Absolute Quantification of Oncogene Expression by qPCR: A Comprehensive Guide for Biomarker Validation and Precision Oncology
This article provides a comprehensive guide for researchers and drug development professionals on implementing absolute quantification methods in qPCR for precise measurement of oncogene expression.
Absolute Quantification of Oncogene Expression by qPCR: A Comprehensive Guide for Biomarker Validation and Precision Oncology
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on implementing absolute quantification methods in qPCR for precise measurement of oncogene expression. It covers the foundational principles that distinguish absolute from relative quantification, detailing why this is critical for reproducible cancer biomarker research. The content explores established and novel methodological approaches, including the standard curve and Single Standard for Marker and Reference (SSMR) methods, alongside digital PCR. A strong emphasis is placed on troubleshooting and optimization to overcome common pitfalls like amplification efficiency variability and suboptimal primer design. Finally, the article validates these techniques through comparative analysis with RNA-Seq and digital PCR, highlighting their indispensable role in clinical applications such as minimal residual disease monitoring and the development of robust prognostic models.
Why Absolute Quantification? Establishing the Need for Precision in Oncology
In the field of oncology research, accurate quantification of gene expression is paramount. Quantitative real-time polymerase chain reaction (qPCR) serves as a gold standard technique for detecting and quantifying nucleic acids, with its utility broadly divided into two methodological approaches: absolute and relative quantification [1] [2]. The choice between these methods has profound implications for data comparability, interpretation, and subsequent scientific conclusions, particularly in sensitive applications like oncogene expression profiling. Absolute quantification provides concrete copy numbers of a target sequence, while relative quantification expresses changes relative to a control sample [3] [4]. This article delineates the core concepts, applications, and practical protocols for both methods, framed within the context of oncogene research.
Core Conceptual Distinctions
The fundamental distinction between absolute and relative quantification lies in the nature of the result. Absolute quantification determines the exact number of target DNA or RNA molecules in a sample, expressed as copy number, mass, or concentration [3] [4]. This method requires a calibration curve from standards of known concentration. In contrast, relative quantification measures the change in target gene expression in a test sample relative to a reference sample (calibrator), typically normalized to one or more reference genes, and reports the result as a fold-change [3] [5].
This core difference dictates their respective applications. Absolute quantification is indispensable in virology (e.g., determining viral load), microbiology (e.g., quantifying microbial adulterants), and clinical diagnostics where a precise numerical value is critical [1]. Relative quantification is the preferred method in functional genomics and transcriptomics, where the primary interest lies in understanding changes in gene expression under different experimental conditions, such as comparing oncogene expression levels between tumor and normal tissue [3] [1].
Table 1: Fundamental Distinctions Between Absolute and Relative Quantification
| Feature | Absolute Quantification | Relative Quantification |
|---|---|---|
| Output | Exact copy number or concentration | Fold-change relative to a calibrator |
| Standard Requirement | External standards with known concentration | Internal reference/reference gene(s) |
| Primary Application | Viral load, microbiological count, copy number variation | Differential gene expression studies |
| Key Assumption | Standard and target amplify with equal efficiency | Reference gene expression is stable across samples |
| Data Normalization | Not required; result is intrinsic | Essential; normalized to reference gene(s) |
Methodological Approaches and Protocols
Absolute Quantification Protocols
Protocol 1: Absolute Quantification Using the Standard Curve Method
This is the most common method for absolute quantification in qPCR [3] [4].
- Standard Preparation: Generate a serial dilution (at least 5 points, 10-fold recommended) of a standard with a known concentration. The standard must be identical or highly similar to the target in terms of amplicon sequence, length, and amplification efficiency [4] [6]. Common standards include:
- Plasmid DNA: Clone the target amplicon into a plasmid. Linearize the plasmid before use. Quantify concentration via spectrophotometry (A260) and calculate copy number using the formula: (X g/µl DNA / [plasmid length in bp x 660]) x 6.022 x 10²³ = Y molecules/µl [4].
- In vitro Transcribed RNA: For mRNA quantification, use RNA standards to account for reverse transcription efficiency. After DNase treatment to remove plasmid DNA, quantify by A260 and calculate copy number with: (X g/µl RNA / [transcript length in nucleotides x 340]) x 6.022 x 10²³ = Y molecules/µl [4].
- qPCR Run: Amplify the standard dilution series and the unknown samples in the same run.
- Standard Curve Generation: Plot the Ct values of the standards against the logarithm of their known concentrations. The software will generate a line of best fit with a slope and R² value.
- Calculation of Unknowns: The qPCR software interpolates the Ct value of the unknown sample against the standard curve to determine its absolute concentration.
Critical Guidelines: The DNA/RNA standard must be pure and accurately quantified. Pipetting for serial dilutions must be precise. Diluted standards should be aliquoted and stored at -80°C to avoid degradation [3].
Protocol 2: The One-Point Calibration (OPC) Method
The standard curve method assumes the amplification efficiency (E) of the standard and the sample are identical, which can lead to significant quantification errors if false [6]. The OPC method corrects for these differences.
- Efficiency Determination: Determine the amplification efficiency (E) for each sample from the fluorescence data using the linear regression method on the dilution series or other software algorithms [5] [6].
- Calibration Point: Use a single known standard concentration.
- Calculation: The target concentration in the sample (N0,sample) is calculated relative to the standard (N0,standard) using the formula: N0,sample = N0,standard × (Estandard)^(Ct,standard) / (Esample)^(Ct,sample) This method has been shown to provide higher accuracy when sample and standard efficiencies differ [6].
Relative Quantification Protocols
Protocol 3: The Comparative Cт (ΔΔCт) Method
This method, popularized by Livak and Schmittgen, is simple but relies on a critical assumption [5] [7].
- Validation Experiment: Before proceeding, it must be verified that the amplification efficiencies of the target gene and the reference gene are approximately equal (within 5%) and close to 100% (E ≈ 2) [5]. This is done by generating a standard curve for both genes with a serial dilution of cDNA. The slope of the curve should be <-3.1 or >-3.6, with an efficiency between 90-110% [5].
- qPCR Run: Amplify the target gene and reference gene for all test and calibrator samples. This can be done in separate wells or, with optimization, in a multiplex reaction.
- Calculation:
Protocol 4: The Pfaffl Method (Efficiency-Corrected Calculation)
When the amplification efficiencies of the target (Etarget) and reference (Eref) genes are not equal, the Pfaffl method must be employed for accurate results [5] [7].
- Efficiency Determination: Calculate the amplification efficiency for both the target and reference gene primers from standard curves as described in Protocol 3 [5].
- qPCR Run: As in Protocol 3.
- Calculation:
This method is more robust as it incorporates actual reaction efficiencies, making it the preferred choice for rigorous gene expression studies, including oncogene research [7].
Table 2: Comparison of Relative Quantification Calculation Methods
| Aspect | Comparative Cт (ΔΔCт) Method | Pfaffl Method |
|---|---|---|
| Core Formula | 2^–ΔΔCт | (Etarget)^(ΔCтtarget) / (Eref)^(ΔCтref) |
| Key Assumption | Etarget = Eref ≈ 2 (100% efficiency) | Accounts for different Etarget and Eref |
| Validation Need | Must demonstrate equal efficiency | Must determine precise efficiency for both genes |
| Advantage | Simplicity, no standard curve needed | Accuracy, flexible for suboptimal efficiencies |
| Limitation | Inaccurate if efficiency assumption is violated | Requires more initial validation and setup |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for qPCR Quantification
| Reagent/Material | Function | Critical Considerations |
|---|---|---|
| Reference Gene Primers | Normalization control in relative quantification. | Must exhibit stable expression across all experimental conditions. Validation with algorithms like geNorm or NormFinder is crucial [5] [2]. |
| Validated Primers/Probes | Specific amplification and detection of the target oncogene. | High amplification efficiency (90-110%), specificity confirmed by melt curve or probe detection. |
| Standard (Plasmid DNA/RNA) | For generating the standard curve in absolute quantification. | Sequence identity to target, accurate initial quantification, and purity are essential [4]. |
| Reverse Transcription Kit | Converts RNA to cDNA for gene expression studies. | Consistent efficiency is critical; use the same kit and protocol for all samples in a study. |
| qPCR Master Mix | Contains enzymes, dNTPs, and buffer for amplification. | Choice of chemistry (SYBR Green vs. TaqMan) depends on need for specificity and multiplexing. |
| Low-Binding Tubes & Tips | Handling of standards and samples, especially for digital PCR. | Prevents loss of low-concentration nucleic acids by adhesion to plastics, reducing variability [3]. |
Experimental Workflow and Data Analysis Visualization
The following diagram illustrates the logical decision-making process for selecting and executing the appropriate quantification method in a qPCR experiment, such as one designed to measure oncogene expression.
qPCR Quantification Method Decision Workflow
Implications for Data Comparability in Oncogene Research
The choice of quantification method directly impacts the nature and comparability of data in oncogene research. Absolute quantification provides a concrete value, such as the number of HER2 transcripts per nanogram of RNA, which can be directly compared across laboratories if standards are harmonized [4]. However, its accuracy is entirely dependent on the quality and accuracy of the external standards.
Relative quantification, while not providing an absolute number, is often more practical for studying expression changes, for instance, in response to a drug treatment. The fold-change result is robust against variations in sample input and RNA quality, as these are normalized by the reference gene [3] [5]. The primary challenge for data comparability here lies in the selection and validation of stable reference genes. Using an inappropriate reference gene whose expression varies with the experimental condition (e.g., a housekeeping gene affected by the cancer phenotype) is a major source of error and invalidates any comparative analysis [5] [2]. It is therefore strongly recommended to validate multiple candidate reference genes and use a geometric mean of several stable genes for normalization [5].
Furthermore, the mathematical approach (ΔΔCт vs. Pfaffl) influences comparability. Data generated using the ΔΔCт method under non-validated efficiency conditions are not comparable to data generated with the more rigorous, efficiency-corrected Pfaffl method [5] [7] [6]. For credible and comparable results in oncogene research, the Pfaffl method is generally advised. Modern statistical packages in R, such as the rtpcr package, facilitate the implementation of these efficiency-corrected models and robust statistical testing, thereby enhancing data reliability and comparability [7].
Both absolute and relative quantification are powerful tools in the molecular oncologist's arsenal. Absolute quantification is unmatched when a definitive copy number is required, but demands scrupulous attention to standard preparation. Relative quantification, particularly the efficiency-corrected Pfaffl method, offers a robust and practical framework for the majority of gene expression studies, such as tracking oncogene modulation. The key to generating comparable, high-quality data lies in transparent reporting of the methods, rigorous validation of reagents (especially reference genes), and the use of appropriate, efficiency-informed calculation models. By adhering to these detailed protocols and understanding the implications of each methodological choice, researchers can ensure their qPCR data on oncogene expression is both reliable and meaningful.
The pursuit of personalized oncology hinges on the discovery and rigorous validation of molecular biomarkers that can accurately predict disease progression and therapeutic response. Within this paradigm, quantitative polymerase chain reaction (qPCR) has emerged as a cornerstone technology for the absolute quantification of oncogene expression, enabling the transition of research findings from the laboratory to the clinical setting. The establishment of reliable prognostic models is critically dependent on the initial identification of candidate genes and their subsequent validation through precise experimental methodologies. This document outlines detailed application notes and protocols for this multi-faceted process, providing researchers with a structured framework for biomarker development. The following sections detail the integrative bioinformatics and experimental pipeline, from high-throughput data analysis to final qPCR validation, with a specific focus on hepatocellular carcinoma (HCC) and breast cancer (BC) as exemplars [8] [9].
Biomarker Discovery & Integrative Bioinformatics Analysis
The initial phase of prognostic model development involves the identification of candidate genes from large-scale transcriptomic datasets.
Data Acquisition and Processing
- Data Sources: Transcriptomic, clinical, survival, and mutation data should be sourced from public repositories such as The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) [8] [9]. For example, the TCGA-HCC dataset includes 363 HCC and 49 normal samples, while the TCGA-BRCA dataset includes 1,096 BC and 113 normal samples [8] [9].
- Gene Lists: Curate disease-specific gene sets from published literature, such as 200 epithelial-mesenchymal transition-related genes (EMTRGs) and 35 anoikis-related genes (ARGs) for HCC, or 781 cytochrome c-related genes (CCRGs) for breast cancer [8] [9].
Identification of Candidate Genes
A multi-step analytical approach is employed to filter and identify robust candidate genes, as visualized in the workflow below.
Diagram 1: Bioinformatics workflow for biomarker discovery.
The key steps involve:
- Differential Expression Analysis: Utilizing tools like DESeq2 to identify genes significantly differentially expressed between tumor and normal tissues (e.g., |log2 Fold Change| > 0.5 and adjusted p-value < 0.05) [8] [9].
- Weighted Gene Co-expression Network Analysis (WGCNA): Constructing a co-expression network to identify modules of highly correlated genes significantly associated with clinical traits of interest, such as EMT or anoikis scores [8].
- Candidate Gene Selection: Selecting the overlapping genes from the differential expression analysis and the key modules identified by WGCNA as high-priority candidates for further analysis [8].
- Prognostic Model Construction: Applying univariate Cox regression and Least Absolute Shrinkage and Selection Operator (LASSO) analysis to the candidate genes to build a multi-gene signature for prognosis prediction [9].
Functional Enrichment Analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses are performed on the candidate genes using the clusterProfiler package to elucidate their biological functions and involvement in cancer-related pathways [8] [9].
Absolute quantification by RT-qPCR is a critical technique for validating the expression levels of identified biomarkers, providing a precise measure of transcript copy number in a sample.
Core Principles and Workflow
The process involves converting RNA to cDNA, followed by qPCR amplification with reference to an absolute standard curve. The key components of the qPCR amplification curve are illustrated below.
Diagram 2: Phases of a qPCR amplification curve.
- Baseline: The initial cycles where fluorescence is background noise [10].
- Exponential Phase: The phase where amplification is most efficient and reproducible [10].
- Threshold: The fluorescence level set above the baseline to define the Ct value [10].
- Ct (Cycle Threshold): The cycle number at which the sample's fluorescence intersects the threshold; it is inversely proportional to the starting quantity of the target transcript [10].
- Plateau Phase: The final phase where reaction components become limited and amplification efficiency drops [10].
Absolute vs. Relative Quantification
The choice between absolute and relative quantification depends on the research question.
Table 1: Comparison of qPCR Quantification Methods
| Feature | Absolute Quantification | Relative Quantification |
|---|---|---|
| Primary Question | How many target transcripts are in the sample? [10] | What is the fold-change in gene expression between samples? [10] |
| Requirement | A standard curve with known copy numbers [10] | A stable reference gene (e.g., Actin, GAPDH) [10] |
| Key Output | Exact copy number of the target gene [10] | Fold-change (e.g., 2.5x upregulation) [10] |
| Common Use Cases | Viral load testing, determining gene copy number [10] | Gene expression studies, developmental biology, diagnostic research [10] |
Experimental Protocol: Absolute Quantification for Biomarker Validation
This protocol is designed to validate the expression of prognostic genes, such as STMN1 and SF3B4 in HCC or CETP and PLAU in breast cancer, using absolute quantification by RT-qPCR [8] [9].
Sample Preparation and RNA Extraction
- Obtain matched tumor and adjacent normal tissue samples from patients, with informed consent.
- Extract total RNA using a commercial kit, ensuring an RNA Integrity Number (RIN) > 8.0 for high-quality samples.
- Quantify RNA concentration and purity using a spectrophotometer (A260/A280 ratio ~2.0).
cDNA Synthesis
- Use 1 µg of total RNA for reverse transcription with a High-Capacity cDNA Reverse Transcription Kit.
- Include a no-reverse transcriptase control (-RT) for each sample to assess genomic DNA contamination.
Standard Curve Preparation for Absolute Quantification
- Clone Target Sequence: Clone the PCR amplicon of the target gene (e.g., STMN1) into a plasmid vector.
- Linearize Plasmid: Linearize the purified plasmid to facilitate in vitro transcription.
- In Vitro Transcription: Synthesize RNA from the linearized template using a T7 or SP6 RNA polymerase kit.
- Purify and Quantify RNA: Purify the transcript and determine its concentration accurately using a fluorometer. Calculate the copy number using the formula:
Copy number/µL = (Concentration (g/µL) / (Transcript length (bp) × 660)) × 6.022 × 10^23 - Create Serial Dilutions: Perform a 10-fold serial dilution of the RNA standard (e.g., from 10^7 to 10^1 copies/µL) in nuclease-free water. Use these to generate the standard curve in the qPCR run.
qPCR Reaction Setup and Data Acquisition
- Reaction Mix: Prepare reactions in triplicate for each standard, sample, and no-template control (NTC). A typical 20 µL reaction contains: 10 µL of 2X SYBR Green Master Mix, 1 µL of forward and reverse primer (10 µM each), 2 µL of cDNA (or standard RNA dilution), and 6 µL of nuclease-free water.
- Thermocycling Conditions:
- Step 1: 95°C for 10 minutes (polymerase activation)
- Step 2: 40 cycles of:
- 95°C for 15 seconds (denaturation)
- 60°C for 1 minute (annealing/extension)
- Melting Curve Analysis: 65°C to 95°C, increment 0.5°C for 5 seconds.
Data Analysis and Validation
- Standard Curve and Efficiency: The qPCR software will generate a standard curve by plotting the Ct values against the log of the starting copy number. The slope of the curve is used to calculate PCR efficiency:
Efficiency (%) = (10^(-1/slope) - 1) × 100. An efficiency between 90% and 105% is considered optimal [10]. - Determine Unknowns: The software will interpolate the copy number in unknown samples from the standard curve.
- Statistical Analysis: Compare the absolute copy numbers of the target genes between tumor and normal groups using an unpaired t-test or Mann-Whitney U test, with statistical significance set at p < 0.05 [8] [9].
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for qPCR-based Biomarker Validation
| Item | Function/Description |
|---|---|
| RNA Extraction Kit | For high-purity, intact total RNA isolation from tissue samples (e.g., TRIzol-based or column-based kits). |
| High-Capacity cDNA Reverse Transcription Kit | Contains all components (reverse transcriptase, buffers, dNTPs, random hexamers) for efficient synthesis of first-strand cDNA from RNA templates. |
| SYBR Green qPCR Master Mix (2X) | A ready-to-use mix containing hot-start DNA polymerase, dNTPs, SYBR Green dye, and optimized buffer for robust and sensitive qPCR amplification. |
| Validated Primer Pairs | Target-specific forward and reverse primers designed to amplify a unique 75-200 bp region of the prognostic gene. Must be tested for specificity and efficiency. |
| In Vitro Transcription Kit | For generating large amounts of specific RNA transcripts from a DNA template to be used as standards for absolute quantification. |
| Nuclease-Free Water | Certified free of nucleases to prevent degradation of RNA and cDNA samples during reaction setup. |
| Plasmid Vector System | For cloning the PCR amplicon to create a template for generating the absolute standard. |
Construction and Application of a Prognostic Risk Model
The validated gene expression data is instrumental in building models to predict patient outcomes.
Risk Score Calculation
A risk score for each patient is calculated using a formula derived from the multivariate Cox regression or LASSO analysis [9]:
Risk score = Σ (Coef_i × Expr_i)
Where Coef_i is the risk coefficient for gene i, and Expr_i is its expression level (e.g., the absolute copy number determined by qPCR).
Model Validation
- Patients are stratified into high-risk and low-risk groups based on the median risk score or an optimal cutoff value.
- Kaplan-Meier survival analysis and log-rank tests are performed to assess the difference in overall survival between the two groups [8] [9].
- The predictive power of the model is evaluated using time-dependent Receiver Operating Characteristic (ROC) curve analysis for 1, 3, and 5-year survival [9].
Integration with Clinical Variables
A nomogram can be constructed to integrate the genetic risk score with key clinical parameters (e.g., TNM stage, age) to provide a quantitative tool for predicting individual patient survival probability [8] [9].
Signaling Pathways and Functional Mechanisms
The prognostic genes often cluster within key oncogenic pathways. For instance, in breast cancer, cytochrome c-related genes may be enriched in pathways like "cytokine-cytokine receptor interaction," which influences apoptosis, immune response, and cell proliferation [9]. The diagram below illustrates a simplified network of transcriptional regulation involving prognostic genes.
Diagram 3: Transcriptional network regulating prognostic genes.
In the field of oncology research, the absolute quantification of oncogene expression via quantitative polymerase chain reaction (qPCR) is a cornerstone for understanding tumorigenesis, patient stratification, and therapy development. The translational value of this research, however, is critically dependent on the achievement of two key types of comparability: sample-to-sample comparability within a single study and cross-study comparability across different laboratories and research initiatives. Inconsistencies in methodology and data analysis can lead to significant discrepancies in published results, undermining the reliability of molecular findings and their application in drug development. This application note details the standardized protocols and analytical frameworks essential for ensuring rigorous, reproducible, and comparable qPCR data in oncogene research.
The Critical Role of Housekeeping Gene Selection
A foundational element for achieving sample-to-sample comparability is the accurate normalization of target gene expression (e.g., oncogenes) against stably expressed internal controls, known as housekeeping genes (HKGs). The improper selection of HKGs is a major source of error and variability in qPCR data [11].
Pitfalls of Commonly Used Housekeeping Genes
Historically, genes like Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin (ACTB) have been used as HKGs based on the assumption of constitutive expression. However, extensive evidence now shows that this assumption is often flawed, particularly in cancer biology [11] [12]. GAPDH, for instance, is not a neutral control but a "pan-cancer marker" whose transcription is influenced by a multitude of factors including insulin, growth hormone, oxidative stress, and tumor protein p53 [11]. Its use for normalization in endometrial cancer and many other tissues is strongly discouraged [11]. Similarly, the expression of ACTB (a cytoskeletal protein) and genes encoding ribosomal proteins (e.g., RPS23, RPS18, RPL13A) can undergo dramatic changes in specific experimental conditions, such as in cancer cells treated with mTOR inhibitors, rendering them categorically inappropriate for normalization in those contexts [12].
Guidelines for Optimal HKG Selection
To ensure robust sample-to-sample comparability, the following practices are recommended:
- Validation is Mandatory: The stability of candidate HKGs must be empirically validated for each specific tissue type, cancer model, and experimental treatment [11] [12].
- Use Multiple HKGs: Relying on a single HKG is a high-risk strategy. Using a combination of at least two validated HKGs for normalization significantly improves the accuracy and reliability of gene expression recalculation [11].
- Identify Condition-Specific Optimal HKGs: Research has demonstrated that the optimal HKG can vary between different cancer cell lines. For example, in mTOR-inhibited A549 lung adenocarcinoma cells, B2M and YWHAZ were the most stable, whereas TUBA1A and GAPDH were best for T98G glioblastoma cells under the same conditions [12]. No single optimal HKG was identified for PA-1 ovarian teratocarcinoma cells, highlighting the necessity for line-specific validation [12].
Table 1: Stability of Candidate Housekeeping Genes in mTOR-Inhibited Cancer Cell Lines
| Cell Line | Most Stable HKGs | Unstable HKGs (to avoid) |
|---|---|---|
| A549 (Lung) | B2M, YWHAZ | ACTB, RPS23, RPS18, RPL13A |
| T98G (Brain) | TUBA1A, GAPDH | ACTB, RPS23, RPS18, RPL13A |
| PA-1 (Ovary) | Not determined (validation required) | ACTB, RPS23, RPS18, RPL13A |
Technical Considerations for qPCR Data Analysis
The method of data analysis following qPCR amplification is another critical determinant of data quality and cross-study comparability.
Accurate Baseline and Threshold Setting
The initial cycles of a qPCR reaction establish the baseline fluorescence, which must be correctly defined to avoid distorting the quantification cycle (Cq) values. The baseline should be set within the early cycles where the fluorescence signal is stable and above any initial reaction stabilization artifacts [13]. The threshold, set within the exponential phase of all amplification plots, must be at a fixed fluorescence intensity for all samples in an experiment. Incorrect baseline or threshold settings can lead to significant inaccuracies in Cq values and, consequently, in fold-change calculations [13].
Choosing a Quantification Method
While the comparative Cq (2−ΔΔCT) method is widely used, its assumption of 100% amplification efficiency for all assays is a major limitation. Variability in amplification efficiency can introduce substantial errors [14]. Alternative methods offer improved accuracy:
- Efficiency-Adjusted Model (Pfaffl Model): This method incorporates the actual, experimentally determined PCR efficiency for each assay, leading to more accurate relative quantification [13].
- Analysis of Covariance (ANCOVA): A flexible linear modeling approach that offers greater statistical power and robustness compared to the 2−ΔΔCT method and is not affected by variability in qPCR amplification efficiency [15].
- Relative Standard Curve Method: This method uses a serial dilution of a reference sample to create a standard curve, against which unknown samples are quantified. It has been shown to provide high accuracy [14].
Table 2: Comparison of qPCR Relative Quantification Methods
| Method | Key Principle | Advantages | Limitations |
|---|---|---|---|
| Comparative Cq (2−ΔΔCT) | Assumes perfect (100%) and equal PCR efficiency for all assays. | Simple, fast, no standard curve needed. | Prone to error if efficiency deviates from 100%. |
| Efficiency-Adjusted (Pfaffl) | Uses actual, calculated PCR efficiencies for target and reference genes. | More accurate than 2−ΔΔCT when efficiencies differ. | Requires determination of individual assay efficiencies. |
| Relative Standard Curve | Quantifies unknowns against a serial dilution standard curve. | High accuracy, does not assume equal efficiency. | Requires running additional standard curves. |
| ANCOVA | Uses a flexible multivariable linear model on raw fluorescence data. | High statistical power, robust to efficiency variance. | Requires raw fluorescence data and more complex analysis. |
Experimental Protocols
Protocol: Validation of Housekeeping Gene Stability
This protocol is designed to identify the most stable HKGs for a specific experimental system.
I. Materials and Reagents
- Research Reagent Solutions:
- RNA Extraction Kit: For high-quality, genomic DNA-free total RNA.
- Reverse Transcription Kit: Includes reverse transcriptase, primers, and buffers for cDNA synthesis.
- qPCR Master Mix: Contains DNA polymerase, dNTPs, buffer, and fluorescent dye (e.g., SYBR Green).
- Primers: Validated, sequence-specific primers for all candidate HKGs and target oncogenes.
II. Procedure
- Experimental Design: Include a range of biological replicates that represent all experimental conditions (e.g., control vs. treated, different cancer cell lines, various tumor grades).
- RNA Extraction and cDNA Synthesis: Isolate total RNA from all samples, ensuring high purity (A260/A280 ratio ~1.8-2.0). Treat with DNase I to remove genomic DNA. Synthesize cDNA using a fixed amount of total RNA (e.g., 1 µg) per reaction.
- qPCR Run: Perform qPCR for all candidate HKGs (e.g., GAPDH, ACTB, B2M, YWHAZ, HPRT, etc.) across all cDNA samples. Include no-template controls (NTCs) for each primer set. Run reactions in technical duplicate or triplicate.
- Data Analysis: Calculate the Cq values for each reaction. Use dedicated algorithms such as geNorm, NormFinder, or BestKeeper to analyze the Cq values and rank the candidate genes based on their expression stability (M-value in geNorm). A lower M-value indicates greater stability.
- Selection: Select the top two or three most stable genes for use in subsequent normalization of oncogene expression.
Protocol: Absolute Quantification of Oncogene Expression Using a Standard Curve
This protocol enables the determination of the exact copy number of an oncogene transcript.
I. Materials and Reagents
- Standard Template: A known quantity of purified PCR product, in vitro transcript, or plasmid containing the oncogene amplicon sequence.
II. Procedure
- Standard Curve Preparation: Prepare a serial dilution (e.g., 5-6 logs) of the standard template with known concentrations (e.g., copies/µL).
- qPCR Run: Amplify the standard dilutions alongside the experimental cDNA samples in the same qPCR run.
- Data Analysis:
- Generate a standard curve by plotting the Cq values of the standards against the logarithm of their known concentrations.
- Determine the PCR efficiency from the slope of the standard curve: Efficiency = [10(−1/slope)] - 1. Ideal efficiency is 90-110%.
- Use the linear equation from the standard curve to calculate the initial concentration (copy number) of the oncogene in each unknown sample.
- Normalization: Normalize the absolute oncogene copy number to the quantity and quality of input RNA. This is optimally achieved by also performing absolute quantification for the validated HKGs and expressing the oncogene data as a ratio (e.g., oncogene copies per copy of HKG).
Visualization of Workflows and Pathways
The following diagrams, generated with Graphviz DOT language, illustrate the logical workflows for the key protocols described.
Title: Workflow for validating housekeeping gene stability
Title: Workflow for absolute quantification using a standard curve
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for qPCR in Oncogene Research
| Reagent / Material | Function / Purpose |
|---|---|
| DNase I Treatment Kit | Degrades contaminating genomic DNA during RNA preparation, preventing false-positive amplification. |
| Reverse Transcription Kit | Converts purified mRNA into stable complementary DNA (cDNA) for use in qPCR amplification. |
| qPCR Master Mix (SYBR Green) | Provides all components necessary for the PCR reaction, including the fluorescent dye that intercalates with double-stranded DNA, allowing for real-time detection. |
| Sequence-Specific Primers | Short oligonucleotides designed to flank the target oncogene or HKG amplicon; critical for specificity and efficiency. |
| Validated HKGs | A panel of pre-tested primer sets for common housekeeping genes to facilitate the initial stability validation screen. |
| Standard Curve Template | A plasmid or purified amplicon of known concentration for generating standard curves in absolute quantification. |
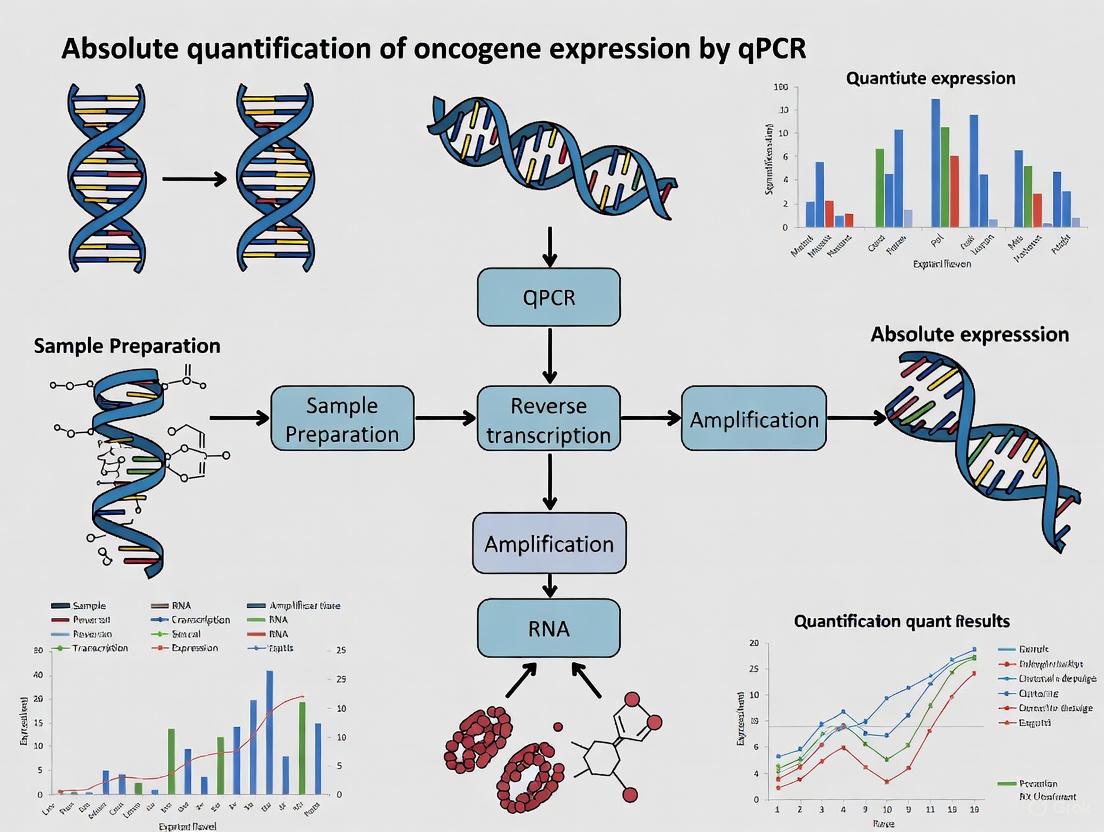
Absolute quantification by qPCR is a powerful method that determines the exact copy number or concentration of a specific nucleic acid target, making it indispensable in oncogene expression studies for precise biomarker quantification and therapeutic monitoring [3] [4]. Unlike relative quantification, which expresses target amount as a ratio to a reference gene, absolute quantification relies on external standards of known concentration to generate a standard curve, enabling direct calculation of target copy numbers in experimental samples [3]. This precision is crucial in oncogene research where subtle expression differences may have significant clinical implications. However, two fundamental challenges critically impact the accuracy and reliability of this approach: significant variability in amplification efficiency across reactions and the complex, resource-intensive process of standard preparation [16]. This article addresses these challenges within the context of oncogene expression analysis, providing detailed protocols and analytical frameworks to enhance data quality and experimental reproducibility.
Challenges in Amplification Efficiency and Standard Preparation
Variability in Amplification Efficiency
Amplification efficiency (E) represents the proportion of template molecules amplified during each PCR cycle, ideally approaching 100% (doubling with each cycle) [17]. In practice, efficiency varies significantly due to multiple factors:
Primary Causes of Efficiency Variation:
- Sample-specific inhibitors that compromise polymerase activity [16]
- Reaction component quality including master mix performance and primer quality [17]
- Template quality and integrity, particularly critical for RNA templates in reverse transcription [4]
- Instrumentation and thermal uniformity across the sample block [18]
The exponential model of PCR amplification has been fundamentally challenged by sigmoidal analysis, which demonstrates that amplification efficiency is not constant but dynamically decreases as amplicon DNA accumulates throughout the reaction [16]. This understanding reframes efficiency as the maximal efficiency (Emax) generated at the onset of thermocycling, requiring revised approaches to efficiency determination and quantification mathematics.
Standard Preparation Complexities
Absolute quantification relies critically on the accuracy and stability of quantitative standards, presenting multiple preparation challenges:
Key Standard Preparation Challenges:
- Accurate initial quantification of stock nucleic acids, particularly vulnerable to contaminants that inflate spectrophotometric measurements [3]
- Precise serial dilution over several orders of magnitude (typically 106-1012-fold), requiring meticulous pipetting technique [3]
- Standard stability, especially for RNA standards which are prone to degradation [3] [4]
- Sequence identity between standards and target to ensure equivalent amplification efficiency [4]
The choice of standard type introduces specific considerations. Plasmid DNA offers production convenience but may not reflect reverse transcription efficiency when quantifying RNA targets. In vitro transcribed RNA standards address this limitation but require stringent DNase treatment to remove template DNA contamination [4]. Each standard type demands specific handling protocols to maintain quantitative integrity throughout the experimental workflow.
Quantitative Comparison of Efficiency Determination Methods
Table 1: Comparison of Amplification Efficiency Determination Methods
| Method | Principle | Key Advantages | Key Limitations | Resource Requirements |
|---|---|---|---|---|
| Standard Curve Method [16] | Positional analysis of serially diluted standards | Current gold standard; high quantitative reliability | Assumes equivalent efficiency in samples; resource intensive | High (multiple reactions per standard) |
| Log-Linear Region Analysis [16] | Exponential mathematics applied to amplification profile | Assesses individual reaction kinetics | Potentially large efficiency underestimations | Low (uses existing amplification data) |
| Linear Regression of Efficiency (LRE) [16] | Sigmoidal analysis of amplification profiles | Accounts for dynamic efficiency; correlates with standard curve | Requires high-quality fluorescence data; less established | Moderate (specialized analysis tools) |
Experimental Protocols
Protocol 1: Determining Amplification Efficiency via Standard Curve
Principle: This method determines amplification efficiency by analyzing the relationship between threshold cycle (Ct) values and the logarithm of known template concentrations in a serial dilution series [16] [17].
Procedure:
- Standard Preparation: Prepare a 5-10 point serial dilution (typically 10-fold dilutions) of your standard material (plasmid DNA containing oncogene insert or in vitro transcribed RNA)
- qPCR Amplification: Amplify the entire dilution series using the same thermal profile as experimental samples
- Data Analysis: Plot average Ct values against the logarithm of the initial template concentration
- Efficiency Calculation: Calculate efficiency using the slope of the standard curve: Efficiency (%) = (10-1/slope - 1) × 100 [17]
Validation Criteria:
- Efficiency Range: 90-110% considered acceptable [17]
- Linearity: Correlation coefficient (R²) ≥ 0.985 [18]
- Slope: -3.1 to -3.6 representing 90-110% efficiency
Protocol 2: Preparation of Plasmid DNA Standards for Absolute Quantification
Principle: Generate quantifiable standards with identical sequence composition to the target oncogene to ensure equivalent amplification efficiency [4].
Procedure:
- Vector Preparation: Clone the target oncogene amplicon into a standard cloning vector containing an RNA polymerase promoter (T7, SP6, or T3)
- Linearization: Linearize the plasmid upstream or downstream of the target sequence to mimic amplification efficiency of genomic DNA or cDNA
- Quantification: Measure DNA concentration by spectrophotometry (A260)
- Copy Number Calculation: Calculate molecular copies using the formula: (X g/μl DNA / [plasmid length in bp × 660]) × 6.022 × 1023 = Y molecules/μl [4]
- Dilution Series: Prepare 5-10 serial dilutions covering the expected target concentration range in experimental samples
- Aliquoting and Storage: Divide diluted standards into small single-use aliquots and store at -80°C to prevent degradation
Critical Considerations:
- Verify insert identity by sequencing
- Ensure spectrophotometric measurements are not inflated by RNA or chemical contaminants
- Use low-binding tubes and tips during dilution to minimize sample loss
Research Reagent Solutions for Absolute Quantification
Table 2: Essential Research Reagents and Materials
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Cloning Vector with Promoter | Standard production | Enables in vitro transcription for RNA standards; provides primer binding sites identical to target [4] |
| RNA Polymerase (T7, SP6, T3) | In vitro transcription | Generates RNA standards for gene expression analysis; accounts for reverse transcription efficiency [4] |
| RNase-free DNase | DNA removal | Eliminates template DNA contamination from in vitro transcribed RNA standards [4] |
| SYBR Green Master Mix | Fluorescent detection | Binds double-stranded DNA during amplification; requires optimization of primer concentrations [7] |
| TaqMan Hydrolysis Probes | Target-specific detection | Provides enhanced specificity through hybridization; suitable for multiplex reactions [7] |
| Low-Binding Plasticware | Sample handling | Minimizes nucleic acid loss during serial dilution preparation; critical for accurate quantification [3] |
Workflow Visualization
Absolute Quantification Workflow
Addressing variability in amplification efficiency and standard preparation represents a foundational requirement for reliable absolute quantification of oncogene expression by qPCR. The integration of robust efficiency determination methods, particularly the standard curve approach as the current gold standard, with meticulous standard preparation protocols provides a framework for enhancing data quality and experimental reproducibility. The implementation of sigmoidal analysis approaches, such as LRE analysis, offers promising alternatives to traditional exponential models by accounting for the dynamic nature of amplification efficiency throughout the reaction profile. For oncogene research applications, where quantitative accuracy directly impacts biological interpretation and potential clinical applications, rigorous attention to these fundamental methodological elements remains essential for generating scientifically valid and translationally relevant data.
Implementing Absolute Quantification: From Standard Curves to Digital PCR
Absolute quantification by quantitative PCR (qPCR) is a powerful method that determines the exact copy number of a specific nucleic acid sequence in a sample, providing crucial data for oncogene expression studies in cancer research and drug development. This approach relies on constructing a standard curve using samples of known concentration, against which unknown samples are quantified [3] [4]. Unlike relative quantification, which expresses gene expression as a fold-change relative to a reference sample, absolute quantification provides concrete copy numbers, enabling direct comparison of results across different experiments and laboratories [3]. This is particularly valuable in preclinical drug efficacy testing, where precise measurement of oncogene expression changes in response to therapeutic compounds is essential [19].
The reliability of absolute quantification hinges entirely on the accuracy and integrity of the standards used. Two of the most common standards are plasmid DNA and in vitro transcribed RNA, each with distinct advantages and appropriate applications [4]. When properly designed and validated, standard curves enable researchers to quantify gene expression with high sensitivity over a wide dynamic range, as demonstrated in drug discovery assays against Leishmania tropica, where standard curves showed linearity over a 9-log concentration range [19]. This technical note details best practices for preparing and using these standards to generate publication-quality data in oncogene expression studies.
Principles of Standard Curve Design
The fundamental principle underlying the standard curve method is the relationship between the quantification cycle (Cq) value and the initial template concentration in the qPCR reaction [3]. During amplification, the point at which the fluorescence signal crosses a predetermined threshold (Cq) is inversely proportional to the logarithm of the initial template concentration. A series of standard dilutions with known concentrations is amplified alongside experimental samples, generating a standard curve from which the concentration of unknowns can be extrapolated [4].
Critical parameters for a reliable standard curve include:
- Linear Dynamic Range: The standard curve should cover the entire expected concentration range of the target in experimental samples [4].
- Amplification Efficiency: Ideally between 90-110%, indicating that the template doubles with each amplification cycle [20] [5].
- Coefficient of Determination (R²): Should be ≥0.985, demonstrating a strong linear relationship between Cq and log concentration [19].
For optimal results, standard curves should consist of at least five data points spanning several orders of magnitude (typically 5- to 10-fold dilutions) to establish a robust linear relationship [21]. Each standard dilution should be run in multiple replicates to account for technical variability, with a minimum of three replicates recommended for reliable quantification [22].
Table 1: Optimal Parameters for Standard Curve Performance
| Parameter | Optimal Value | Acceptable Range | Calculation Method |
|---|---|---|---|
| Amplification Efficiency | 100% | 90-110% | E = 10^(-1/slope) |
| Slope | -3.32 | -3.1 to -3.6 | Linear regression of standard curve |
| R² Value | 1.000 | ≥0.985 | Linear regression of standard curve |
| Dynamic Range | >6 logs | Minimum 5 logs | Concentration range of standards |
| Number of Data Points | 5-7 | Minimum 5 | Serial dilutions of standard |
Plasmid DNA Standards
Preparation of Plasmid DNA Standards
Plasmid DNA is a widely used standard for absolute quantification due to its stability, ease of production, and ability to be accurately quantified [4]. The preparation process begins with cloning the target sequence, including the amplicon and flanking regions, into an appropriate vector. For optimal accuracy, the insert should be generated by RT-PCR from total RNA or mRNA, or by PCR from cDNA, ensuring it includes at least 20 bp upstream and downstream of the primer binding sites used in the qPCR assay [4].
A critical step in preparation is the linearization of plasmid DNA. Supercoiled plasmid conformation can exhibit different amplification efficiency compared to genomic DNA or cDNA, potentially leading to quantification inaccuracies [20] [4]. Linearization with restriction enzymes upstream or downstream of the target sequence produces a template that more closely mimics natural DNA substrates and provides more consistent amplification [4]. After linearization, plasmid concentration should be determined by spectrophotometry at A260, ensuring the A260/A280 ratio is between 1.8-2.0, indicating pure nucleic acid preparation free from protein or RNA contamination [20].
Copy Number Calculation and Dilution Scheme
The copy number of plasmid DNA standards is calculated from the spectrophotometric concentration measurement using the formula:
Copy number/μL = (X g/μL DNA / [plasmid length in bp × 660]) × 6.022 × 10^23 [4]
Where X g/μL is the mass concentration determined by spectrophotometry, plasmid length is in base pairs, 660 Da is the average molecular weight of one base pair, and 6.022 × 10^23 is Avogadro's number. This calculation is essential for converting mass-based concentration measurements to copy numbers relevant for molecular quantification [4].
Serial dilutions should be prepared over a range covering the expected target concentrations in experimental samples, typically spanning 5-6 orders of magnitude [21]. To minimize pipetting error in large dilution series, prepare an initial high-concentration stock and perform sequential dilutions rather than diluting from the original stock each time. Dilution stability must be considered, with aliquots stored at -80°C and subjected to a single freeze-thaw cycle to prevent degradation [3].
Table 2: Troubleshooting Plasmid DNA Standards
| Problem | Potential Cause | Solution |
|---|---|---|
| Poor amplification efficiency | Supercoiled plasmid conformation | Linearize plasmid before use |
| Inconsistent standard curve | Residual RNA contamination | Treat with RNase during purification |
| 10-fold discrepancy in copy number | Using plasmid size instead of amplicon size in calculation | Use amplicon length for copy number calculation [20] |
| High variability between replicates | Plasmid carryover contamination | Use dedicated areas for pre- and post-PCR work |
| Non-linear standard curve | Primer-dimer formation | Redesign primers or add dissociation curve step |
In Vitro Transcribed RNA Standards
Preparation of RNA Standards
In vitro transcribed RNA standards are essential for absolute quantification of gene expression when measuring RNA targets, as they account for the efficiency of the reverse transcription step [4]. To create these standards, the target sequence is first cloned into a vector containing a bacteriophage RNA polymerase promoter (T7, SP6, or T3). The orientation of the insert must be verified to ensure that in vitro transcription produces the sense transcript [4].
Following in vitro transcription, complete removal of template DNA is critical, as residual plasmid DNA will lead to overestimation of RNA concentration and serve as an efficient template in subsequent PCR reactions, skewing quantification results [4]. Treatment with RNase-free DNase is essential, followed by purification of the transcript. RNA integrity should be confirmed by gel or capillary electrophoresis, showing a single discrete band without degradation products or aberrant transcripts [4].
Quantification and Stability of RNA Standards
The concentration of purified RNA is determined by spectrophotometry, and the copy number is calculated using a modified formula that accounts for the different molecular weight of RNA:
Copy number/μL = (X g/μL RNA / [transcript length in nucleotides × 340]) × 6.022 × 10^23 [4]
Here, 340 Da represents the average molecular weight of one ribonucleotide, and transcript length is in nucleotides. RNA standards are particularly labile compared to DNA, requiring careful handling to prevent degradation. Diluted standards should be divided into small single-use aliquots and stored at -80°C to preserve integrity [3]. The inclusion of RNase inhibitors in dilution buffers may enhance stability, though this should be validated to ensure it doesn't interfere with subsequent reverse transcription or PCR reactions.
Alternative Standard Templates
gBlocks Gene Fragments as Standards
Double-stranded gBlocks Gene Fragments offer a versatile alternative to traditional plasmid standards, particularly when rapid standard development or multiplex quantification is required [21]. These synthetic DNA fragments, up to 3000 bp in length, can be designed to incorporate multiple control amplicon sequences into a single construct, enabling quantification of several targets from the same standard [21].
Key advantages of gBlocks Gene Fragments include:
- Design Flexibility: Sequences can be customized with necessary overlaps or restriction sites for downstream applications [21].
- Multi-Target Standards: Multiple control sequences can be combined on a single fragment, reducing pipetting steps and experimental variability in multiplex experiments [21].
- Rapid Production: Compared to cloning, gBlocks fragments can be produced quickly, facilitating assay optimization [21].
- Contamination Control: Artificial sequences distinguishable from wild-type sequences can be designed, enabling detection of contamination through melt curve analysis [21].
When designing gBlocks with multiple targets, sequences should be separated by several intervening T bases, but not more than 9, as this may interfere with manufacturing [21].
PCR Products and Genomic DNA as Standards
Purified PCR products can serve as practical alternatives to plasmid standards, particularly when rapid standard preparation is prioritized [4]. The PCR product should include the target amplicon plus at least 20 bp of flanking sequence on either side to ensure native amplification efficiency [4]. While quicker to produce than cloned standards, PCR products may contain unidentified sequence errors that could affect amplification efficiency and quantification accuracy [21].
Genomic DNA is appropriate as a standard only when the target is present as a single-copy gene per haploid genome and amplification of pseudogenes or related sequences can be excluded [4]. For example, in mouse models, 1 µg of genomic DNA corresponds to approximately 3.4 × 10^5 copies of a single-copy gene, based on the known genome size of Mus musculus (2.7 × 10^9 bp) [4].
Experimental Protocol: Absolute Quantification of Oncogene Expression
Standard Curve Setup and Validation
This protocol outlines the steps for implementing plasmid DNA and RNA standards for absolute quantification of oncogene expression in drug efficacy studies, adapting approaches validated in anti-parasitic drug discovery [19].
Materials Required:
- Linearized plasmid DNA or in vitro transcribed RNA standard
- RNase-free water for dilutions
- qPCR master mix with appropriate chemistry (SYBR Green or probe-based)
- Primers validated for efficiency and specificity
- Nuclease-free low-binding tubes and pipette tips
Procedure:
- Prepare Stock Solution: Resuspend standard in appropriate buffer and quantify by spectrophotometry. Verify purity (A260/A280 ratio of 1.8-2.0).
- Calculate Copy Number: Use the appropriate formula for DNA or RNA standards as described in sections 3.2 and 4.2.
- Serial Dilutions: Prepare a 10-fold serial dilution series covering at least 5 orders of magnitude, with concentrations spanning the expected range in experimental samples.
- qPCR Setup: Aliquot appropriate volume of each standard dilution into qPCR plates, with a minimum of three replicates per dilution.
- Amplification: Run qPCR with optimized cycling conditions including a dissociation curve step for SYBR Green assays.
- Standard Curve Analysis: Plot Cq values against log copy number to generate standard curve. Validate using parameters in Table 1.
Data Analysis and Quality Control
For absolute quantification, the standard curve is used to determine the copy number of unknown samples by interpolating their Cq values against the curve [4]. Several quality control measures must be implemented:
- Efficiency Validation: Amplification efficiency should be calculated from the slope of the standard curve (Efficiency = 10^(-1/slope)) and fall within 90-110% [5].
- Negative Controls: Include no-template controls to detect contamination and no-reverse-transcriptase controls for RNA quantification to assess genomic DNA contamination [20].
- Melt Curve Analysis: For SYBR Green assays, perform dissociation curve analysis to verify amplification of a single specific product and absence of primer-dimers [20].
When quantifying oncogene expression in response to drug treatments, include appropriate controls such as untreated cells and calibrator samples to enable both absolute and relative comparison of expression levels [19].
Table 3: Research Reagent Solutions for Absolute Quantification
| Reagent Type | Specific Examples | Function in Experiment |
|---|---|---|
| Standard Templates | gBlocks Gene Fragments, Plasmid Vectors | Provide known copy number references for standard curve generation [21] [4] |
| Polymerase Systems | Hot Start Taq Polymerase, One-Step RT-qPCR Kits | Enzymatic amplification of target sequences with minimal background |
| Detection Chemistries | SYBR Green, TaqMan Probes | Fluorescent detection of amplified DNA in real-time |
| Nucleic Acid Purification Kits | Plasmid Prep Kits, RNA Extraction Kits | Isolation of high-quality nucleic acids for standard preparation |
| Quality Control Tools | DNase I (RNase-free), RNase Inhibitors | Maintain integrity of standards and prevent degradation |
Application in Oncogene Expression Studies
In oncogene expression research within drug development, absolute quantification provides critical data on transcript copy number changes in response to therapeutic compounds [19]. The standard curve method enables precise measurement of even small expression differences, which is particularly valuable when assessing drug efficacy against low-abundance targets [22]. The high sensitivity of properly validated standard curves allows detection of expression changes earlier in treatment, accelerating preclinical decision-making.
The selection of appropriate standards depends on the specific application: plasmid DNA standards are suitable for DNA targets or when reverse transcription efficiency is accounted for separately, while RNA standards are essential when quantifying RNA expression levels and incorporating reverse transcription efficiency into the quantification [4]. For multiplex studies assessing multiple oncogenes or resistance markers, gBlocks Gene Fragments incorporating multiple target sequences provide consistent quantification while reducing experimental variability [21].
Regardless of the standard chosen, rigorous validation following the best practices outlined in this document ensures generation of reproducible, publication-quality data that meets the stringent requirements of drug development pipelines and regulatory submissions.
The imperative for precise, comparable gene expression data in oncology research is paramount, particularly in the development of prognostic and predictive models for cancer patients. Traditional relative quantification by quantitative reverse transcription PCR (qRT-PCR), which relies on separate standards for marker and reference genes, introduces variability that compromises data integrity and cross-study comparability. This Application Note details the methodology and advantages of the Single Standard for Marker and Reference genes (SSMR) approach, an absolute quantification system designed to overcome these critical limitations. Framed within the context of absolute quantification of oncogene expression, we provide a detailed protocol for implementing SSMR-based qRT-PCR, present quantitative data demonstrating its superior comparability, and equip researchers with the necessary tools to enhance the rigor of their gene expression analyses.
In the shift towards personalized cancer medicine, molecular variables such as gene expression levels are increasingly used alongside clinical variables to model prognosis and explain variations in survival and therapeutic response [23]. While genome-wide studies identify candidate genes, their validation and inclusion in robust prognostic models often employ qRT-PCR due to its sensitivity and quantitative nature [23].
A pivotal challenge in qRT-PCR analysis is data normalization to account for variations in sample input. Traditional relative quantification methods use a reference gene (e.g., ACTB, GAPDH) for normalization, with the expression of both the marker gene and the reference gene quantified using separate, independent standard curves [23] [2]. This approach is fundamentally flawed because it relies on the assumption that the molar concentrations of the two external standards are accurately known and equal. Any inequality, as pointed out in foundational SSMR research, directly translates into incomparable gene expression data, akin to "taking a half-foot ruler as one foot to measure the same person’s height" [23]. This lack of comparability is a significant barrier to establishing prognostic models, a process that often requires combining independent datasets to achieve statistically meaningful sample sizes [23].
The SSMR approach addresses this by enabling absolute quantification through a multigene DNA standard containing the amplicon sequences for both the marker and reference genes ligated together in a single fragment at a one-to-one ratio [23]. This ensures that any variation in the quantity of the standard or the sample affects both genes equally, thereby eliminating a major source of bias and ensuring that normalized gene expression data are directly comparable across different experiments and laboratories.
Theoretical Foundation and Advantages of the SSMR Approach
Pitfalls of Relative Quantification and Conventional Normalization
Traditional relative quantification is susceptible to several sources of error that can distort biological conclusions:
- Inequality of Standards: The core issue is the use of two distinct physical standards for quantification. Any inaccuracy in the quantification or dilution of either standard introduces a systematic error into the final normalized expression value [23].
- Instability of Reference Genes: The expression of commonly used reference genes (e.g., ACTB, GAPDH, various ribosomal proteins) can be highly variable under different experimental conditions, such as pharmacological inhibition of key cellular pathways. For instance, the expression of ACTB, RPS23, RPS18, and RPL13A undergoes dramatic changes in cancer cells treated with dual mTOR inhibitors, rendering them "categorically inappropriate" for normalization [12]. Similar instability has been observed for reference miRNAs like SNORD48 and U6 in endometrial cancer studies [24].
- Assumption of 100% PCR Efficiency: Early relative quantification models assumed perfect PCR efficiency, which is often not the case in practice. While efficiency-adjusted models (e.g., Pfaffl method) exist, they still rely on the integrity of separate standard curves [25].
The SSMR Solution: Conceptual Workflow
The following diagram illustrates the core conceptual difference between the traditional relative quantification method and the SSMR approach, highlighting how SSMR ensures a fixed 1:1 ratio for quantification.
The SSMR method provides two key mechanistic advantages:
- Elimination of Standard-Derived Error: By using a single standard, the quantitative relationship between the marker and reference gene is fixed and known, removing the variability introduced by preparing and quantifying two independent standards [23].
- Independence from Sample and Standard Variation: SSMR-based quantification produces normalized gene expression data that are independent of variations in both the concentration of the cDNA sample and the absolute quantity of the standard used to generate the standard curve. The relative expression ratio remains constant even if the nominal concentration of the standard is miscalculated [23].
Detailed SSMR Protocol for Absolute Quantification of Oncogenes
Research Reagent Solutions
The following table details the essential materials required for implementing the SSMR protocol.
Table 1: Key Research Reagents for SSMR-based qRT-PCR
| Item | Function/Description | Example/Comment |
|---|---|---|
| SSMR DNA Standard | Multigene DNA fragment for absolute quantification. | Clone PCR amplicons of target oncogenes (e.g., PAX6, PTEN) and reference genes (e.g., ACTB, RPS9) into a single plasmid. Amplify, purify, and quantify accurately [23]. |
| Primer Design Software | To ensure specific amplification. | Use software (e.g., PrimerDesigner) to design primers that avoid genomic DNA amplification and processing pseudogenes [23]. |
| qPCR Instrument | Real-time fluorescence detection. | Platforms such as Roche LightCycler or Applied Biosystems StepOne are suitable [23]. |
| qPCR Master Mix | Contains polymerase, dNTPs, buffer, and fluorescent dye. | FAST-START DNA Master SYBR Green I mix or equivalent TaqMan master mixes [23] [26]. |
| Template cDNA | Reverse-transcribed RNA from tumor samples. | Synthesize from high-quality, DNase I-treated total RNA. Use consistent reverse transcription protocols across samples [23]. |
Step-by-Step Experimental Workflow
The complete experimental procedure, from standard preparation to data analysis, is outlined below.
Step 1: SSMR Construct Generation
- Procedure: Select target oncogenes (e.g., VEGFA, EGFR, MMP2) and a validated reference gene (see Section 3.3). Generate PCR amplicons for each gene and ligate them sequentially into a single plasmid vector using standard molecular cloning techniques. The final construct should contain all amplicon sequences in a contiguous fragment.
- Critical Note: The accuracy of the entire assay depends on the precise quantification of this SSMR standard. Use high-precision methods like spectrophotometry (NanoDrop) or fluorometry (Qubit) to determine DNA concentration and calculate molar concentration based on the molecular weight of the construct [23].
Step 2: Primer Design and Validation
- Procedure: Design primers with software to produce amplicons 75-200 bp in length. Crucially, design primers to span exon-exon junctions where possible to prevent amplification of genomic DNA. Validate primer specificity by analyzing melt curves for a single peak and ensure PCR efficiency is between 90% and 110% (slope of -3.6 to -3.1) using a dilution series of the SSMR standard [23] [26].
Step 3: SSMR Standard Dilution
- Procedure: Serially dilute the SSMR stock in 10 mM Tris-HCl (pH 7.5) or a similar low-buffer solution to create a standard curve. A typical 6-point curve from 1x10^6 to 1x10^1 molecules per 4 μL is recommended. Prepare fresh dilutions for each run or aliquot and store at -20°C to avoid freeze-thaw cycles.
Step 4: qPCR Plate Setup
- Reaction Mix (Example for 10 μL):
- 4 μL SSMR standard, cDNA sample, or Nuclease-Free Water (for NTC)
- 1 μL 10X Primer-MgCl2 mix (final: 0.5 μM primers, 2.5-4 mM MgCl2)
- 1 μL FAST-START DNA Master SYBR Green I mix (2X)
- 4 μL Nuclease-Free Water
- Plate Layout: Include the SSMR standard curve in duplicate on every plate. Run all cDNA samples in at least duplicate. Include NTCs for each primer set to check for primer-dimer or contamination [23] [26].
Step 5: qPCR Run
- Cycling Conditions (SYBR Green I, Roche LightCycler):
- Enzyme Activation: 95°C for 10 min
- Amplification (40 cycles): 95°C for 15 sec (denaturation), 60°C for 30-60 sec (annealing/extension with fluorescence acquisition)
- Melt Curve Analysis: 65°C to 95°C with continuous fluorescence acquisition.
Step 6: Data Analysis
- The qPCR software generates a standard curve for each gene by plotting the Cq value against the logarithm of the known starting quantity.
- The absolute copy number for both the marker gene and the reference gene in each cDNA sample is calculated directly from their respective Cq values using the formula:
Quantity (copies) = 10^( (Cq value - Y-intercept) / Slope )[26]. - The final, normalized oncogene expression value is expressed as the ratio of the absolute copy number of the oncogene to the absolute copy number of the reference gene.
Selection and Validation of Reference Genes
The SSMR method, while correcting for standard-related errors, does not negate the need for a stably expressed reference gene. The choice of reference gene is critical and must be empirically validated for your specific cancer model and experimental conditions.
- Commonly Unstable Genes: Genes like ACTB (cytoskeleton) and RPS23, RPS18, RPL13A (ribosomal proteins) have been shown to be unstable in dormant cancer cells induced by mTOR inhibition [12].
- Candidate Genes: Potential stable genes for cancer studies include B2M, YWHAZ, TUBA1A, GAPDH, TBP, and CYC1, but their stability is context-dependent [12].
- Validation: Use algorithms such as geNorm, NormFinder, or BestKeeper to evaluate the expression stability of multiple candidate reference genes across all experimental conditions before selecting the most stable one for your SSMR construct [2].
Experimental Validation and Data Comparison
Quantitative Demonstration of SSMR Robustness
To demonstrate the robustness of the SSMR approach, a foundational study quantified gene expression under two different conditions: using a truthfully diluted SSMR standard and a "falsely" diluted SSMR standard (with half the denoted quantity). The results, summarized below, validate the method's independence from standard concentration errors.
Table 2: SSMR Robustness Against Standard Variation [23]
| Gene Target | SSMR Standard Condition | Calculated Gene Expression in cDNA Sample | Observed Fold-Change | Expected Fold-Change |
|---|---|---|---|---|
| PAX6 | Truthful (T) Dilution | Value = X | 2.0 | 2.0 |
| False (F) Dilution (½ Quantity) | Value = 2X | |||
| RPS9 | Truthful (T) Dilution | Value = Y | 2.0 | 2.0 |
| False (F) Dilution (½ Quantity) | Value = 2Y | |||
| PAX6/RPS9 Ratio | Truthful (T) Dilution | X / Y | 1.0 (Equivalent) | 1.0 (Equivalent) |
| False (F) Dilution (½ Quantity) | 2X / 2Y |
As illustrated, while the absolute expression values for individual genes doubled with the erroneous standard, the normalized ratio (PAX6/RPS9) remained identical. This confirms that the final, biologically relevant result is independent of inaccuracies in the absolute quantification of the standard itself [23].
Cross-Study Comparability in Glioma Research
The critical advantage of the SSMR approach was demonstrated in a study analyzing glioblastoma multiforme (GBM) samples processed in two different laboratories (Lab I and Lab II). Gene expression data were generated using both traditional relative quantification and the SSMR-based absolute quantification. The comparability of the normalized data (log10 ratios) between the two labs was evaluated using statistical equivalence testing (TOST procedure).
Table 3: Comparability of GBM Gene Expression Data Between Two Labs [23]
| Gene Target | Quantification Method | Statistical Equivalence Between Labs? | Notes |
|---|---|---|---|
| Oncogene A | Relative qRT-PCR | No | Incomparable results due to inequality in molar concentration of two separate standards. |
| Oncogene B | Relative qRT-PCR | No | Incomparable results due to inequality in molar concentration of two separate standards. |
| Oncogene C | SSMR-based qRT-PCR | Yes | Approximate comparability achieved. Normalized data is independent of variations in sample and standard quantity. |
The study concluded that while relative quantification failed to produce comparable data, the SSMR-based system ensured the comparability of gene expression data, which is essential for combining independent datasets to build powerful prognostic models with large sample sizes [23].
The SSMR approach represents a significant methodological advancement for the absolute quantification of gene expression in oncology research. By replacing the multiple, independent standards of relative quantification with a single, unified standard, it eliminates a fundamental source of technical variability. This protocol provides researchers with a detailed roadmap to implement this robust method, thereby enhancing the integrity, reliability, and cross-study comparability of their qPCR data. The adoption of SSMR is particularly crucial for the rigorous validation of oncogene expression and the development of robust, clinically relevant prognostic and predictive models in cancer.
The precise measurement of oncogene expression levels is a cornerstone of modern cancer research and therapeutic development. For years, quantitative PCR (qPCR) has been the established method for nucleic acid quantification, providing both relative and absolute quantification approaches. Relative quantification determines the ratio between the amount of target gene and a reference gene, typically a stably expressed endogenous control present in all samples [4]. This method is invaluable for comparing gene expression across different tissues or experimental conditions, such as stimulated versus unstimulated cells. In contrast, absolute quantification measures the exact amount of a target nucleic acid sequence, expressed as copy number or concentration, rather than a ratio [4]. This approach relies on external standards of known concentration to generate a standard curve, against which unknown samples are compared.
While qPCR has proven immensely valuable, absolute quantification using this method presents significant challenges for oncogene research. The requirement for precisely calibrated standard curves introduces variability, and quantification accuracy is highly dependent on logarithmic amplification efficiency during each PCR cycle [27] [28]. When amplification is inhibited by sample impurities or when primer/probe binding is suboptimal due to sequence mismatches, quantification can be significantly underestimated [27]. These limitations become particularly problematic when measuring low-abundance oncogene transcripts or rare mutations, where the highest level of precision is required.
Digital PCR (dPCR) represents a transformative approach that addresses these fundamental limitations. By partitioning a sample into thousands of individual reactions, dPCR enables absolute quantification without standard curves, offering superior precision and sensitivity for critical applications in cancer research and molecular diagnostics [27] [28]. This application note explores the technical foundations, performance advantages, and practical implementation of dPCR for absolute quantification of oncogene expression, providing researchers with comprehensive protocols and analytical frameworks to enhance their quantitative gene expression studies.
Fundamental Principles of Digital PCR
Core Technological Framework
Digital PCR operates on a fundamentally different principle than qPCR. Rather than monitoring amplification in real-time through a bulk reaction, dPCR partitions the PCR mixture into thousands to millions of individual reactions [27]. Through this partitioning, the template molecules are randomly distributed across the reactions such that each partition contains zero, one, or a few target molecules. Following end-point PCR amplification, each partition is analyzed for fluorescence [27]. Partitions containing the target sequence (positive) fluoresce, while those without it (negative) do not. The ratio of positive to negative partitions enables absolute quantification of the target molecules in the original sample through Poisson statistics [27].
This partitioning-based approach provides dPCR with several inherent advantages. First, by eliminating the need for standard curves, dPCR removes a major source of inter-laboratory variability and eliminates the requirement for well-characterized reference materials [27]. Second, because quantification is based on endpoint detection rather than amplification efficiency during the exponential phase, dPCR is less affected by PCR inhibitors that may be present in clinical samples [27] [29]. Third, the partitioning effect enhances the detection of rare mutations by effectively enriching low-abundance targets within individual partitions [29].
Practical Workflow Implementation
The typical dPCR workflow involves several key steps that distinguish it from conventional qPCR. First, nucleic acids are extracted from patient samples using standard methods. The sample is then prepared in a master mix containing primers, probes, and PCR reagents, similar to qPCR preparations. This mixture is partitioned into thousands of individual reactions using microfluidic technology—either through water-in-oil droplet systems (ddPCR) or nanoplate-based systems (ndPCR) [27] [30]. The partitioned samples undergo conventional PCR amplification to endpoint. Following amplification, each partition is analyzed for fluorescence using specialized readers that detect positive versus negative reactions [27]. Finally, the results are analyzed using Poisson statistics to determine the absolute copy number of the target sequence in the original sample [27].
Figure 1: Digital PCR Workflow. The sample undergoes partitioning, amplification, and fluorescence reading before final analysis using Poisson statistics.
Comparative Performance Analysis: dPCR vs. qPCR
Technical Performance Metrics
Direct comparative studies demonstrate distinct performance advantages of dPCR over qPCR, particularly for applications requiring high precision and sensitivity. A 2025 study comparing dPCR and Real-Time RT-PCR for respiratory virus detection during the 2023-2024 tripledemic found that dPCR demonstrated superior accuracy, especially for high viral loads of influenza A, influenza B, and SARS-CoV-2, and for medium loads of RSV [31]. The study also reported greater consistency and precision than Real-Time RT-PCR, particularly in quantifying intermediate viral levels [31]. This enhanced performance is attributed to dPCR's reduced susceptibility to amplification efficiency variations and sample inhibitors.
Further evidence comes from a comparative analysis of different dPCR platforms, which examined the precision of two digital PCR applications for copy number comparisons. The study found that both nanoplate-based (QIAcuity One) and droplet-based (QX200) dPCR platforms demonstrated similar detection and quantification limits and yielded high precision across most analyses [30]. The Limit of Detection (LOD) for the nanoplate-based system was approximately 0.39 copies/μL input, while the droplet-based system showed an LOD of approximately 0.17 copies/μL input [30]. The Limit of Quantification (LOQ) was determined at 1.35 copies/μL input for the nanoplate-based system and 4.26 copies/μL input for the droplet-based system [30].
Application-Specific Advantages in Oncology Research
In the context of oncogene research, dPCR's capabilities align perfectly with the field's most demanding requirements. The technology's exceptional sensitivity enables detection of rare oncogenic mutations present at very low allele frequencies in complex biological samples [29]. This capability is particularly valuable for liquid biopsy applications, where researchers must identify and quantify circulating tumor DNA (ctDNA) fragments that are typically short and present in very low concentrations amid abundant wild-type DNA [32]. Digital PCR can detect down to 0.1% variant allele frequency, and in some specialized applications, as low as 0.001% mutant allele frequency, making it indispensable for cancer early detection, therapeutic response monitoring, and minimal residual disease assessment [29].
Table 1: Comparative Analysis of qPCR and dPCR Performance Characteristics
| Parameter | qPCR | dPCR |
|---|---|---|
| Quantification Method | Relative or absolute via standard curves | Absolute without standard curves |
| Precision | Moderate | High, especially for low-abundance targets [31] |
| Sensitivity | Moderate | High (detection down to 0.1% VAF or lower) [29] |
| Effect of Inhibitors | Significant impact on quantification | Reduced impact [27] |
| Dynamic Range | Large (~7-8 logs) | Smaller due to partition count limitation [27] |
| Multiplexing Capability | High (4-6 colors) | Limited (typically 2 colors) [27] |
| Throughput | High | Moderate to high (increasing with newer systems) |
| Cost per Sample | Lower | Higher |
For copy number variation (CNV) analysis, dPCR provides more sensitive quantification of small fold changes and enables better discrimination between complex somatic copy number variations compared to qPCR [29]. While qPCR offers sufficient resolution for variants with 0 to 5 genomic copies, dPCR excels in quantifying more subtle variations that may have significant clinical implications in oncology [29].
Platform Selection and Experimental Design
dPCR Platform Comparison
Researchers considering dPCR implementation have multiple platform options, each with distinct characteristics and advantages. The two primary technologies are droplet-based digital PCR (ddPCR) and nanoplate-based digital PCR (ndPCR). Droplet-based systems, such as the Bio-Rad QX200, generate thousands of nanoliter-sized water-in-oil droplets that function as independent reaction vessels [31]. In contrast, nanoplate-based systems, such as the QIAcuity from QIAGEN, employ fixed nanowells on a microfluidic chip, facilitating high-throughput processing and seamless integration with automated workflows [31].
A 2025 comparative study of these platforms found that while both offer comparable sensitivity and precision, the QIAcuity system allows for faster setup and reduced sample handling, making it particularly well-suited to high-throughput laboratory environments [31] [30]. The study also highlighted that precision can be influenced by factors such as restriction enzyme selection, noting that HaeIII generally provided higher precision than EcoRI, especially for the QX200 system [30].
Table 2: Digital PCR Platform Comparison
| Platform | Partitioning Method | Partition Number | Reaction Volume | Key Features |
|---|---|---|---|---|
| QX200 (Bio-Rad) | Droplet-based | 20,000 per reaction | 20 μL | Established system, broad application base |
| QIAcuity (QIAGEN) | Nanoplate-based | Up to 26,000 nanowells | 40 μL | Automated, high-throughput, integrated system |
| QuantStudio Absolute Q (Thermo Fisher) | Chip-based | Up to 20,000 partitions | Not specified | Integrated partitioning and thermal cycling |
| RainDrop (RainDance) | Droplet-based | Millions of picodroplets | Not specified | High dynamic range, rare target detection |
Implementation Considerations for Oncogene Research
When implementing dPCR for oncogene quantification studies, several factors require careful consideration. The limited dynamic range of dPCR, constrained by the number of partitions, means that samples with expected high target concentrations may require dilution to achieve accurate quantification [27]. For rare mutation detection, the input DNA amount is critical—higher inputs increase the probability of detecting low-frequency mutations [33]. One validated protocol for ultrasensitive detection of chimeric antigen receptor (CAR) constructs utilized dual-input reactions (20 ng and 500 ng) with a combined analysis approach to achieve consistent target detection around 1 × 10⁻⁵ (0.001%) while maintaining excellent specificity and reproducibility [33].
Assay design principles for dPCR largely follow those for qPCR, with particular attention to avoiding sequence polymorphisms in binding regions that could cause allele dropout [27]. For RNA quantification, the reverse transcription step introduces variability, as not all RNA molecules may be converted to cDNA [27]. The limited multiplexing capacity of most dPCR platforms (typically two colors) necessitates careful panel design, potentially requiring multiple singleplex reactions to interrogate multiple targets [27].
Applications in Oncology Research and Biomarker Development
Liquid Biopsy and Circulating Tumor DNA Analysis
Liquid biopsy represents one of the most impactful applications of dPCR in oncology research. The ability to non-invasively detect and monitor cancer through analysis of circulating tumor DNA (ctDNA) has transformed cancer management and therapeutic development. dPCR enhances liquid biopsy research by providing highly sensitive and precise quantification of DNA molecules, enabling detection of low-abundance genetic mutations and tracking of tumor dynamics [32]. The characteristically short length and low concentration of ctDNA fragments in circulation make dPCR's sensitivity particularly valuable for this application [32].
Commercial dPCR assays specifically designed for liquid biopsy applications, such as the Absolute Q dPCR assays, enable reproducible, specific detection of known somatic mutations with sensitivity down to 0.1% variant allele frequency in genes relevant for cancer research [32]. These preformulated assays offer minimal hands-on time and rapid results, typically within 90 minutes when used with compatible dPCR systems [32]. The robust performance of dPCR in detecting rare mutants against a background of wild-type DNA makes it ideal for monitoring treatment response, quantifying residual tumor burden, and detecting emerging resistance mutations during targeted therapy.
CAR T-Cell Therapy Monitoring
Digital PCR has proven particularly valuable in the emerging field of cell and gene therapy, especially for monitoring chimeric antigen receptor (CAR) T-cell therapies. The in vivo engraftment, expansion, and persistence of CAR T-cells are pivotal components of treatment efficacy, with peak expansion in the first 28 days and subsequent long-term persistence correlating with overall response and durability in hematologic malignancies [33]. Monitoring CAR T-cell levels also helps manage serious adverse effects, as the highest levels observed in clinical cohorts have correlated strongly with the temporal diagnosis of grade 2 and 3 cytokine release syndrome [33].
A validated clinical dPCR assay for CAR T-cell detection demonstrated consistent target detection around 0.001% with excellent specificity and reproducibility, enabling monitoring across multiple time points of early expansion (day 6 to 28) and long-term persistence (up to 479 days) [33]. The assay detected CAR vectors at levels ranging from 0.005% to 74% (vector versus reference gene copies), highlighting the exceptional dynamic range achievable with optimized dPCR protocols [33]. This application exemplifies how dPCR's precision at both high and low target concentrations provides unique insights into therapeutic mechanisms and clinical outcomes.
Figure 2: Oncology Applications of Digital PCR. dPCR enables multiple applications in cancer research and clinical management, leading to improved patient outcomes.
Detailed Experimental Protocol for Oncogene Quantification
Sample Preparation and Assay Design
Nucleic Acid Extraction:
- Extract genomic DNA or total RNA from patient samples using standardized extraction methods compatible with your sample type (blood, tissue, FFPE, etc.).
- For liquid biopsy applications, use specialized circulating nucleic acid extraction kits to maximize yield from plasma samples.
- Quantify extracted nucleic acids using fluorometric methods (e.g., Qubit dsDNA HS Assay) for accurate concentration measurement [33].
- Assess nucleic acid quality through spectrophotometric ratios (A260/A280 ~1.8-2.0) or automated electrophoresis systems.
Reverse Transcription (for RNA Targets):
- For gene expression analysis, convert RNA to cDNA using reverse transcriptase with random hexamers and/or gene-specific primers.
- Use uniform reaction conditions across all samples to minimize technical variability in reverse transcription efficiency.
- Include no-reverse transcriptase controls (-RT controls) to monitor for genomic DNA contamination.
Assay Design Considerations:
- Design primer and probe sets to span exon-exon junctions when working with RNA to specifically amplify spliced transcripts and avoid genomic DNA amplification.
- Validate assay specificity using positive and negative control samples.
- Optimize primer and probe concentrations empirically to achieve optimal separation between positive and negative partitions.
- For multiplex assays, ensure minimal spectral overlap between fluorophores and verify detection specificity for each channel.
dPCR Reaction Setup and Thermal Cycling
Reaction Preparation:
- Prepare master mix according to manufacturer recommendations for your specific dPCR system.
- Include reference gene assay for normalization in oncogene expression studies.
- Include positive and negative controls in each run to ensure assay performance.
- For rare mutation detection, consider using restriction enzymes to improve precision, as studies have shown enzymes like HaeIII can provide higher precision compared to EcoRI [30].
Partitioning and Amplification:
- Load samples into appropriate partitioning devices (cartridges, chips, or plates) according to manufacturer instructions.
- Ensure proper partition formation by verifying uniform droplet/nanowell generation.
- Perform PCR amplification using optimized thermal cycling conditions:
- Initial denaturation: 95°C for 10 minutes
- 40 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing/Extension: 55-60°C for 1 minute (optimize based on assay)
- Final extension: 72°C for 5-10 minutes
- Signal stabilization: 98°C for 10-15 minutes (for probe-based assays)
- Note: Specific cycling conditions may vary by instrument and assay design.
Data Acquisition and Analysis
Fluorescence Reading:
- After amplification, read partitions using the appropriate instrument reader.
- Verify data quality by assessing:
- Separation between positive and negative partitions
- Total partition count
- Background fluorescence levels
Data Analysis:
- Use manufacturer's software to automatically classify positive and negative partitions.
- Apply Poisson correction to account for multiple targets per partition, especially in samples with high target concentration.
- For absolute quantification, calculate copies per microliter of input sample using the formula derived from Poisson statistics:
- Concentration = −ln(1 − p) × (total partitions / reaction volume)
- Where p is the fraction of positive partitions
- Normalize oncogene expression levels to reference genes for expression studies.
- For rare mutation detection, calculate variant allele frequency as (mutant copies / total copies) × 100.
Quality Control Measures:
- Monitor partition count consistency across samples (e.g., >10,000 partitions for reliable quantification).
- Verify negative control samples show minimal false positives (<3 positive partitions typically acceptable).
- Confirm positive control samples fall within expected concentration ranges.
- Assess technical replicate consistency (CV < 10% typically acceptable).
Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for dPCR Experiments
| Reagent/Material | Function | Considerations |
|---|---|---|
| dPCR Master Mix | Provides enzymes, nucleotides, and buffer for amplification | Select probe-based or EvaGreen chemistry based on application |
| Primer/Probe Sets | Target-specific amplification and detection | Optimize concentrations; FAM/HEX/VIC common for multiplexing |
| Nucleic Acid Extraction Kits | Isolation of high-quality DNA/RNA from samples | Select based on sample type (blood, tissue, FFPE, etc.) |
| Restriction Enzymes | Improve DNA accessibility and precision | HaeIII may provide better precision than EcoRI [30] |
| Digital PCR Plates/Cartridges | Sample partitioning and amplification | Platform-specific consumables |
| Positive Control Templates | Assay performance verification | Synthetic oligonucleotides or characterized reference materials |
| Reference Gene Assays | Normalization of sample input | Select stably expressed genes appropriate for your sample type |
Digital PCR represents a significant advancement in nucleic acid quantification technology, offering absolute quantification without standard curves and exceptional precision for detecting low-abundance targets. In oncogene research, these capabilities translate to more reliable measurement of gene expression, more sensitive detection of rare mutations, and improved monitoring of therapeutic interventions like CAR T-cell therapy. While factors such as throughput, cost, and dynamic range may still make qPCR the preferred choice for some applications, dPCR has established itself as the gold standard for applications demanding the highest levels of sensitivity and precision. As the technology continues to evolve with improvements in multiplexing, automation, and data analysis, dPCR is poised to play an increasingly central role in cancer research, biomarker development, and clinical diagnostics.
The absolute quantification of gene expression by quantitative polymerase chain reaction (qPCR) is a foundational technique in molecular biology, playing a critical role in oncogene research and drug development. This method determines the exact copy number of a specific nucleic acid sequence within a sample, providing essential data for understanding gene dosage effects in cancer pathways [3] [4]. Absolute quantification can be performed using different methodological approaches, primarily the standard curve method and digital PCR, each with distinct advantages and requirements [3]. The core principle underlying qPCR quantification is the mathematical relationship between the original amount of target nucleic acid and the cycle threshold (Ct) value at which amplification is detected, expressed as Quantity ~ e^(-Ct), where 'e' represents the geometric amplification efficiency [34].
Calibration is the process that establishes the relationship between the instrument's signal response and the analyte's known concentration, forming the basis for accurate quantification [35]. In multi-point calibration, a series of standards with known concentrations are analyzed to construct a calibration curve that spans the expected concentration range of unknown samples. This approach minimizes the effect of errors in individual standards and does not assume a perfectly linear response across all concentrations [35]. In contrast, single-point calibration utilizes just one standard of known concentration to determine the response factor, which is then applied to calculate unknown concentrations. While simpler and more rapid, this method carries significant limitations, as any error in characterizing the single standard propagates directly into all subsequent calculations, and it inherently assumes a perfectly linear relationship between signal and concentration across the entire measurement range [35]. The concept of one-point calibration has shown utility in other analytical fields, such as laser-induced breakdown spectroscopy, where it serves as a compromise between full calibration curves and completely standard-less approaches [36].
The Critical Role of PCR Efficiency
Understanding Amplification Efficiency
PCR amplification efficiency is a fundamental parameter in qPCR that significantly impacts quantification accuracy. Efficiency is defined as the ratio of target molecules at the end of a PCR cycle to the number at the start of that cycle, expressed as a value between 1 and 2 or as a percentage between 0% and 100% [34]. Ideal amplification efficiency of 100% (corresponding to a value of 2) indicates perfect doubling of the target sequence each cycle during the exponential amplification phase. Deviations from this ideal efficiency introduce substantial errors in quantification; for instance, a Ct value of 20 calculated with 80% efficiency versus 100% efficiency results in an 8.2-fold difference in calculated quantity [34]. This dramatic effect underscores why precise efficiency assessment is non-negotiable for reliable absolute quantification, particularly in oncogene research where subtle expression differences may have clinical significance.
Efficiencies exceeding the theoretical maximum of 100% are mathematically possible when calculated from standard curves but are physically impossible in practice. These anomalous results typically indicate methodological problems, most commonly polymerase inhibition in more concentrated samples, where contaminants from nucleic acid isolation (e.g., ethanol, phenol, heparin, or hemoglobin) interfere with enzyme activity [37]. As inhibitors become diluted, efficiency measurements often normalize, revealing the true amplification efficiency. Other factors contributing to apparent efficiencies over 100% include pipetting errors, enzyme activators, reverse transcriptase inhibition, inaccurate dilution series, and formation of unspecific products or primer dimers [37].
Assessing PCR Efficiency Accurately
Robust efficiency assessment requires strategic experimental design to minimize imprecision. Research demonstrates that efficiency estimation varies significantly across different qPCR instruments, with uncertainty potentially as high as 42.5% (95% confidence interval) if standard curves with only a single qPCR replicate are used across multiple plates [38]. Key recommendations for precise efficiency assessment include: (1) generating one robust standard curve with at least 3-4 qPCR replicates at each concentration; (2) recognizing that efficiency is instrument-dependent but reproducibly stable on a single platform; and (3) using larger transfer volumes (2-10μL) when constructing serial dilution series to reduce sampling error and enable calibration across wider dynamic ranges [38].
The most common method for efficiency determination utilizes standard curves with the formula: E = 10^(-1/slope), where the slope is derived from a plot of Ct values against the logarithm of known concentrations [34]. A slope of -3.32 corresponds to 100% efficiency, with steeper slopes indicating lower efficiencies [34]. Alternative assessment methods include the User Bulletin #2 approach, which corrects for potential pipette calibration error by subtracting slopes of two standard curves from the same dilution series, and visual assessment of amplification plot parallelism, which offers the advantage of not requiring standard curves while providing a qualitative efficiency evaluation [34].
Table 1: Methods for Assessing qPCR Amplification Efficiency
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| Standard Curve Efficiency | Calculated from slope of Ct vs. log(quantity) plot | Provides quantitative efficiency value | Prone to errors from inhibitors, pipetting inaccuracies, and dilution errors |
| Visual Assessment | Examination of parallelism in logarithmic amplification plots | No standard curves needed; not impacted by pipette calibration errors | Qualitative assessment without numerical efficiency value |
| User Bulletin #2 | Subtraction of slopes from two standard curves from same dilution series | Corrects for pipette calibration error | Does not correct for other error sources; still error-prone |
One-Point Calibration Methodology
Theoretical Basis and Workflow
The One-Point Calibration method represents a strategic compromise in qPCR quantification, positioned between the comprehensive multi-point calibration and the efficiency-assuming ΔΔCt method. Its fundamental principle involves using a single calibration standard of known concentration and matrix composition similar to the test samples to empirically determine essential experimental parameters that are often difficult to characterize precisely [36]. In the context of absolute quantification for oncogene research, this approach can compensate for efficiency differences between standard and sample that would otherwise introduce systematic errors in copy number determination.
The OPC method operates by establishing a response factor from the single calibration point, which is then applied to unknown samples analyzed under identical conditions. This approach implicitly captures the effective amplification efficiency for the specific assay conditions, reagent batch, and instrument platform at the time of analysis. For oncogene expression studies, this can be particularly valuable when working with difficult sample matrices or when standardized reference materials are limited. The method shares conceptual similarities with approaches used in other analytical fields where a single reference material calibrates the system response while maintaining adaptability to variations in analytical parameters [36].
Experimental Protocol for OPC in Oncogene Quantification
Protocol: Absolute Quantification of Oncogene Expression Using One-Point Calibration
I. Calibration Standard Preparation
- Standard Selection: Utilize a certified reference material for your target oncogene (e.g., plasmid DNA with cloned oncogene sequence, in vitro transcribed RNA, or genomic DNA with known copy number) [4]. For plasmid DNA standards, linearize the vector upstream or downstream of the insert to more closely mimic genomic DNA amplification characteristics [4].
- Concentration Verification: Precisely determine standard concentration using spectrophotometry (A260 measurement). Calculate copy number using the formula: (X g/μl DNA / [plasmid length in base pairs × 660]) × 6.022 × 10²³ = Y molecules/μl [4].
- Dilution Scheme: Prepare a single working dilution of the standard that falls within the expected concentration range of test samples. While OPC uses one point for calibration, verification dilutions are recommended to confirm appropriate concentration.
II. Sample Preparation and Nucleic Acid Extraction
- Sample Types: Process test samples (cell lines, tumor tissues, liquid biopsies) alongside calibration standard using identical extraction methods.
- Quality Control: Assess nucleic acid purity spectrophotometrically (A260/A280 ratios >1.8 for DNA, >2.0 for RNA) to identify potential PCR inhibitors [37].
- Reverse Transcription: For RNA targets, use consistent reverse transcription conditions across all samples with sufficient controls.
III. qPCR Setup and Execution
- Reaction Composition: Prepare master mix containing all qPCR components (polymerase, dNTPs, buffer, primers, probe) to minimize pipetting variability.
- Plate Layout: Include calibration standard in triplicate, test samples in duplicate or triplicate, no-template controls (NTC), and potentially a dilution series for efficiency verification.
- Cycling Conditions: Apply optimized thermal cycling parameters consistent with assay validation data.
IV. Data Analysis and Quantification
- Ct Acquisition: Determine Ct values using consistent threshold settings across all runs.
- Response Factor Calculation: Calculate the response factor (RF) from the calibration standard: RF = Copy Number of Standard / 2^(-Ct_standard).
- Unknown Sample Quantification: Apply the response factor to test samples: Copy Numberunknown = RF × 2^(-Ctunknown).
V. Quality Assurance Measures
- Acceptance Criteria: Establish predefined criteria for standard deviation of calibration standard replicates (<0.2 Ct), NTC contamination checks, and amplification curve shape assessment.
- Efficiency Tracking: Monitor the implicit efficiency calculated from the standard: E_implicit = 10^(-1/slope), where slope is derived from the relationship between the log copy number and Ct of the standard.
Table 2: Troubleshooting Common Issues in One-Point Calibration
| Problem | Potential Causes | Solutions |
|---|---|---|
| High variability in standard replicates | Pipetting inaccuracies, poor mixing, bubble formation | Use calibrated pipettes, mix thoroughly, centrifuge plates before run |
| Discrepancy between expected and calculated standard quantity | Standard degradation, inaccurate initial quantification, dilution error | Verify standard stability, use multiple quantification methods, prepare fresh dilutions |
| Inhibition in test samples | Carryover of contaminants from extraction | Purify samples, use inhibitor-tolerant master mixes, dilute samples |
| Efficiency differences between standard and sample | Matrix effects, secondary structures | Use matrix-matched standards, optimize primer design, include SPUD assay |
Comparative Analysis of Quantification Methods
Method Selection Framework
Choosing the appropriate quantification approach requires careful consideration of experimental goals, resource constraints, and required precision levels. The One-Point Calibration method occupies a specific niche in this landscape, offering a balanced solution for certain research scenarios commonly encountered in oncogene studies and drug development workflows.
Table 3: Comparison of qPCR Quantification Methods for Oncogene Research
| Parameter | One-Point Calibration | Multi-Point Calibration | ΔΔCt Method (Relative) | Digital PCR (Absolute) |
|---|---|---|---|---|
| Quantification Type | Absolute | Absolute | Relative | Absolute |
| Standards Required | One point of known concentration | Multiple points (≥5 recommended) | No standards for quantification | No external standards |
| Efficiency Compensation | Empirical determination from standard | Calculated from curve slope | Assumed equal or separately determined | Not required |
| Throughput | Medium | Low | High | Low |
| Precision | Moderate | High | High for relative changes | Very High |
| Best Applications | Medium-throughput absolute quantification with limited standard material | High-precision absolute quantification | Gene expression fold-changes in well-defined systems | Rare allele detection, complex mixtures, precise copy number variation |
The selection of an appropriate quantification method must align with research objectives. Absolute quantification methods, including OPC and multi-point calibration, are essential when determining exact copy numbers of oncogenes or when establishing diagnostic thresholds [4]. In contrast, relative quantification suffices for comparing gene expression across different treatment conditions or tissue types when the focus is on fold-change differences rather than absolute values [3] [4]. Digital PCR provides the highest precision without requiring external calibration but with lower throughput and higher cost [3].
Practical Implementation Considerations
Successful implementation of the One-Point Calibration method requires attention to several critical practical aspects. The accuracy of quantification fundamentally depends on the quality and characterization of the calibration standard. Plasmid DNA standards should be linearized to better mimic the amplification characteristics of genomic DNA or cDNA targets, as supercoiled plasmid conformation can exhibit different amplification efficiency [4]. For RNA quantification, RNA standards are preferred over DNA standards because they account for variable reverse transcription efficiency, a significant source of quantification error [4].
The single calibration point should be positioned within the linear dynamic range of the assay and ideally near the center of expected sample concentrations to minimize extrapolation error. While the OPC method uses a single point for the calibration calculation, including additional verification points at different concentrations strengthens the quality assurance by confirming appropriate assay performance across the measurement range. This approach provides some validation of the linearity assumption without requiring a full standard curve.
When applying the OPC method across multiple assay runs, consistency in reagent lots, instrument calibration, and technical personnel improves result comparability. For long-term studies, establishing a quality control system with archived calibration standards tracked over time helps monitor and correct for assay drift. In regulated drug development environments, more comprehensive validation including full standard curves may be necessary to meet regulatory standards, though OPC can serve as a valuable routine monitoring tool once assays are properly validated.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Materials for One-Point Calibration qPCR
| Reagent/Material | Function | Implementation Notes |
|---|---|---|
| Certified Reference Standards | Calibrator for absolute quantification | Plasmid DNA, in vitro transcribed RNA, or genomic DNA with known copy number; verify sequence identity to target |
| Nuclease-Free Water | Diluent for standards and reactions | Use ultra-pure grade to prevent enzymatic degradation and contamination |
| Low-Binding Tubes and Tips | Liquid handling for standards | Minimizes adsorption losses, critical for accurate dilution of low-concentration standards |
| qPCR Master Mix | Enzymatic amplification | Select inhibitor-tolerant formulations for complex biological samples; verify compatibility with detection chemistry |
| Target-Specific Primers/Probes | Sequence-specific amplification | Design following universal guidelines (e.g., amplicon size 50-150 bp, Tm optimization) for 100% efficiency |
| RNA Integrity Assessment Tools | Quality control for RNA samples | Bioanalyzer, RIN assessment; critical for gene expression studies to ensure comparable sample quality |
| Digital PCR System (Optional) | Independent quantification verification | Validates calibration standards and provides orthogonal confirmation for critical results |
The One-Point Calibration method represents a pragmatically balanced approach for absolute quantification of oncogene expression in qPCR applications. By combining the simplicity of single-standard calibration with empirical determination of amplification efficiency, OPC addresses a critical need in research and diagnostic settings where resource constraints or sample limitations preclude full multi-point calibration, yet absolute quantification is necessary. The method's effectiveness hinges on rigorous standard characterization, consistent experimental execution, and appropriate quality control measures. When implemented with careful attention to the protocols and considerations outlined herein, OPC provides researchers and drug development professionals with a reliable tool for generating quantitatively accurate oncogene expression data that can inform biological understanding and therapeutic development.
The absolute quantification of key oncogenes and tumor suppressor genes, such as EGFR and p53, via quantitative PCR (qPCR) represents a cornerstone of modern molecular oncology. This approach provides a precise count of specific nucleic acid targets, offering critical insights into tumor biology, prognostication, and therapeutic decision-making [4]. Unlike relative quantification, which expresses changes as a ratio to a reference gene, absolute quantification determines the exact copy number of a target sequence, making it indispensable for applications requiring precise thresholds, such as measuring viral loads, determining gene copy number variations, or quantifying circulating tumor DNA (ctDNA) for minimal residual disease monitoring [4] [39].
The emergence of ctDNA as a non-invasive biomarker for tracking tumor burden and treatment response has further underscored the need for rigorous absolute quantification techniques [40]. Accurate measurement of ctDNA presents a significant challenge, as it requires discriminating rare, tumor-derived mutations from a vast background of wild-type DNA [41]. This protocol details the application of absolute qPCR for the sensitive and reliable quantification of mutations in the EGFR and p53 genes, which are pivotal in numerous cancer types, including non-small cell lung cancer (NSCLC) and a wide spectrum of other malignancies [42] [41].
Key Principles of Absolute Quantification
Absolute quantification in qPCR relies on the construction of a standard curve using known concentrations of a reference standard to determine the exact quantity of an unknown target in a sample [4]. The process involves several key steps and considerations:
- Standard Curve Generation: A dilution series of at least five different concentrations of a standard is amplified alongside the unknown samples [4]. The standard must have identical primer and probe binding sites, a highly similar or identical sequence between the primers, and equivalent amplification efficiency to the target molecule [4].
- Calculation of Copy Number: The Cycle Threshold (Ct) values of the standard dilutions are plotted against the logarithm of their known concentrations. The Ct value of the unknown sample is then interpolated from this curve to determine its initial quantity [4]. For DNA standards, such as linearized plasmids, the copy number can be calculated using the formula: (X g/µl DNA / [plasmid length in base pairs x 660]) x 6.022 x 10^23 = Y molecules/µl [4].
- Data Analysis Fundamentals: Accurate quantification is contingent on proper setup of the qPCR run. This includes baseline correction, which accounts for background fluorescence during the initial cycles, and appropriate threshold setting, which should be placed within the logarithmic, linear phase of amplification where the curves for all samples are parallel [43].
Application Note: Detection of EGFR T790M Mutation
Background
The EGFR T790M mutation is a leading mechanism of acquired resistance to first-generation EGFR tyrosine kinase inhibitors in NSCLC patients [41]. Detecting and quantifying this mutation in ctDNA allows for non-invasive therapy monitoring and timely intervention with third-generation inhibitors. The challenge lies in achieving ultra-sensitive detection against a high background of wild-type DNA.
Methodology: TEAM-PCR
An ultrasensitive method termed Triple Enrichment Amplification of Mutation PCR (TEAM-PCR) has been developed to address this challenge [41]. This qPCR-based method was validated to quantify the EGFR T790M mutation over an assay range of 25-1,000,000 copies per reaction, even in the presence of 1,000,000 copies of wild-type DNA [41]. The Limit of Detection (LOD) for this assay was established at five copies per reaction, demonstrating exceptional sensitivity [41].
Performance Data
Table 1: Performance characteristics of the TEAM-PCR assay for EGFR T790M detection.
| Parameter | Result | Description |
|---|---|---|
| Assay Range | 25 - 1,000,000 copies/reaction | Linear range of quantification. |
| Wild-type DNA Background | Up to 1,000,000 copies | Robust performance in high wild-type background. |
| Limit of Detection (LOD) | 5 copies/reaction | The lowest quantity reliably detected. |
| Validation | Followed essential bioanalysis guidance | Ensures reliability for clinical applications. |
Application Note: Detection of p53 Mutation
Background
The p53 tumor suppressor gene is mutated in approximately 50% of all human cancers, making it one of the most critical genetic drivers of oncogenesis [42]. Its detection is therefore highly valuable for early cancer screening and diagnosis.
Methodology: NATR-NER/Cas12a System
To overcome limitations of traditional nucleic acid amplification technologies, which can suffer from nonspecific amplification, a novel fluorescent platform was developed [42]. This method couples a Nicking endonuclease-assisted target recycling (NATR)-triggered no-nonspecific exponential rolling circle amplification (NER) reaction with a CRISPR/Cas12a system (NATR-NER/Cas12a) [42].
- Workflow: The presence of the target p53 gene triggers the endonuclease Nt.BstNBI to cleave circular single-stranded DNA (ssDNA) preprimers into linear fragments. These fragments then act as primers to initiate the NER reaction, generating abundant short ssDNA fragments. These fragments are recognized by the CRISPR/Cas12a system, activating its trans-cleavage activity and producing a fluorescence signal [42].
- Performance: This strategy demonstrated a wide linear range (10 fM–1 nM) and an exceptionally low detection limit of 0.77 fM. It also specifically recognized single-base mismatched DNA, highlighting its high fidelity. In serum samples, results showed good agreement with standard qPCR but at a lower cost [42].
Performance Data
Table 2: Performance characteristics of the NATR-NER/Cas12a assay for p53 mutation detection.
| Parameter | Result | Description |
|---|---|---|
| Linear Range | 10 fM – 1 nM | Broad dynamic range for quantification. |
| Limit of Detection (LOD) | 0.77 fM | Ultra-high sensitivity. |
| Specificity | Recognized single mismatched DNA | High fidelity for point mutations. |
| Validation in Serum | Good agreement with qPCR | Cost-effective and clinically applicable. |
Detailed Experimental Protocol
Workflow for Absolute qPCR Quantification
The following diagram outlines the core workflow for absolute quantification of oncogenes using qPCR, from sample preparation to data analysis.
Step-by-Step Procedure
This protocol provides a generalized guide for absolute quantification of targets like EGFR or p53. Specific reagent optimization will be required.
Standard Preparation
- Type: For DNA targets (e.g., mutant EGFR), use a linearized plasmid containing the target sequence. For RNA/cDNA targets, use in vitro transcribed RNA standards [4].
- Quantification: Precisely determine the concentration of the standard via spectrophotometry (e.g., Nanodrop) [4].
- Calculation: Calculate the copy number/µL using the appropriate formula. For plasmid DNA: (X g/µl DNA / [plasmid length in base pairs x 660]) x 6.022 x 10^23 = Y molecules/µl [4].
- Dilution Series: Perform a serial dilution (e.g., 1:10) of the standard in nuclease-free water to create at least 5 data points covering the expected concentration range of your samples.
Sample Nucleic Acid Extraction
- Extract DNA from your sample source (e.g., tumor tissue, plasma for ctDNA) using a validated method [41] [40]. For ctDNA, use kits specifically designed for cell-free DNA to maximize yield of short fragments.
- If using plasma, spike in a known concentration of quantification standards (QSs) before extraction to correct for sample loss during purification, as is done in quantitative NGS (qNGS) protocols [40].
qPCR Reaction Setup
- Perform reactions in triplicate for both the standard curve dilutions and the test samples.
- Use a reaction mix that includes a master mix (polymerase, dNTPs, buffer), sequence-specific primers and probes (e.g., TaqMan), and template DNA (standard or sample) [4] [41].
- Use a thermal cycler protocol appropriate for your primers, probe, and polymerase. A typical two-step protocol might be: 1. Initial Denaturation: 95°C for 2-10 minutes; 2. Amplification (40-50 cycles): Denature at 95°C for 15 seconds, Anneal/Extend at 60°C for 1 minute.
Data Analysis
- Baseline and Threshold: In the qPCR software, set the baseline cycles (e.g., 5-15) and place the threshold within the exponential phase of amplification where all curves are parallel [43] [39].
- Standard Curve: The software will generate a standard curve by plotting the Ct values of the standards against the log of their known concentrations. A slope of -3.3 indicates 100% PCR efficiency. Efficiencies between 85-110% are generally acceptable [39].
- Interpolation: The software will interpolate the concentration of the unknown samples based on their Ct values and the standard curve equation, providing an absolute quantity (e.g., copies/µL) for each sample [4].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents and materials required for absolute qPCR of oncogenes.
| Reagent/Material | Function | Examples & Notes |
|---|---|---|
| Standard (Plasmid DNA or RNA) | Serves as the calibrator for generating the standard curve. Must be of known concentration and sequence-identical to the target [4]. | Linearized plasmid with cloned target sequence (e.g., EGFR T790M) [4]. |
| Sequence-Specific Primers & Probes | Enables specific amplification and detection of the target oncogene or mutation. | TaqMan probes are commonly used for high specificity [4] [41]. |
| qPCR Master Mix | Contains core components for amplification: DNA polymerase, dNTPs, and optimized buffer. | Should be selected for high efficiency and robustness, especially for ctDNA analysis. |
| Nucleic Acid Extraction Kit | Isulates DNA/RNA from complex biological samples. | For ctDNA, use specialized cell-free DNA kits [40]. |
| Digital PCR (dPCR) System | (Optional) For ultra-sensitive quantification and independent validation. Can be used for absolute quantification without a standard curve [40] [44]. | Used in advanced methods like TEAM-PCR for validation [41]. |
| CRISPR/Cas12a System | (For novel methods) Provides high-fidelity detection and signal amplification when coupled with pre-amplification methods like RCA [42]. | Components include Lba Cas12a enzyme and specific crRNA [42]. |
Advanced Techniques and Comparison
The field of absolute quantification is evolving beyond traditional qPCR. Digital PCR (dPCR) offers absolute quantification without a standard curve by partitioning a sample into thousands of individual reactions, and is renowned for its high sensitivity and precision in detecting rare mutations [40]. Furthermore, the integration of pre-amplification strategies, such as the NATR-NER system for p53, with highly specific detection methods like CRISPR/Cas12a, pushes the limits of sensitivity and specificity, enabling detection down to the attomolar range [42].
Another advanced approach is Quantitative Next-Generation Sequencing (qNGS), which combines the broad mutation screening capability of NGS with absolute quantification. This is achieved by using Unique Molecular Identifiers (UMIs) to correct for PCR amplification bias and Quantification Standards (QSs)—synthetic DNA spikes of known concentration added to the sample before processing—to correct for sample loss and enable conversion of variant allele frequency (VAF) into absolute copy numbers per volume of plasma [40]. This technique is particularly powerful for simultaneously monitoring multiple ctDNA variants in a single assay.
Absolute quantification by qPCR remains a powerful, accessible, and validated method for the precise measurement of critical cancer genes like EGFR and p53. The protocols and application notes detailed herein, from the ultrasensitive TEAM-PCR for EGFR T790M to the high-fidelity NATR-NER/Cas12a system for p53, provide a robust framework for researchers and clinical scientists. Adherence to optimized protocols, rigorous validation of standards and assays, and proper data analysis as outlined will ensure the generation of reliable, reproducible data. These quantitative insights are fundamental for advancing our understanding of tumor biology, monitoring disease progression, and ultimately guiding the development and application of targeted cancer therapies.
Optimizing Your Assay: Overcoming Pitfalls for Publication-Quality Results
The absolute quantification of oncogene expression via qPCR represents a powerful technique for understanding cancer biology and evaluating therapeutic efficacy. However, the accurate measurement of specific transcript copies in a background of complex genomic DNA is a significant challenge. A primary obstacle is the presence of single nucleotide polymorphisms (SNPs) within primer and probe binding sites, which can drastically reduce annealing efficiency and lead to inaccurate quantification [45]. Even a single base mismatch can compromise assay performance, particularly when located near the 3' end of a primer where extension initiation occurs [46]. This application note details a comprehensive strategy for incorporating SNP awareness into primer design protocols, ensuring specific and reliable amplification for absolute quantification of oncogene expression in complex genomes.
Critical Design Parameters for SNP-Aware Primers
Foundational Primer Design Principles
Effective primer design begins with establishing core parameters that govern binding specificity and amplification efficiency, particularly crucial when working with oncogene targets prone to sequence variations.
Table 1: Essential Parameters for Standard PCR Primer Design
| Parameter | Optimal Range | Rationale |
|---|---|---|
| Primer Length | 18–30 bases [47] | Balances specificity with adequate melting temperature. |
| Melting Temperature (Tm) | 60–64°C [47] | Ideal for standard cycling conditions and enzyme function. |
| Tm Difference Between Primers | ≤ 2°C [45] [47] | Ensures both primers bind simultaneously and efficiently. |
| GC Content | 35–65% (ideal: 50%) [45] [47] | Provides sufficient sequence complexity while avoiding overly stable structures. |
| Amplicon Length | 70–150 bp [45] [47] | Optimal for qPCR efficiency, especially with fragmented samples. |
Strategic Considerations for SNP Avoidance and Utilization
Beyond standard parameters, specific strategies must be employed to manage genomic variability.
- Check for SNPs in Binding Sites: Before finalizing designs, utilize genome databases like UCSC Genome Browser to ensure primer and probe binding sites do not contain common SNPs [45]. This is critical for avoiding false negatives in genotyping or expression analysis.
- 3'-End Specificity for Allele Discrimination: In assays designed to distinguish alleles (e.g., for mutant-specific oncogene detection), the penultimate base at the 3' end of an allele-specific primer should be positioned at the SNP site. This dramatically increases the discriminative power, as the polymerase has reduced efficiency to extend a mismatched primer [46].
- Design Over Exon-Exon Junctions: To ensure amplification specifically from cDNA and not contaminating genomic DNA, design primers to span an exon-exon junction. This is particularly valuable in oncogene research. The ideal design places the 3' end of one primer across the junction, with only 3–4 bases at its extreme 3' end located in the adjacent exon [45] [48].
Figure 1: A strategic workflow for designing SNP-aware PCR primers, highlighting critical checks for binding sites and the option for allele-specific design.
Computational Tools for Validation and Specificity Checking
In Silico Analysis of Primer Properties
Once a candidate primer pair is designed, rigorous computational validation is a non-negotiable step.
- Check for Secondary Structures: Use tools like IDT's OligoAnalyzer to screen primers for self-dimers, hairpins, and heterodimers. The ΔG value for any secondary structure should be weaker (more positive) than –9.0 kcal/mol to prevent formation during the reaction [47].
- Assess Thermodynamic Stability: Calculate Tm using nearest-neighbor thermodynamics and the actual reaction conditions (e.g., 50 mM K+, 3 mM Mg2+) rather than simplistic formulas. This provides a more accurate prediction of annealing behavior [48] [47].
- Validate Specificity with BLAST: Perform a BLAST search against the entire host genome to ensure the primers are unique to the target oncogene sequence and will not generate off-target amplicons [45] [47]. This can be done directly from tools like the OligoAnalyzer.
Ensuring Specificity with Primer-BLAST
NCBI's Primer-BLAST is an indispensable tool for combining primer design with specificity analysis [48]. Key parameters to set for oncogene assays include:
- Specificity Checking: Enable the option to check primer specificity against an appropriate database (e.g., "Refseq RNA" or a specific genomic assembly).
- Organism Selection: Always specify the target organism to limit the search and increase speed and relevance.
- Exon Junction Span: For mRNA quantification, use the parameter that requires primers to span an exon-exon junction to prevent genomic DNA amplification [48].
- Mismatch Tolerance: Adjust the maximum number of mismatches allowed to unintended targets; requiring at least 2-3 mismatches, especially near the 3' end, increases specificity [48].
Table 2: Key In-Silico Validation Tools and Their Applications
| Tool Name | Primary Function | Application in SNP-Aware Design |
|---|---|---|
| UCSC Genome Browser | Genome Visualization | Identifying common SNPs within potential primer binding sites [45]. |
| NCBI Primer-BLAST | Primer Design & Specificity Check | Designing primers and verifying they are unique to the target in the presence of a complex genome [48]. |
| IDT OligoAnalyzer | Oligo Thermodynamic Analysis | Calculating precise Tm, checking for self-complementarity, and running BLAST analysis [47]. |
| FastPCR | Assay Design for Genotyping | Specialized software for designing competitive allele-specific PCR (KASP) assays [46]. |
Experimental Protocol: Absolute Quantification with SNP-Verified Primers
Protocol: qPCR Setup for Absolute Quantification of Oncogene Expression
This protocol is designed for use with hydrolysis (TaqMan) probes and a standard curve for absolute quantification, incorporating checks for SNP-related artifacts.
Materials:
- Template: High-quality, DNase-treated total RNA from tissue or cell lines.
- Reverse Transcription Kit: e.g., ZymoScript RT PreMix Kit for high-efficiency cDNA synthesis [45].
- qPCR Master Mix: A hot-start, probe-based mix suitable for absolute quantification (e.g., kits containing dUTP and UDG for carryover prevention) [49].
- Primers & Probes: SNP-verified, HPLC-purified primers and dual-labeled probes.
- Standard Curve Material: A serially diluted plasmid containing the cloned target oncogene sequence [50].
Procedure:
- cDNA Synthesis: Convert 1 µg of total RNA to cDNA in a 20 µL reaction according to the manufacturer's instructions. Include a no-reverse-transcriptase (-RT) control to detect genomic DNA contamination.
- Prepare Standard Curve: Linearize the plasmid stock and quantify it spectrophotometrically. Calculate the copy number and prepare a 6-point, 10-fold serial dilution series in TE buffer. Aliquot and store at -80°C to avoid degradation [51] [50].
- qPCR Reaction Setup: Prepare reactions in triplicate for both standards and unknown cDNA samples.
Table 3: qPCR Reaction Setup for a 20 µL Reaction
Component Volume per Reaction (µL) Final Concentration 2X qPCR SuperMix 10.0 1X Forward Primer (10 µM) 0.4 200 nM Reverse Primer (10 µM) 0.4 200 nM TaqMan Probe (10 µM) 0.2 100 nM Template (cDNA or Standard) 2.0 - Nuclease-free Water to 20.0 - - qPCR Cycling Conditions:
- UDG Incubation: 50°C for 2 minutes (if using dUTP/UDG systems) [49].
- Initial Denaturation: 95°C for 2 minutes.
- Amplification (40 cycles):
- Denature: 95°C for 15 seconds.
- Anneal/Extend: 60°C for 30-60 seconds (optimize based on primer Tm).
- Data Analysis:
- The instrument's software will generate a standard curve from the dilutions (Ct vs. log copy number).
- Use this curve to interpolate the absolute copy number of the target oncogene in each unknown cDNA sample.
The Scientist's Toolkit: Essential Reagents for Reliable qPCR
Table 4: Key Research Reagent Solutions for Oncogene qPCR
| Reagent / Kit | Function | Key Feature |
|---|---|---|
| ZymoScript RT PreMix Kit [45] | cDNA Synthesis | Provides high-yield, full-length cDNA in less than 15 minutes. |
| Zymo DNA Clean & Concentrator Kits [45] | PCR Product Purification | Maximizes DNA concentration and removes contaminants for sensitive downstream applications. |
| Platinum qPCR SuperMix [49] | qPCR Amplification | Contains a hot-start polymerase and UDG carryover prevention, ideal for SNP genotyping. |
| Bisulfite Conversion Kits [45] | Methylation Analysis | For complete bisulfite conversion of DNA, enabling methylation-specific PCR (MSP) studies in cancer. |
| Pre-designed TaqMan SNP Genotyping Assays [49] | Allele Discrimination | Validated, ready-to-use assays for specific SNP targets, saving development time. |
Data Analysis and Interpretation
For absolute quantification, the standard curve method is the gold standard. The stability of your standard is critical for reproducible results. Studies show that cloned plasmid DNA (linear or circular) is significantly more stable during storage at -20°C than purified PCR products, leading to more reliable copy number assignments over time [50]. Always run the standard curve on the same plate as the unknowns to control for inter-run variation.
When analyzing results, a significant reduction in amplification efficiency or failure to amplify in a subset of samples may indicate a polymorphic variant interfering with primer or probe binding. This underscores the necessity of the pre-design SNP screening steps outlined in this document. For allele-specific PCR, successful genotyping is visualized by clear clustering of samples into homozygous and heterozygous groups based on their fluorescence signals [46].
Meticulous, SNP-aware primer design is not merely a best practice but a fundamental requirement for obtaining biologically meaningful data from the absolute quantification of oncogene expression in complex genomes. By integrating in-silico SNP screening, stringent specificity checks with tools like Primer-BLAST, and adherence to robust thermodynamic principles, researchers can develop assays that are both highly specific and sensitive. The protocols and strategies detailed herein provide a reliable framework for advancing cancer research and drug development by ensuring that qPCR results accurately reflect the underlying molecular reality.
In the context of oncogene research, the absolute quantification of gene expression levels using quantitative polymerase chain reaction (qPCR) is a powerful technique for understanding cancer biology and evaluating therapeutic efficacy. The accuracy of this quantification hinges on a robustly optimized qPCR assay, where key performance metrics include an amplification efficiency of 100% ± 5% and a standard curve linearity (R²) ≥ 0.99 [52] [53]. Achieving these parameters is not trivial and requires a meticulous, stepwise optimization protocol to ensure that the data generated are reliable, reproducible, and suitable for supporting critical decisions in drug development. This Application Note provides a detailed protocol for researchers and scientists to systematically optimize qPCR assays for the absolute quantification of oncogene expression.
Stepwise Optimization Protocol
A sequential approach to optimization is critical for isolating and correcting variables that affect qPCR performance. The protocol below outlines the key stages.
Primer and Probe Design
The foundation of a specific and efficient qPCR assay is careful in silico design.
- Sequence-Specificity for Homologous Genes: For oncogenes with homologous family members or pseudogenes, design primers based on single-nucleotide polymorphisms (SNPs) that are unique to the target sequence. This prevents off-target amplification and false-positive results [52].
- In Silico Design Tools: Utilize tools such as Primer3Plus, Primer-BLAST, or NCBI's Primer Blast to design primers and probes. These tools help test for off-target binding within the host genome [52] [54].
- Empirical Testing: Design and empirically test at least three candidate primer/probe sets, as in silico predictions do not always translate to optimal performance in the lab [54].
- Amplicon Characteristics: Amplicons should typically be 85–200 bp in length. The probe (for hydrolysis probe assays) should be labeled with a 5' fluorophore (e.g., FAM) and a 3' quencher (e.g., BHQ1) [52] [55].
Experimental Optimization of Reaction Conditions
After design, primer and probe sets require experimental fine-tuning.
Table 1: Key Components for Optimization
| Component | Typical Optimization Range | Function & Goal |
|---|---|---|
| Primer Concentration | 0.2 – 1.0 µM | To ensure sufficient primer binding without promoting dimerization or non-specific amplification [55]. |
| Probe Concentration | 62.5 – 250 nM | To provide a clear fluorescent signal above background without inhibiting the reaction [55]. |
| Annealing Temperature | Gradient from 51°C to 59°C | To identify the temperature that yields the lowest Cq (quantification cycle) and highest fluorescence intensity [55]. |
| cDNA Input Range | Serial dilutions (e.g., 5-6 logs) | To establish the dynamic range and ensure linearity of the standard curve [52]. |
Validation of Assay Performance
Once optimal conditions are determined, the assay must be rigorously validated.
- Standard Curve and Efficiency: Prepare a serial dilution (at least 5 logs) of a known quantity of target, such as a synthetic oligo or plasmid containing the insert [53]. Run the dilution series in triplicate. Plot the log of the initial template quantity against the Cq value. Perform linear regression analysis.
- Specificity: For SYBR Green assays, perform a melt curve analysis post-amplification to confirm a single, specific amplification product. For probe-based assays, ensure no amplification in no-template controls (NTCs) [56] [53].
- Sensitivity and Precision: Determine the limit of detection (LOD). Assess intra- and inter-assay precision by calculating the coefficient of variation (% CV) of Cq values for replicates; it should typically be < 5% [55].
The following workflow summarizes the entire optimization and validation process:
The Scientist's Toolkit: Research Reagent Solutions
Selecting the right reagents is fundamental to a successful qPCR assay. The table below lists essential materials and their functions.
Table 2: Essential Research Reagents for qPCR Optimization
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| Sequence-Specific Primers | Amplification of the target oncogene sequence. | Must be designed based on SNPs to distinguish between homologous genes; HPLC-purified for accuracy [52]. |
| Hydrolysis Probes (e.g., TaqMan) | Sequence-specific detection of the amplicon. | Provides higher specificity than intercalating dyes; must be validated alongside primers [54] [55]. |
| SYBR Green I Dye | Binds double-stranded DNA for detection. | Cost-effective; requires melt curve analysis to confirm specificity [56] [53]. |
| qPCR Master Mix | Contains Taq polymerase, dNTPs, and buffer. | Optimized formulations can improve efficiency and consistency; choose one compatible with your platform [56]. |
| Standard Template (Plasmid DNA/Synthetic Oligo) | Used for generating the standard curve. | Must be a pure, quantifiable species for absolute quantification; serial dilutions define the dynamic range [3] [53]. |
| No-Template Control (NTC) | Control for contamination and primer-dimer formation. | A critical quality control step; should yield no amplification [56]. |
Data Analysis and Quality Assessment
Following optimization and data acquisition, a rigorous quality assessment is imperative.
- Quality Score Metrics: Adopt a scoring system (e.g., 1-5) to evaluate key assay characteristics such as PCR efficiency, dynamic range (R²), precision (Cq variation), fluorescence signal consistency, and curve shape. This allows for a rapid, visual assessment of overall assay health [56].
- The "Dots in Boxes" Method: This high-throughput analysis method plots PCR efficiency (y-axis) against ΔCq (Cq(NTC) - Cq(Lowest Input)) (x-axis). High-quality assays (efficiency 90-110%, ΔCq ≥ 3) will appear as solid dots within a defined box on the graph, facilitating quick comparison of multiple targets or conditions [56].
The decision process for quality control is summarized below:
A meticulously optimized qPCR assay is non-negotiable for the absolute quantification of oncogene expression in drug development. The stepwise protocol outlined herein—encompassing strategic primer design, empirical optimization of reaction conditions, and rigorous validation against the benchmarks of R² ≥ 0.99 and efficiency of 100% ± 5%—provides a robust framework for generating reliable and reproducible data. Adherence to this protocol and the associated quality control measures will ensure that qPCR results accurately reflect biological reality, thereby strengthening research outcomes and regulatory submissions in the field of oncology.
In the context of absolute quantification of oncogene expression by qPCR, the accuracy of results is fundamentally dependent on the reliability of standard curves. This method relies on a standard curve generated from serial dilutions of a standard with a known concentration to calculate the absolute quantity of the target nucleic acid in experimental samples [57] [58]. Any variability or inaccuracy in the preparation and stability of these standards propagates through the quantification process, potentially leading to misleading conclusions about oncogene expression levels that could impact drug development decisions. Adherence to strict protocols for standard preparation is therefore not merely a procedural step but a critical foundation for data integrity in molecular research and diagnostics.
Quantitative Evidence: Documenting Variability in Standard Curves
A systematic evaluation of inter-assay variability is essential to justify the necessity of rigorous standard preparation. A 2025 study investigating RT-qPCR standard curves for virus quantification, relevant to assay design in other areas like oncology, provides compelling quantitative evidence. The study conducted thirty independent standard curve experiments for different viral targets and observed significant variability in key performance parameters [57].
Table 1: Inter-assay Variability of RT-qPCR Standard Curves for Different Targets
| Viral Target | Average Efficiency (%) | Observed Variability | Key Findings |
|---|---|---|---|
| SARS-CoV-2 (N2 gene) | 90.97% | Highest heterogeneity (CV 4.38–4.99%) | Demonstrated the lowest efficiency and highest variability |
| Norovirus GII (NoVGII) | >90% | Higher inter-assay efficiency variability | Better sensitivity but inconsistent performance |
| Hepatitis A (HAV) | >90% | Lower variability | More consistent performance |
This observed heterogeneity occurred despite optimal efficiency rates (>90%), underscoring that efficiency alone is not a sufficient indicator of a robust standard curve [57]. The findings led to the conclusion that including a standard curve in every experiment is essential to obtain reliable results, rather than relying on historical or master curves to reduce time and costs [57]. This is particularly critical in oncology research, where small changes in oncogene expression levels can be biologically significant.
Further compounding this, a 2025 study highlighted that measurement uncertainty increases markedly at low input concentrations, often exceeding the magnitude of biologically meaningful differences [59]. This is due to technical variability, stochastic amplification, and efficiency fluctuations that confound quantification, making rigorous standard preparation and replication non-negotiable for accurate absolute quantification, especially for low-abundance oncogene transcripts.
Experimental Protocols for Standard Preparation and Validation
Protocol: Preparation of Serial Dilutions for Standard Curves
Principle: This protocol describes the creation of a precise serial dilution series from a stock of a known concentration (e.g., gBlock gene fragment, plasmid, or synthetic RNA) for absolute standard curve generation in oncogene quantification [57] [59].
Materials:
- Nucleic Acid Standard: Cloned target oncogene sequence (e.g., in plasmid) or synthetic oligonucleotide (gBlock) with known concentration.
- Diluent: Nuclease-free water or TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0).
- Labware: Nuclease-free, low-retention microcentrifuge tubes and pipette tips.
- Equipment: calibrated precision pipettes, microcentrifuge, spectrophotometer or fluorometer for nucleic acid quantification.
Procedure:
- Stock Solution Quantification: Accurately determine the concentration of the standard stock solution using a UV spectrophotometer (measuring A260) or, more accurately, a fluorometer-based method (e.g., Qubit). Convert this concentration to copies/µL using the molecular weight of the standard.
- Initial Dilution: Perform a primary dilution in nuclease-free water to create a high-concentration working stock (e.g., 10^9 copies/µL). This minimizes the impact of volumetric errors in subsequent dilutions.
- Serial Dilution Setup: Prepare at least five (recommended: six or seven) 10-fold serial dilutions.
- Label nuclease-free tubes for each dilution point (e.g., 10^8, 10^7, ..., 10^1 copies/µL).
- Pipette the appropriate volume of diluent into each tube.
- To create the 10^8 copies/µL dilution, transfer a precise volume of the 10^9 copies/µL stock into the first tube, mixing thoroughly by pipetting up and down or vortexing. Avoid foaming.
- Change pipette tips after every transfer.
- Continue this process sequentially to create the entire dilution series.
- Aliquoting and Storage: To prevent freeze-thaw degradation, aliquot each dilution point into single-use volumes before storage at recommended temperatures (e.g., -20°C or -80°C for RNA). This ensures the standard is thawed only once [57].
Protocol: Validation of Standard Curve Parameters
Principle: This protocol ensures that the prepared standard curve meets the minimum performance criteria for use in absolute quantification, as per MIQE guidelines.
Materials:
- Prepared serial dilutions of the standard.
- qPCR Master Mix (e.g., TaqMan Fast Virus 1-Step Master Mix for RNA targets).
- Primers and probe specific to the target oncogene.
- Real-time PCR instrument and associated analysis software.
Procedure:
- qPCR Run: Amplify each dilution of the standard curve in duplicate or triplicate on the same qPCR plate as the unknown samples.
- Data Analysis: The instrument's software will generate a standard curve by plotting the Quantification Cycle (Cq) against the logarithm of the initial concentration.
- Parameter Validation: Assess the following key parameters [57] [60]:
- Amplification Efficiency: Calculate from the slope of the standard curve (Efficiency = 10^(-1/slope) - 1). The ideal efficiency is 100% (slope of -3.32), with an acceptable range of 90–110% (slope between -3.6 and -3.1) [60].
- Linearity (R²): The coefficient of determination (R²) should be ≥ 0.990, indicating a strong linear relationship.
- Y-Intercept: Represents the Cq at one copy/reaction. It should be consistent between runs for the same assay.
- Acceptance Criteria: Only standard curves meeting the above efficiency and linearity criteria should be used for quantifying oncogene expression in unknown samples. If criteria are not met, troubleshoot the dilution series, primer design, or reaction conditions.
The Scientist's Toolkit: Essential Reagents for Reliable Standard Curves
Table 2: Key Research Reagent Solutions for Standard Preparation
| Reagent / Material | Function & Importance | Recommendations for Use |
|---|---|---|
| Synthetic Nucleic Acid Standards (gBlocks, synthetic RNA) | Provides a consistent and pure quantifiable standard free from biological contaminants; essential for absolute quantification. | Obtain from a certified biological resource center [57]. Validate sequence upon receipt. |
| Nuclease-Free Water | Serves as the diluent for standards and controls; prevents degradation of nucleic acids by environmental nucleases. | Use certified nuclease-free water. Aliquot to maintain sterility. |
| TaqMan Fast Virus 1-Step Master Mix | An optimized ready-to-use mix for RT-qPCR, reducing handling steps and associated variability [57]. | Suitable for one-step RT-qPCR assays. Includes all components except primers, probe, and template. |
| ROX Passive Reference Dye | Included in some master mixes to normalize for non-PCR-related fluorescence fluctuations between wells, reducing well-to-well variation [60]. | Use according to the manufacturer's instructions for your specific instrument platform. |
| DNA/RNA Stabilization Reagent (e.g., RNAlater) | Preserves the integrity of nucleic acid standards, especially labile RNA, by inhibiting nucleases [60]. | Use for temporary storage of RNA standards or tissue samples if immediate processing is not possible. |
Workflow and Pathway Visualizations
Standard Curve Generation and Validation Workflow
The following diagram illustrates the critical steps and decision points in the process of creating and validating a qPCR standard curve for absolute quantification.
Pathway from Poor Standards to Experimental Error
This diagram outlines the logical sequence of how inadequately prepared standards lead to quantification errors and ultimately flawed research conclusions, particularly in an oncogene expression context.
The absolute quantification of oncogene expression demands meticulous attention to the preparation and stability of standard curves. As evidenced by recent studies, inter-assay variability is a significant source of error that can be mitigated by including a validated, well-prepared standard curve in every qPCR run [57] [59]. By adhering to the detailed protocols for serial dilution and validation, utilizing the appropriate reagents outlined in the toolkit, and integrating these practices into a rigorous workflow, researchers can significantly enhance the reliability of their data. This rigorous approach provides the accuracy required for robust drug development and meaningful scientific discovery in oncology research.
The absolute quantification of oncogene expression represents a pivotal frontier in molecular oncology and therapeutic development. Accurate measurement of these often low-abundance transcripts is crucial for cancer staging, minimal residual disease detection, and monitoring treatment efficacy. However, conventional reverse transcription-quantitative real-time PCR (RT-qPCR) faces significant limitations when quantifying targets with high cycle threshold (Cq) values, typically above 30-35 cycles, where results become unreliable due to poor reproducibility and sensitivity constraints [61] [22]. According to MIQE guidelines, Cq values exceeding this range are often considered unreliable, creating a critical technological gap for researchers investigating faint but biologically significant oncogenic signals [61]. This application note synthesizes current methodologies and provides detailed protocols to overcome these limitations, enabling robust absolute quantification of low-abundance oncogenes.
The fundamental challenge stems from both biological and technical factors. Biologically, many oncogenic transcripts and regulatory RNAs exist at low copy numbers per cell. Technically, conventional RT-qPCR encounters limitations related to primer-dimer artifacts, differential primer efficiency, and susceptibility to inhibitors present in complex biological samples [62] [22]. Digital PCR (dPCR) technologies address some limitations but require specialized instrumentation [61]. This document outlines targeted amplification strategies, advanced detection chemistries, and optimized experimental designs to push detection boundaries while maintaining data integrity essential for publication-quality research and diagnostic applications.
Technological Landscape: Comparison of Quantification Platforms
Performance Characteristics of Major Detection Platforms
Table 1: Comparative analysis of qPCR and ddPCR for low-abundance target detection
| Parameter | Conventional qPCR | Droplet Digital PCR (ddPCR) | Targeted Pre-amplification Methods |
|---|---|---|---|
| Limit of Detection | ~10-20 copies/reaction (highly dependent on reaction efficiency) [22] | <10 copies/reaction; precise detection at single-copy level [63] [22] | Enhances qPCR detection by 100-1000x for transcripts with Cq >30 [61] |
| Quantification Principle | Relative to standard curve (Cq-based) [64] | Absolute counting via end-point detection (Poisson distribution) [63] | Target-specific pre-amplification followed by qPCR quantification [61] |
| Impact of Inhibitors | High susceptibility; causes Cq shifts and efficiency reduction [63] [22] | Reduced susceptibility; binary readout minimizes impact [63] [22] | Variable; depends on pre-amplification efficiency and purification steps [61] |
| Reaction Efficiency Considerations | Critical (85-110% acceptable); affects all Cq values and quantification accuracy [64] | Less critical; end-point detection efficiency-independent within range [22] | Critical for pre-amplification step; requires validation [61] |
| Dynamic Range | 6-7 logs with optimal efficiency [64] | 4-5 logs linear dynamic range [63] | Extends qPCR dynamic range for low-abundance targets [61] |
| Data Reproducibility | Moderate to high for medium-abundance targets; poor for low-abundance (Cq≥29) [22] | High precision for low-abundance targets (<5% CV) [63] [22] | High when optimized; enables detection of low-abundance isoforms [61] |
| Best Applications | Medium to high abundance targets; well-characterized samples; relative quantification [64] | Absolute quantification; rare targets; inhibitor-containing samples; copy number variation [63] [22] | Low-abundance transcripts; isoform quantification; samples with known 5'-end sequences [61] |
Advanced Detection Chemistries for Enhanced Sensitivity
Beyond platform selection, detection chemistry significantly impacts sensitivity. While SYBR Green and TaqMan probes represent standard approaches, recent advances in metal-sensitive dyes offer enhanced visual detection capabilities. Pyridylazophenol dyes (5-Bromo-PAPS and 5-Nitro-PAPS) complexed with manganese ions enable robust colorimetric detection compatible with both isothermal and PCR amplification [65]. These dyes undergo dramatic color shifts from red to yellow upon amplification, providing high contrast between positive and negative reactions. This approach enables detection without sophisticated instrumentation while maintaining sensitivity in fully buffered reactions, overcoming limitations of pH-based detection methods [65].
Methodological Approaches: Protocols for Enhanced Detection
STALARD: Selective Target Amplification for Low-Abundance RNA Detection
The STALARD method provides a targeted pre-amplification approach to overcome sensitivity limitations of conventional RT-qPCR for known low-abundance transcripts [61]. This protocol is particularly valuable for quantifying specific oncogene isoforms or splice variants that share known 5'-end sequences.
Protocol: STALARD Workflow
Step 1: Primer Design
- Design a gene-specific primer (GSP) complementary to the 5'-end of the target RNA (substituting T for U).
- Design parameters: Tm = 62°C, GC content 40-60%, no predicted hairpin or self-dimer structures [61].
- Synthesize a GSP-tailed oligo(dT) primer (GSoligo(dT)) consisting of the GSP sequence followed by (dT)24VN (where V = A, G, or C; N = any base).
Step 2: First-Strand cDNA Synthesis
- Use 1 µg of total RNA and 1 µL of 50 µM GSoligo(dT) primer.
- Perform reverse transcription using HiScript IV 1st Strand cDNA Synthesis Kit or equivalent.
- The resulting cDNA contains the GSP sequence at both ends.
Step 3: Targeted Pre-amplification
- Prepare 50 µL PCR reaction with 1 µL cDNA, 1 µL of 10 µM GSP, and SeqAmp DNA Polymerase.
- Thermal cycling: 95°C for 1 min; 9-18 cycles of 98°C for 10s, 62°C for 30s, 68°C for 1 min/kb; 72°C for 10 min.
- Critical: Limit cycles to 9-18 to maintain quantitative representation while achieving sufficient amplification.
Step 4: Purification and Quantification
- Purify PCR products using AMPure XP beads at 1.0:0.7 product:beads ratio.
- Quantify using standard qPCR with isoform-specific primers or analyze via nanopore sequencing for isoform discovery [61].
Absolute Quantification Using Single Standard for Marker and Reference Genes
For absolute quantification in oncogene research, the SSMR approach enables direct comparison of results across experiments and laboratories—a critical requirement for multi-center clinical trials and biomarker validation [23].
Protocol: SSMR Workflow for Absolute Quantification
Step 1: Standard Construction
- Clone PCR amplicons of target oncogenes and reference genes into a single plasmid vector.
- Critical: Ensure equimolar representation of all amplicons in the final construct.
- Alternatively, synthesize a gBlock gene fragment containing all targets and references in a 1:1 ratio.
Step 2: Standard Quantification and Dilution
- Precisely quantify the SSMR DNA using fluorometric methods (Qubit dsDNA HS Assay).
- Calculate molecular concentration based on molecular weight.
- Prepare 10-fold serial dilutions from 1×10^10 to 1×10^2 molecules/μL in 10 mM Tris-HCl (pH 7.5).
Step 3: qPCR Setup and Analysis
- Run unknown samples and SSMR standard dilutions on the same plate.
- Use identical reaction conditions for both standards and samples.
- Calculate absolute copy numbers in unknown samples by interpolating Cq values against the SSMR standard curve [23].
Table 2: Essential research reagents for low-abundance target detection
| Reagent Category | Specific Examples | Function & Importance | Optimization Notes |
|---|---|---|---|
| Reverse Transcriptase | HiScript IV | High efficiency cDNA synthesis; critical for representative cDNA libraries | Use gene-specific or oligo(dT) priming depending on target characteristics [61] |
| DNA Polymerase | SeqAmp DNA Polymerase, QX200 ddPCR EvaGreen Supermix | Efficient amplification with minimal bias; essential for pre-amplification and digital PCR | Match polymerase characteristics to application (e.g., hot-start for specificity) [61] [63] |
| Specialized Primers | GSP-tailed oligo(dT) primers, SSMR constructs | Enable targeted amplification and absolute quantification | Verify specificity and efficiency empirically; in silico design insufficient [61] [23] [62] |
| Detection Chemistries | SYBR Green I, EvaGreen, 5-Bromo-PAPS/Mn²⁺, TaqMan probes | Signal generation for quantification; choice impacts sensitivity and specificity | SYBR Green I offers higher precision than TaqMan for absolute quantification [65] [66] |
| Nucleic Acid Purification | AMPure XP beads, DNeasy PowerSoil Pro Kit | Remove inhibitors and size selection; critical for reaction efficiency | SPRI bead ratios affect size selection; optimize for application [61] [63] |
| Quantification Standards | SSMR, gBlock gene fragments, linear DNA standards | Enable absolute quantification and cross-experiment comparisons | Plasmid-free standards reduce cloning steps while maintaining accuracy [23] [67] |
Implementation Framework: Optimized Workflows for Reliable Results
Primer Design and Validation Strategy
Robust primer design is paramount for sensitive detection, especially for low-abundance targets where non-specific amplification can overwhelm the true signal [62].
Target Identification: Precisely define the amplification target using curated reference sequences (e.g., NCBI RefSeq). For oncogenes, consider transcript variants, pseudogenes, and homologous sequences that might confound specificity [62].
Empirical Validation: Never rely solely on in silico predictions. Perform temperature gradient PCR (56-65°C) to determine optimal annealing temperature. Verify with melt curve analysis for SYBR Green-based assays or probe validation for hydrolysis probes [62].
Efficiency Determination: Prepare a 10-fold serial dilution of template (SSMR standard or validated positive control). Calculate efficiency using the formula: Efficiency (%) = (10^(-1/slope) - 1) × 100. Acceptable efficiency ranges from 90-110% [64].
Experimental Design for Maximized Sensitivity
Sample Quality Assessment: Use fluorometric methods (Qubit) rather than spectrophotometry for nucleic acid quantification. Require A260/280 ratios ≥1.8 for RNA and ≥1.6 for DNA [63].
Inhibition Testing: For qPCR, include a sample dilution series to identify inhibition effects. For ddPCR, assess droplet separation quality and interface droplets as inhibition indicators [63] [22].
Technical Replication: Perform minimum three technical replicates for qPCR. For ddPCR, increased sample volume partitioning provides inherent replication [68] [63].
Controls: Include no-template controls (NTC), no-reverse transcription controls, and positive controls in every run. For absolute quantification, include the standard curve on the same plate [23] [63].
Pushing the detection limits for low-abundance oncogenic targets requires integrated strategies combining appropriate technology selection, optimized experimental design, and rigorous validation. The STALARD method provides exceptional sensitivity for known transcripts, while ddPCR offers robust absolute quantification in challenging samples. The SSMR approach enables cross-experiment comparability essential for biomarker validation. Implementation of these methodologies, coupled with stringent quality control measures, empowers researchers to reliably investigate the faint molecular signatures driving oncogenesis and treatment response, ultimately advancing precision oncology through enhanced molecular detection capabilities.
The Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines provide a standardized framework for conducting, documenting, and reporting quantitative PCR (qPCR) experiments to ensure their technical robustness, reproducibility, and transparency [69] [70]. First published in 2009 and recently updated as MIQE 2.0, these guidelines were established in response to widespread concerns about inadequate experimental reporting and flawed qPCR practices in scientific literature [69] [70]. The fundamental premise is simple yet powerful: no matter how powerful the technique, without methodological rigor, data cannot be trusted [70].
qPCR remains one of the most ubiquitous techniques in molecular biology laboratories, playing a critical role in diverse applications from basic research to clinical diagnostics. However, the technique is often mistakenly viewed as straightforward, leading to complacency in experimental design and validation [70]. This has resulted in a proliferation of publications with insufficient technical information for work to be reproduced, frequently revealing flawed practices that likely yield erroneous conclusions [69] [70]. Within the specific context of oncogene expression studies, where precise quantification can directly impact diagnostic, prognostic, or therapeutic decisions, adherence to these guidelines becomes particularly crucial for generating clinically relevant data.
The MIQE guidelines encompass every aspect of the qPCR workflow, from sample acquisition and assay design to data analysis and normalization strategies [69]. Their adoption helps standardize experimental protocols, maximizes efficient utilization of resources, and enhances the impact of this essential technology by ensuring that published results are both technically sound and independently verifiable [71].
Core Principles and Requirements
The Evolution from MIQE to MIQE 2.0
The original MIQE guidelines, published in 2009, provided researchers with a comprehensive checklist of minimum information required for publishing qPCR experiments [70]. Building on this foundation, MIQE 2.0 represents a significant evolution that addresses emerging applications and incorporates lessons learned from over a decade of implementation [70]. These updated guidelines reflect current technological advances while maintaining the core principle that proper validation and transparent reporting are non-negotiable for scientific credibility.
Despite widespread awareness of MIQE, compliance remains problematic, with serious deficiencies persisting in experimental transparency, assay validation, and data reporting [70]. Common failures include poorly documented sample handling, absent assay validation, inappropriate normalization, missing PCR efficiency calculations, and insufficient statistical justification [70]. The consequences extend beyond academic concerns – when qPCR results underpin decisions in biomedical research or clinical diagnostics, methodological flaws carry real-world consequences for patient care and public health policy [70].
Key Reporting Requirements
The MIQE guidelines outline specific information that must be reported across all phases of a qPCR experiment. Table 1 summarizes the essential requirements for different stages of the qPCR workflow, with particular emphasis on aspects critical for oncogene quantification studies.
Table 1: Essential MIQE Reporting Requirements for qPCR Experiments
| Experimental Stage | Required Information | Specific Application to Oncogene Studies |
|---|---|---|
| Sample/Template | Tissue source, storage conditions, nucleic acid quality/quantity, genomic DNA contamination assessment [69] | Tumor tissue provenance, processing delays, RNA integrity numbers (RIN >8.0 recommended), demonstration of minimal genomic DNA contamination (ΔCq >5 between +RT and -RT) |
| Assay Design | Primer/probe sequences or database accession numbers, amplicon size, genomic location [69] [71] | For TaqMan assays, provide Assay ID and amplicon context sequence; verify specificity for oncogene transcript variants of interest |
| Assay Validation | PCR efficiency (from calibration curve), linear dynamic range, limit of detection, specificity confirmation (melting curve or sequencing) [69] [72] | Efficiency of 90-110% with r² >0.98; demonstrate specificity for oncogene target against related family members; test for absence of inhibitors in tumor samples |
| Reverse Transcription | cDNA synthesis protocol, priming method (oligo-dT, random hexamers, or gene-specific), RNA input amount [69] | Use consistent priming strategy across all samples; document RNA input (typically 10-1000ng); include no-RT controls for each sample |
| Data Analysis | Cq determination method, normalization strategy, statistical methods, outlier identification [69] [15] | Use multiple validated reference genes; avoid 2−ΔΔCT method when efficiency ≠ 100%; consider ANCOVA for improved statistical power [15] |
| Experimental Controls | No template controls (NTC), no amplification controls (NAC), positive controls, inter-run calibrators [69] | Include NTC for contamination monitoring; use positive controls with known oncogene copy numbers; implement inter-run calibrators for multi-plate experiments |
Implementing MIQE in Oncogene Expression Studies
Sample Acquisition and Quality Assessment
The preanalytical phase represents the most vulnerable stage for introducing variability in oncogene expression studies. Sample integrity must be rigorously controlled from the moment of collection, particularly when working with clinical tumor specimens that may exhibit varying degrees of necrosis or degradation. The MIQE guidelines emphasize that "sample handling affects experimental variability," requiring detailed reporting of tissue source, stabilization method, and storage conditions [69]. For longitudinal studies spanning extended periods, consistent storage conditions become particularly critical.
RNA quality assessment is non-negotiable for reliable oncogene quantification. The guidelines recommend using microfluidics-based systems (e.g., Bioanalyzer or TapeStation) to generate RNA Integrity Numbers (RIN) or RNA Quality Indicators (RQI) [69]. Authors are explicitly cautioned against "quantitatively comparing RNAs of widely dissimilar quality, e.g., RIN/RQI values of 4.5 versus 9.5" [69]. Furthermore, the guidelines note that "rRNAs yielding similar RIN/RQI numbers can contain mRNAs that differ significantly in their integrity," highlighting the importance of also assessing mRNA quality specifically when measuring oncogene expression [69]. For formalin-fixed paraffin-embedded (FFPE) tumor samples, which often yield partially degraded RNA, additional validation is essential to demonstrate that amplification efficiency remains acceptable.
Assay Design and Validation for Oncogene Targets
Proper assay design requires reporting database accession numbers, amplicon size, and primer sequences for each target [69]. When using pre-designed assays such as TaqMan assays, providing the unique Assay ID is typically sufficient, as manufacturers ensure that "the primer/probe sequence within a TaqMan Assay ID will never change" [71]. However, for full MIQE compliance, researchers should also provide the amplicon context sequence, which can be generated using the TaqMan Assay Search Tool and NCBI resources as outlined by Thermo Fisher Scientific [71].
Assay validation represents perhaps the most critical component for reliable oncogene quantification. The guidelines require empirical confirmation of primer specificity (via sequencing or melt curve analysis), determination of PCR efficiency using a dilution series, and establishment of the linear dynamic range [69]. PCR efficiency should be reported for each assay, with values between 90-110% generally considered acceptable [69]. For oncogene targets that may be rare or have low expression levels, establishing the limit of detection (LOD) and limit of quantification (LOQ) becomes particularly important. The recent MIQE 2.0 guidelines reinforce that "assay efficiencies are assumed, not measured" represents a fundamental methodological failure that can lead to exaggerated fold-change calculations [70].
Data Normalization and Analysis Strategies
Appropriate normalization is "an essential component of a reliable qPCR assay" according to the MIQE guidelines [69]. For mRNA quantification, including oncogene expression studies, the guidelines state that "normalisation should be performed against multiple reference genes, chosen from a sufficient number of candidate reference genes tested from independent pathways using at least one algorithm (e.g., GeNorm)" [69]. The use of fewer than three reference genes is generally not advisable unless specifically validated [69].
Regarding data analysis methods, recent evidence suggests moving beyond the traditional 2−ΔΔCT method. Studies indicate that "ANCOVA enhances statistical power compared to 2−ΔΔCT" and that "ANCOVA P-values are not affected by variability in qPCR amplification efficiency" [15]. This approach offers particular advantages for oncogene studies where precise quantification of fold-changes directly impacts biological interpretations. Furthermore, the guidelines encourage researchers to "share raw qPCR fluorescence data along with detailed analysis scripts that start from raw input and produce final figures and statistical tests" to enhance reproducibility [15].
The following workflow diagram illustrates the key stages of implementing MIQE-compliant qPCR for oncogene expression studies:
MIQE-Compliant qPCR Workflow for Oncogene Studies
Advanced Applications and Future Directions
MIQE for Clinical Research Applications
The principles outlined in the MIQE guidelines form the foundation for more specialized applications, particularly in clinical research settings where qPCR-based tests for oncogene expression face significant standardization challenges [72]. The noticeable lack of technical standardization remains a huge obstacle in the translation of qPCR-based tests from research use only (RUO) to in vitro diagnostics (IVD) [72]. To address this gap, recent consensus guidelines propose a clinical research (CR) assay validation level, which occupies an intermediate position between RUO and fully certified IVD assays [72].
For oncogene expression tests with potential clinical applications, researchers must consider both analytical performance characteristics (trueness, precision, analytical sensitivity, and specificity) and clinical performance (diagnostic sensitivity, specificity, and predictive values) [72]. The validation rigor should follow a "fit-for-purpose" concept where "the level of validation associated with a medical product development tool is sufficient to support its context of use" [72]. This approach is particularly relevant for oncogene expression panels that might guide treatment decisions or patient stratification in clinical trials.
Digital PCR and Emerging Technologies
While initially developed for quantitative real-time PCR, the MIQE principles have been extended to emerging technologies, including digital PCR (dPCR) through the publication of digital MIQE (dMIQE) guidelines [73]. Digital PCR offers potential advantages for oncogene quantification, particularly for rare mutations or low-level expression changes, as it "allows the precise quantification of nucleic acids, facilitating the measurement of small percentage differences and quantification of rare variants" [73].
The dMIQE guidelines address the unique requirements of digital PCR platforms while maintaining the core MIQE principle that "comprehensive disclosure of all relevant experimental details is required" to enable independent evaluation of experimental data [73]. As the field moves toward increasingly precise quantification requirements for oncogene expression, particularly in liquid biopsy applications or minimal residual disease monitoring, these dMIQE guidelines will become increasingly relevant.
Successful implementation of MIQE-compliant qPCR requires careful selection of reagents and resources. Table 2 outlines essential components for oncogene expression studies with specific quality control considerations.
Table 2: Research Reagent Solutions for MIQE-Compliant Oncogene qPCR
| Reagent/Resource | Function | Quality Control Requirements |
|---|---|---|
| RNA Isolation Kits | Nucleic acid extraction from tumor samples | Assess yield and purity (A260/A280 ~1.8-2.0); verify integrity (RIN >7.0 for fresh tissue) |
| Reverse Transcriptase | cDNA synthesis from RNA templates | Document enzyme source, priming method, and reaction conditions; include no-RT controls |
| qPCR Master Mix | Provides enzymes, nucleotides, buffer for amplification | Report manufacturer and formulation; verify compatibility with detection chemistry (SYBR Green vs. probe-based) |
| Validated Primers/Probes | Target-specific amplification | Provide sequences or assay IDs; document validation data including efficiency and specificity |
| Reference Gene Panels | Normalization of expression data | Validate stability for specific tumor type; use multiple genes (≥3) from independent pathways |
| Quality Control Materials | Monitoring assay performance | Include positive controls, negative controls, and inter-run calibrators |
The MIQE guidelines provide an essential framework for ensuring the reliability and reproducibility of qPCR data, particularly in methodologically sensitive applications such as oncogene expression quantification. By addressing all phases of the qPCR workflow—from sample acquisition to data analysis—these guidelines help researchers avoid common pitfalls that compromise data integrity. The recent introduction of MIQE 2.0 reinforces that "guidelines alone are not enough" and calls for cultural change among researchers, reviewers, and journal editors to treat qPCR with the same rigor expected of other molecular techniques [70].
For researchers focused on oncogene quantification, adherence to MIQE principles represents more than a publication requirement—it establishes a foundation for generating clinically relevant data that can reliably inform diagnostic, prognostic, and therapeutic decisions. As the field advances toward increasingly precise molecular measurements, the MIQE framework will continue to evolve, but its core mission will remain constant: to ensure that qPCR results are not just published, but are robust, reproducible, and reliable.
Validating Your Findings: Ensuring Accuracy and Clinical Relevance
Orthogonal validation, the process of confirming results from one experimental method with another, independent technique, is a cornerstone of rigorous gene expression analysis. In transcriptomics, quantitative real-time PCR (qPCR) is widely regarded as the gold standard for validating results obtained from high-throughput platforms like RNA-Sequencing (RNA-Seq) and microarrays [74]. This verification is particularly critical in oncogene research and drug development, where accurately quantifying expression changes directly impacts therapeutic discovery and clinical decision-making. While RNA-Seq and microarrays provide powerful, genome-wide screening capabilities, each platform has inherent technical variations that can affect quantitative accuracy. RNA-Seq offers a higher dynamic range and can detect novel transcripts, whereas microarrays provide a cost-effective solution for well-characterized genomes, but both may yield inconsistent results for specific genes, especially those with low expression levels or small fold-changes [74] [75] [76]. The integration of qPCR validation strengthens data reliability, enhances reproducibility, and builds confidence in gene signatures used for patient stratification and biomarker identification in oncology.
The Critical Need for Validation
Concordance and Discordance Between Platforms
Comparative studies reveal a complex landscape of agreement between transcriptomic platforms. A comprehensive analysis comparing five RNA-Seq pipelines to wet-lab qPCR results across over 18,000 protein-coding genes found that 15–20% of genes showed 'non-concordant' results, defined as instances where methods yielded differential expression in opposing directions or one method detected change while the other did not [74]. However, this discordance is not random; approximately 93% of non-concordant genes exhibited fold changes lower than 2, and about 80% showed fold changes below 1.5 [74]. This indicates that disagreements most frequently affect genes with subtle expression changes, while strongly differentially expressed genes typically show good concordance across platforms.
The situation is particularly important for studies where the entire biological conclusion rests on differential expression of only a few genes, especially if these genes have low expression levels or small expression differences [74]. In such cases, orthogonal validation becomes not merely recommended but essential for verifying key findings. Furthermore, transforming high-dimensional gene expression data into gene set enrichment scores has been shown to increase correlation between RNA-Seq and microarray platforms, suggesting that biological interpretation at the pathway level may be more reproducible than individual gene measurements [75].
Platform-Specific Technical Considerations
RNA-Seq offers advantages including a wide dynamic range, ability to detect novel transcripts, and no requirement for pre-defined probes [76] [77]. However, its analysis involves complex bioinformatic pipelines encompassing trimming, alignment, counting, and normalization steps, with each stage introducing potential variability. A systematic evaluation of 192 alternative analytical pipelines demonstrated that methodological choices significantly impact results, emphasizing the need for verification [77].
Microarrays, while having a more established analytical framework and lower cost, suffer from limitations such as background hybridization noise, probe saturation effects, and a narrower dynamic range that compromises detection of both lowly and highly expressed genes [75] [76]. These technical constraints can affect quantitative accuracy, particularly for genes expressed at extreme levels.
Table 1: Key Comparison Between Transcriptomic Platforms
| Feature | RNA-Seq | Microarray | qPCR |
|---|---|---|---|
| Dynamic Range | High (>10⁵ fold) [76] | Limited (∼10³ fold) [76] | Very High (up to 10⁹ fold) [19] |
| Throughput | Genome-wide | Genome-wide | Limited targets (typically <100) |
| Detection Capability | Novel transcripts, splice variants | Pre-defined transcripts only | Pre-designed targets only |
| Technical Sensitivity | High for most genes | Reduced for low-abundance transcripts | Extremely high |
| Primary Application | Discovery | Profiling | Validation/Quantification |
Validation Workflows and Experimental Design
Core Validation Workflow
The following diagram illustrates the comprehensive workflow for orthogonal validation of transcriptomics data, from initial screening to final confirmed gene list:
Candidate Gene Selection Strategy
Selecting appropriate candidates for validation is a critical decision point that significantly impacts resource allocation and conclusion validity. The following decision tree guides this selection process:
Prioritize genes for validation when your biological conclusion depends heavily on a small number of genes, when observed fold changes are small (<2), when genes have low expression levels, or when targeting known oncogenes for therapeutic development [74]. For studies reporting large gene signatures, select a representative subset encompassing different expression ranges and functional categories.
Detailed qPCR Validation Protocols
Protocol 1: Relative Quantification for RNA-Seq Validation
This protocol describes how to validate RNA-Seq results using relative quantification with reference genes, suitable for most research scenarios.
4.1.1 RNA Quality Control and cDNA Synthesis
- Input Material: Use the same RNA samples originally analyzed by RNA-Seq to eliminate sample-to-sample variability.
- RNA Quality Assessment: Verify RNA integrity using Agilent Bioanalyzer or similar systems. RIN > 8.0 is recommended for optimal results [78] [77].
- Genomic DNA Removal: Perform on-column or solution-based DNase digestion to eliminate genomic DNA contamination [77].
- Reverse Transcription: Use 100 ng–1 μg total RNA with reverse transcriptase and oligo(dT) or random hexamer primers. Include controls without reverse transcriptase (-RT) to detect genomic contamination.
4.1.2 qPCR Assay Design and Validation
- Amplicon Design: Design amplicons 70–150 bp in length, preferably spanning exon-exon junctions to avoid genomic DNA amplification.
- Primer Validation: Test primer efficiency using standard curves with serial dilutions (typically 5–10 points). Acceptable efficiency ranges from 90–110% (R² > 0.99) [79].
- Reaction Conditions: Use 10–20 μL reaction volumes with SYBR Green or probe-based chemistry. Run samples in technical triplicates to account for pipetting error.
4.1.3 Data Normalization and Analysis
- Reference Gene Selection: Select multiple validated housekeeping genes (see Section 5.1). Using a single unverified gene like GAPDH can introduce significant bias [11] [79].
- Stability Assessment: Use algorithms like GeNorm or NormFinder to evaluate reference gene stability in your specific experimental system.
- Expression Calculation: Calculate relative expression using the ΔΔCt method or equivalent model-based approaches.
Table 2: Troubleshooting Common qPCR Validation Issues
| Problem | Potential Cause | Solution |
|---|---|---|
| Poor correlation with RNA-Seq | Inappropriate reference genes | Validate HKG stability; use global mean normalization if profiling many genes [79] |
| High variability between replicates | RNA degradation or pipetting errors | Check RNA quality (RIN); use robotic liquid handling systems |
| Non-specific amplification | Primer-dimer or mispriming | Optimize annealing temperature; design new primers; switch to probe chemistry |
| Efficiency outside 90–110% | Poor primer design or reaction inhibitors | Redesign primers; purify RNA template; dilute cDNA |
Protocol 2: Absolute Quantification for Microarray Validation in Oncogene Research
This protocol describes absolute quantification approaches, particularly valuable for therapeutic development where precise copy number determination is required.
4.2.1 Standard Curve Preparation
- Template Generation: Clone target amplicon into plasmid vector or obtain commercially available gBlocks gene fragments.
- Quantification and Dilution: Precisely quantify standard material using spectrophotometry (NanoDrop) and fluorometry (Qubit). Prepare serial 10-fold dilutions covering 6–8 orders of magnitude (from ~10⁷ to 10¹ copies) to encompass expected target concentrations [19].
- Storage Considerations: Aliquot and store standards at –80°C to prevent freeze-thaw degradation.
4.2.2 Absolute qPCR Setup
- Parallel Reactions: Run unknown samples and standard curves on the same plate to minimize inter-assay variability.
- Copy Number Calculation: Determine sample copy number by interpolating Ct values against the standard curve. Include no-template controls to detect contamination.
- Quality Control: Standard curves must demonstrate linearity (R² > 0.99) across the quantification range [19].
4.2.3 Data Reporting for Therapeutic Development
- Normalization to Cell Number: Express results as copies per cell using cell counts during RNA isolation or reference to endogenous controls.
- Precision Assessment: Report intra- and inter-assay coefficients of variation.
- Statistical Analysis: Apply appropriate statistical tests comparing absolute copy numbers between experimental conditions.
Critical Methodological Considerations
Selection and Validation of Housekeeping Genes
The choice of internal control genes represents one of the most critical factors in obtaining accurate qPCR results. Many traditionally used housekeeping genes show significant variability in cancer contexts:
GAPDH Pitfalls: While commonly used, GAPDH is unsuitable as a pan-cancer marker and shows substantial variability in endometrial cancer and other malignancies [11]. Its expression is regulated by multiple factors including insulin, growth hormone, oxidative stress, and hypoxia, making it particularly unreliable in cancer studies [11].
Ribosomal Protein Genes: Genes such as RPS5, RPL8, and RPS19 demonstrate high stability in gastrointestinal cancers and represent better alternatives to traditional controls [79].
Validation Requirement: Always validate potential reference genes using stability algorithms like GeNorm or NormFinder. Use a minimum of two validated HKGs for normalization [11] [79].
Global Mean Alternative: When profiling larger gene sets (>55 genes), the global mean of all expressed genes can outperform single reference genes in normalization accuracy [79].
Experimental Design and Statistical Considerations
- Biological Replicates: Prioritize biological over technical replication. Include sufficient replicates (typically n ≥ 3) to ensure statistical power.
- Blinding and Randomization: Perform sample processing and analysis blinded to experimental conditions when possible.
- Correlation Analysis: Assess concordance using Pearson or Spearman correlation comparing log₂ fold-changes between platforms.
- Acceptance Criteria: Define pre-established criteria for successful validation (e.g., concordant direction of change, fold-change difference < 30%).
Research Reagent Solutions
Table 3: Essential Reagents and Kits for Orthogonal Validation
| Reagent Category | Specific Examples | Application Notes |
|---|---|---|
| RNA Isolation Kits | AllPrep DNA/RNA (Qiagen), RNeasy Plus (Qiagen) [78] [77] | Simultaneous DNA/RNA preservation; genomic DNA elimination |
| RNA Quality Assessment | Agilent 2100 Bioanalyzer [78] [77] | Critical for RIN determination; essential for FFPE samples |
| Reverse Transcription Kits | SuperScript First-Strand Synthesis (Thermo Fisher) [77] | Include -RT controls for genomic DNA contamination |
| qPCR Master Mixes | SYBR Green, TaqMan assays (Applied Biosystems) [77] | Probe-based for specificity; SYBR Green for cost-effectiveness |
| Reference Gene Panels | Commercially available HKG panels | Pre-validated for specific tissue types; reduces optimization time |
Orthogonal validation of transcriptomics data using qPCR remains an essential component of rigorous gene expression analysis, particularly in oncogene research and drug development. While RNA-Seq and microarray technologies continue to advance, the precision, sensitivity, and quantitative reliability of qPCR make it indispensable for confirming key findings before drawing biological conclusions or making therapeutic decisions. By implementing the standardized protocols and considerations outlined in this application note, researchers can significantly enhance the reliability and reproducibility of their gene expression studies, ultimately accelerating the translation of genomic discoveries into clinical applications.
The absolute quantification of oncogene expression is a critical component of modern cancer research, particularly in the management of hematological malignancies. Minimal residual disease (MRD) refers to the small number of cancer cells that persist in patients after treatment, typically at levels undetectable by conventional microscopy [80]. In clinical practice, MRD serves as a pivotal biomarker for assessing treatment efficacy, predicting relapse risk, and guiding therapeutic decisions [80] [81]. The ability to accurately detect and quantify these residual cancer cells has profound implications for patient outcomes, as the presence of MRD often precedes clinical relapse [80] [82].
The evolution of MRD detection methodologies has progressed from traditional morphological assessments to increasingly sophisticated molecular techniques. Each advancement has brought improvements in sensitivity and specificity, enabling researchers and clinicians to detect progressively lower levels of residual disease [80]. Among these techniques, polymerase chain reaction (PCR)-based methods have emerged as particularly valuable tools. The transition from conventional PCR to real-time quantitative PCR (qPCR) represented a significant step forward, allowing for the monitoring of amplification reactions in real time [83]. More recently, digital PCR (dPCR) has introduced a fundamentally different approach to nucleic acid quantification, promising enhanced precision and sensitivity for low-abundance targets [83] [84].
This application note provides a comprehensive comparison of qPCR and dPCR technologies within the context of MRD studies, with a specific focus on their application in the absolute quantification of oncogene expression. We present structured experimental data, detailed protocols, and practical guidance to inform method selection for research and clinical applications in oncology and drug development.
Technical Comparison of qPCR and Digital PCR
Fundamental Principles and Methodologies
Real-time quantitative PCR (qPCR) operates on the principle of monitoring PCR amplification in real time using fluorescence detection. This technology employs either DNA-binding dyes or target-specific probes that generate fluorescent signals as the target DNA amplifies. The point at which the fluorescence crosses a predetermined threshold is known as the cycle threshold (CT), which correlates inversely with the initial quantity of the target nucleic acid [3] [4]. For absolute quantification, qPCR requires comparison to a standard curve generated from samples of known concentration, introducing potential variables in quantification accuracy [3] [4].
In contrast, digital PCR (dPCR) utilizes a fundamentally different approach based on sample partitioning and Poisson statistics. The reaction mixture is partitioned into thousands of individual reactions, each containing zero, one, or a few target molecules. Following end-point PCR amplification, each partition is analyzed for fluorescence, and the fraction of negative partitions is used to calculate the absolute target concentration without the need for a standard curve [83] [84]. This partitioning process reduces the effects of amplification efficiency variations and provides direct absolute quantification of target molecules [3] [84].
The following diagram illustrates the fundamental workflow differences between these two technologies:
Performance Characteristics for MRD Detection
The performance characteristics of qPCR and dPCR differ significantly, particularly in the context of detecting low-abundance targets characteristic of MRD. The following table summarizes key performance metrics based on current literature:
Table 1: Performance Comparison of MRD Detection Methods
| Parameter | qPCR | Digital PCR | References |
|---|---|---|---|
| Sensitivity | 10⁻⁴ to 10⁻⁶ (depending on target) | 10⁻⁴ to 10⁻⁶ (potentially higher for specific mutations) | [80] [81] [82] |
| Quantification Approach | Relative (requires standard curve) | Absolute (no standard curve needed) | [3] [83] [84] |
| Precision at Low Target Concentrations | Moderate (higher variability) | High (lower coefficient of variation) | [85] [30] [81] |
| Tolerance to PCR Inhibitors | Moderate | High | [85] [84] |
| Dynamic Range | 6-7 logarithmic decades | 5-6 logarithmic decades | [30] |
| Multiplexing Capability | Moderate (spectral overlap challenges) | High (independent partition analysis) | [85] [83] |
Recent studies have demonstrated that dPCR offers superior precision, particularly at low target concentrations. In a 2025 study comparing dPCR and qPCR for periodontal pathogen detection, dPCR showed lower intra-assay variability (median CV%: 4.5%) compared to qPCR, with particularly improved detection of low bacterial loads [85]. This enhanced precision at low concentrations translates directly to MRD applications, where accurate quantification of rare targets is essential.
In a blinded prospective study focusing on acute lymphoblastic leukemia (ALL), ddPCR outperformed qPCR with a significantly better quantitative limit of detection and sensitivity. The number of critical MRD estimates below the quantitative limit was reduced by sixfold in a retrospective cohort and by threefold in a prospective cohort, demonstrating the practical advantages of dPCR for clinical MRD monitoring [81].
Clinical Applications in Hematological Malignancies
Both qPCR and dPCR have been extensively applied to MRD detection in various hematological malignancies, with each technology offering distinct advantages for specific applications:
Table 2: Clinical Applications in Hematological Malignancies
| Application | qPCR Performance | Digital PCR Performance | References |
|---|---|---|---|
| ALL MRD Monitoring | Gold standard for Ig/TCR rearrangement detection | Superior sensitivity and precision; reduces unquantifiable results | [81] [82] |
| AML Mutation Detection | Limited for immature Ig/TCR rearrangements | Effective for somatic SNVs (e.g., IDH2, NPM1); sensitivity to 0.002% | [84] [82] |
| MPN Driver Mutations (JAK2, CALR) | Sensitivity ~0.01-1% | Sensitivity ~0.01-0.1%; precise allele burden quantification | [84] |
| Lymphoma MRD | Applicable with specific targets | Effective for universal SNV-based monitoring | [82] |
The application of dPCR for somatic single nucleotide variant (SNV) detection represents a particularly significant advancement in MRD monitoring. A 2023 study demonstrated that ddPCR-based MRD detection targeting SNVs achieved sensitivity up to 1×10⁻⁴ and successfully detected micro-residual disease that was missed by conventional PCR-MRD in a T-ALL patient [82]. This approach offers universality, as it can be applied to various malignant diseases regardless of tumor-specific Ig/TCR or surface antigen patterns.
Experimental Protocols for MRD Detection
Digital PCR Protocol for Mutation-Specific MRD Detection
The following protocol provides a detailed methodology for MRD detection using droplet digital PCR (ddPCR) targeting somatic single nucleotide variants (SNVs), based on established procedures with demonstrated clinical utility [82]:
Principle: This protocol enables absolute quantification of mutant allele frequency in patient samples using a droplet digital PCR system. The approach involves partitioning the PCR reaction into thousands of nanoliter-sized droplets, followed by end-point amplification and counting of positive and negative droplets for absolute quantification without standard curves.
Materials & Equipment:
- QX200 Droplet Digital PCR System (Bio-Rad) or equivalent nanoplate-based system
- ddPCR Supermix for Probes (No dUTP)
- Target-specific primers and fluorescent probes (FAM and HEX/VIC labeled)
- Restriction enzyme (e.g., Hind III, HaeIII, or EcoRI)
- DG8 Cartridges and Droplet Generator
- C1000 Touch Thermal Cycler with 96-Deep Well Reaction Module
- PX1 PCR Plate Sealer
- QX200 Droplet Reader
Procedure:
- Assay Design:
- Identify tumor-specific SNVs through whole-exome sequencing of tumor specimens compared to matched normal DNA.
- Design primer pairs to amplify 50-150 bp products encompassing the mutation site.
- Design two allele-specific probes: one labeled with FAM for the mutant allele and one labeled with HEX/VIC for the wild-type allele.
Reaction Setup:
- Prepare 22 μL reaction mixtures containing:
- 11 μL ddPCR Supermix for Probes (No dUTP)
- Primers at a final concentration of 0.25 μM each
- Probes at a final concentration of 1 μM each
- 10 units of restriction enzyme (Hind III or equivalent)
- 150 ng of genomic DNA (or equivalent for other sample types)
- Gently mix reactions by pipetting; avoid introducing bubbles.
- Prepare 22 μL reaction mixtures containing:
Droplet Generation:
- Transfer 20 μL of each reaction mixture to DG8 Cartridge wells.
- Add 70 μL of Droplet Generation Oil to the appropriate wells.
- Place the DG8 Cartridge into the Droplet Generator.
- After droplet generation (approximately 20,000 droplets per sample), carefully transfer the droplets to a 96-well PCR plate.
PCR Amplification:
- Seal the PCR plate using the PX1 PCR Plate Sealer at 180°C for 5 seconds.
- Perform amplification using the following thermal cycling conditions:
- Enzyme activation: 95°C for 10 minutes
- 40-45 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing/Extension: 55-60°C (assay-specific) for 60 seconds
- Enzyme deactivation: 98°C for 10 minutes
- Final hold: 12°C
- Include a ramp rate of 2°C/second between steps.
Droplet Reading and Analysis:
- Place the PCR plate in the QX200 Droplet Reader.
- Analyze each well sequentially, measuring fluorescence in both FAM and HEX/VIC channels.
- Use QuantaSoft software to count positive and negative droplets for each channel.
- Apply Poisson statistics to calculate the absolute concentration of mutant and wild-type alleles.
- Calculate mutant allele frequency as: [mutant copies/μL] / ([mutant copies/μL] + [wild-type copies/μL]) × 100
Critical Considerations:
- Restriction Enzyme Selection: The choice of restriction enzyme can significantly impact precision, particularly for targets with high copy numbers. In comparative studies, HaeIII demonstrated higher precision compared to EcoRI, especially for the QX200 system [30].
- DNA Input Quality: Use high-quality DNA with minimal fragmentation. For FFPE samples, consider specialized extraction kits and potentially increase DNA input.
- Partitioning Efficiency: Ensure adequate droplet generation (typically >10,000 droplets per sample) for reliable quantification.
- Background Signal: Establish limits of blank (LoB) using negative control samples from healthy individuals.
qPCR Protocol for Absolute Quantification in MRD Studies
Principle: This protocol describes absolute quantification of specific genetic targets using real-time qPCR with a standard curve approach. The method relies on comparing CT values of unknown samples to a standard curve generated from samples with known concentrations.
Materials & Equipment:
- Real-time PCR instrument (e.g., Applied Biosystems 7500, Roche LightCycler 480)
- qPCR Master Mix (e.g., TaqMan Gene Expression Master Mix)
- Target-specific primers and probes
- DNA standards of known concentration
- Optical 96-well reaction plates or strips
- Microseal adhesives
Procedure:
- Standard Preparation:
- Prepare serial dilutions (at least 5 points) of DNA standards with known concentrations.
- Use plasmid DNA, PCR fragments, or genomic DNA as standards, ensuring they contain the target sequence.
- For plasmid standards, linearize before use to better match amplification efficiency of genomic DNA.
- Calculate copy number using the formula: (X g/μL DNA / [length in bp × 660]) × 6.022 × 10²³ = Y molecules/μL [4]
Reaction Setup:
- Prepare 20-25 μL reactions containing:
- 1× qPCR Master Mix
- Primers and probes at optimized concentrations (typically 0.1-0.9 μM for primers, 0.1-0.2 μM for probes)
- DNA template (typically 50-100 ng per reaction)
- Include no-template controls and standards in each run.
- Perform each reaction in triplicate for improved accuracy.
- Prepare 20-25 μL reactions containing:
PCR Amplification:
- Use the following thermal cycling conditions:
- Enzyme activation: 95°C for 10 minutes
- 40-50 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 60 seconds
- Monitor fluorescence during the annealing/extension step of each cycle.
- Use the following thermal cycling conditions:
Data Analysis:
- Determine CT values for each reaction using the instrument software.
- Generate a standard curve by plotting CT values against the logarithm of the known input copy number.
- Use the linear regression equation from the standard curve to calculate the copy number in unknown samples.
- Apply any necessary correction factors for sample normalization.
Critical Considerations:
- Amplification Efficiency: Ensure the efficiency of the target amplification is between 90-110% with R² > 0.99 for the standard curve.
- Sample Quality: Assess DNA quality and quantity before analysis; degraded samples may yield inaccurate results.
- Inhibition Testing: Test for PCR inhibitors by spiking samples with known quantities of target and assessing recovery.
- Assay Validation: Validate each assay for specificity, sensitivity, and linear dynamic range before clinical application.
The Scientist's Toolkit: Essential Reagents and Materials
The following table outlines key reagents and materials essential for implementing MRD detection protocols using both qPCR and dPCR technologies:
Table 3: Essential Research Reagents for MRD Detection
| Category | Specific Items | Function/Application | Technical Notes |
|---|---|---|---|
| Nucleic Acid Extraction | QIAamp DNA Mini Kit, GeneRead DNA FFPE Kit | Isolation of high-quality DNA from various sample types | FFPE-specific kits crucial for archival tissue analysis [82] |
| dPCR Systems | QX200 Droplet Digital PCR (Bio-Rad), QIAcuity (QIAGEN) | Partitioning-based absolute quantification | QIAcuity uses nanoplate technology; QX200 employs droplet generation [30] [83] |
| dPCR Master Mixes | ddPCR Supermix for Probes (No dUTP), QIAcuity Probe PCR Kit | Optimized reaction mixtures for digital PCR applications | No dUTP recommended for restriction enzyme digestion steps [82] |
| qPCR Master Mixes | TaqMan Gene Expression Master Mix, various probe-based mixes | Fluorescence-based real-time monitoring of amplification | Contains all components except primers/probes/template [4] |
| Detection Chemistry | TaqMan hydrolysis probes, Primers for SNV detection | Target-specific amplification and detection | Dual-labeled probes (FAM/HEX) enable multiplex detection [82] |
| Enzymes for Digestion | Hind III, HaeIII, EcoRI restriction enzymes | Improve target accessibility, especially in repetitive regions | Enzyme choice affects precision; HaeIII recommended for some systems [30] [82] |
| Reference Materials | Plasmid standards, synthetic oligonucleotides, control DNA | Standard curve generation (qPCR) and assay validation | Characterized cell lines useful as qualitative controls [4] |
Comparative Data Analysis and Interpretation
Sensitivity and Precision in Clinical Samples
Direct comparisons between qPCR and dPCR in clinical studies have demonstrated meaningful differences in performance characteristics relevant to MRD detection:
In a 2025 study comparing dPCR and qPCR for pathogen detection, dPCR demonstrated superior sensitivity for detecting low bacterial loads, particularly for P. gingivalis and A. actinomycetemcomitans. The Bland-Altman plots highlighted good agreement between the methods at medium/high loads but significant discrepancies at low concentrations (< 3 log₁₀Geq/mL), resulting in qPCR false negatives and a 5-fold underestimation of prevalence for A. actinomycetemcomitans in periodontitis patients [85]. While this study focused on microbial detection, the findings translate directly to MRD applications where low target abundance is common.
A cross-platform comparison of dPCR systems published in 2025 evaluated the precision of the QX200 droplet digital PCR (ddPCR) from Bio-Rad and the QIAcuity One nanoplate-based digital PCR (ndPCR) from QIAGEN. Both platforms demonstrated similar detection and quantification limits and yielded high precision across most analyses. The LOQ for ndPCR was approximately 1.35 copies/μL input (54 copies/reaction), while ddPCR showed an LOQ of approximately 4.26 copies/μL input (85.2 copies/reaction) [30]. This study highlights the importance of platform-specific validation for MRD assays.
Practical Implementation Considerations
When implementing MRD detection protocols, several practical considerations influence method selection:
Throughput and Workflow Efficiency: qPCR typically offers faster run times and higher throughput capabilities, while dPCR provides more robust quantification without standard curves. For laboratories with established standard curves and validated assays, qPCR may remain efficient for high-volume testing. However, for new targets or when standard materials are unavailable, dPCR offers significant advantages.
Cost Considerations: dPCR reactions typically have higher per-sample reagent costs compared to qPCR. However, this must be balanced against reduced requirements for replicate reactions and standard curves in dPCR, as well as potential improvements in detection accuracy that may prevent costly false negatives in clinical decision-making.
Data Interpretation Guidelines:
- For dPCR, establish threshold settings consistently based on negative controls
- Apply Poisson correction for volume adjustments in dPCR quantification
- For qPCR, ensure standard curves meet efficiency and linearity criteria (R² > 0.98, efficiency 90-110%)
- Implement rigorous quality control measures, including negative and positive controls in each run
The following diagram illustrates a decision framework for method selection based on experimental requirements:
The comparison between qPCR and dPCR for MRD detection reveals a nuanced landscape where each technology offers distinct advantages depending on the specific application requirements. qPCR remains a well-established, cost-effective technology suitable for high-throughput applications where standardized controls are available. Its extensive validation history and familiar workflow make it appropriate for many routine clinical applications.
In contrast, dPCR provides significant advantages for applications requiring absolute quantification without standard curves, detection of very low-abundance targets, and analysis of challenging sample types containing PCR inhibitors. The technology's enhanced precision at low concentrations and ability to detect rare mutations at frequencies as low as 0.001% make it particularly valuable for monitoring residual disease after treatment [84] [82].
For researchers focused on the absolute quantification of oncogene expression, dPCR represents a powerful tool that complements and extends existing qPCR capabilities. The technology's ability to provide direct absolute quantification without reference standards makes it especially valuable for novel targets where well-characterized control materials are unavailable. As MRD detection continues to evolve toward increasingly sensitive and precise measurements, dPCR is poised to play an expanding role in both research and clinical applications, ultimately contributing to more personalized and effective cancer therapies.
Absolute quantification by quantitative PCR (qPCR) provides a critical framework for accurately determining oncogene expression levels by measuring exact copy numbers of nucleic acid sequences, thereby preventing the over- or under-estimation of biologically significant effect sizes in drug development research. Unlike relative quantification methods that rely on reference genes and assume optimal amplification efficiency, absolute quantification utilizes standardized curves from samples of known concentration, directly addressing the profound impact that variations in PCR efficiency can have on calculated expression ratios [86] [3]. This application note details robust protocols for implementing absolute quantification, complete with standardized materials and data visualization workflows, to generate precise and reproducible oncogene expression data essential for confident therapeutic decision-making.
In molecular oncology and drug development, accurately measuring changes in oncogene expression is paramount for assessing therapeutic efficacy, understanding resistance mechanisms, and defining biomarkers. The quantification cycle (Cq) in qPCR is inversely related to the starting concentration of the target nucleic acid [86]. However, the Cq value is not determined by target concentration alone; it is also a function of PCR amplification efficiency (E) and the level at which the quantification threshold (Nq) is set, as described by the equation: Cq = log(Nq) - log(N0) / log(E) [86]. When these factors are not accounted for, the interpretation of Cq values can be severely biased, leading to inaccurate conclusions.
Relative quantification, often using the comparative Cq (ΔΔCq) method, is popular for its simplicity but operates under the critical assumption that the amplification efficiencies of the target and reference genes are equal and near 100% [5]. When this assumption is violated—which is frequently the case—the resulting fold-change calculations can be dramatically inaccurate. One review notes that interpreting Cq values while assuming 100% efficient PCR can lead to assumed gene expression ratios that are 100-fold off from the true value [86]. Absolute quantification circumvents these pitfalls by providing an instrument-independent measure of exact copy number, establishing a firm foundation for assessing true effect size.
Comparative Analysis of Quantification Methods
The choice between quantification methods has a direct impact on the accuracy, reproducibility, and interpretability of gene expression data. The following table summarizes the core differences.
Table 1: Core Differences Between Absolute and Relative Quantification Methods
| Feature | Absolute Quantification (Standard Curve Method) | Relative Quantification (ΔΔCq Method) |
|---|---|---|
| Output | Exact copy number or concentration of the target [3] | Fold-change in gene expression relative to a calibrator sample [3] [5] |
| Standard Requirement | Requires a standard curve from samples of known concentration (e.g., plasmid DNA, in vitro transcribed RNA) [3] | Requires a stable endogenous control/reference gene (e.g., GAPDH, β-actin) [5] |
| Key Assumption | The standard curve accurately reflects the amplification behavior of the unknown samples. | The amplification efficiencies of the target and reference gene are approximately equal and near 100% [5]. |
| Impact of Efficiency (E) | Efficiency is directly incorporated into the standard curve, correcting for inefficiencies [38]. | Efficiency differences are not accounted for in the ΔΔCq calculation, leading to potential miscalculation [86] [5]. |
| Data Comparability | Results are absolute and can be compared across different plates, runs, and laboratories with proper standardization. | Results are relative to a specific experiment, making cross-laboratory comparisons difficult. |
The mathematical basis for the risk of miscalculation with the ΔΔCq method is evident in the standard formula for calculating the expression ratio (RQ):
RQ = 2^(-ΔΔCq) [5].
This formula is a special case of the more robust Pfaffl method, RQ = (E_target)^(ΔCt_target) / (E_reference)^(ΔCt_reference), where the efficiencies (E) of both assays are assumed to be 2 [5]. If the true efficiency of an assay is 1.8 (90%) instead of 2, the calculated ratio using the ΔΔCq method will be substantially inaccurate. In contrast, absolute quantification using a standard curve inherently incorporates the actual amplification efficiency of the assay, as the slope of the standard curve is used to determine efficiency (E = 10^(-1/slope)) and is directly used to interpolate the starting quantity of unknown samples [5].
Protocols for Absolute Quantification in Oncogene Research
Protocol 1: Establishing a Standard Curve for Absolute Quantification
This protocol describes the generation of a precise standard curve using serial dilutions of a target of known concentration, a cornerstone of absolute quantification.
1. Research Reagent Solutions Table 2: Essential Materials for Standard Curve Generation
| Item | Function & Critical Guidelines |
|---|---|
| Standard Template | Plasmid DNA or in vitro transcribed RNA containing the target oncogene sequence. Must be a single, pure species; contamination with RNA can inflate copy number determination [3]. |
| Spectrophotometer | For accurately measuring the concentration (A260) of the standard template to determine its absolute quantity [3]. |
| Low-Binding Plastics | Low-retention tubes and tips are critical to prevent sample loss during serial dilution, which is a major source of error [3]. |
2. Detailed Workflow
- Quantify Standard Stock: Precisely measure the concentration of the purified standard (plasmid DNA or RNA) via A260 spectrophotometry.
- Calculate Copy Number: Convert the mass concentration to molecular copy number/μL using the molecular weight of the standard.
- Perform Serial Dilutions: Perform a minimum of 5-log serial dilutions (e.g., 10-fold dilutions) to create a standard curve spanning the expected concentration range of your experimental samples. Critical Guideline: Using a larger volume (e.g., 10 μL) when constructing serial dilutions reduces sampling error [38]. Prepare small aliquots of diluted standards to avoid freeze-thaw cycles [3].
- Run qPCR Plate: Amplify each dilution in a minimum of 3-4 technical replicates to ensure a robust estimation of PCR efficiency [38].
- Generate Standard Curve: Plot the mean Cq value of each standard dilution against the logarithm of its known starting quantity. The slope, y-intercept, and correlation coefficient (R²) of the curve are used for subsequent calculations.
Protocol 2: Absolute Quantification of Oncogene Expression in Tissue Samples
This protocol applies the standard curve to determine the absolute copy number of an oncogene in extracted RNA samples.
1. Research Reagent Solutions Table 3: Essential Materials for Oncogene Quantification
| Item | Function & Critical Guidelines |
|---|---|
| RNA Extraction Kit | For high-quality, intact RNA isolation from tissue samples. RNA integrity (RIN) should be assessed. |
| Reverse Transcription Kit | For synthesizing cDNA from RNA samples. Use a consistent priming method (e.g., oligo-dT, random hexamers) across all samples. |
| qPCR Master Mix | A precision-formulated mix containing DNA polymerase, dNTPs, and optimized buffer. |
| Target-Specific Primers | Validated primers for the oncogene of interest. Efficiency should be pre-determined and fall between 90-110% [5]. |
2. Detailed Workflow
- Sample Preparation: Extract total RNA from treated and control tissue samples. Assess RNA quality and quantity.
- Reverse Transcription: Synthesize first-strand cDNA from a fixed amount of RNA (e.g., 1 μg) for all samples in a single run to minimize technical variation.
- qPCR Setup: In the same plate as the standard curve dilutions, run the experimental cDNA samples alongside a no-template control (NTC).
- Data Interpolation: For each experimental sample, use the mean Cq value to interpolate its starting quantity (in copy number) from the standard curve.
- Data Normalization (Optional): The absolute copy number of the oncogene can be normalized to the absolute copy number of a reference gene per sample, or to the total mass of RNA input, to account for variations in sample loading.
Workflow and Data Analysis Visualization
Diagram 1: Absolute Quantification Workflow
Diagram 2: Factors Influencing Cq Values
Absolute quantification by qPCR is a powerful and non-negotiable tool for precisely assessing the effect size of oncogene expression changes in critical research and drug development. By directly measuring copy number and incorporating PCR efficiency into its calculations, it eliminates the systematic errors inherent in relative quantification methods that rely on unstable assumptions. The protocols and frameworks provided herein empower scientists to generate data that truly reflects biological reality, thereby de-risking the pipeline from biomarker discovery to therapeutic development.
The precision of oncogene expression data obtained through quantitative polymerase chain reaction (qPCR) is fundamental to advancements in cancer research and therapeutic development. Achieving consistent and comparable results across different laboratories presents a significant challenge, primarily due to technical variations in sample processing, RNA extraction, cDNA synthesis, and data normalization [79]. Absolute quantification provides a fundamental unit of measure, such as the exact number of DNA or RNA molecules, which allows for direct gene-to-gene and sample-to-sample comparisons, making it particularly valuable for cross-laboratory studies [3] [87]. This case study addresses the critical need for a standardized workflow that ensures the rigor and reproducibility of absolute quantification data, thereby enabling reliable biomarker validation and robust drug discovery efforts.
Core Challenge: Normalization and Variability
A primary obstacle in multi-laboratory studies is the selection of an appropriate normalization strategy to minimize technical variability. Normalization is crucial for eliminating variation introduced during sample processing, RNA extraction, and cDNA synthesis, ensuring that the final analysis reflects true biological variation [79]. The most common strategy employs internal reference genes (RGs). However, the expression of classic "housekeeping" genes can be unstable under different pathological conditions, such as cancer, which may skew normalized data and lead to incorrect biological interpretations [79]. An alternative method is the global mean (GM) expression, which uses the average expression of a large set of profiled genes for normalization [79]. The performance of these strategies can be evaluated by measuring the coefficient of variation (CV), where a lower mean CV indicates superior reduction of intra-group variability [79].
Table 1: Comparison of qPCR Quantification Methods
| Method | Principle | Key Requirement | Advantages | Disadvantages |
|---|---|---|---|---|
| Absolute Quantification (Standard Curve) [3] | Quantitates unknowns based on a known quantity (e.g., plasmid DNA) via a standard curve. | Standards of known concentration, accurate pipetting. | Provides fundamental units (e.g., copy number); allows direct sample and gene comparisons [87]. | Requires pure standards; prone to dilution errors; DNA standards do not control for reverse transcription efficiency [3]. |
| Absolute Quantification (Digital PCR) [3] | Partitions a sample into many reactions and counts positive vs. negative partitions. | Partitioning mechanism, low-binding plastics to prevent sample loss. | No need for standards or endogenous controls; highly tolerant to inhibitors; provides absolute count [3]. | Requires specialized equipment; optimal sample concentration must be predetermined. |
| Relative Quantification [3] | Analyzes gene expression changes relative to a reference sample (e.g., untreated control). | Stable reference genes and/or a calibrator sample. | Does not require standards of known concentration; high throughput with comparative CT method [3]. | Results are unitless and cannot be directly compared between genes; reference gene stability is critical. |
Proposed Workflow for Absolute Quantification
The following workflow is designed to maximize accuracy, precision, and consistency for the absolute quantification of oncogenes across multiple laboratories.
Experimental Protocol
Step 1: Sample Preparation and Incorporation of Exogenous Control
- Purpose: To normalize for inevitable losses of genetic material during the extraction process, which are more pronounced at lower concentrations [88].
- Procedure: Prior to nucleic acid extraction, spike all samples with a fixed, known concentration of an exogenous bacterial culture (e.g., Escherichia coli) or synthetic oligonucleotide that is absent in the study samples. This serves as a calibrator to account for losses during centrifugation and extraction [88].
Step 2: gDNA Extraction and Standard Curve Preparation
- gDNA Extraction: Perform gDNA extraction using a standardized, validated kit across all participating laboratories. The exogenous control added in Step 1 will be co-extracted with the sample gDNA.
- Standard Curve Preparation: For absolute quantification, a standard curve is required [3].
- Standard Material: Use a plasmid containing the cloned oncogene sequence or a synthetic gBlock gene fragment.
- Quantification: Precisely measure the concentration of the standard stock solution by A260 and calculate the copy number using its molecular weight [3].
- Serial Dilution: Perform a log-fold serial dilution (e.g., 10-fold) of the standard to create a concentration series spanning the expected dynamic range of the target in the samples (e.g., from 10^1 to 10^8 copies/μL) [89]. Dilute in a solution that mimics the sample matrix (e.g., yeast tRNA) [89]. Aliquot diluted standards to avoid freeze-thaw cycles [3].
Step 3: qPCR Run
- Reaction Setup: Run all samples, standards, and no-template controls (NTCs) in triplicate (technical replicates) to improve precision and allow for outlier detection [87].
- Master Mix: Use a precision qPCR master mix containing a passive reference dye to correct for pipetting variations and optical anomalies [87].
- Plate Preparation: Ensure good pipetting technique and plate loading practices. Visually confirm consistent volume delivery, seal the plate, and centrifuge it to collect liquid at the bottom of wells and remove air bubbles [87].
- Cycling Conditions: Follow manufacturer-recommended cycling protocols for the chosen master mix and instrument.
Data Analysis Protocol
Step 4: Data Curation and Preprocessing
- Baseline Correction: Manually set the baseline to the early cycles representing the background fluorescence (e.g., cycles 5-15), avoiding the initial cycles with reaction stabilization artifacts [90]. Incorrect baseline setting can significantly distort Cq values [90].
- Threshold Setting: Set the threshold at a fluorescence intensity within the exponential phase of all amplification curves where the curves are parallel [90]. This ensures that ΔCq values between samples are not affected by the chosen threshold.
- Curation Filters: Exclude data that does not meet quality criteria:
Step 5: Absolute Quantification and Normalization
- Calculate Absolute Copy Number: For each sample, the qPCR software will interpolate the Cq value from the standard curve to determine the absolute copy number of the target oncogene.
- Normalize with Exogenous Control: To account for extraction efficiency and technical losses, normalize the oncogene copy number using the exogenous control [88].
Normalized Oncogene Copy No. = (Raw Oncogene Copy No.) / (Exogenous Control Copy No.)
Step 6: Statistical Analysis
- Precision Measurement: Calculate the Coefficient of Variation (CV) for technical replicates. CV = (Standard Deviation / Mean) * 100%. A lower CV indicates higher precision and more consistent results [87].
- Statistical Significance: Use Analysis of Covariance (ANCOVA) to compare normalized oncogene levels between experimental groups (e.g., tumor vs. normal). ANCOVA offers greater statistical power and robustness compared to the traditional 2^−ΔΔCT method, and its P-values are not affected by variability in qPCR amplification efficiency [15].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Absolute qPCR
| Item | Function | Considerations for Use |
|---|---|---|
| Exogenous Control (e.g., E. coli culture, synthetic oligo) | Calibrator to normalize for gDNA loss during extraction, improving accuracy at low concentrations [88]. | Must be absent in the biological samples under study; added at a fixed concentration before extraction. |
| Cloned Plasmid or Synthetic Gene Fragment | Serves as the standard of known concentration for generating the absolute quantification standard curve [3]. | Must be a single, pure species; concentration must be accurately measured via A260. |
| Passive Reference Dye (e.g., ROX) | Normalizes for pipetting volume variations and optical anomalies across the plate well [87]. | Included in many commercial master mixes; required for some instrument optics. |
| Validated Reference Genes (RPS5, RPL8, HMBS) | Stable endogenous controls for normalizing sample input, though stability must be verified per experimental condition [79]. | For canine GI tissue, RPS5, RPL8, and HMBS were identified as stable; optimal for small gene sets [79]. |
| qPCR Master Mix with UNG | Contains enzymes, dNTPs, and buffer for efficient amplification. UNG prevents carryover contamination. | Select a mix with high efficiency and precision; suitability for multiplexing if required. |
Visualization of the Exogenous Control Normalization Strategy
The following diagram illustrates the logical workflow of the exogenous control normalization strategy, which is key to accounting for technical losses.
This case study outlines a comprehensive workflow for achieving consistent and reproducible absolute quantification of oncogene expression. The core of the strategy lies in combining a spike-in exogenous control to normalize for technical losses with a rigorously prepared standard curve for absolute quantification. Adherence to detailed protocols for data curation, including proper baseline and threshold setting, and the use of robust statistical methods like ANCOVA, further enhances data reliability. By implementing this standardized approach, independent laboratories can generate comparable and high-quality qPCR data, thereby accelerating collaborative research in cancer biology and drug development.
In the pursuit of personalized cancer medicine, researchers and drug development professionals increasingly recognize the limitations of single-technology approaches. While next-generation sequencing (NGS) provides an unparalleled broad-scale discovery platform for identifying novel mutations, fusion genes, and expression signatures, it faces challenges in absolute quantification, especially for low-abundance targets critical in minimal residual disease monitoring and oncogene expression studies. Conversely, absolute quantitative PCR (qPCR) offers precise, calibration-free nucleic acid quantification but within a limited target scope. The integration of these technologies creates a synergistic workflow that leverages the comprehensive profiling capabilities of sequencing with the precise, sensitive quantification of absolute qPCR, providing a complete molecular picture essential for advancing oncogene research and therapeutic development.
This application note demonstrates how combining absolute qPCR with sequencing technologies enhances the validation and quantification of clinically relevant biomarkers across various cancer research applications. We provide detailed protocols and data demonstrating how this integrated approach improves analytical sensitivity, validates NGS findings, and enables precise monitoring of treatment response and disease progression, ultimately supporting more accurate diagnostic and therapeutic decisions in oncology.
Integrated Workflow: From Discovery to Validation
The synergistic relationship between sequencing and absolute qPCR technologies enables a comprehensive workflow from initial biomarker discovery to final validation and clinical application. Sequencing technologies, particularly RNA and DNA exome sequencing, provide an unbiased discovery platform for identifying a wide spectrum of molecular alterations in tumor samples, including single nucleotide variants (SNVs), insertions/deletions (INDELs), copy number variations (CNVs), gene fusions, and expression signatures across thousands of genes simultaneously [78]. This broad-scale analysis is indispensable for uncovering novel therapeutic targets and understanding complex tumor biology.
Following target discovery, absolute qPCR delivers essential orthogonal validation and precise quantification of prioritized biomarkers. As emphasized by Dr. Christopher Mason of Weill Cornell Medicine, "We use RNA sequencing extensively... However, qPCR is the most sensitive method we use to validate gene fusion events, expression changes, or isoform variations" [91]. This validation step is critical for confirming NGS findings before progressing to clinical application or therapeutic decision-making. The integrated workflow ensures that discoveries made through comprehensive sequencing are translated into robust, quantitatively validated biomarkers suitable for monitoring treatment response, assessing minimal residual disease, and guiding personalized therapy.
The following diagram illustrates this complementary relationship and standard workflow:
Key Applications in Oncology Research
Validation of Sequencing-Derived Biomarkers
The transition from NGS-based discovery to clinically applicable biomarkers requires rigorous validation. Absolute qPCR serves as this critical bridge, providing orthogonal confirmation of NGS findings through fundamentally different technical principles. In cancer research, this is particularly valuable for validating expression changes in oncogenes and tumor suppressor genes, fusion events, and specific mutations identified through sequencing. Dr. Mason's team utilizes qPCR specifically "to validate gene fusion events, expression changes, or isoform variations" discovered through their RNA-seq workflows, establishing it as their "high bar for validation" [91]. This practice ensures that observations from sequencing are not artifacts of the complex NGS workflow but represent bona fide biological signals worthy of further investigation and potential clinical application.
Minimal Residual Disease (MRD) Monitoring
The extreme sensitivity and absolute quantification capabilities of qPCR make it indispensable for MRD monitoring, where detecting minute populations of residual cancer cells is critical for predicting relapse and guiding treatment decisions. "One really exciting application of qPCR is in monitoring MRD," notes Dr. Mason. "It allows us to track mutations like EGFR in a patient's blood after therapy" [91]. While NGS can identify the specific mutations present in a patient's tumor, absolute qPCR provides the precise, sensitive quantification needed to monitor these mutations over time, enabling clinicians to detect recurrence weeks or months before clinical manifestation. This capability is "fundamentally changing how we look at cancer" by allowing researchers to "monitor it, look for variations of it, and search for new mutations that arise" throughout the treatment course [91].
Absolute Quantification of Oncogene Expression
Understanding the precise expression levels of oncogenes is crucial for cancer biology research, particularly when studying gene amplification, targeted therapy response, and resistance mechanisms. Absolute qPCR provides copy number quantification that is essential for these applications. Unlike relative quantification methods, absolute qPCR "measures the exact amount of a target, such as its copy number or concentration, offering precise data crucial for various analyses" [4]. This is achieved through the use of external standards with known concentrations, enabling researchers to determine the exact copy number of oncogene transcripts in a sample, a capability particularly valuable when studying genes with amplified copy numbers in cancer, such as MYC, ERBB2, or EGFR.
Experimental Protocols
Protocol 1: Orthogonal Validation of NGS-Derived Fusion Genes by Absolute qPCR
Purpose: To validate novel gene fusions identified by RNA sequencing using absolute qPCR quantification.
Materials:
- RNA samples used for original RNA-seq analysis
- High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher)
- TaqMan Fast Advanced Master Mix (Thermo Fisher)
- Custom-designed TaqMan assays spanning fusion junctions
- Digital PCR system (QX200 Droplet Digital PCR System, Bio-Rad) or real-time PCR instrument (QuantStudio 5, Thermo Fisher)
Procedure:
- cDNA Synthesis: Convert 500 ng–1 µg of total RNA to cDNA using the High-Capacity cDNA Reverse Transcription Kit according to manufacturer's protocol.
- Assay Design: Design TaqMan assays specifically targeting the fusion junction identified by RNA-seq. Ensure amplicons span the breakpoint with the probe covering the junction sequence.
- Standard Curve Preparation: For absolute quantification, prepare a standard curve using gBlock gene fragments containing the exact fusion sequence. Serially dilute from 10^7 to 10^1 copies/µL.
- qPCR Setup:
- Reaction volume: 20 µL
- Template: 2 µL cDNA (or standard)
- Master mix: 10 µL TaqMan Fast Advanced Master Mix
- Primers/Probe: 1 µL custom TaqMan assay (final concentration: 900 nM primers, 250 nM probe)
- Nuclease-free water: 7 µL
- Amplification Parameters:
- UDG incubation: 50°C for 2 minutes (if using pre-amp with UDG)
- Polymerase activation: 95°C for 20 seconds
- 40 cycles of: 95°C for 1 second (denaturation), 60°C for 20 seconds (annealing/extension)
- Data Analysis: Generate standard curve from dilution series. Calculate absolute copy numbers of fusion transcripts in unknown samples using the standard curve equation.
Troubleshooting Notes:
- For low-abundance fusions, consider using digital PCR for enhanced sensitivity and absolute quantification without standard curves [83].
- Always include no-template controls and positive controls (if available) in each run.
- Verify assay specificity by Sanger sequencing of qPCR products for novel fusions.
Protocol 2: Absolute Quantification of Oncogene Expression in Tumor Samples
Purpose: To absolutely quantify expression levels of oncogenes identified as dysregulated by RNA-seq.
Materials:
- RNA extracted from tumor and matched normal tissues
- MiScript Reverse Transcription Kit (Qiagen)
- QuantiNova Probe PCR Kit (Qiagen)
- Validated PrimeTime qPCR assays for target oncogenes and reference genes (Integrated DNA Technologies)
- Optical 96-well plates and sealing films
- Real-time PCR instrument with multiplex capability
Procedure:
- RNA Quality Control: Assess RNA integrity using TapeStation 4200 (Agilent Technologies). Use only samples with RNA Integrity Number (RIN) >7.0 [78].
- cDNA Synthesis: Convert 1 µg of total RNA to cDNA using the MiScript Reverse Transcription Kit with oligo-dT and random hexamers.
- Reference Gene Selection: Select multiple reference genes (e.g., GAPDH, ACTB, RPLP0) based on stable expression across sample types. "We typically use multiple reference genes like GAPDH or ribosomal genes and include both biological and technical replicates," advises Dr. Mason [91].
- Standard Curve Preparation: Create absolute standard curves using known quantities of in vitro transcribed RNA for each target. Calculate copy numbers using the formula: (X g/µl RNA / [transcript length in nucleotides × 340]) × 6.022 × 10^23 = Y molecules/µl [4].
- Multiplex qPCR Setup:
- Reaction volume: 25 µL
- Template: 2 µL cDNA (1:10 dilution)
- Master mix: 12.5 µL QuantiNova Probe PCR Master Mix
- Target assay: 1.25 µL (final 250 nM probe, 900 nM primers)
- Reference assay: 1.25 µL (final 250 nM probe, 900 nM primers)
- Nuclease-free water: 8 µL
- Amplification Parameters:
- Activation: 95°C for 2 minutes
- 45 cycles of: 95°C for 5 seconds, 60°C for 30 seconds (with acquisition)
- Data Analysis: Calculate absolute copy numbers of target and reference genes from respective standard curves. Normalize oncogene expression as copies per 1000 copies of reference gene.
Critical Considerations:
- Account for reverse transcription efficiency variations by using RNA standards rather than DNA standards [4].
- For formalin-fixed paraffin-embedded (FFPE) samples, include pre-amplification steps and account for potential RNA fragmentation.
- Validate assay efficiency (90–110%) for both target and reference genes.
Comparative Performance Data
Analytical Sensitivity Across Molecular Applications
Table 1: Performance Characteristics of Integrated Sequencing and qPCR Approaches
| Application | Technology | Limit of Detection | Quantification Capability | Key Advantage |
|---|---|---|---|---|
| Fusion Gene Detection | RNA-seq | Varies by coverage | Relative | Unbiased discovery of novel fusions [78] |
| Absolute qPCR | 1-10 copies | Absolute | High-sensitivity validation of known fusions [91] | |
| Oncogene Expression | RNA-seq | Dependent on depth | Relative | Genome-wide expression profiling |
| Absolute qPCR | <5 copies/reaction | Absolute | Precise copy number quantification [4] | |
| MRD Monitoring | NGS Panels | 0.1%-1% VAF | Relative | Multi-target screening |
| Digital PCR | 0.001%-0.01% VAF | Absolute | Ultra-sensitive single molecule counting [83] | |
| Variant Validation | WES/WGS | 2%-5% VAF | Relative | Exome/genome-wide variant calling [78] |
| qPCR/dPCR | 0.1%-1% VAF | Absolute | Orthogonal confirmation of hotspots |
Integrated Assay Performance in Clinical Validation Study
Table 2: Validation Metrics from Combined RNA/DNA Exome Sequencing with qPCR Confirmation
| Parameter | DNA WES Only | RNA-seq + WES | qPCR-Validated |
|---|---|---|---|
| Actionable Alterations Detected | 89% of cases | 98% of cases [78] | >99% of validated targets |
| Fusion Genes Identified | Limited | Comprehensive recovery | 100% specificity confirmed |
| Sensitivity for Low VAF Variants | 2-5% VAF | 1-2% VAF (via RNA) | 0.1-1% VAF [91] |
| Turnaround Time | 7-10 days | 8-11 days | 2-4 hours post-assay design |
| Cost Per Sample | $$ | $$-$$$ | $-$$ |
Data adapted from validation of integrated RNA-seq and WES assay applied to 2230 clinical tumor samples, which demonstrated "direct correlation of somatic alterations with gene expression, recovery of variants missed by DNA-only testing, and improves detection of gene fusions" [78].
Research Reagent Solutions
Table 3: Essential Materials for Integrated Sequencing and qPCR Workflows
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Nucleic Acid Extraction | AllPrep DNA/RNA Mini Kit (Qiagen) [78] | Simultaneous co-extraction of DNA and RNA maintains sample parity for integrated analysis |
| RNA Quality Assessment | TapeStation 4200 (Agilent) [78] | Critical for determining RIN, especially valuable for FFPE-derived samples |
| Library Preparation | SureSelect XTHS2 (Agilent) [78] | Hybridization capture-based exome enrichment for both DNA and RNA |
| Absolute Quantification Standards | gBlock Gene Fragments (IDT) | Synthetic DNA standards for absolute qPCR calibration without molecular clones |
| qPCR Master Mixes | TaqMan Fast Advanced Master Mix (Thermo Fisher) | Optimized for fast cycling protocols, excellent for high-throughput applications |
| Digital PCR Systems | QIAcuity (Qiagen) [83] | Nanoplatform technology enabling absolute quantification without standard curves |
| Multiplex Assays | PrimeTime qPCR Assays (IDT) | Predesigned and validated assays for both target and reference genes |
Implementation Workflow and Technology Decision Guide
Successful implementation of an integrated sequencing and qPCR workflow requires strategic planning and understanding the complementary roles of each technology. The following decision pathway guides researchers in selecting the appropriate workflow based on their specific research questions and applications:
The integration of absolute qPCR with sequencing technologies represents a paradigm shift in molecular oncology research, combining the comprehensive discovery power of NGS with the precise quantification capabilities of qPCR. This synergistic approach enables researchers to move from biomarker discovery to validated, quantitative assays with clear clinical and research applications. As demonstrated through the protocols and data presented, this integrated workflow enhances the detection of actionable alterations, improves diagnostic accuracy, and enables monitoring of treatment response and disease progression with unprecedented sensitivity.
For drug development professionals, this approach streamlines the transition from basic research findings to clinically applicable biomarkers, supporting the development of targeted therapies and companion diagnostics. The validation frameworks and implementation guidelines provided here offer practical pathways for adopting this integrated approach, ultimately contributing to more personalized and effective cancer treatment strategies. As the field continues to evolve, the tight coupling of discovery and validation technologies will undoubtedly play an increasingly central role in advancing both cancer biology knowledge and clinical practice.
Conclusion
Absolute quantification by qPCR transcends being a mere technical method; it is a foundational pillar for precision in oncology research. By providing comparable and standardized measurements of oncogene expression, it directly enables the development of reliable prognostic models and the validation of actionable biomarkers. As the field advances, the synergy between highly optimized absolute qPCR assays and emerging technologies like digital PCR and next-generation sequencing will be crucial. This integrated approach is poised to fundamentally enhance clinical practice, particularly in monitoring minimal residual disease and personalizing therapeutic strategies, ultimately leading to more predictable and successful patient outcomes.
References
- 1. Real-time quantitative PCR: A tool for absolute and relative ... [https://pubmed.ncbi.nlm.nih.gov/34132460/]
- 2. Reference genes in real-time PCR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3825189/]
- 3. Absolute vs. Relative Quantification for qPCR [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/absolute-vs-relative-quantification-real-time-pcr.html]
- 4. Absolute Quantification qPCR | Relative ... [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/pcr-quantification/pcr-quantification]
- 5. Relative Quantification of Gene Expression Using qPCR [https://bitesizebio.com/29322/methods-relative-quantification-qpcr-data/]
- 6. Simple Absolute Quantification Method Correcting for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3370567/]
- 7. rtpcr: a package for statistical analysis and graphical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530193/]
- 8. Identification of prognostic biomarkers related to epithelial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12223776/]
- 9. Identification and experimental validation of prognostic ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1627134/full]
- 10. How To Interpret RT-qPCR Results [https://www.goldbio.com/blogs/articles/how-to-interpret-rt-qpcr-results?srsltid=AfmBOorVsoDU7HqNJrIYpl2hgtfLJV-_tVpoAASWoqinLaN3kR5URRK3]
- 11. Selecting Optimal Housekeeping Genes for RT-qPCR in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12429465/]
- 12. Selection and validation of reference genes for RT-qPCR ... [https://www.nature.com/articles/s41598-025-02951-7]
- 13. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOorMmrBJ5Jh2LWTYFaIrN2dNg2hgphjbx1rpp_PAAMY7FO6t9K8c]
- 14. Relative quantification of mRNA: comparison of methods ... [https://bmcmolbiol.biomedcentral.com/articles/10.1186/1471-2199-8-113]
- 15. Analyzing qPCR data: Better practices to facilitate rigor and ... [https://www.sciencedirect.com/science/article/pii/S2405580825004431]
- 16. Critical evaluation of methods used to determine amplification ... [https://bmcmolbiol.biomedcentral.com/articles/10.1186/1471-2199-9-96]
- 17. How To Interpret RT-qPCR Results [https://www.goldbio.com/blogs/articles/how-to-interpret-rt-qpcr-results?srsltid=AfmBOoqjLJvwriRNHVsYxNqZ8JTOkiJT8QeIksqoC0xIW-9yWVEJdv5T]
- 18. Amplification efficiency and thermal stability of qPCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4578049/]
- 19. Case of a drug used in the treatment of leishmaniasis [https://www.sciencedirect.com/science/article/pii/S0034528822003319]
- 20. Tech Clinic #5: Copy Number Determination for Plasmid ... [https://bitesizebio.com/13515/tech-clinic-4-copy-number-determination-for-plasmid-standard-curves/]
- 21. Designing standard curves for qPCR | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/easily-designed-standard-curves-for-qpcr/]
- 22. Droplet Digital PCR versus qPCR for gene expression ... [https://www.nature.com/articles/s41598-017-02217-x]
- 23. Standardization of Gene Expression Quantification by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2935814/]
- 24. Absolute Quantification of Selected microRNAs Expression ... [https://www.mdpi.com/1422-0067/25/6/3286]
- 25. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOopytOTWrS1O5T5btWqP4zz2zfO6uJw1nZqlI27JUN7yjs9TIeKn]
- 26. qPCR and qRT-PCR analysis: Regulatory points to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7786041/]
- 27. Applications of Digital PCR for Clinical Microbiology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5442518/]
- 28. Digital PCR (dPCR) vs Real-Time PCR (qPCR) [https://firalismolecularprecision.com/2025/04/09/dpcr-qpcr-differences/]
- 29. Digital PCR Applications | Thermo Fisher Scientific - ES [https://www.thermofisher.com/us/en/home/life-science/pcr/digital-pcr/applications.html]
- 30. Comparing the precision of two digital PCR applications for ... [https://www.nature.com/articles/s41598-025-13143-8]
- 31. Comparative Performance of Digital PCR and Real-Time RT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12474457/]
- 32. Digital PCR for Liquid Biopsy [https://www.thermofisher.com/us/en/home/life-science/pcr/digital-pcr/oncology-research/liquid-biopsy.html]
- 33. Validation of a High-Sensitivity Assay for Detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10488325/]
- 34. Efficiency of Real-Time PCR [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/efficiency-real-time-pcr-qpcr.html]
- 35. 1.3: Methods of Calibration [https://chem.libretexts.org/Courses/Duke_University/CHEM_401L%3A_Analytical_Chemistry_Lab/06%3A_Instrument_Facilities_for_CHEM401L/01%3A_Analytical_Equiptment_and_Methods_for_Calibration/1.03%3A_Methods_of_Calibration]
- 36. One-point calibration for calibration-free laser-induced ... [https://www.sciencedirect.com/science/article/abs/pii/S0584854713001195]
- 37. Understanding qPCR Efficiency and Why It Exceeds 100% [https://biosistemika.com/blog/qpcr-efficiency-over-100/]
- 38. How good is a PCR efficiency estimate: Recommendations ... [https://www.sciencedirect.com/science/article/pii/S2214753515000169]
- 39. How To Interpret RT-qPCR Results [https://www.goldbio.com/blogs/articles/how-to-interpret-rt-qpcr-results?srsltid=AfmBOoqmnhpdjLFrAUfnG8xFf6CSf7aZM73NXpEGkCzeJRzMPcl87WlJ]
- 40. Absolute Quantification of Nucleotide Variants in Cell-Free ... [https://www.mdpi.com/2072-6694/17/5/783]
- 41. Development and validation of an ultrasensitive qPCR method to... [https://pubmed.ncbi.nlm.nih.gov/39812332/]
- 42. Programmable no-nonspecific genetic analytical system ... [https://www.nature.com/articles/s41598-025-24565-9]
- 43. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOootKWVlgTsTMkzvi-4d5uJxZT48ZcIMCP-CEJH9OHmtM4ijg_gz]
- 44. of prokaryotes in the... | Nature Absolute quantification Protocols [https://www.nature.com/articles/s41596-025-01165-5?error=cookies_not_supported&code=b99561c0-aedf-419b-835a-32160fc656cb]
- 45. How to Design Primers for PCR Experiments [https://www.zymoresearch.com/blogs/blog/how-to-design-primers-for-pcr-experiments?srsltid=AfmBOorEghYqfhhs28zhmaYdh8tPmC0K2U8cNdxKiw-xp4W1D_iMj4Q1]
- 46. Designing Allele-Specific Competitive-Extension PCR-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8921500/]
- 47. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 48. Primer designing tool - NCBI - NIH [https://www.ncbi.nlm.nih.gov/tools/primer-blast/]
- 49. qPCR Protocol for SNP Genotyping [https://www.thermofisher.com/us/en/home/references/protocols/nucleic-acid-amplification-and-expression-profiling/pcr-protocol/qpcr-for-snp-genotyping.html]
- 50. Comparison of different standards for real-time PCR-based ... [https://www.sciencedirect.com/science/article/abs/pii/S0022175910000062]
- 51. Absolute vs. Relative Quantification for qPCR [https://www.thermofisher.com/cl/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/absolute-vs-relative-quantification-real-time-pcr.html]
- 52. An optimized protocol for stepwise optimization of real-time ... [https://www.nature.com/articles/s41438-021-00616-w]
- 53. Article 4: Optimisation of the PCR step of a qPCR assay [https://www.europeanpharmaceuticalreview.com/article/756/article-4-optimisation-of-the-pcr-step-of-a-qpcr-assay/]
- 54. Recommendations for Method Development and ... [https://link.springer.com/article/10.1208/s12248-023-00880-9]
- 55. One-step and two-step qPCR assays for CAPRV2023 [https://www.frontiersin.org/journals/veterinary-science/articles/10.3389/fvets.2025.1620997/full]
- 56. High-Throughput Data Analysis for qPCR [https://www.neb.com/en-us/tools-and-resources/feature-articles/development-of-a-high-throughput-data-analysis-method-for-quantitative-real-time-pcr?srsltid=AfmBOopV-ggQuVjQ3JQk7Kr6sSvxqcbbfntMUgGGg3ko15p6bOqHmQTN]
- 57. The Impact of the Variability of RT-qPCR Standard Curves ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12029521/]
- 58. a reliable and accurate approach for qPCR analysis | BMC ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05630-y]
- 59. Quantifying Uncertainty in Low-Copy qPCR [https://www.mdpi.com/1422-0067/26/16/7796]
- 60. Top Ten Most Common Real-Time qRT-PCR Pitfalls [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rtpcr-analysis/general-articles/ten-most-common-real-time-qrt-pcr-pitfalls.html]
- 61. STALARD: Selective Target Amplification for Low- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12482509/]
- 62. qPCR primer design revisited - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC5702850/]
- 63. Comparative Study Between Droplet Digital PCR and Real ... [https://www.mdpi.com/2076-2607/13/11/2426]
- 64. How To Interpret RT-qPCR Results [https://www.goldbio.com/blogs/articles/how-to-interpret-rt-qpcr-results?srsltid=AfmBOop5Yq9N4DcSQKb7IruNi_eRaSYYgKhqNCmKSpg1VLAgMTVojpKv]
- 65. Improved visual detection of DNA amplification using ... [https://www.nature.com/articles/s42003-022-03973-x]
- 66. A novel procedure for absolute real-time quantification of gene ... [https://plantmethods.biomedcentral.com/articles/10.1186/1746-4811-8-9]
- 67. Absolute quantification of gene expression in biomaterials ... [https://pubmed.ncbi.nlm.nih.gov/17034848/]
- 68. Optimization of five qPCR protocols toward the detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8563462/]
- 69. MIQE précis: Practical implementation of minimum standard ... [https://bmcmolbiol.biomedcentral.com/articles/10.1186/1471-2199-11-74]
- 70. MIQE 2.0 and the Urgent Need to Rethink qPCR Standards [https://pmc.ncbi.nlm.nih.gov/articles/PMC12154065/]
- 71. MIQE Guidelines | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/why-choose-taqman-assays/publish-real-time-pcr-results.html]
- 72. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 73. The digital MIQE guidelines: Minimum Information for ... [https://pubmed.ncbi.nlm.nih.gov/23570709/]
- 74. Do results obtained with RNA-sequencing require ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7823214/]
- 75. Increased comparability between RNA-Seq and microarray ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7549825/]
- 76. An updated comparison of microarray and RNA-seq for ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11548-3]
- 77. Systematic comparison and assessment of RNA-seq ... [https://www.nature.com/articles/s41598-020-76881-x]
- 78. Clinical and analytical validation of a combined RNA ... [https://www.nature.com/articles/s43856-025-00934-3]
- 79. Evaluation of normalisation strategies for qPCR data ... [https://www.nature.com/articles/s41598-025-06657-8]
- 80. Minimal Residual Disease Detection: Implications for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12079024/]
- 81. A New View on Minimal Residual Disease Quantification in ... [https://www.sciencedirect.com/science/article/pii/S1525157822001453]
- 82. Minimal residual disease detection by mutation-specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10225364/]
- 83. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 84. Digital PCR: A Reliable Tool for Analyzing and Monitoring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7247671/]
- 85. Digital PCR outperforms quantitative real-time PCR for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288185/]
- 86. Use and Misuse of Cq in qPCR Data Analysis and Reporting [https://pmc.ncbi.nlm.nih.gov/articles/PMC8229287/]
- 87. Quantitative PCR (qPCR) Data Analysis [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/gene-expression-analysis-real-time-pcr-information/precision-qpcr.html]
- 88. Optimized bacterial absolute quantification method by ... [https://www.sciencedirect.com/science/article/pii/S0167701224000071]
- 89. Evaluation validation of a qPCR curve analysis method and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8596907/]
- 90. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOoq0gsfX2-sp0CE8J8I16aWy0UUsf0XgFu7CorJPqzAWlJlsIRBu]
- 91. Cancer Research to Microbes: Harnessing the Power of ... [https://www.thermofisher.com/blog/behindthebench/harnessing-the-power-of-qpcr/]