scoring functions directly impacts the success of structure-based drug discovery campaigns, particularly in complex fields like anticancer research where targeting diverse protein families requires robust and accurate computational protocols [11] [128]. Despite decades of development and refinement, scoring functions face significant challenges in consistently predicting binding affinities across different target classes, creating an ongoing need for systematic comparison and optimization [125] [128]. This application note provides a structured framework for evaluating docking software and scoring functions, with specific consideration for anticancer drug discovery applications.
Key Concepts and Scoring Function Types
Scoring functions are generally categorized into three main approaches, each with distinct theoretical foundations and practical implications for virtual screening:
Empirical Scoring Functions: These functions evaluate binding affinity using a weighted sum of interaction terms (e.g., hydrogen bonding, hydrophobic interactions) derived through statistical regression against experimental affinity data [125]. Examples include the London dG, Alpha HB, and Affinity dG functions implemented in MOE software [125]. Their computational efficiency makes them particularly suitable for high-throughput virtual screening.
Force-Field Based Functions: These methods employ classical molecular mechanics force fields using Lennard-Jones and Coulomb potentials to describe van der Waals and electrostatic interactions [125]. The GBVI/WSA dG function in MOE represents this category, offering a more physics-based approach to binding affinity prediction [125] [129].
Machine Learning-Based Functions: Recently developed scoring functions utilize algorithms such as random forest, support vector machines, and neural networks to learn complex relationships between structural descriptors and binding affinities from large datasets of protein-ligand complexes [129] [128]. The DockTScore platform exemplifies this approach, combining physics-based terms with machine learning for improved accuracy [128].
Performance Challenges and Considerations
A critical challenge in scoring function development and application is the heterogeneous performance across different target proteins and ligand chemotypes [128]. Functions demonstrating excellent predictive power for one protein family may perform poorly for others, necessitating careful selection and validation for specific research contexts. Additionally, recent studies indicate that machine learning-based functions may exhibit overoptimistic performance in benchmark tests due to data biases, with significantly reduced accuracy when predicting affinities for proteins not included in training datasets (vertical tests) [129].
Table 1: Classification and Characteristics of Scoring Functions
| Type | Theoretical Basis | Advantages | Limitations | Representative Examples |
|---|---|---|---|---|
| Empirical | Weighted sum of interaction terms calibrated to experimental data | Fast computation, suitable for virtual screening | Limited physical basis, dependent on training set | London dG, Alpha HB [125] |
| Force-Field | Molecular mechanics force fields | Physics-based description of interactions | Requires solvation corrections, computationally intensive | GBVI/WSA dG [125] |
| Knowledge-Based | Statistical potentials from structural databases | No need for experimental affinity data | Limited to frequently observed interactions | - |
| Machine Learning | Algorithms trained on structural and affinity data | Can capture complex patterns, high potential accuracy | Risk of overfitting, limited interpretability | DockTScore [128] |
Comparative Performance Analysis
Methodology for Comparison
Standardized benchmark datasets enable objective comparison of scoring function performance. The CASF-2013 benchmark subset of the PDBbind database, containing 195 high-quality protein-ligand complexes with binding affinity data, serves as a widely-adopted reference for comparative assessments [125]. Similarly, the DUD-E (Directory of Useful Decoys: Enhanced) dataset provides a framework for evaluating virtual screening performance through enrichment metrics [128].
Performance evaluation typically focuses on multiple docking outputs:
- Best Docking Score (BestDS): The most favorable binding energy among predicted poses [125]
- Best RMSD (BestRMSD): The lowest root-mean-square deviation between predicted poses and the experimental ligand structure [125]
- RMSD_BestDS: The RMSD between the pose with the best docking score and the experimental structure [125]
- DS_BestRMSD: The docking score of the pose with the lowest RMSD to the experimental structure [125]
Comparative Performance of MOE Scoring Functions
A recent pairwise comparison of five scoring functions implemented in MOE software using InterCriteria Analysis (ICrA) revealed significant performance variations [125]. The study evaluated London dG, ASE, Affinity dG, Alpha HB, and GBVI/WSA dG functions across the CASF-2013 dataset, measuring their agreement based on different docking outputs.
Table 2: Performance Comparison of MOE Scoring Functions Based on ICrA Analysis [125]
| Scoring Function | Type | BestDS Performance | BestRMSD Performance | RMSD_BestDS Performance | DS_BestRMSD Performance |
|---|---|---|---|---|---|
| London dG | Empirical | Dissonance | Variable/Positive Consonance | Dissonance | Dissonance |
| Alpha HB | Empirical | Dissonance | Variable/Positive Consonance | Dissonance | Dissonance |
| ASE | Empirical | Dissonance | Variable/Positive Consonance | Dissonance | Dissonance |
| Affinity dG | Empirical | Dissonance | Variable/Positive Consonance | Dissonance | Dissonance |
| GBVI/WSA dG | Force-Field | Dissonance | Variable/Positive Consonance | Dissonance | Dissonance |
The analysis identified BestRMSD as the most discriminating docking output for comparing scoring function performance, with only this metric producing "varicolored" ICrA results (combinations of positive consonance and dissonance) between the five functions [125]. London dG and Alpha HB demonstrated the highest comparability among the evaluated functions, suggesting potential complementarity in virtual screening workflows [125].
Advancements in Machine Learning Scoring Functions
Recent research has produced next-generation scoring functions combining physics-based descriptors with machine learning algorithms. The DockTScore platform exemplifies this approach, incorporating optimized MMFF94S force-field terms, solvation and lipophilic interaction terms, and improved estimation of ligand torsional entropy contributions [128]. The development of target-specific scoring functions for particular protein classes such as proteases and protein-protein interactions (PPIs) represents a promising direction for improving predictive accuracy [128].
Machine learning-based scoring functions face particular challenges regarding generalizability and training data requirements. Studies comparing performance on experimental structures versus computer-generated complexes found similar accuracy levels, suggesting the potential for expanding training datasets through computational approaches [129]. However, significant performance reductions occur when these functions are applied to protein targets not represented in training data, highlighting the importance of appropriate validation protocols [129].
Experimental Protocols for Performance Evaluation
Protocol 1: Benchmarking with CASF-2013
Purpose: To evaluate and compare the performance of scoring functions using a standardized benchmark dataset.
Materials:
- CASF-2013 dataset (195 protein-ligand complexes from PDBbind database)
- Molecular docking software (MOE, AutoDock, or similar)
- Hardware: Multi-core workstation with sufficient memory for docking calculations
Procedure:
- Dataset Preparation: Download the CASF-2013 benchmark set from the PDBbind database (http://www.pdbbind.org.cn/). Prepare protein structures by adding hydrogen atoms, assigning protonation states, and energy minimization using appropriate software (e.g., MOE Protein Preparation Wizard) [128].
- Re-docking Experiments: For each protein-ligand complex, extract the crystallographic ligand and perform re-docking into the prepared protein structure. Generate multiple poses (typically 30) per ligand [125].
- Pose Evaluation: Calculate RMSD values between predicted poses and the experimental ligand conformation to assess docking accuracy.
- Scoring Function Assessment: Apply each scoring function to the generated poses and record:
- Best docking score among all poses (BestDS)
- Lowest RMSD among all poses (BestRMSD)
- RMSD of the pose with the best docking score (RMSDBestDS)
- Docking score of the pose with lowest RMSD (DSBestRMSD) [125]
- Performance Analysis: Evaluate scoring function performance using InterCriteria Analysis or correlation statistics with experimental binding affinities.
Protocol 2: Virtual Screening Validation with DUD-E
Purpose: To assess the ability of scoring functions to distinguish active compounds from decoys in a virtual screening context.
Materials:
- DUD-E dataset (http://dude.docking.org/) containing active compounds and property-matched decoys for specific target proteins
- Computing cluster or cloud resources for high-throughput docking
Procedure:
- Target Selection: Identify relevant anticancer targets (e.g., kinases, proteases) available in the DUD-E database.
- Library Preparation: Prepare both active compounds and decoy molecules by generating 3D structures, enumerating tautomers, and optimizing geometry.
- Virtual Screening: Dock each compound from both active and decoy sets into the target protein binding site using multiple scoring functions.
- Enrichment Analysis: Calculate enrichment factors (EF) at different percentiles of the screened library (e.g., EF1%, EF5%) to quantify the early recognition of active compounds.
- Statistical Evaluation: Generate receiver operating characteristic (ROC) curves and calculate area under curve (AUC) values to compare overall screening performance across different scoring functions [128].
Protocol 3: Development of Target-Specific Scoring Functions
Purpose: To create customized scoring functions optimized for specific anticancer targets.
Materials:
- Curated dataset of protein-ligand complexes for the target of interest
- Machine learning libraries (scikit-learn, XGBoost, or similar)
- Molecular descriptor calculation software
Procedure:
- Data Curation: Collect high-quality protein-ligand complexes for the target protein class (e.g., proteases, protein-protein interactions) from the PDBbind database or literature sources. Include binding affinity data (Kd, Ki, or IC50 values) [128].
- Structure Preparation: Manually curate each complex structure, paying particular attention to protonation states, tautomeric forms, and binding site water molecules. Perform energy minimization to optimize hydrogen bonding networks [128].
- Descriptor Calculation: Compute physics-based interaction descriptors including van der Waals energy, electrostatic interactions, solvation effects, and entropic contributions [128].
- Model Training: Split the dataset into training (75%) and test (25%) sets. Employ multiple linear regression, support vector machines, or random forest algorithms to develop scoring functions [128].
- Validation: Evaluate the target-specific scoring function on an independent test set not used during training. Compare performance against general-purpose scoring functions using correlation coefficients and mean absolute error metrics.
Research Reagent Solutions
Table 3: Essential Computational Tools for Docking and Virtual Screening
| Tool Name | Type | Key Features | Application in Anticancer Research |
|---|---|---|---|
| MOE (Molecular Operating Environment) | Commercial Software Suite | Multiple scoring functions (London dG, Alpha HB, GBVI/WSA dG), structure preparation tools | Comprehensive docking simulations and scoring function comparison [125] |
| AutoDock Suite | Open-Source Software | AutoDock Vina with improved scoring function, support for flexible receptor docking | Virtual screening of natural product libraries against cancer targets [126] [11] |
| PDBbind Database | Curated Database | Collection of protein-ligand complexes with binding affinity data | Benchmarking and training set for scoring function development [125] [128] |
| DUD-E Dataset | Benchmark Database | Active compounds and decoys for target proteins | Validation of virtual screening protocols [128] |
| DockTScore | Machine Learning Scoring Function | Physics-based terms combined with ML algorithms, target-specific variants | Enhanced binding affinity prediction for challenging targets [128] |
| GOLD | Docking Software | Genetic algorithm for pose prediction, multiple scoring functions | Protein-ligand docking in structure-based drug design [129] |
Workflow Visualization
Diagram 1: Virtual screening workflow for anticancer drug discovery.
The comparative analysis of docking software and scoring functions reveals a complex landscape where no single approach universally outperforms others across all target classes and application contexts. Empirical scoring functions offer computational efficiency for large-scale virtual screening, while machine learning-based methods show promising accuracy improvements, particularly when developed for specific target classes relevant to anticancer drug discovery [125] [128].
The integration of physics-based descriptors with advanced machine learning algorithms represents the current state-of-the-art in scoring function development, addressing limitations of traditional methods while maintaining physicochemical interpretability [128]. Furthermore, the development of target-specific scoring functions for important anticancer target classes such as proteases and protein-protein interactions shows particular promise for improving virtual screening success rates [128].
For researchers engaged in anticancer drug discovery, a consensus approach combining multiple scoring functions with careful validation against experimental data provides the most robust strategy for identifying promising therapeutic candidates. The protocols outlined in this application note offer a structured framework for conducting such evaluations, enabling more effective implementation of computational docking in the ongoing search for novel anticancer agents.
The journey from a computational prediction to a viable anticancer drug candidate is complex and fraught with high attrition rates. Experimental validation serves as the critical bridge between in silico predictions and clinical application, providing the necessary biological context to prioritize candidates for further development. While computational methods like virtual screening enable researchers to efficiently sift through billions of compounds and identify potential hits based on structural compatibility with cancer-related targets, these predictions remain theoretical without experimental confirmation [51] [52]. The integration of in vitro (test tube) and in vivo (whole living organism) studies creates a robust, iterative feedback loop that progressively validates drug efficacy and safety, ultimately reducing the risk of late-stage failures in oncology drug development [11] [8].
This integrated approach is particularly vital in cancer research due to the disease's complexity. Cancer involves not just mutated oncogenes and tumor suppressor genes, but also complex tumor microenvironment interactions, immune system evasion, and metastatic processes that are difficult to model entirely in silico [11] [52]. The multidisciplinary nature of modern anticancer drug discovery requires seamless coordination between computational chemists, cell biologists, and in vivo pharmacologists to establish a conclusive link between target engagement and therapeutic effect. As evidenced by research on VRK family genes in hepatocellular carcinoma, this validation pipeline can reveal novel therapeutic targets and biomarkers while providing insights into drug resistance mechanisms [130].
Integrated Experimental Workflow: From Computation to Validation
The following diagram illustrates the comprehensive workflow for experimental validation following virtual screening in anticancer drug discovery:
Figure 1: Integrated workflow for experimental validation of computationally identified anticancer compounds.
Research Reagent Solutions: Essential Materials for Experimental Validation
Table 1: Key research reagents and their applications in experimental validation
| Reagent/Category | Specific Examples | Function in Validation | Application Context |
|---|---|---|---|
| Cell-Based Assay Kits | CCK-8, MTT, ATP-based viability assays | Quantify cell viability and compound cytotoxicity | In vitro screening of anticancer activity [130] |
| Invasion/Migration Tools | Transwell chambers, Matrigel, wound healing assays | Assess metastatic potential and anti-migration effects | In vitro metastasis models [130] |
| Gene Modulation Reagents | siRNA, shRNA lentivirus, CRISPR-Cas9 systems | Target validation through knockdown/knockout | Functional genomics studies [130] |
| Animal Models | Mouse xenograft models, PDX (Patient-Derived Xenografts) | In vivo efficacy and toxicity evaluation | Preclinical therapeutic validation [130] |
| Molecular Biology Tools | qRT-PCR reagents, Western blot materials, IHC kits | Mechanism of action and biomarker analysis | Target engagement and pathway modulation [130] |
Quantitative Data Presentation in Experimental Validation
Effective presentation of quantitative data is essential for interpreting experimental results and making informed decisions in the drug discovery pipeline. Statistical comparison between experimental groups relies on appropriate data visualization to convey complex relationships efficiently [131].
Table 2: Representative in vitro validation data for VRK2 knockdown in hepatocellular carcinoma
| Experimental Assay | Control Group | VRK2 Knockdown Group | P-value | Significance |
|---|---|---|---|---|
| CCK-8 Proliferation (48h) | 100.0% ± 5.2% | 62.3% ± 4.8% | < 0.001 | * |
| Colony Formation (count) | 45.7 ± 3.2 | 18.9 ± 2.4 | < 0.001 | * |
| Wound Healing Closure (%) | 85.3% ± 6.1% | 41.2% ± 5.3% | < 0.001 | * |
| Transwell Invasion (cells) | 132.5 ± 8.7 | 67.3 ± 6.2 | < 0.001 | * |
| Apoptosis Rate (%) | 4.8% ± 1.1% | 18.9% ± 2.3% | < 0.001 | * |
Table 3: In vivo efficacy data for VRK2 targeting in xenograft models
| Parameter | Control Group | Treatment Group | Statistical Significance | Effect Size |
|---|---|---|---|---|
| Tumor Volume (mm³) | 852.6 ± 125.3 | 412.8 ± 89.7 | P < 0.001 | Cohen's d = 1.84 |
| Tumor Weight (g) | 0.86 ± 0.15 | 0.41 ± 0.11 | P < 0.001 | Cohen's d = 1.72 |
| Metastatic Nodules | 8.3 ± 1.5 | 3.2 ± 1.1 | P < 0.001 | Cohen's d = 1.93 |
| Proliferation Index (%) | 68.5% ± 7.2% | 35.2% ± 6.3% | P < 0.001 | Cohen's d = 2.01 |
| Animal Body Weight (g) | 22.3 ± 1.1 | 21.8 ± 1.3 | P = 0.32 | Not Significant |
Detailed Experimental Protocols
Protocol 1: In Vitro Cytotoxicity and Proliferation Assays
Purpose: To evaluate the direct anticancer effects of computationally identified compounds on cancer cell viability and proliferative capacity.
Materials:
- Cancer cell lines (e.g., MHCC97H hepatocellular carcinoma cells) [130]
- CCK-8 cell counting kit or MTT reagent
- Compound solutions (serially diluted)
- 96-well cell culture plates
- Microplate reader
Procedure:
- Cell Seeding: Harvest exponentially growing cells and seed into 96-well plates at 2,000 cells/well in 100μL complete medium. Include blank wells (medium only) and control wells (cells without treatment) [130].
- Compound Treatment: After 24 hours incubation (37°C, 5% CO₂), add serially diluted compounds to treatment wells. Maintain untreated controls with equivalent DMSO concentration.
- Incubation: Incubate plates for 24, 48, 72, and 96 hours to establish time-response relationships.
- Viability Assessment: Add 10μL CCK-8 solution to each well and incubate for 2 hours at 37°C [130].
- Absorbance Measurement: Measure absorbance at 450nm using a microplate reader. Calculate cell viability relative to untreated controls.
- Data Analysis: Generate dose-response curves and calculate ICâ‚…â‚€ values using nonlinear regression analysis.
Technical Notes: Ensure cells are in logarithmic growth phase throughout the experiment. Perform at least three biological replicates with technical triplicates for statistical robustness.
Protocol 2: Gene Knockdown Validation Using siRNA/shRNA
Purpose: To functionally validate potential anticancer targets identified through computational approaches.
Materials:
- Validated siRNA or shRNA constructs targeting gene of interest
- Lipofectamine 3000 or similar transfection reagent
- Appropriate cell lines
- qRT-PCR reagents for knockdown confirmation
- Functional assay materials (based on target)
Procedure:
- Cell Preparation: Seed cells (1-2 × 10âµ/well) in 6-well plates and incubate for 24 hours to reach 60-70% confluence [130].
- Transfection Complex Preparation: For each well, dilute 5μL Lipofectamine 3000 in 125μL serum-free medium. In separate tubes, dilute 50-100nM siRNA in 125μL serum-free medium. Combine diluted siRNA with diluted Lipofectamine 3000 (1:1 ratio) and incubate for 15 minutes at room temperature [130].
- Transfection: Add transfection complexes dropwise to cells. Gently swirl plates to ensure even distribution.
- Incubation: Incubate cells for 48-72 hours at 37°C with 5% CO₂.
- Knockdown Confirmation: Harvest cells for qRT-PCR analysis to verify target gene expression reduction.
- Functional Assessment: Proceed with relevant functional assays (proliferation, apoptosis, migration, etc.) to characterize phenotypic effects of gene knockdown.
Technical Notes: Include appropriate controls (non-targeting siRNA, mock transfection). Optimize siRNA concentration and transfection time for each cell line.
Protocol 3: In Vivo Xenograft Efficacy Studies
Purpose: To evaluate anticancer efficacy of validated compounds or genetic targets in a physiologically relevant context.
Materials:
- Immunocompromised mice (e.g., nude or NSG strains)
- Cancer cells for implantation
- Test compounds or lentivirus for stable gene modulation
- Calipers for tumor measurement
- Institutional Animal Care and Use Committee (IACUC) approval
Procedure:
- Xenograft Establishment: Harvest exponentially growing cancer cells and resuspend in PBS:Matrigel (1:1 ratio). Subcutaneously inject 1-5 × 10ⶠcells into the flanks of 6-8 week old mice [130].
- Randomization: When tumors reach 100-150mm³, randomize animals into treatment and control groups (n=6-10 per group).
- Treatment Administration: Administer test compounds via appropriate route (oral gavage, intraperitoneal injection, etc.) at predetermined schedule. For genetic studies, utilize stably transfected cells or inducible systems.
- Tumor Monitoring: Measure tumor dimensions 2-3 times weekly using digital calipers. Calculate tumor volume using the formula: Volume = (Length × Width²)/2.
- Endpoint Analysis: After 3-6 weeks, euthanize animals and collect tumors for weight measurement and molecular analysis (histopathology, protein extraction, etc.).
- Metastasis Assessment: For metastasis models, quantify metastatic nodules in target organs (lung, liver) through visual inspection or histological examination.
Technical Notes: All procedures must follow institutional animal care guidelines. Monitor animal weight and overall health as indicators of treatment toxicity.
Signaling Pathway Analysis in Experimental Validation
Understanding the molecular mechanisms underlying anticancer activity is crucial for target validation and biomarker identification. The following diagram illustrates a representative signaling pathway analysis for VRK2 in hepatocellular carcinoma:
Figure 2: Signaling pathway analysis for VRK2 knockdown in hepatocellular carcinoma, demonstrating multiple mechanisms of action.
The integration of in vitro and in vivo studies represents an indispensable component of the anticancer drug discovery pipeline, transforming computational predictions into biologically validated therapeutic candidates. This multidisciplinary approach enables researchers to establish causal relationships between target modulation and therapeutic efficacy while assessing physiological relevance in complex biological systems. The iterative nature of this validation process—where in vivo findings inform refined in vitro models and computational analyses—creates a powerful feedback loop that enhances the efficiency of oncology drug development [130] [8].
As virtual screening methodologies continue to advance, enabling the interrogation of ultra-large chemical libraries encompassing billions of compounds [51], the role of robust experimental validation becomes increasingly critical for prioritizing the most promising candidates. The future of anticancer drug discovery lies in the seamless integration of these complementary approaches, leveraging the strengths of each methodology while acknowledging their respective limitations. Through this coordinated strategy, researchers can accelerate the translation of computational hits into clinically viable anticancer therapies, ultimately addressing the pressing need for more effective cancer treatments.
Within modern anticancer drug discovery, the hit identification process has been revolutionized by structure-based virtual screening (VS), which computationally predicts how small molecules interact with therapeutic targets [51]. However, the ultimate value of these predictions hinges on their accuracy, making experimental validation a critical step. X-ray crystallography provides the definitive method for this validation by revealing the atomic-level three-dimensional structure of a protein-ligand complex [132]. Confirming a computationally predicted binding mode with an experimental crystal structure verifies the screening methodology and provides invaluable insights for lead optimization [3] [115]. This case study details the protocol and application of X-ray crystallography in validating hits from a virtual screen, framed within a broader research thesis on computational protocols for anticancer drug development.
Representative Case Studies in Anticancer Discovery
Virtual Screening Against the KLHDC2 Ubiquitin Ligase
Researchers developed an open-source, AI-accelerated virtual screening platform (OpenVS) to screen multi-billion compound libraries [3]. The platform utilized a two-tiered docking protocol: a high-speed initial screen (Virtual Screening Express, VSX) followed by a more accurate, flexible-receptor method (Virtual Screening High-precision, VSH) for top hits [3]. This campaign targeted KLHDC2, a human ubiquitin ligase, and successfully identified a hit compound with single-digit micromolar binding affinity.
Validation by X-ray Crystallography: The critical validation step involved solving the X-ray crystallographic structure of the KLHDC2 protein in complex with the identified hit compound [3]. The solved structure demonstrated remarkable agreement with the binding pose predicted by the RosettaVS docking program, confirming the platform's predictive power. This experimental validation provided the confidence to proceed with a focused screen, which ultimately yielded six additional hit compounds with similar affinity, underscoring the value of structural validation in an iterative drug discovery cycle [3].
AI-Driven Discovery of an AKR1C3 Inhibitor
In a separate study targeting the enzyme AKR1C3—a target in prostate and breast cancers—researchers employed a deep learning neural network (AtomNet) for the initial virtual screen of a synthesizable chemical library [115]. From 87 potential inhibitors selected by AI, biological screening identified a hit compound (designated "compound 4") featuring a novel scaffold not previously reported in the literature [115].
Validation by X-ray Crystallography: To understand the binding mechanism, the research team determined the 3D structure of AKR1C3 in complex with its cofactor (NADP+) and compound 4 using X-ray diffraction data collected at the MAX IV synchrotron [115]. The structure revealed that the 7-hydroxy group of the compound's coumarin scaffold interacted specifically with the enzyme's oxyanion site. This atomic-level detail, unobtainable through computation alone, provided a clear rationale for the inhibitory activity and a structural blueprint for designing a new series of inhibitors [115].
Table 1: Summary of Key Experimental Outcomes from Case Studies
| Target Protein | Target Role in Cancer | Virtual Screening Method | Key Experimental Outcome | Validated Binding Affinity |
|---|---|---|---|---|
| KLHDC2 [3] | Ubiquitin Ligase | RosettaVS (Physics-based) | X-ray structure confirmed predicted pose; 7 total hits found. | Single-digit µM |
| AKR1C3 [115] | Enzyme (Overexpressed) | AtomNet (AI-based) | X-ray revealed novel binding mode for a new scaffold. | Not Specified |
Integrated Experimental Protocol
This section provides a detailed methodology for the key experiments cited, from computational hit identification to experimental structural validation.
Protocol Part I: Structure-Based Virtual Screening
This protocol is adapted from the RosettaVS workflow used in the KLHDC2 case study [3].
Step 1: Target Protein Preparation
- Obtain a high-resolution 3D structure of the target protein (e.g., from the Protein Data Bank, PDB) or a robust homology model.
- Using molecular visualization software, define the binding site coordinates for docking.
- Prepare the protein structure by adding hydrogen atoms, correcting protonation states of residues, and optimizing side-chain conformations.
Step 2: Ligand Library Preparation
- Select a chemical library for screening (e.g., ZINC, Enamine, or a bespoke library).
- Generate credible 3D conformers for each compound.
- Assign appropriate bond orders and formal charges, and minimize the structures using a molecular mechanics force field.
Step 3: Hierarchical Docking and Scoring
- Initial Triage (VSX Mode): Perform rapid, rigid-receptor docking of the entire library. This step prioritizes speed to filter out obvious non-binders.
- Refined Docking (VSH Mode): Take the top-ranking compounds from the initial screen and subject them to high-precision docking that incorporates receptor flexibility (e.g., flexible side chains and limited backbone movement).
- Ranking: Score the final poses using a robust scoring function that combines enthalpy (∆H) and entropy (∆S) terms to predict binding affinity accurately.
Protocol Part II: X-ray Crystallographic Validation
This protocol outlines the general workflow for structural validation, as demonstrated in both case studies [3] [115] [133].
Step 1: Protein Expression, Purification, and Complex Formation
- Express and purify the target protein to high homogeneity.
- Form the protein-ligand complex by incubating the purified protein with a molar excess of the hit compound.
- Use size-exclusion chromatography to isolate the pure complex and ensure the ligand is bound.
Step 2: Crystallization and Data Collection
- Screen for crystallization conditions using robotic systems and commercial sparse-matrix screens.
- Optimize initial hits to produce large, well-ordered single crystals suitable for diffraction.
- Flash-cool the crystal in liquid nitrogen and collect X-ray diffraction data at a synchrotron beamline.
Step 3: Structure Solution and Analysis
- Index and integrate the diffraction images to obtain a set of structure factor amplitudes.
- Solve the phase problem by molecular replacement (MR) using a known related protein structure as a search model.
- Build and refine the atomic model iteratively. The initial model will have the protein and any known cofactors. The difference electron density map (e.g., Fo - Fc) will reveal clear density for the bound ligand.
- Build the ligand into the observed density, refine its geometry, and analyze the specific protein-ligand interactions (hydrogen bonds, hydrophobic contacts, etc.).
The following workflow diagram illustrates the complete integrated protocol from virtual screening to structural validation.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for VS and Crystallographic Validation
| Item / Reagent | Function / Application | Examples / Specifications |
|---|---|---|
| Target Protein | The macromolecule (e.g., kinase, enzyme) against which drugs are designed. | KLHDC2 Ubiquitin Ligase [3]; AKR1C3 Enzyme [115] |
| Chemical Library | A collection of small molecules for virtual screening. | ZINC, Enamine REAL, Mcule library; multi-billion compound scale [3] [115] |
| Virtual Screening Software | Computationally docks and scores ligands against the target. | RosettaVS [3]; AtomNet (AI-based) [115]; Autodock Vina, Schrödinger Glide |
| Crystallization Kit | Sparse-matrix screens to identify initial crystal growth conditions. | JCSG+, Morpheus, MemGold (commercial screens) |
| Synchrotron Beamline | High-intensity X-ray source for diffraction data collection. | MAX IV BioMAX [115]; Other national synchrotron facilities |
| Crystallography Software Suite | For processing diffraction data, model building, and refinement. | CCP4 Suite [133]; PHENIX [133]; Coot |
The integration of sophisticated computational virtual screening with rigorous experimental validation by X-ray crystallography represents a powerful paradigm in rational anticancer drug design. The case studies of KLHDC2 and AKR1C3 demonstrate that this synergy is not merely confirmatory but is, in fact, a generative process. It builds a cycle of prediction and validation that enhances the reliability of computational models and provides the critical structural insights needed to transform initial hits into promising lead compounds. As computational methods and AI continue to advance, their coupling with high-resolution structural biology techniques will remain a cornerstone of efficient and effective oncology drug discovery.
Success Rates and Hit Identification Across Different Cancer Targets
Computer-Aided Drug Design (CADD) has become a cornerstone in modern anticancer drug discovery, dramatically cutting down the time and resources required in the early stages of the drug development pipeline [134]. Virtual screening (VS), a core computational technique within CADD, enables researchers to search libraries of small molecules to identify structures most likely to bind to specific cancer drug targets [31]. This application note details standardized protocols for implementing virtual screening strategies focused on various cancer targets, with particular emphasis on assessing success rates and improving hit identification efficiency. The content is framed within a broader thesis on computational protocols for virtual screening in anticancer drug discovery research, providing drug development professionals with practical methodologies for target selection, screening execution, and results validation.
Current Landscape of Cancer Targets
The Evolving Burden of Cancer
The continued need for improved cancer therapeutics is underscored by recent statistics. In 2025, approximately 2,041,910 new cancer cases and 618,120 cancer deaths are projected to occur in the United States alone [135]. While overall cancer mortality rates have declined steadily since the 1990s—averting over 4.5 million deaths—significant challenges remain, including stark disparities among population groups and increasing incidence of certain cancers among younger adults and women [136] [135]. These statistics highlight the critical need for more effective, targeted therapies.
Success Rates of Targeted Therapies and Companion Diagnostics
The success of targeted cancer therapies depends heavily on accurate companion diagnostics to identify eligible patients. Next-generation sequencing (NGS) tests like the Oncomine Dx Target Test (ODxTT) enable comprehensive genetic profiling from limited tissue samples, but their performance varies based on sample quality and cancer type.
Table 1: Analysis Success Rates of Companion Diagnostics Across Different Sample Types
| Test Type | Cancer Type | Sample Requirements | Success Rate | Key Limitations |
|---|---|---|---|---|
| Oncomine Dx Target Test | NSCLC | Tissue surface area >1.04 mm², Tumor cells >375 [137] | 75.6% (98/119 cases) [137] | Failure due to insufficient nucleic acid concentration [138] |
| Oncomine Dx Target Test | NSCLC | Tumor content ≥20% after trimming [138] | 90% (104/116 cases) [138] | 8% invalid results, 2% failure to pass nucleic acid threshold [138] |
| PNA-LNA PCR Clamp Test (EGFR) | NSCLC | Standard formalin-fixed paraffin-embedded samples [138] | 100% (116/116 cases) [138] | Limited to single-gene analysis [138] |
The data reveals that while comprehensive NGS panels offer multi-gene analysis capability, their success rates (75.6%-90%) remain lower than conventional single-gene tests (100%) due to higher sample quality requirements and analytical sensitivity issues [137] [138]. This underscores the importance of optimal sample selection and processing protocols for reliable target identification.
Computational Methodologies for Virtual Screening
Virtual Screening Approaches
Virtual screening methods are broadly categorized into structure-based and ligand-based approaches, with hybrid methods combining elements of both [31].
Structure-Based Virtual Screening (SBVS) requires the three-dimensional structure of the target protein, determined experimentally through X-ray crystallography or NMR, or generated computationally through homology modeling [139]. The primary SBVS method is molecular docking, which predicts how small molecules bind to a target protein and calculates binding affinity using scoring functions [31]. Molecular dynamics simulations assess the stability of ligand-receptor complexes under physiological conditions [31].
Ligand-Based Virtual Screening (LBVS) is employed when the 3D protein structure is unknown but information about active ligands is available [31]. Techniques include pharmacophore modeling, which identifies essential steric and electronic features necessary for molecular recognition [31] [139]; shape-based screening, which identifies compounds with similar three-dimensional shapes to known active molecules [31]; and Quantitative Structure-Activity Relationship (QSAR) modeling, which correlates chemical structural descriptors with biological activity [31].
Hybrid Methods leverage both structural information and ligand similarity to overcome limitations of individual approaches [31]. These methods utilize evolutionary ligand-binding information and can employ global structural similarity combined with pocket-specific analysis [31].
Specialized Computational Techniques for Cancer Targets
Pathway Activation Analysis: For cancer targets, the OncoFinder algorithm calculates Pathway Activation Strength (PAS) to analyze intracellular signaling pathway activity [140]. The PAS value is calculated as:
PASp = ∑n NIInp × ARRnp × BTIFn × lg(CNRn)
Where p represents the pathway index, n represents the protein index, NIInp indicates protein involvement in the pathway, ARRnp represents the activator/repressor role, BTIFn indicates if expression exceeds the confidence interval, and CNRn is the case-to-normal expression ratio [140]. This approach helps identify abnormally activated pathways in specific cancer types that can be targeted therapeutically.
Scaffold-Focused Screening: Natural product scaffolds like anthraquinones have shown particular promise in anticancer drug discovery [134]. The 9,10-anthraquinone moiety serves as a privileged chemical scaffold for developing analogues with diverse pharmaceutical properties [134]. Several anthraquinone-based drugs including anthracyclines (daunorubicin, doxorubicin) and synthetic anthraquinones (mitoxantrone, pixantrone) are already clinically approved for various cancers [134].
Experimental Protocols
General Virtual Screening Workflow
Diagram 1: Virtual screening workflow for cancer drug discovery.
Protocol 1: Structure-Based Virtual Screening for Kinase Targets
Objective: Identify novel kinase inhibitors using molecular docking approaches.
Materials and Reagents:
- Target: 3D structure of kinase domain (PDB format)
- Software: Molecular docking program (AutoDock, GOLD, or Glide)
- Compound Library: Chemspace Lead-Like Compound Library (1.3M compounds) [139]
- Computing Infrastructure: Linux cluster with batch queue processor
Procedure:
- Target Preparation:
- Obtain crystal structure from Protein Data Bank
- Remove water molecules and add hydrogen atoms
- Define binding site coordinates based on known kinase inhibitors
- Assign partial charges using appropriate force field
Compound Library Preparation:
- Download structures in SMILES or SDF format
- Generate 3D conformations using molecular modeling software
- Optimize geometry using molecular mechanics force fields
- Calculate partial atomic charges
Molecular Docking:
- Set docking parameters and scoring function
- Perform high-throughput docking of entire library
- Generate multiple binding poses for each compound
- Calculate binding scores for all poses
Post-Docking Analysis:
- Cluster compounds based on binding scores
- Analyze binding interactions of top-ranked compounds
- Select diverse chemotypes for further evaluation
Validation: Include known kinase inhibitors as positive controls to validate docking protocol. Compounds showing better binding scores than controls proceed to experimental testing.
Protocol 2: Ligand-Based Virtual Screening for GPCR Targets
Objective: Identify novel GPCR ligands using similarity searching and pharmacophore modeling.
Materials and Reagents:
- Reference Ligands: Known active compounds for target GPCR
- Software: ROCS (shape-based screening), Phase (pharmacophore modeling)
- Compound Library: MCE Bioactive Compound Library (28,621 compounds) [139]
- Computing Infrastructure: Workstation with multi-core processor
Procedure:
- Reference Ligand Preparation:
- Collect known active compounds with experimental IC50 values
- Generate multiple low-energy conformations
- Align structures based on common pharmacophore features
Pharmacophore Model Development:
- Identify common chemical features among active compounds
- Define spatial relationships between features
- Validate model using known active and inactive compounds
- Optimize model parameters to maximize enrichment
Shape-Based Screening:
- Use ROCS for rapid 3D shape similarity searching [31]
- Align database compounds to reference shape
- Calculate shape Tanimoto coefficients
- Rank compounds based on shape complementarity
Compound Selection:
- Apply drug-like filters (Lipinski's Rule of Five)
- Remove compounds with potential toxicity liabilities
- Select diverse chemotypes from top-ranked compounds
Validation: Use decoy sets containing known actives and inactives to calculate enrichment factors and validate screening protocol.
Protocol 3: Pathway-Centric Screening Using Transcriptomic Data
Objective: Identify compounds that reverse cancer-specific pathway activation patterns.
Materials and Reagents:
- Gene Expression Data: Tumor vs. normal tissue RNA-seq data
- Software: OncoFinder algorithm for pathway analysis [140]
- Compound Database: DrugBank or Connectivity Map
- Computing Infrastructure: R/Python environment with statistical packages
Procedure:
- Pathway Activation Analysis:
- Obtain gene expression data for cancer type of interest
- Calculate Pathway Activation Strength (PAS) using OncoFinder algorithm [140]
- Identify significantly upregulated pathways in tumor samples
- Select pathways with clinical relevance to cancer progression
Drug Scoring:
- Map molecular targets of available compounds to dysregulated pathways
- Calculate Drug Score (DS) based on ability to inhibit activated pathways [140]
- Rank compounds based on predicted efficacy against specific cancer type
Multi-Target Prioritization:
- Identify compounds targeting multiple dysregulated pathways
- Evaluate potential for combination therapies
- Assess selectivity for cancer pathways vs. normal physiological pathways
Validation: Compare predicted efficacy with clinical trial results for known drugs to validate scoring algorithm [140].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents and Resources for Anticancer Virtual Screening
| Resource Category | Specific Examples | Key Features/Functions | Application Context |
|---|---|---|---|
| Commercial Compound Libraries | MCE Bioactive Compound Library (28,621 compounds) [139] | Preclinical/clinical stage bioactive compounds | Hit identification, lead optimization |
| Chemspace Lead-Like Library (1.3M compounds) [139] | Lead-like compounds with favorable properties | Large-scale virtual screening | |
| MCE Virtual Screening Library (10M compounds) [139] | Ultra-large library for AI/ML screening | AI-driven drug discovery | |
| Specialized Libraries | Protein-Protein Interaction Modulators Library (2,906 compounds) [139] | Compounds targeting PPIs, challenging drug targets | Disruption of protein-protein interactions |
| Asinex Macrocycles Library (10,091 compounds) [139] | Diverse macrocyclic compounds with enhanced properties | Targeting difficult binding sites | |
| Covalent Inhibitors Library (942 compounds) [139] | Compounds with mild electrophilic moieties | Irreversible inhibition strategies | |
| Computational Tools | ROCS (Rapid Overlay of Chemical Structures) [31] | Shape-based molecular similarity searching | Ligand-based virtual screening |
| OncoFinder Algorithm [140] | Pathway Activation Strength calculation | Pathway-centric drug discovery | |
| Molecular Docking Software (AutoDock, GOLD) [31] | Structure-based binding pose prediction | Structure-based drug design | |
| Data Resources | Protein Data Bank (PDB) | Experimentally determined protein structures | Target preparation for SBVS |
| Cancer Genomics Data (TCGA) | Multi-omics cancer genomics data | Target identification and prioritization |
Signaling Pathways in Cancer Targeted Therapy
Diagram 2: Key cancer signaling pathways and targeted therapies.
This application note has detailed computational protocols and experimental considerations for successful virtual screening campaigns against cancer targets. The integrated approach combining structure-based methods, ligand-based techniques, and pathway-centric analysis provides a comprehensive framework for identifying novel anticancer compounds with higher success rates. As the field advances, several emerging trends promise to further enhance hit identification efficiency:
The growing integration of artificial intelligence and machine learning in virtual screening pipelines enables more accurate prediction of compound activity and optimization of screening libraries [134]. Additionally, the rise of ultra-large virtual screening libraries containing 10+ million compounds provides unprecedented chemical space coverage while requiring sophisticated computational infrastructure for efficient exploration [139]. The development of more sophisticated pathway analysis tools like OncoFinder allows for patient-specific drug scoring based on individual tumor pathway activation profiles, moving toward personalized virtual screening approaches [140].
By implementing the standardized protocols and utilizing the research reagents outlined in this document, drug discovery researchers can systematically approach virtual screening for anticancer drug development with greater predictability and higher potential for identifying clinically relevant compounds.
Analysis of False Positives and Strategies for Mitigation
In the field of anticancer drug discovery, virtual screening (VS) has become an indispensable computational technique for identifying novel lead compounds by rapidly evaluating massive chemical libraries [31]. However, a significant challenge that undermines its efficiency is the prevalence of false-positive hits—compounds predicted computationally to have desirable activity but which fail to show efficacy in subsequent biological assays [141]. These false positives consume substantial resources, misdirect medicinal chemistry efforts, and prolong development cycles. In a typical virtual screen, only about 12% of the top-scoring compounds actually demonstrate activity when tested experimentally, highlighting the severity of this issue [142]. Within the specific context of anticancer research, where targets are often complex protein-protein interactions and chemical libraries must be expansive to find novel chemotypes, the risk of false positives is particularly acute [143]. This Application Note analyzes the origins of false positives in structure-based and ligand-based virtual screening and provides detailed, actionable protocols for their mitigation, framed within a comprehensive computational workflow for anticancer drug discovery.
Understanding the common sources of false positives is the first step toward their mitigation. The following table categorizes these sources, their underlying causes, and their typical impact on the virtual screening process.
Table 1: Major Sources of False Positives in Virtual Screening
| Source Category | Specific Cause | Impact on Screening |
|---|---|---|
| Methodological Limitations | Overly simplistic scoring functions [144] | Poor affinity prediction, leading to the prioritization of non-binders |
| Inadequate treatment of protein flexibility [145] | Identification of compounds that do not fit the true conformational state of the target | |
| Chemical & Compound Issues | Promiscuous, "frequent-hitter" compounds [141] | Non-specific binding or assay interference, yielding false activity readings |
| Non-druglike compound properties [141] | Compounds are active in vitro but cannot be developed into drugs | |
| Data & Model Integrity | Poor decoy set design in model training [142] | Over-optimistic performance metrics and poor real-world predictive power |
| Algorithm overtraining on limited data [142] | Models fail to generalize to new chemical scaffolds outside the training set |
Integrated Protocols for False Positive Mitigation
This section outlines three advanced protocols designed to minimize false positive rates in virtual screening campaigns for anticancer drug discovery.
Protocol 1: Machine Learning Classification
The use of machine learning (ML) classifiers, specifically trained to distinguish true binders from decoys, has proven highly effective in reducing false positives. A key to success is the construction of a robust training dataset.
Detailed Methodology:
Dataset Construction (D-COID Strategy):
- Objective: Create a training set of known active complexes matched with highly compelling, target-specific decoys [142].
- Procedure:
- Curate a set of experimentally confirmed active complexes from sources like the Protein Data Bank (PDB) for your target of interest (e.g., tubulin for anticancer agents) [146].
- Generate matched decoys for each active compound by selecting molecules with similar physicochemical properties (e.g., molecular weight, logP) but different 2D topology to ensure they are not likely to bind.
- Perform molecular docking for all actives and decoys to generate predicted binding poses. This ensures the ML model learns from docking artifacts that often lead to false positives.
Model Training & Implementation (vScreenML):
- Framework: Train a classifier using the XGBoost framework on the D-COID dataset [142].
- Features: The model uses a combination of features describing the protein-ligand complex, including energy terms, interaction fingerprints, and shape complementarity.
- Prospective Application: Screen large chemical libraries (e.g., ZINC). Prioritize compounds based on the classifier's predicted probability of activity rather than traditional docking scores alone.
Validation: A prospective application of this protocol against acetylcholinesterase resulted in nearly all candidate inhibitors showing detectable activity, with 10 out of 23 compounds having an IC50 better than 50 µM, a significant enrichment over traditional methods [142].
Protocol 2: Multi-Objective Optimization in Docking
Traditional molecular docking relies on a single scoring function, which is often inadequate. The Multi-Objective Scoring Function Optimization Methodology (MOSFOM) simultaneously optimizes multiple, potentially conflicting objectives during the docking search itself.
Detailed Methodology:
- Define Objectives: Select two or more diverse scoring functions. A proven combination is the energy score (AMBER force field) and the contact score (shape complementarity) from DOCK [144].
- Multi-Objective Optimization (MO) Setup:
- Algorithm: Employ a Multi-Objective Evolutionary Algorithm (MOEA) or Genetic Algorithm (GA) to find a set of Pareto-optimal solutions—poses that represent the best compromises between the chosen objectives [144].
- Implementation: During the conformational search for each ligand, the algorithm does not seek a single "best" pose but a population of poses that are non-dominated in terms of both energy and contact scores.
- Pose Selection & Ranking: From the final Pareto-optimal set of poses, select the one that best satisfies a predefined prioritization rule (e.g., best energy score among the top 10% for shape complementarity). Rank all compounds in the library based on this selection.
Advantage: This method yields more reasonable binding conformations by balancing different interaction criteria, which enhances the hit rate and greatly reduces the false-positive rate compared to consensus scoring, which merely re-scores a limited number of top poses from a primary screen [144].
Protocol 3: Combined and Hybrid Screening Strategies
Leveraging the strengths of different VS approaches in a combined or hybrid workflow can overcome the limitations inherent in any single method.
Detailed Methodology:
- Structure-Based & Ligand-Based Hybrid:
- Step 1 (Ligand-Based Filtering): Use known active compounds for a target (e.g., a tubulin inhibitor [146]) to perform a shape-based similarity search (e.g., using ROCS) or a pharmacophore search against a large library to create a focused subset [31].
- Step 2 (Structure-Based Refinement): Subject the focused subset to rigorous molecular docking and the ML classification described in Protocol 1.
- Multi-Structure Docking: To account for protein flexibility, dock the library into multiple representative protein conformations (e.g., from molecular dynamics simulations or multiple crystal structures). Compounds that consistently score well across multiple conformations are more likely to be true binders [145].
- Experimental Triangulation: Integrate orthogonal computational data, such as from omics technologies (genomics, proteomics), to validate target engagement hypotheses before experimental testing [147].
Diagram 1: Integrated VS workflow for mitigating false positives.
The Scientist's Toolkit: Research Reagent Solutions
The following table details key computational and experimental reagents essential for implementing the protocols described in this note.
Table 2: Essential Research Reagents and Resources
| Reagent / Resource | Type | Function in False Positive Mitigation |
|---|---|---|
| D-COID Dataset [142] | Computational Dataset | Provides a high-quality benchmark for training ML models to recognize and filter false-positive binding modes. |
| vScreenML Classifier [142] | Software/ML Model | A pre-trained, general-purpose ML classifier that scores protein-ligand complexes for a high probability of true activity. |
| Multi-Objective Optimization Algorithm (MOSFOM) [144] | Computational Method | Enables simultaneous optimization of multiple scoring criteria during docking, leading to more robust pose prediction. |
| ROCS (Rapid Overlay of Chemical Structures) [31] | Software | Performs shape-based ligand screening to prioritize compounds with high 3D shape similarity to known actives, a strong initial filter. |
| Tubulin Protein Structure (e.g., PDB 1SA1) [146] | Structural Biology Resource | A well-defined target structure for docking in anticancer discovery; using multiple such structures addresses flexibility. |
| ACD/MDDR Databases [144] | Chemical Database | Sources of known active compounds and decoy molecules for model training and validation. |
| GC-MS (Gas Chromatography-Mass Spectrometry) [148] | Analytical Instrument | The "gold-standard" for confirmatory testing in experimental validation, used here as an analogy for rigorous final verification of computational hits. |
The problem of false positives in virtual screening represents a significant bottleneck in anticancer drug discovery. By moving beyond single-method approaches and adopting integrated strategies—such as machine learning classifiers trained on carefully curated data, multi-objective optimization in docking, and hybrid workflows—researchers can significantly enrich the quality of their computational hits. The protocols and tools detailed in this Application Note provide a clear roadmap for deploying these advanced strategies, ultimately leading to more efficient identification of novel, potent, and druglike anticancer agents with a higher probability of success in preclinical development.
Conclusion
Computational virtual screening has emerged as a cornerstone technology in modern anticancer drug discovery, dramatically accelerating the identification of promising therapeutic candidates while reducing development costs. The integration of AI and machine learning with traditional physics-based methods has enabled researchers to navigate billion-compound libraries with unprecedented efficiency. Recent successes in identifying potent inhibitors for targets including tubulin, PAK2, and various kinases demonstrate the tangible impact of these approaches. As the field advances, future developments will likely focus on improved modeling of complex biological systems, enhanced prediction of drug resistance mechanisms, and tighter integration of multi-omics data. The continued evolution of these computational protocols promises to further bridge the gap between in silico predictions and clinical success, ultimately delivering more effective and personalized cancer treatments to patients worldwide.
References
- 1. Techniques in Virtual Screening Drug Discovery [https://www.azolifesciences.com/article/Virtual-Screening-Techniques-in-Drug-Discovery.aspx]
- 2. Artificial Intelligence-Driven Innovations in Oncology Drug Discovery [https://www.dovepress.com/artificial-intelligence-driven-innovations-in-oncology-drug-discovery--peer-reviewed-fulltext-article-DDDT]
- 3. An artificial intelligence accelerated virtual platform for... screening [https://www.nature.com/articles/s41467-024-52061-7?error=cookies_not_supported&code=9862c916-3d94-4575-a0d8-0c0c265cb767]
- 4. Docking and Virtual in Screening | SpringerLink Drug Discovery [https://link.springer.com/protocol/10.1007/978-1-4939-7201-2_18]
- 5. Machine Learning Techniques Revolutionizing... - DEV Community [https://dev.to/clairlabs/machine-learning-techniques-revolutionizing-target-identification-in-drug-discovery-1h8c]
- 6. AI ( Artificial intelligence) Assisted Drug Platform Discovery [https://www.aurigeneservices.com/services/discovery/ai-assisted-drug-discovery]
- 7. Current AI technologies in cancer diagnostics and treatment | Molecular... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02369-9]
- 8. Application of computational methods for anticancer drug discovery, design, and optimization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7110968/]
- 9. Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, ADMET, and Molecular Dynamics (MD) Simulation of Potential Inhibitors of PD-L1 from the Library of Marine Natural Products - PubMed [https://pubmed.ncbi.nlm.nih.gov/35049884/]
- 10. Frontiers | Pharmacophore screening, molecular docking, and MD simulations for identification of VEGFR-2 and c-Met potential dual inhibitors [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1534707/full]
- 11. Anticancer Drug Discovery Based on Natural Products: From Computational Approaches to Clinical Studies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10813144/]
- 12. Pharmacophore modeling in drug design - PubMed [https://pubmed.ncbi.nlm.nih.gov/40175047/]
- 13. of natural MCL1 inhibitors using pharmacophore modelling... Discovery [https://pubmed.ncbi.nlm.nih.gov/40120151/]
- 14. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein | Scientific Reports [https://www.nature.com/articles/s41598-021-83626-x]
- 15. Top 5 Drug Trends Discovery Driving Breakthroughs 2025 [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 16. Target Identification and Validation (Small Molecules) | UCL Therapeutic Innovation Networks - UCL – University College London [https://www.ucl.ac.uk/therapeutic-innovation-networks/target-identification-and-validation-small-molecules]
- 17. The Path to Oncology Drug Target : An... | SpringerLink Validation [https://link.springer.com/protocol/10.1007/978-1-62703-311-4_1]
- 18. A Pipeline for Drug Target and Identification Validation [https://pubmed.ncbi.nlm.nih.gov/28057848/]
- 19. Systematic identification of cancer pathways and potential drugs for intervention through multi-omics analysis | The Pharmacogenomics Journal [https://www.nature.com/articles/s41397-025-00361-6]
- 20. Emerging Promise of Computational Techniques in Anti - Cancer ... [https://pubmed.ncbi.nlm.nih.gov/35892749/]
- 21. Application of computational methods for anticancer ... drug discovery [https://www.elsevier.es/en-revista-boletin-medico-del-hospital-infantil-201-articulo-application-computational-methods-for-anticancer-S2444340917001145]
- 22. Accelerating Molecular Docking using Machine Learning Methods - PubMed [https://pubmed.ncbi.nlm.nih.gov/38850231/]
- 23. Target Validation - Improving and Accelerating Therapeutic Development for Nervous System Disorders - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK195039/]
- 24. Accelerating Cancer Drug Discovery with Digital Pathology-Based Biomarker Analysis [https://blog.crownbio.com/accelerating-cancer-drug-discovery-with-digital-pathology-based-biomarker-analysis]
- 25. & Target for Identification Immunotherapies... Validation Cancer [https://explicyte.com/cro-services/target-identification-validation-services-cancer-immunotherapies/]
- 26. Frontiers | Editorial: Repurposed targeting cancer signaling... drugs [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1628842/full]
- 27. ReDO_DB: the repurposing drugs in oncology database - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6345075/]
- 28. Drug Repurposing in Oncology: A Systematic Review of Randomized Controlled Clinical Trials [https://www.mdpi.com/2072-6694/15/11/2972]
- 29. Frontiers | Structure-Based Virtual Screening: From Classical to ... [https://www.frontiersin.org/articles/10.3389/fchem.2020.00343/full]
- 30. Research Business Report Drug Repurposing Strategic 2025 [https://www.globenewswire.com/news-release/2025/05/28/3089241/0/en/Drug-Repurposing-Strategic-Research-Business-Report-2025-Global-Market-to-Reach-37-3-Billion-by-2030-Surging-Demand-for-Personalized-Therapies-Promotes-Indication-Specific-Repurpos.html]
- 31. Virtual screening - Wikipedia [https://en.wikipedia.org/wiki/Virtual_screening]
- 32. Strategies in Computational : Anticancer ... Drug Discovery Virtual [https://healthinformaticsjournal.com/index.php/IJMI/article/view/2352]
- 33. Computational repurposing of oncology drugs through offâ€target drug binding interactions from pharmacological databases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11022299/]
- 34. Predictive Oncology and Every Cure Announce a Strategic ... [https://www.barchart.com/story/news/34597261/predictive-oncology-and-every-cure-announce-a-strategic-collaboration-to-pursue-drug-repurposing-for-cancer-patients]
- 35. , pharmacokinetic, and DFT studies of Virtual ... screening anticancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC9976450/]
- 36. analyses of structural features and scaffold diversity for... Comparative [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-017-0212-4]
- 37. Chemoinformatics-based enumeration of chemical libraries: a tutorial | Journal of Cheminformatics | Full Text [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-020-00466-z]
- 38. Screening Compound Anticancer | Life Libraries Chemicals [https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/anticancer-library]
- 39. CCSMD: A Database Built from Smart Reaction Modules and... :: SSRN [https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4239455]
- 40. Platforms – Discovery Bioscience Anticancer [https://anticancerbio.com/discovery-platforms/]
- 41. DNA-encoded chemical libraries: foundations and applications in lead discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5214759/]
- 42. A Comprehensive Survey of Prospective Structure - Based ... Virtual [https://pmc.ncbi.nlm.nih.gov/articles/PMC9781938/]
- 43. Molecular Docking: A powerful approach for structure-based drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3151162/]
- 44. Docking (molecular) - Wikipedia [https://en.wikipedia.org/wiki/Docking_(molecular)]
- 45. Key Topics in Molecular Docking for Drug Design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6769580/]
- 46. Practices in Molecular and Docking - Structure ... Based Virtual [https://experiments.springernature.com/articles/10.1007/978-1-4939-7756-7_3?error=cookies_not_supported&code=b00611f4-0a8d-426a-b62e-189e5f71257e]
- 47. Fundamentals of Molecular Docking and Comparative Analysis of Protein–Small-Molecule Docking Approaches | IntechOpen [https://www.intechopen.com/chapters/82586]
- 48. Improving the Virtual Screening Ability of Target-Specific Scoring ... [https://ncbi.nlm.nih.gov/pmc/articles/PMC6713720]
- 49. A statistical framework to evaluate virtual screening | BMC Bioinformatics | Full Text [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-10-225]
- 50. Application of computational methods for anticancer drug discovery, design, and optimization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7154613/]
- 51. Perspectives on current approaches to virtual screening in drug discovery - PubMed [https://pubmed.ncbi.nlm.nih.gov/39132881/]
- 52. Virtual screening of potential anticancer drugs based on microbial products - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1044579X21002078]
- 53. T009 · Ligand - based — TeachOpenCADD... pharmacophores [https://projects.volkamerlab.org/teachopencadd/talktorials/T009_compound_ensemble_pharmacophores.html]
- 54. Applications and Limitations of Pharmacophore Modeling – Protac [https://drugdiscoverypro.com/applications-and-limitations-of-pharmacophore-modeling/]
- 55. Ligand-based Pharmacophore Modeling, Virtual Screening and Molecular Docking Studies for Discovery of Potential Topoisomerase I Inhibitors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6396084/]
- 56. Exploring conformational search protocols for ligand - based virtual... [https://pubmed.ncbi.nlm.nih.gov/25408244/]
- 57. An artificial intelligence accelerated virtual screening platform for drug discovery | Nature Communications [https://www.nature.com/articles/s41467-024-52061-7]
- 58. NVIDIA NIM Agent Blueprint Redefines Hit Identification... | NVIDIA Blog [https://blogs.nvidia.com/blog/nvidia-nim-agent-blueprint-virtual-screening/]
- 59. Advances of Artificial Intelligence in Anti-Cancer Drug Design: A Review of the Past Decade - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9963982/]
- 60. Applications in Machine - Learning Anti cancer Drug Discovery [https://link.springer.com/chapter/10.1007/978-3-030-67051-1_10]
- 61. Receptor-based virtual screening protocol for drug discovery - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S000398611500257X]
- 62. Advancing Anticancer : Leveraging Metabolomics and... Drug Discovery [https://pubmed.ncbi.nlm.nih.gov/39431333/]
- 63. Docking and Molecular Studies Reveal the... Molecular Dynamics [https://pubmed.ncbi.nlm.nih.gov/37764441/]
- 64. docking and Molecular study of... | PLOS One dynamics simulation [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0254035]
- 65. Network Pharmacology-Guided Discovery and Computational... :: SSRN [https://papers.ssrn.com/sol3/papers.cfm?abstract_id=5551479]
- 66. Using molecular to explore the binding of the... dynamics simulation [https://link.springer.com/article/10.1007/s00894-019-4024-5]
- 67. and docking studies on the Molecular ... dynamics simulation binding [https://mbrc.shirazu.ac.ir/article_628.html]
- 68. studies for DNA sequence... Molecular dynamics simulation [https://pubmed.ncbi.nlm.nih.gov/24446378/]
- 69. Leveraging machine learning models in evaluating ADMET properties for drug discovery and development - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12205928/]
- 70. Frontiers | Recent advances in AI-based toxicity prediction for drug... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1632046/full]
- 71. A review of the current trends in computational approaches in drug design and metabolism | Discover Public Health [https://link.springer.com/article/10.1186/s12982-024-00229-3]
- 72. ® - Simulations Plus - Machine Learning- ADMET ... Predictor ADMET [https://www.simulations-plus.com/software/admetpredictor/]
- 73. FP-ADMET: a compendium of fingerprint-based ADMET prediction models | Journal of Cheminformatics | Full Text [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-021-00557-5]
- 74. , Virtual profiling, PASS screening , and bioactivity... ADMET prediction [https://pubmed.ncbi.nlm.nih.gov/34107799/]
- 75. Computer-aided anti - cancer of EGFR protein based... drug discovery [https://pubmed.ncbi.nlm.nih.gov/37676262/]
- 76. , Docking, Virtual and System Pharmacology... screening ADMET [https://www.nature.com/articles/s41598-018-23768-7?error=cookies_not_supported&code=4e394090-a0bf-4e91-a81e-c9f7e07b24fb]
- 77. of a novel potent Discovery through tubulin ... inhibitor virtual [https://pubmed.ncbi.nlm.nih.gov/40830098/]
- 78. Mitotic inhibitor - Wikipedia [https://en.wikipedia.org/wiki/Mitotic_inhibitor]
- 79. A review of research progress of antitumor drugs based on tubulin targets - Cheng - Translational Cancer Research [https://tcr.amegroups.org/article/view/40172/html]
- 80. Novel Tubulin-Targeting Therapies Make Headway [https://www.onclive.com/view/novel-tubulin-targeting-therapies-make-headway]
- 81. Exploring drug for repurposing : a systematic virtual... PAK 2 inhibition [https://pubmed.ncbi.nlm.nih.gov/41021189/]
- 82. PAK2 - Wikipedia [https://en.wikipedia.org/wiki/PAK2]
- 83. Phosphoproteomics identifies determinants of PAK inhibitor sensitivity in leukaemia cells | Cell Communication and Signaling | Full Text [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-025-02107-0]
- 84. Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking | Nature Protocols [https://www.nature.com/articles/s41596-021-00659-2]
- 85. Molecular docking-based computational platform for high-throughput virtual screening - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8754542/]
- 86. The protein flexibility in receptor-ligand docking simulations | Journal of Cheminformatics | Full Text [https://jcheminf.biomedcentral.com/articles/10.1186/1758-2946-2-S1-O11]
- 87. Frontiers | Molecular docking and dynamics in protein serine/threonine... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1696204/full]
- 88. Exploring the Role of Receptor Flexibility in Structure-Based Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4459653/]
- 89. Assessing an ensemble docking -based virtual strategy for... screening [https://pubmed.ncbi.nlm.nih.gov/25233367/]
- 90. Computational protein-ligand docking and virtual with... drug screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC4868550/]
- 91. Incorporating backbone flexibility in MedusaDock improves ligand-binding pose prediction in the CSAR2011 docking benchmark - PubMed [https://pubmed.ncbi.nlm.nih.gov/23237273/]
- 92. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2760638/]
- 93. Interactive Flexible-Receptor Molecular Docking in Virtual Reality Using DockIT - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9749019/]
- 94. - Ultra screening with an evolutionary algorithm in Rosetta... large library [https://www.nature.com/articles/s42004-025-01758-x?error=cookies_not_supported&code=a0a2744a-37f8-4e7f-99a1-abefe2300aa8]
- 95. Spaces • Chemical - Ultra Compound Collections by BioSolveIT Large [https://www.biosolveit.de/chemical-spaces/]
- 96. A guideline for reporting experimental protocols in life sciences - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5978404/]
- 97. analysis of Computational - speed - PMC accuracy tradeoff [https://pmc.ncbi.nlm.nih.gov/articles/PMC9768160/]
- 98. for an automated Protocol pipeline including library... virtual screening [https://pubmed.ncbi.nlm.nih.gov/41206870/]
- 99. Frontiers | Discovering Anti- Cancer via Computational Methods Drugs [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2020.00733/full]
- 100. Force field (chemistry) - Wikipedia [https://en.wikipedia.org/wiki/Force_field_(chemistry)]
- 101. Scoring Functions | SpringerLink [https://link.springer.com/chapter/10.1007/1-4020-4407-0_9]
- 102. Scoring and lessons learned with the CSAR benchmark using an improved iterative knowledge-based scoring function - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3190652/]
- 103. Predicting Biological Activities through QSAR Analysis and Docking-based Scoring - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3445294/]
- 104. targets | Nature Validating cancer drug [https://www.nature.com/articles/nature04873?error=cookies_not_supported&code=e2025a43-35de-4449-9a3d-517ff7e75b25]
- 105. MovableType Software for Fast Free Energy-Based Virtual ... Screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC7781189/]
- 106. Quantitative scoring of differential drug sensitivity for individually... [https://www.nature.com/articles/srep05193?error=cookies_not_supported&code=834a3b54-4791-4256-9ab3-4308ccec5ed3]
- 107. protein–ligand docking and Computational drug virtual ... screening [https://experiments.springernature.com/articles/10.1038/nprot.2016.051?error=cookies_not_supported&code=1e357c3d-8ad5-47a8-9406-169214d9d52a]
- 108. PayloadGenX, a multi - stage virtual hybrid for... screening approach [https://pubmed.ncbi.nlm.nih.gov/40168837/]
- 109. PayloadGenX, a multi-stage hybrid virtual screening approach for payload design: A microtubule inhibitor case study - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1476927125000994]
- 110. Multi-stage structure-based virtual screening approach combining 3D pharmacophore, docking and molecular dynamic simulation towards the identification of potential selective PARP-1 inhibitors | BMC Chemistry | Full Text [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-025-01389-2]
- 111. AI-driven High Throughput Screening for Targeted Drug Discovery [https://oxfordglobal.com/discovery-development/resources/ai-driven-high-throughput-screening-for-targeted-drug-discovery]
- 112. [2309.11687] Large-scale Pretraining Improves Sample Efficiency of... [https://arxiv.org/abs/2309.11687]
- 113. Large-Scale Pretraining Improves Sample Efficiency of Active ... [https://pubmed.ncbi.nlm.nih.gov/38442000/]
- 114. AI viable alternative to traditional small-molecule drug discovery process | Chapman Newsroom [https://news.chapman.edu/2024/04/02/ai-viable-alternative-to-traditional-small-molecule-drug-discovery-process/]
- 115. New anti - cancer candidate revealed with AI and... drug screening [https://www.maxiv.lu.se/article/new-anti-cancer-candidate-revealed-with-ai-drug-screening-and-x-rays/]
- 116. Reducing false positive rate of docking- based by... virtual screening [https://colab.ws/articles/10.1093/bib/bbac626]
- 117. Frontiers | Editorial: Advancing drug with discovery : AI –target... drug [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1721323/full]
- 118. Multimodal AI Predicts Clinical Outcomes of Drug ... - Zitnik Lab [https://zitniklab.hms.harvard.edu/projects/Madrigal/]
- 119. One size does not fit all: revising traditional paradigms for assessing... [https://link.springer.com/article/10.1186/s13321-025-00948-y]
- 120. Probing the Structure-Based Virtual of... Screening Performance [https://www.dovepress.com/benchmarking-the-structure-based-virtual-screening-performance-of-wild-peer-reviewed-fulltext-article-DDDT]
- 121. An Improved Metric and Benchmark for Assessing the Performance ... [https://arxiv.org/html/2403.10478v1]
- 122. Docking and Virtual in Screening | Springer Nature... Drug Discovery [https://experiments.springernature.com/articles/10.1007/978-1-4939-7201-2_18?error=cookies_not_supported&code=3219c06e-17d9-41f4-a23a-f018ea1bbe4b]
- 123. Docking and Virtual Screening in Drug Discovery - PubMed [https://pubmed.ncbi.nlm.nih.gov/28809009/]
- 124. Docking and scoring in virtual screening for drug discovery: methods and applications - PubMed [https://pubmed.ncbi.nlm.nih.gov/15520816/]
- 125. Pairwise Performance Comparison of Docking Scoring ... Functions [https://www.mdpi.com/1420-3049/30/13/2777]
- 126. Computational protein-ligand docking and virtual drug screening with the AutoDock suite - PubMed [https://pubmed.ncbi.nlm.nih.gov/27077332/]
- 127. Docking and Virtual Screening in Drug Discovery: Finding the Right Hit | Deep Origin [https://www.deeporigin.com/blog/docking-and-virtual-screening-in-drug-discovery-finding-the-right-hit]
- 128. New machine learning and physics-based scoring functions for drug discovery | Scientific Reports [https://www.nature.com/articles/s41598-021-82410-1]
- 129. Machine Learning Scoring Functions for Drug Discovery from Experimental and Computer-Generated Protein–Ligand Structures: Towards Per-Target Scoring Functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9966217/]
- 130. Frontiers | Integrating bioinformatics and experimental to... validation [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1614702/full]
- 131. Statistical data presentation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5453888/]
- 132. What is X ? Uses, How It Works & Top... Ray Crystallography [https://www.linkedin.com/pulse/what-x-ray-crystallography-uses-how-works-top-companies-40iqf]
- 133. Structural analysis of human ADAR2-RNA complexes by X - ray ... [https://pubmed.ncbi.nlm.nih.gov/39870445/]
- 134. Insights into the computer -aided drug design and discovery based on... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10473489/]
- 135. statistics, Cancer 2025 [https://pubmed.ncbi.nlm.nih.gov/39817679/]
- 136. in Cancer | AACR 2025 Progress Report Cancer 2025 [https://cancerprogressreport.aacr.org/progress/cpr25-contents/cpr25-cancer-in-2025/]
- 137. Tissue surface area and tumor cell count affect the success rate of the Oncomine Dx Target Test in the analysis of biopsy tissue samples - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7812068/]
- 138. Comparison of the analytical performance between the Oncomine Dx Target Test and a conventional single gene test for epidermal growth factor receptor mutation in nonâ€small cell lung cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7882374/]
- 139. Virtual Screening - MedChemExpress [https://www.medchemexpress.com/virtual-screening.html]
- 140. A method for predicting target drug efficiency in cancer based on the analysis of signaling pathway activation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4745731/]
- 141. False positives in the early stages of drug discovery - PubMed [https://pubmed.ncbi.nlm.nih.gov/20939815/]
- 142. Machine learning classification can reduce false positives in structure-based virtual screening - PubMed [https://pubmed.ncbi.nlm.nih.gov/32669436/]
- 143. Improving small molecule virtual screening strategies for the next generation of therapeutics - PubMed [https://pubmed.ncbi.nlm.nih.gov/29920436/]
- 144. An effective docking strategy for virtual screening based on multi-objective optimization algorithm | BMC Bioinformatics | Full Text [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-10-58]
- 145. Combined strategies in structure-based virtual screening - PubMed [https://pubmed.ncbi.nlm.nih.gov/31995074/]
- 146. A Decade of Discovery [https://www.hws.edu/news/2025/a-decade-of-discovery.aspx]
- 147. Frontiers | Strategies for the drug development of cancer therapeutics [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1656012/full]
- 148. Can a Drug Test Lead to a false ? - Positive .com Drugs [https://www.drugs.com/article/false-positive-drug-tests.html]