A Comprehensive CoMFA and CoMSIA Protocol for Designing Pteridinone Derivatives as PLK1 Inhibitors in Prostate Cancer
This article provides a detailed computational protocol for applying Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) to design and optimize pteridinone-based Polo-like kinase 1 (PLK1)...
A Comprehensive CoMFA and CoMSIA Protocol for Designing Pteridinone Derivatives as PLK1 Inhibitors in Prostate Cancer
Abstract
This article provides a detailed computational protocol for applying Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) to design and optimize pteridinone-based Polo-like kinase 1 (PLK1) inhibitors. Overexpression of PLK1 is a hallmark of numerous cancers, including prostate cancer, making it a promising broad-spectrum anti-cancer target. The protocol covers the foundational principles of 3D-QSAR, a step-by-step methodological workflow for model construction using software like SYBYL-X, strategies for troubleshooting and optimizing model performance, and robust validation techniques integrating molecular docking, dynamics simulations, and ADMET profiling. Aimed at researchers and drug development professionals, this guide demonstrates how these in silico methods can efficiently identify critical structural features for activity, predict new candidates with high potency and desirable drug-like properties, and accelerate the discovery of novel anti-cancer therapies.
PLK1 as an Anti-Cancer Target and the Promise of Pteridinone Scaffolds
PLK1 Structure and Functional Domains
Polo-like Kinase 1 (PLK1) is a 603-amino acid serine/threonine kinase with a molecular mass of approximately 66 kDa, playing pivotal roles in cell cycle regulation [1]. Its structure comprises two primary functional domains connected by an inter-domain linker.
Table 1: Structural Domains of PLK1
| Domain | Location | Key Features | Functional Role |
|---|---|---|---|
| Kinase Domain (KD) | N-terminal (residues 49-310) | Catalytic activity; contains critical phosphorylation site Thr210 [1]; ATP-binding pocket with residues Lys82, Glu131, Cys133, Asp194 [1]. | Phosphorylates downstream substrates to drive mitotic events [2] [1]. |
| Polo-Box Domain (PBD) | C-terminal (residues ~345-603) | Two polo-box motifs (PB1, PB2) forming a phosphopeptide-binding site; recognizes consensus motif S-pT-P [2] [1]. | Subcellular localization; substrate recognition; autoinhibition of KD [2] [1]. |
The activation of PLK1 is a tightly regulated process. The kinase domain is initially maintained in an autoinhibited state through interactions with the PBD. Full activation requires the phosphorylation of Thr210 in the kinase domain's activation loop by upstream kinases, such as Aurora A, and the binding of the PBD to primed substrates, which disrupts the autoinhibitory conformation [2] [1].
PLK1's Central Role in Cell Cycle and Cancer Proliferation
PLK1 expression is low in non-dividing cells but peaks during the G2 and M phases of the cell cycle, where it acts as a master regulator of mitosis [2] [1]. Its essential functions include centrosome maturation, bipolar spindle formation, kinetochore-microtubule attachment, chromosome segregation, and cytokinesis [2] [3] [1].
Dysregulation of PLK1 is a hallmark of many cancers. Its overexpression is frequently observed in various tumors and is often correlated with poor prognosis, increased tumor aggressiveness, and drug resistance [3] [1] [4]. PLK1 drives tumorigenesis through multiple mechanisms:
- Interaction with Tumor Suppressors and Oncogenes: PLK1 can phosphorylate and inactivate the tumor suppressor PTEN, thereby activating the PI3K/AKT signaling pathway to promote cell survival and growth [3]. It also enhances the stability of the MYC oncoprotein and regulates the p53 signaling axis [3].
- Promotion of Epithelial-Mesenchymal Transition (EMT): PLK1 is a key regulator of EMT, a process critical for cancer metastasis, via pathways like TGF-β and β-catenin in cancers such as non-small cell lung cancer and gastric cancer [3] [5].
- Induction of Genomic Instability: Overexpression of PLK1 can lead to mitotic errors, chromosomal missegregation, and aneuploidy, fueling cancer initiation and progression [6].
Computational Analysis: CoMFA and CoMSIA for Pteridinone Derivatives
Computational methods like 3D-QSAR (Quantitative Structure-Activity Relationship) are crucial in modern drug discovery for identifying potent and selective PLK1 inhibitors. The following protocol outlines the application of Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) on a series of pteridinone derivatives, which are known PLK1 inhibitors [7].
Protocol for 3D-QSAR Model Development
Objective: To build predictive 3D-QSAR models correlating the molecular structure of pteridinone derivatives with their PLK1 inhibitory activity (pIC₅₀).
Materials & Software:
- Dataset: A set of synthesized pteridinone derivatives with experimentally determined IC₅₀ values [7].
- Software: Molecular modeling software (e.g., SYBYL-X).
Methodology:
Data Preparation and Molecular Alignment:
- Divide the dataset into a training set (~80% of molecules) to build the model and a test set (~20%) for external validation [7].
- Minimize the energy of all molecules using a standard force field (e.g., Tripos).
- Perform molecular alignment, a critical step where all molecules are superimposed based on a common scaffold or pharmacophore, using a rigid body method [7].
Descriptor Calculation (Field Generation):
- Place the aligned molecules into a 3D grid with a default spacing of 1 Å.
- For CoMFA, calculate steric (Lennard-Jones potential) and electrostatic (Coulombic potential) field energies at each grid point using a probe atom [7].
- For CoMSIA, calculate similarity indices, which can include additional fields: steric, electrostatic, hydrophobic, and hydrogen-bond donor and acceptor [7].
Statistical Analysis and Model Validation:
- Use the Partial Least Squares (PLS) regression method to correlate the CoMFA/CoMSIA field descriptors with the biological activity (pIC₅₀) [7].
- Validate the model's robustness and predictive power using:
Table 2: Key Statistical Parameters for Validated 3D-QSAR Models [7]
| Model Type | Q² | R² | R²ₚᵣₑ𝒹 | Notes |
|---|---|---|---|---|
| CoMFA | 0.67 | 0.992 | 0.683 | High conventional correlation coefficient (R²) indicates excellent model fit. |
| CoMSIA/SEAH | 0.66 | 0.975 | 0.767 | Combines Steric, Electrostatic, Acceptor, Hydrophobic fields; shows strong predictive R². |
The resulting contour maps from CoMFA and CoMSIA visually guide chemical optimization by indicating regions where specific steric bulk, electropositive/negative groups, or hydrophobic features can enhance or diminish biological activity.
Essential Research Reagents and Experimental Protocols
Table 3: Research Reagent Solutions for PLK1 Inhibition Studies
| Reagent / Assay | Function / Application | Example / Specification |
|---|---|---|
| PLK1 Inhibitors | Tool compounds for probing PLK1 function; therapeutic candidates. | BI-2536, Volasertib (ATP-competitive) [1] [5]. PBD-targeting inhibitors (disrupt protein-protein interactions) [2] [1]. |
| Cell Lines | In vitro models for studying PLK1 biology and inhibitor efficacy. | Cancer cell lines with high PLK1 expression (e.g., SW982 synovial sarcoma, prostate cancer lines) [7] [5]. |
| Antibodies | Detection of PLK1 expression, localization, and phosphorylation. | Anti-PLK1, anti-phospho-T210 PLK1, apoptosis markers (Bax, Bcl-2) [3] [5]. |
| Cell-Based Assays | Functional assessment of inhibitor effects on cancer phenotypes. | Cell proliferation (CCK-8), cell cycle analysis (flow cytometry), apoptosis (caspase-3), migration/invasion (Transwell) [5]. |
| Molecular Docking Software | Predicting ligand binding modes and interactions with PLK1. | AutoDock Vina, with PLK1 crystal structure (e.g., PDB: 2RKU) [7]. |
| Molecular Dynamics (MD) Simulation | Assessing stability of ligand-protein complexes over time. | GROMACS, AMBER; simulation duration ~50-100 ns [8] [7]. |
Protocol:In VitroValidation of PLK1 Inhibitor Efficacy
Objective: To evaluate the anti-cancer effects of a PLK1 inhibitor (e.g., BI2536) on cell proliferation, cell cycle, and apoptosis.
Materials:
- Cancer cell lines (e.g., SW982, SSX1) [5].
- PLK1 inhibitor (e.g., BI2536, dissolved in DMSO).
- Cell culture medium and supplements.
- CCK-8 kit, Propidium Iodide, Annexin V staining kit, Transwell chambers.
Methodology:
Cell Proliferation Assay (CCK-8):
Cell Cycle Analysis by Flow Cytometry:
- Treat cells with the inhibitor for 48 hours.
- Harvest, fix in ethanol, and stain with Propidium Iodide.
- Analyze DNA content using a flow cytometer. PLK1 inhibition is expected to cause a G2/M phase arrest [5].
Apoptosis Assay:
- Treat cells and harvest after 48 hours.
- Stain with Annexin V and Propidium Iodide.
- Analyze by flow cytometry. Effective PLK1 inhibition should increase the percentage of cells in early and late apoptosis, often correlated with up-regulation of pro-apoptotic proteins like Bax [5].
Migration and Invasion Assay (Transwell):
- For invasion assays, coat the upper chamber of a Transwell insert with Matrigel.
- Seed serum-starved cells into the upper chamber with the inhibitor. Place medium with serum in the lower chamber as a chemoattractant.
- After 24-48 hours, fix, stain, and count the cells that have migrated/invaded through the membrane. PLK1 inhibition typically reduces migration and invasion capabilities [5].
Polo-like kinase 1 (PLK1) is a serine/threonine kinase that functions as a crucial regulator of cell cycle progression, with essential roles in centrosome maturation, spindle assembly, kinetochore-microtubule attachment, and cytokinesis [1]. The overexpression of PLK1 has been documented in numerous cancer types, including prostate, lung, and colon cancers, and this dysregulation correlates strongly with increased tumor aggressiveness, metastatic potential, and poor clinical prognosis [9] [1]. Importantly, while PLK1 is abundant in proliferating cells, its expression remains low or undetectable in most differentiated adult tissues, making it an attractive therapeutic target with a potential favorable therapeutic window [1]. The structural organization of PLK1 comprises two primary functional domains: an N-terminal kinase domain (KD) that contains the ATP-binding catalytic site, and a C-terminal polo-box domain (PBD) responsible for substrate recognition and subcellular localization [1]. Current inhibitor development strategies target either of these domains, with KD inhibitors representing the most advanced class to date [1].
Pteridinone derivatives have emerged as a promising novel class of PLK1 inhibitors with demonstrated potential for prostate cancer therapy [9] [10]. These compounds exhibit strong binding affinity to the kinase domain of PLK1 and have shown potent inhibitory activity in both enzymatic and cellular assays [9]. Recent computational studies have provided structural insights into the molecular interactions responsible for their activity, revealing key binding residues including R136, R57, Y133, L69, L82, and Y139 within the PLK1 active site [10] [11]. The application of advanced computational approaches, particularly three-dimensional quantitative structure-activity relationship (3D-QSAR) studies using CoMFA and CoMSIA methodologies, has enabled researchers to establish robust models that correlate the structural features of pteridinone derivatives with their biological activity, providing valuable guidance for the rational design of more potent and selective PLK1 inhibitors [9].
Computational Methodology and Protocols
Molecular Modeling and Alignment Protocol
Objective: To generate properly aligned and energetically optimized molecular structures for 3D-QSAR analysis.
Procedure:
- Structure Preparation: Begin by constructing or importing the two-dimensional structures of pteridinone derivatives into molecular modeling software such as SYBYL-X. Convert these to three-dimensional representations and add hydrogen atoms.
- Energy Minimization: Optimize the geometry of each molecule using the Tripos force field with Gasteiger-Hückel atomic partial charges. Apply the Powell gradient algorithm with a convergence criterion of 0.005 kcal/mol Å and a maximum of 1000 iterations to ensure stable molecular configurations [9].
- Molecular Alignment: Employ a rigid body alignment technique using the "distill" function in SYBYL-X. Select the most biologically active compound as the template structure and align all other molecules to this reference based on their common molecular framework [9]. This alignment is critical as it directly influences the quality and predictive power of subsequent 3D-QSAR models.
CoMFA and CoMSIA Modeling Protocol
Objective: To establish quantitative relationships between molecular fields and PLK1 inhibitory activity.
Procedure:
- Field Calculation: After molecular alignment, place each compound within a 3D grid with 1-2 Å spacing in all Cartesian directions. Calculate steric and electrostatic field energies for CoMFA using a sp³ carbon probe atom with a +1 charge, truncating energy values at 30 kcal/mol. For CoMSIA, calculate additional similarity indices including hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields [9] [12].
- Partial Least-Squares (PLS) Analysis: Correlate the calculated field descriptors with experimental biological activities (pIC₅₀ values) using the PLS regression algorithm. Begin with leave-one-out (LOO) cross-validation to determine the optimal number of components (ONC) that yields the highest cross-validated correlation coefficient (Q²) [9].
- Model Validation: Validate the final model using both internal (cross-validation) and external validation approaches. Divide the dataset into a training set (approximately 80% of compounds) to build the model and a test set (remaining 20%) to evaluate its predictive power. A robust model should exhibit Q² > 0.5 and R²ₚᵣₑ𝒹 > 0.6 [9] [10].
Table 1: Statistical Parameters of 3D-QSAR Models for Pteridinone Derivatives [9] [10]
| Model Type | Q² | R² | SEE | NOC | R²ₚᵣₑ𝒹 | Field Contributions |
|---|---|---|---|---|---|---|
| CoMFA | 0.67 | 0.992 | 0.059 | 6 | 0.683 | Steric: 53.8%, Electrostatic: 46.2% |
| CoMSIA/SHE | 0.69 | 0.974 | 0.116 | 6 | 0.758 | Steric: 14.3%, Electrostatic: 31.8%, Hydrophobic: 53.9% |
| CoMSIA/SEAH | 0.66 | 0.975 | 0.113 | 6 | 0.767 | Steric: 12.3%, Electrostatic: 20.6%, Hydrophobic: 41.1%, Acceptor: 26.0% |
Molecular Docking Protocol
Objective: To predict binding orientations and interactions between pteridinone derivatives and the PLK1 kinase domain.
Procedure:
- Protein Preparation: Obtain the crystal structure of PLK1 (e.g., PDB ID: 2RKU or 3FC2) from the Protein Data Bank. Remove water molecules and co-crystallized ligands, add polar hydrogen atoms, assign partial charges, and minimize the protein structure using appropriate force fields [9] [13].
- Ligand Preparation: Generate three-dimensional structures of pteridinone derivatives and optimize their geometries using molecular mechanics force fields.
- Docking Execution: Perform docking simulations using software such as AutoDock Vina or MOE Dock. Define the binding site around known active site residues with a grid box of sufficient size to accommodate ligand binding. Utilize scoring functions to evaluate binding affinities [9] [13].
- Interaction Analysis: Visually inspect the resulting ligand-protein complexes to identify key interactions including hydrogen bonds, hydrophobic interactions, and ionic contacts. Pay particular attention to residues R136, R57, Y133, L69, L82, and Y139 which have been identified as critical for ligand binding [10].
Molecular Dynamics Simulation Protocol
Objective: To evaluate the stability and conformational dynamics of PLK1-inhibitor complexes under simulated physiological conditions.
Procedure:
- System Setup: Solvate the docked complexes in an explicit water model (e.g., SPC/E) within a cubic box with a minimum 1.0 nm distance between the protein and box edge. Add ions to neutralize the system charge [13].
- Energy Minimization: Perform energy minimization using steepest descent or conjugate gradient algorithms to remove steric clashes and bad contacts.
- Equilibration: Conduct equilibration in two phases: (1) NVT ensemble for 100-500 ps to stabilize temperature, and (2) NPT ensemble for 100-500 ps to stabilize pressure.
- Production Run: Perform production MD simulations for at least 50-100 ns at constant temperature (300 K) and pressure (1 bar) using software such as GROMACS. Apply periodic boundary conditions and the AMBER99SB-ILDN force field for proteins and ligands [9] [13].
- Trajectory Analysis: Analyze the resulting trajectories to calculate root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and hydrogen bonding patterns to assess complex stability over time.
Key Research Findings and Data Interpretation
Structural Insights from 3D-QSAR Contour Maps
The CoMFA and CoMSIA models generated for pteridinone derivatives provide valuable visual guidance for molecular optimization through their steric, electrostatic, and hydrophobic contour maps [9]. The CoMFA steric field maps indicate regions where bulky substituents enhance or diminish activity, while electrostatic maps highlight areas favoring positively or negatively charged groups. The CoMSIA hydrophobic maps reveal regions where hydrophobic substituents contribute favorably to PLK1 binding affinity. These contour maps serve as powerful tools for medicinal chemists, allowing them to predict the biological activity of newly designed analogs before synthesis and biological evaluation [9] [10].
Molecular Docking and Binding Mode Analysis
Molecular docking studies of pteridinone derivatives with PLK1 (PDB: 2RKU) have revealed consistent binding patterns characterized by multiple hydrogen bonding and hydrophobic interactions with key active site residues [10]. The most active compounds typically form hydrogen bonds with residues R136 and R57 in the hinge region of the kinase domain, mimicking the adenine moiety of ATP. Additionally, hydrophobic interactions with L69, L82, and Y139 contribute significantly to binding affinity and ligand stabilization within the active site. These computational predictions provide atomic-level insights into the structural determinants of inhibitor potency and selectivity [10] [11].
Table 2: Experimental Biological Activities of Selected Pteridinone Derivatives [9]
| Compound Number | IC₅₀ (nM) | pIC₅₀ | Test Set | Key Structural Features |
|---|---|---|---|---|
| 3 | 8.42 | 5.074 | Training | Optimized substituent at R position |
| 18 | 36.30 | 5.086 | Training | Balanced steric and electronic properties |
| 23 | 9.26 | 4.936 | Training | Favorable hydrophobic interactions |
| 28 | 7.18 | 4.787 | Training | Most active compound in series |
| 2 | 53.59 | 4.270 | Test | Used for model validation |
| 5 | 26.59 | 4.575 | Test | Used for model validation |
| 19 | 9.25 | 4.440 | Test | Used for model validation |
Molecular Dynamics and Binding Stability
Molecular dynamics simulations conducted over 50-100 ns have demonstrated that complexes formed between PLK1 and potent pteridinone inhibitors remain structurally stable throughout the simulation period [9] [13]. The root mean square deviation (RMSD) values of the protein-ligand complexes typically stabilize after an initial equilibration phase, indicating maintained binding poses without significant conformational rearrangements. Consistent hydrogen bonding patterns with key residues such as R136 and R57 further confirm the stability of the predicted binding modes. These simulations provide dynamic validation of the docking results and offer insights into the structural flexibility of both the inhibitor and the protein binding site [9].
ADMET Property Predictions
Assessment of absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties represents a crucial step in early drug discovery. For pteridinone derivatives, computational ADMET predictions have indicated favorable drug-like properties for several lead compounds [9] [10]. Specifically, compound 28 from the series not only exhibited potent PLK1 inhibitory activity but also demonstrated promising ADMET characteristics, suggesting its potential as a viable drug candidate for prostate cancer therapy. These computational predictions require experimental validation but provide valuable guidance for compound prioritization and optimization [9].
Research Toolkit and Implementation Guide
Essential Research Reagents and Computational Tools
Table 3: Key Research Reagents and Computational Resources for PLK1 Inhibitor Development
| Category | Specific Tools/Reagents | Function/Application | Protocol Reference |
|---|---|---|---|
| Software Packages | SYBYL-X 2.1.1 | Molecular modeling, alignment, and 3D-QSAR analysis | [9] |
| Molecular Operating Environment (MOE) | Pharmacophore modeling, molecular docking, and virtual screening | [13] | |
| GROMACS | Molecular dynamics simulations | [13] | |
| AutoDock Tools/Vina | Molecular docking simulations | [9] | |
| Biological Materials | PLK1 Protein (PDB: 2RKU, 3FC2) | Structural studies and inhibitor screening | [9] [13] |
| DU-145 Prostate Cancer Cells | Cellular anti-proliferative assays | [13] | |
| Chemical Resources | Pteridinone Derivatives Library | Structure-activity relationship studies | [9] |
| Combinatorial Chemistry Database | Virtual screening for novel hits | [13] |
The following diagram illustrates the integrated computational and experimental workflow for developing pteridinone-based PLK1 inhibitors:
Critical Implementation Notes
- Data Quality Assurance: Ensure biological activity data (IC₅₀ values) are obtained under consistent experimental conditions to maintain dataset homogeneity for QSAR modeling.
- Model Validation Rigor: Always validate 3D-QSAR models using both internal (cross-validation) and external (test set prediction) methods to ensure predictive reliability.
- Binding Site Consistency: When performing molecular docking, clearly define the binding site around known catalytic residues to enable meaningful comparison across different inhibitor series.
- Dynamic Confirmation: Use molecular dynamics simulations to confirm binding mode stability, as static docking poses may not represent biologically relevant conformations.
- Experimental Correlation: Whenever possible, correlate computational predictions with experimental results to refine and validate computational models iteratively.
The integration of computational approaches including 3D-QSAR, molecular docking, and molecular dynamics simulations has proven invaluable in the development and optimization of pteridinone derivatives as potent PLK1 inhibitors. The robust CoMFA and CoMSIA models established for this chemical series provide strong predictive power for designing novel analogs with enhanced inhibitory activity. Molecular docking and dynamics simulations offer atomic-level insights into binding interactions and complex stability, while ADMET predictions help prioritize compounds with favorable drug-like properties. Compound 28 emerges as a particularly promising lead worthy of further investigation. The protocols and analytical frameworks presented herein provide a comprehensive roadmap for researchers pursuing PLK1-targeted anticancer drug discovery, with potential applications extending to other kinase inhibitor development programs. Future work should focus on expanding the chemical diversity of the training set, exploring alternative binding modes, and validating computational predictions through synthetic and biological studies.
Three-Dimensional Quantitative Structure-Activity Relationship (3D-QSAR) represents a significant advancement over traditional 2D-QSAR methods by incorporating the critical spatial orientation of molecules. While classical QSAR describes molecular properties using descriptors independent of spatial coordinates (e.g., logP, molar refractivity), 3D-QSAR characterizes molecules through a set of values measured at numerous locations in the three-dimensional space surrounding them [14]. This approach is predicated on the fundamental principle that biological activity depends on a ligand's affinity for its receptor, a process that occurs in three dimensions through complex intermolecular forces [14]. The receptor does not perceive a ligand as a simple collection of atoms and bonds, but rather as a specific three-dimensional shape carrying distinct electrostatic and steric force fields [14].
The development of 3D-QSAR has become indispensable in modern drug discovery, particularly in scenarios where the three-dimensional structure of the target receptor remains unknown. By mapping and comparing the molecular interaction fields around sets of ligands, researchers can establish robust correlations between biological activities and spatial molecular features [14]. These methodologies have proven valuable across diverse therapeutic targets, including PLK1 inhibition for cancer therapy [15] [9], glycogen synthase kinase-3 inhibition for diabetes [16], and mTOR inhibition [17], demonstrating their broad applicability in rational drug design.
Theoretical Foundations
Molecular Interaction Fields (MIFs)
The core theoretical concept underlying 3D-QSAR is the Molecular Interaction Field (MIF), which represents the spatial distribution of interaction energies between a molecule and standardized chemical probes [14]. These fields are calculated using a 3D lattice of grid points regularly distributed in space around the molecule of interest. At each grid point, the interaction energy between the molecule and a selected probe is computed using appropriate potential energy functions [14].
The probe concept is fundamental to MIF calculation—just as Earth's magnetic field can only be detected with a compass, molecular interaction fields require appropriate probes for measurement [14]. Probes typically consist of specific atoms or functional groups with associated energy functions, selected according to the field property being investigated:
- Steric fields: Typically probed with an sp³ carbon atom [14]
- Electrostatic fields: Measured using a charged sp³ carbon atom (+1 charge) [14]
- Advanced probes: Extended to molecular fragments (CH₃, NH₂, CONH₂, H₂O, NH₃⁺, COO⁻) for specialized interactions [14]
Fundamental Forces in Molecular Recognition
Molecular recognition between ligands and their receptors depends primarily on two non-covalent interactions:
Electrostatic Interactions: These occur between polar or charged groups and can be either attractive or repulsive. Calculated using Coulomb's law, electrostatic interactions diminish with the inverse of the distance between interacting atoms, meaning they remain significant even at relatively long ranges (10 Å or more) [14].
Steric Interactions: These non-electrostatic interactions between non-bonded atoms encompass both repulsive forces (due to electron cloud interpenetration at short distances) and attractive dispersion forces (at longer distances). Steric potential follows a Lennard-Jones function, where repulsive forces increase with the inverse twelfth power of distance [14].
The interplay between these forces governs molecular binding: electrostatic fields primarily drive the initial approach of ligand to receptor at longer distances, while steric forces dominate the final binding steps at shorter ranges [14].
CoMFA Methodology
Theoretical Principles
Comparative Molecular Field Analysis (CoMFA), the first validated 3D-QSAR approach, establishes correlations between biological activity and steric/electrostatic fields surrounding molecules [14]. The method operates on the fundamental premise that differences in biological activity among structurally related compounds correlate with differences in their steric and electrostatic interaction fields [18].
The CoMFA methodology involves several systematic steps:
- Molecular Modeling: Structures are sketched and converted to 3D conformations [19]
- Geometry Optimization: Energy minimization using standardized force fields (e.g., Tripos) [9] [19]
- Molecular Alignment: Critical step where molecules are superimposed according to a defined rule [9]
- Field Calculation: Steric and electrostatic fields computed at grid points [14]
- Statistical Analysis: Partial Least Squares (PLS) regression correlates field values with biological activity [9]
Field Calculation and Analysis
In CoMFA, the steric field is calculated using a Lennard-Jones 6-12 potential, while the electrostatic field employs Coulomb's law [14]. These interaction energies are computed at each grid point using a carbon sp³ atom as the probe, with typical energy truncation limits set at ±30 kcal/mol to reduce noise [9].
The resulting data matrix, comprising thousands of field values for each compound, is analyzed using Partial Least Squares (PLS) regression to extract latent variables that best explain the variance in biological activity. Model quality is assessed through several statistical parameters:
- q² (cross-validated correlation coefficient): Must be >0.5 for predictive models [9]
- r² (conventional correlation coefficient): Measures goodness of fit [9]
- SEE (standard error of estimate): Should be minimized [9]
- F-value: Measures statistical significance [9]
Table 1: Representative CoMFA Statistical Results from Various Studies
| Study Focus | q² Value | r² Value | r²pred | Number of Components | Reference |
|---|---|---|---|---|---|
| Pyridinone derivatives as HIV-1 RT inhibitors | 0.706 | - | 0.720 | - | [18] |
| Pteridinones as PLK1 inhibitors | 0.67 | 0.992 | 0.683 | - | [9] |
| Triazine morpholinos as mTOR inhibitors | 0.735 | 0.974 | 0.769 | - | [17] |
| Pyrimidinones as TNKS2 inhibitors | 0.584 | 0.944 | 0.741 | 6 | [19] |
CoMSIA Methodology
Theoretical Advancements
Comparative Molecular Similarity Indices Analysis (CoMSIA) extends beyond CoMFA by incorporating additional molecular field types and employing a different potential function. While CoMFA focuses solely on steric and electrostatic fields, CoMSIA typically includes steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields [15] [17].
A significant theoretical improvement in CoMSIA is the use of a Gaussian-type distance-dependent function instead of the Lennard-Jones and Coulomb potentials used in CoMFA. This function eliminates the extreme energy values when the probe atom approaches the molecular surface, resulting in more stable models and eliminating the need for arbitrary energy truncation [15].
Field Descriptors in CoMSIA
CoMSIA calculates five distinct property fields that provide a more comprehensive description of ligand-receptor interactions:
- Steric field: Describes volume requirements
- Electrostatic field: Maps charge distribution
- Hydrophobic field: Characterizes lipophilicity patterns
- Hydrogen bond donor field: Identifies hydrogen-donating capabilities
- Hydrogen bond acceptor field: Maps hydrogen-accepting capabilities
The inclusion of hydrophobic and hydrogen-bonding fields frequently provides superior interpretability compared to CoMFA, as these interactions play crucial roles in biological recognition [15].
Table 2: Representative CoMSIA Statistical Results from Various Studies
| Study Focus | q² Value | r² Value | r²pred | Fields Used | Reference |
|---|---|---|---|---|---|
| Pyridinone derivatives as HIV-1 RT inhibitors | 0.723 | - | 0.750 | - | [18] |
| Dihydro-pyrazolo-quinazolines as PLK1 inhibitors | 0.598 | 0.925 | - | S,E,H,D,A | [15] |
| Pteridinones as PLK1 inhibitors (SEAH) | 0.66 | 0.975 | 0.767 | S,E,A,H | [9] |
| Aminopyrimidines as GSK-3 inhibitors | 0.598 | 0.925 | - | S,E,H,D,A | [16] |
Experimental Protocol for Pteridinone Derivatives as PLK1 Inhibitors
Molecular Preparation and Alignment
For studies on pteridinone derivatives targeting PLK1 inhibition, the following protocol has been established [9]:
Dataset Curation: Collect experimental IC₅₀ values for pteridinone derivatives and convert to pIC₅₀ (−logIC₅₀) values. The activity range should span at least 3 log units for robust model development.
Training/Test Set Division: Typically, 70-80% of compounds form the training set for model development, while 20-30% serve as the test set for external validation [9]. The test set should represent the structural and activity diversity of the entire dataset.
Molecular Sketching and Optimization: Construct 3D molecular structures using sketching functions (e.g., SKETCH in Sybyl-X). Apply Gasteiger-Hückel atomic partial charges and minimize structures using the Tripos force field with Powell gradient algorithm (convergence criterion: 0.005 kcal/mol Å) [9] [19].
Molecular Alignment: Employ a rigid body distill alignment method in Sybyl-X 2.1 software to superimpose all molecules based on their common core structure [9]. Proper alignment is critical for model quality.
Field Calculation and Statistical Analysis
Grid Definition: Create a 3D lattice extending 4Å beyond all aligned molecules in all directions, with grid spacing of 1-2Å [9].
Field Computation: Calculate CoMFA steric (Lennard-Jones) and electrostatic (Coulombic) fields using an sp³ carbon with +1 charge as the probe [9]. For CoMSIA, compute similarity indices for selected field combinations using a Gaussian function with attenuation factor α=0.3 [9].
PLS Regression: Perform Partial Least Squares analysis with leave-one-out (LOO) cross-validation to determine optimal number of components (ONC) [9]. Column filtering (2.0 kcal/mol) can be applied to speed up analysis [9].
Model Validation: Assess predictive power through external validation using the test set. A predictive r² (r²pred) >0.6 indicates good external predictability [9].
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for 3D-QSAR Studies
| Reagent/Software | Function/Purpose | Application Notes |
|---|---|---|
| Sybyl-X 2.1.1 | Molecular modeling and 3D-QSAR analysis | Commercial software platform for CoMFA/CoMSIA studies [9] [20] |
| Tripos Force Field | Molecular mechanics energy minimization | Standardized force field for geometry optimization [9] [19] |
| Gasteiger-Hückel Method | Partial atomic charge calculation | Standard approach for assigning electrostatic charges [9] [19] |
| PLS (Partial Least Squares) | Statistical correlation analysis | Multivariate regression method for field-activity correlation [9] |
| AutoDock Tools/Vina | Molecular docking validation | Validates binding conformations from 3D-QSAR [9] |
| Gaussian-type Function | Distance-dependent potential for CoMSIA | Provides smoother sampling of molecular similarity [15] |
Contour Map Interpretation and Application
Analyzing CoMFA and CoMSIA Contour Maps
The interpretation of contour maps represents a crucial step in extracting meaningful structure-activity relationships from 3D-QSAR models:
CoMFA Map Interpretation:
- Green contours: Regions where bulky groups enhance activity
- Yellow contours: Areas where bulky groups decrease activity
- Blue contours: Zones where positive charge improves activity
- Red contours: Regions where negative charge enhances activity [20]
CoMSIA Map Interpretation:
- Yellow/Gray hydrophobic contours: Yellow indicates hydrophobic-favorable regions; gray indicates hydrophobic-disfavored regions
- Cyan/Purple H-bond contours: Cyan shows H-bond donor favorable areas; purple shows H-bond donor unfavorable regions [15] [20]
Application to Pteridinone PLK1 Inhibitors
In the study of pteridinone derivatives as PLK1 inhibitors, CoMFA and CoMSIA contour maps revealed key structural requirements for optimal activity [9]:
- Steric requirements: Specific regions around the pteridinone core where bulky substituents significantly enhanced PLK1 inhibitory activity
- Electrostatic preferences: Defined zones favoring electron-withdrawing or electron-donating groups
- Hydrophobic interactions: Areas where lipophilic substituents improved binding affinity
- Hydrogen-bonding patterns: Critical donor and acceptor positions for optimal interaction with the PLK1 active site
These insights directly guided the design of novel pteridinone derivatives with improved PLK1 inhibitory potency and selectivity [9].
Validation Methods
Statistical Validation Techniques
Robust 3D-QSAR models require comprehensive validation using multiple approaches:
Internal Validation: Assessed through leave-one-out (LOO) cross-validation, yielding q² value >0.5 [9]
External Validation: Evaluated by predicting test set activities, with r²pred >0.6 indicating good predictive power [9] [19]
Goodness-of-Fit: Measured by conventional r² (>0.8) and standard error of estimate (SEE) [9]
Complementary Validation Methods
Beyond statistical validation, several approaches strengthen 3D-QSAR model reliability:
Molecular Docking: Validates proposed binding modes and confirms bioactive conformations [9] [19]
Molecular Dynamics Simulations: Assesses complex stability and refines interaction patterns [9]
ADMET Prediction: Evaluates drug-like properties of designed compounds [9]
For pteridinone PLK1 inhibitors, the integration of 3D-QSAR with molecular docking and dynamics simulations provided a comprehensive understanding of binding interactions and supported the design of novel inhibitors with improved potency and selectivity profiles [9].
Within the context of developing a robust CoMFA/CoMSIA protocol for Polo-like kinase 1 (PLK1) inhibition research, the initial and most critical step is the meticulous definition of the experimental dataset. PLK1 is a well-validated, broad-spectrum anti-cancer target, and pteridinone derivatives have emerged as a promising class of potent inhibitors [10] [9]. The reliability of subsequent 3D-QSAR models is entirely contingent upon the quality, consistency, and relevance of the underlying data. This application note details a standardized protocol for sourcing and preparing a series of pteridinone derivatives to ensure the generation of predictive and meaningful computational models for anti-cancer drug discovery.
Data Sourcing and Compound Selection
Sourcing Experimental Biological Data
The primary source of data should be peer-reviewed literature reporting the synthesis and biological evaluation of pteridinone derivatives against PLK1. The ideal dataset comprises a congeneric series of compounds with a consistent core scaffold but varying substituents, all tested under comparable experimental conditions to ensure data homogeneity [10] [9].
- Key Data Point: The half-maximal inhibitory concentration (IC50) is the standard experimental measure for inhibitory activity.
- Data Transformation: For QSAR analysis, IC50 values (reported in nM or µM) must be converted to pIC50 values using the formula: pIC50 = -log10(IC50), where IC50 is expressed in molar concentration (M) [9]. This transformation linearizes the relationship between concentration and binding affinity.
Table 1: Example Dataset of Pteridinone Derivatives and Their Biological Activities [9]
| Compound Number | Substituent (R) | IC50 (nM) | pIC50 |
|---|---|---|---|
| 3 | - | 8.42 | 5.074 |
| 28 | - | 7.18 | 4.787 |
| 18 | - | 36.30 | 5.086 |
| 24 | - | 17.50 | 5.033 |
| 5 * | - | 26.59 | 4.575 |
| 19 * | - | 9.25 | 4.440 |
Compounds marked with an asterisk () can be designated as part of an external test set.*
Dataset Division for Validation
A critical step in protocol development is the division of the full dataset into a training set and a test set.
- Training Set: Typically comprises 80% of the compounds and is used to build the CoMFA and CoMSIA models [9].
- Test Set: Comprises the remaining 20% of the compounds and is used to validate the predictive ability of the developed models. The test set molecules should be selected to represent the structural and activity diversity of the entire dataset [9].
Experimental Protocol: Data Preparation and Molecular Modeling
Molecular Structure Preparation and Optimization
Objective: To generate accurate, low-energy 3D structures for all compounds in the dataset.
- Sketching: Draw the 3D structures of all pteridinone derivatives using molecular modeling software such as SYBYL-X.
- Energy Minimization: Minimize the energy of each structure using the Tripos Force Field.
- Apply Gasteiger-Hückel atomic partial charges.
- Use the Powell conjugate gradient algorithm with a convergence criterion of 0.005 kcal/mol Å for 1000 iterations or until convergence is achieved [9] [21].
- This step produces a stable, low-energy conformation for each molecule, which is essential for subsequent alignment.
Molecular Alignment
Objective: To superimpose all molecules in a meaningful way based on their common structural features and presumed binding mode. Molecular alignment is one of the most sensitive parameters in 3D-QSAR [9] [21].
- Method: Rigid Body Distillation
- Identify the shared dihydropteridinone core structure across all derivatives.
- Select the most active molecule in the dataset as the template structure.
- Superimpose the core structure of every other molecule onto the template core by minimizing the root-mean-square deviation (RMSD) of atomic coordinates [9].
- Alternative Method: Docking-Based Alignment
- For a more biologically relevant alignment, dock each molecule into the active site of the PLK1 protein (e.g., PDB code: 2RKU).
- Use the resulting docked conformations for molecular alignment, ensuring the molecules are oriented according to their putative interactions with the target [18].
The following diagram illustrates the key steps and decision points in the dataset preparation workflow.
3D-QSAR Field Calculation
Objective: To compute the molecular fields that describe the physicochemical properties of the aligned molecules.
- Place the set of aligned molecules into a 3D grid with a default spacing of 2.0 Å [21].
- Use a sp3 carbon probe atom with a charge of +1 to calculate interaction energies at each grid point.
- Calculate the following fields for CoMFA and CoMSIA:
- CoMFA: Calculates Steric (Lennard-Jones potential) and Electrostatic (Coulombic) fields. A default energy cutoff of 30 kcal/mol is applied [9] [21].
- CoMSIA: Can calculate additional fields, including Hydrophobic, Hydrogen Bond Donor, and Hydrogen Bond Acceptor fields, providing a more nuanced description of molecular interactions [10] [22].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Components for Dataset Preparation and 3D-QSAR
| Item Name | Function/Application | Specification/Notes |
|---|---|---|
| SYBYL-X Software | Integrated molecular modeling suite for structure building, minimization, alignment, and CoMFA/CoMSIA analysis. | Uses Tripos Force Field; includes modules for partial least-squares (PLS) analysis [9] [21]. |
| Protein Data Bank (PDB) Structure 2RKU | Crystal structure of PLK1 kinase domain. Serves as a template for docking-based alignment and understanding key inhibitor interactions. | Key active site residues for pteridinones: R136, R57, Y133, L69, L82, Y139 [10] [9]. |
| AutoDock Vina / GOLD | Molecular docking software. Used to predict the binding conformation of each pteridinone derivative in the PLK1 active site for docking-based alignment. | Alternative to rigid alignment; provides a more biologically relevant orientation [18]. |
| PLK1 Inhibitor BI-2536 | A well-characterized dihydropteridinone-based PLK1 inhibitor. A useful reference compound and a starting point for structural derivatization. | Co-crystal structure available; IC50 ~ 0.83 nM [23] [24]. |
A rigorously defined dataset is the cornerstone of any successful CoMFA/CoMSIA study. By adhering to this protocol—meticulously sourcing homogeneous biological data, properly transforming activity values, strategically splitting the dataset, and carefully executing molecular preparation and alignment—researchers can establish a solid foundation for building predictive 3D-QSAR models. In the context of PLK1 inhibitor research, this structured approach enables the rational design of novel pteridinone derivatives with optimized potency and improved therapeutic potential for cancer treatment.
A Step-by-Step Protocol for Building Robust CoMFA and CoMSIA Models
Within the framework of three-dimensional quantitative structure-activity relationship (3D-QSAR) studies, such as Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), molecular alignment is a critical preprocessing step that significantly influences the predictive power and robustness of the resulting models [25] [26]. The core objective is to superimpose a set of molecules in a manner that reflects their presumed binding orientation at the target protein's active site. For research focused on discovering Polo-like kinase 1 (PLK1) inhibitors among pteridinone derivatives, selecting an appropriate template and a reliable superposition method is paramount for identifying the key structural features governing biological activity [9]. This protocol details the application of rigid body alignment and template selection, specifically within the context of a CoMFA/CoMSIA study on pteridinone derivatives, to guide the design of novel anti-cancer drug candidates.
Theoretical Background and Key Concepts
The Role of Alignment in 3D-QSAR
Molecular alignment establishes a common reference frame, allowing for the comparison of steric, electrostatic, and other physicochemical fields across a series of compounds. A successful alignment maximizes the overlap of structurally and functionally similar regions, ensuring that subsequent field calculations are biologically relevant. Inconsistent or poor alignment can lead to models with low predictive accuracy and misleading structure-activity interpretations [26]. The choice of the alignment rule is often considered one of the most sensitive steps in the 3D-QSAR workflow [27].
Rigid Body Superposition
Rigid body alignment, also known as rigid superposition, involves translating and rotating entire molecules as inflexible units to achieve optimal overlap with a chosen template. This method relies on identifying a maximum common substructure (MCS) or a common core shared among all molecules in the dataset [27]. The underlying assumption is that this shared substructure is responsible for a common binding mode, while variable substituents account for differences in potency. The primary advantage of this method is its computational efficiency and simplicity, making it a robust first choice for series of congeneric ligands with a well-defined core structure, such as the pteridinone derivatives under investigation [9].
Application Notes: A Case Study on Pteridinone Derivatives
The following application notes are framed within a broader thesis investigating PLK1 inhibition by pteridinone derivatives, a promising class of anti-cancer agents [9].
The diagram below illustrates the integrated protocol for molecular modeling and alignment, from initial template selection through to final model validation, as applied in pteridinone derivative research.
Template Selection Strategies
The choice of template is a foundational decision. The table below summarizes and compares the primary strategies as applied in recent literature.
Table 1: Template Selection Strategies for Molecular Alignment
| Strategy | Protocol Description | Advantages | Considerations | Application Context |
|---|---|---|---|---|
| Most Active Compound | Select the molecule with the highest reported biological activity (e.g., lowest IC50) as the template for alignment [9] [12]. | Provides a model based on the confirmed bioactive conformation of the best inhibitor. | May not be suitable for highly diverse datasets with multiple scaffolds. | Used in the PLK1/pteridinone study, where compound 28 (most active) served as the reference [9]. |
| Common Core Substructure | Identify the maximum common substructure (MCS) shared by all molecules in the dataset using computational algorithms [27]. | Ensures consistent overlap of the fundamental pharmacophore; highly robust for congeneric series. | Requires a significant shared core among all derivatives. | Applied in a PKB/Akt inhibitor study using the Distill module to define the MCS [27]. |
| Pharmacophore-Based | Align molecules based on a common pharmacophore model that identifies key functional features (H-bond donors/acceptors, hydrophobic regions) [25]. | Focuses on functional similarity rather than purely structural overlap; good for diverse scaffolds. | The quality of alignment is dependent on the accuracy of the generated pharmacophore model. | Utilized in a study of α1A-AR antagonists, where alignment was guided by a GALAHAD-generated pharmacophore [25]. |
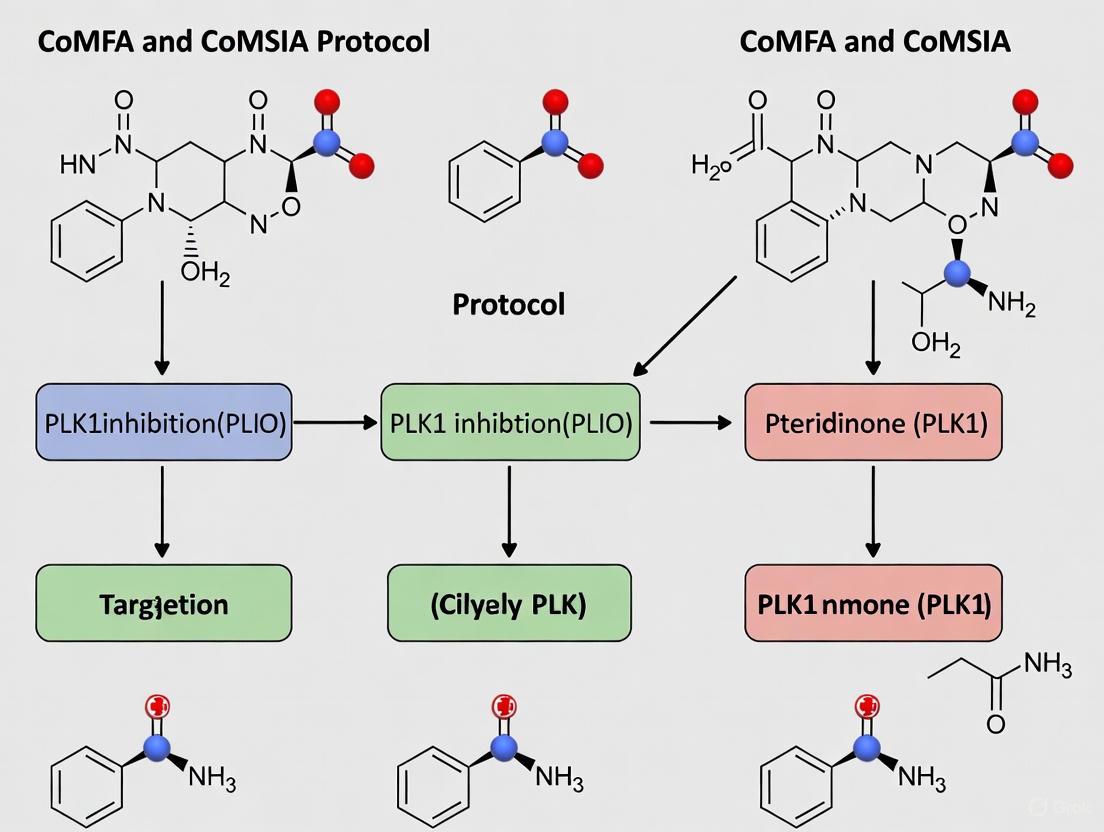
In the specific research on pteridinone derivatives targeting PLK1, the most active compound in the series (Molecular N° 28) was selected as the template structure [9]. All other derivatives were then aligned to this template, ensuring that the model's frame of reference was anchored to the conformation of the most potent inhibitor.
Protocol for Rigid Body Alignment Using the Distill Method
This protocol utilizes the Distill module, available in molecular modeling software suites like SYBYL, to perform a rigid body alignment based on the Maximum Common Substructure (MCS) [12] [27].
Table 2: Step-by-Step Protocol for Rigid Body Alignment with Distill
| Step | Action | Parameters & Notes |
|---|---|---|
| 1. Template Preparation | Sketch the 2D structure of the chosen template molecule (e.g., the most active compound). | For pteridinone derivatives, this is the core pteridinone structure with its invariant substituents [9]. |
| 2. Database Construction | Build the 3D structures of all molecules in the dataset (training and test sets) and save them in a .mol2 database file. |
Ensure all hydrogen atoms are added. Gasteiger-Hückel charges are typically used for QSAR studies [9] [27]. |
| 3. Energy Minimization | Optimize the geometry of all molecules to a stable, low-energy conformation using a molecular mechanics force field. | Force Field: Tripos Standard Force Field.Convergence Criterion: Gradient of 0.05 kcal/mol Å [27]. |
| 4. MCS Identification | Run the Distill module to automatically identify the largest group of connected atoms common to the template and all other molecules. | Set the minimum atom count in MCS fragments (e.g., as low as 3 atoms) to ensure a meaningful core is found [27]. |
| 5. Molecular Superposition | Command the software to fit all molecules in the database to the template coordinates using the best mapping of the MCS atoms. | The algorithm performs a least-squares fit, translating and rotating each molecule as a rigid body to achieve optimal overlap [27]. |
| 6. Alignment Validation | Visually inspect the resulting aligned molecular set to confirm consistent and logical overlap of the common core. | Check for outliers and verify that variable regions are appropriately oriented in 3D space. |
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Research Reagent Solutions for Molecular Modeling and Alignment
| Tool/Software | Type | Primary Function in Alignment & Modeling |
|---|---|---|
| SYBYL-X | Software Suite | An integrated molecular modeling environment used for structure sketching, energy minimization, and running CoMFA/CoMSIA studies [9] [27]. |
| Distill Module | Software Module | A tool within SYBYL-X that performs rigid body alignment by identifying the Maximum Common Substructure (MCS) and superimposing molecules onto a template [27]. |
| Tripos Force Field | Algorithm | A set of mathematical equations and parameters used for molecular mechanics calculations and geometry optimization of organic molecules [9] [12]. |
| Gasteiger-Hückel Charges | Computational Method | A method for calculating partial atomic charges, which are essential for the computation of electrostatic fields in CoMFA [9] [27]. |
| GALAHAD | Software Module | A tool for generating pharmacophore models and molecular alignments based on ligand similarities, useful as an alternative or complementary approach [25]. |
Validation and Integration
Statistical Validation of the Alignment
The ultimate validation of a successful alignment is the statistical quality of the resulting 3D-QSAR model. In the PLK1 study, the rigid body alignment of pteridinone derivatives led to highly predictive CoMFA and CoMSIA models. The models exhibited strong internal consistency (CoMFA R² = 0.992) and predictive power for external test sets (CoMFA R²pred = 0.758), confirming that the chosen template and superposition technique effectively captured the structure-activity relationship [9].
Integration with Broader Research Workflow
As shown in Figure 1, the alignment protocol is not an isolated step. It is preceded by careful molecular preparation and followed by 3D field calculation and statistical analysis using the Partial Least Squares (PLS) algorithm [9] [26]. The insights gained from the contour maps of the validated model, which highlight regions where steric bulk or electronegative groups enhance or diminish activity, can then be used to guide the rational design of new, more potent pteridinone-based PLK1 inhibitors. This creates an iterative cycle of design, synthesis, and testing that is central to modern drug discovery.
In the context of our broader thesis on developing a robust CoMFA/CoMSIA protocol for pteridinone derivatives as PLK1 inhibitors, the precise configuration of molecular field calculations represents a critical methodological foundation. Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) are ligand-based, alignment-dependent 3D-QSAR methods that correlate biological activity with molecular interaction fields [28]. These approaches operate on the fundamental principle that differences in biological activity can be explained by variations in the steric, electrostatic, and hydrophobic fields surrounding a set of aligned molecules [9] [28]. For our research focusing on novel anti-cancer agents targeting polo-like kinase 1 (PLK1), accurate field parameterization is essential for generating predictive models that can guide the rational design of more potent therapeutics.
The calculation of molecular fields involves sampling interaction energies between a probe atom and the molecular structures at numerous grid points throughout a defined region in space [28]. While CoMFA traditionally focuses on steric (Lennard-Jones) and electrostatic (Coulombic) potentials, CoMSIA extends this approach by incorporating additional similarity indices, including hydrophobic fields and hydrogen-bonding descriptors, using a Gaussian-type distance dependence for smoother potential functions [28]. This protocol details the specific parameter configurations for field calculations and descriptor generation, optimized for our series of pteridinone derivatives, to ensure the development of statistically robust and chemically interpretable models.
Theoretical Framework and Key Concepts
Fundamental Differences Between CoMFA and CoMSIA Fields
The core distinction between CoMFA and CoMSIA lies in their mathematical treatment of molecular interactions. CoMFA calculates steric fields using Lennard-Jones potential and electrostatic fields using Coulombic potential, which can lead to sharp changes in energy near molecular surfaces [28]. In practice, this often necessitates the implementation of energy cut-offs (typically 30 kcal/mol) to avoid unrealistic values [9] [29]. Conversely, CoMSIA employs a Gaussian distribution of similarity indices, creating softer potential fields that are less sensitive to minor changes in molecular alignment and grid positioning [28]. This fundamental difference in approach significantly impacts how parameters must be configured for each method.
CoMSIA additionally expands the physicochemical properties considered in the analysis to include hydrophobic fields and explicit hydrogen bond donor and acceptor fields [28]. The hydrophobic field is particularly valuable in drug design, as it encodes the solvent-reliant molecular entropic contributions that influence binding affinity [28]. For our studies on PLK1 inhibitors, which function in aqueous cellular environments, capturing these hydrophobic interactions is essential for accurate activity prediction.
Critical Parameters Governing Field Calculations
Several key parameters directly control the quality and characteristics of the generated molecular fields:
- Grid Spacing: Determines the resolution of field sampling, with smaller spacing (1-2 Å) providing higher resolution but increasing computational load [29].
- Attenuation Factor (α): Specific to CoMSIA, this parameter controls the steepness of the Gaussian fall-off, with a default value of 0.3 providing a reasonable balance between locality and generality [30] [28].
- Probe Atom Characteristics: Defined by charge, radius, and physicochemical properties, the probe atom serves as an artificial representation of receptor interaction sites [28].
- Charge Calculation Method: The algorithm used to compute partial atomic charges significantly impacts electrostatic field calculations and overall model quality [31].
Computational Protocols for Field Calculation
Molecular System Preparation
Prior to field calculation, proper molecular system preparation is essential:
- Structure Optimization: Generate 3D molecular structures and minimize using Tripos force field with Powell method (energy gradient convergence criterion of 0.01 kcal/mol) and Gasteiger-Hückel atomic partial charges [9] [29].
- Molecular Alignment: Align molecules using a common substructure or pharmacophore hypothesis. For pteridinone derivatives, we employed rigid distill alignment using the most active compound as template [9] [20].
- Grid Generation: Create a 3D lattice encompassing all aligned molecules with 2.0 Å grid spacing in x, y, and z directions, extending 4.0 Å beyond molecular dimensions in all directions [9].
CoMFA Field Calculation Protocol
The following protocol details CoMFA field calculation as implemented in SYBYL-X software:
- Field Type Selection: Enable both steric and electrostatic fields.
- Probe Configuration: Utilize an sp³ carbon atom with +1.0 charge and van der Waals radius of 1.52 Å as the probe [29].
- Energy Calculation: Compute steric fields using Lennard-Jones 6-12 potential and electrostatic fields using Coulombic potential with distance-dependent dielectric constant [28].
- Energy Truncation: Apply energy cut-off value of 30 kcal/mol for both field types to avoid extreme values [9] [29].
- Column Filtering: Set filter value to 2.0 kcal/mol to reduce noise and accelerate analysis [9].
CoMSIA Field Calculation Protocol
For CoMSIA calculations, implement this extended protocol:
- Field Selection: Enable five field types: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor [28].
- Probe Configuration: Employ a common probe atom with charge +1, hydrophobicity +1, hydrogen bond donor +1, and hydrogen bond acceptor +1 [28].
- Similarity Indices Calculation: Compute using Gaussian-type distance dependence function: ( A{F,k}^q (j) = -\sum w{probe,k} w{ik} e^{-\alpha r{iq}^2} ) where ( A_{F,k}^q (j) ) is the similarity index at grid point q for molecule j, w are physicochemical properties, α is the attenuation factor (0.3), and r is the distance between grid point and atom [28] [32].
- Grid Definition: Use the same lattice box as for CoMFA analysis with 1-2 Å grid spacing [30].
Table 1: Standard Parameter Settings for Field Calculations in CoMFA and CoMSIA
| Parameter | CoMFA Setting | CoMSIA Setting |
|---|---|---|
| Grid Spacing | 2.0 Å | 2.0 Å |
| Probe Atom | sp³ C⁺ | sp³ C⁺ |
| Probe Charge | +1 | +1 |
| Steric Field | Lennard-Jones potential | Gaussian similarity |
| Electrostatic Field | Coulombic potential | Gaussian similarity |
| Hydrophobic Field | Not available | Gaussian similarity |
| H-bond Donor Field | Not available | Gaussian similarity |
| H-bond Acceptor Field | Not available | Gaussian similarity |
| Attenuation Factor | Not applicable | 0.3 |
| Energy Cut-off | 30 kcal/mol | Not applicable |
Partial Atomic Charge Calculation Methods
The method for calculating partial atomic charges significantly impacts electrostatic field calculations. Our comparative analysis revealed:
Table 2: Comparison of Partial Atomic Charge Calculation Methods
| Method | Theory Basis | Computational Cost | Recommended Use |
|---|---|---|---|
| Gasteiger-Hückel | Empirical | Low | Standard screening |
| AM1-BCC | Semi-empirical | Medium | High-accuracy models |
| AM1 | Semi-empirical | Medium | Balanced approach |
| MMFF94 | Force field based | Low | Molecular mechanics |
| RESP | Ab initio | High | Benchmark studies |
Based on comprehensive comparisons, semi-empirical methods (AM1, AM1-BCC) generally yield superior predictive accuracy in both CoMFA and CoMSIA studies compared to purely empirical methods like Gasteiger-Hückel [31]. For our pteridinone derivative studies, we implemented AM1-BCC charges to optimize model quality while maintaining computational efficiency.
Application to Pteridinone Derivatives as PLK1 Inhibitors
Implementation in Prostate Cancer Research
In our specific research on pteridinone derivatives as PLK1 inhibitors for prostate cancer treatment, we applied the above protocols to establish robust 3D-QSAR models [9]. We calculated fields for 28 pteridinone derivatives with experimentally determined PLK1 inhibition values (IC50), divided into training (22 compounds) and test (6 compounds) sets [9]. The resulting models demonstrated excellent predictive capability, with CoMFA (Q² = 0.67, R² = 0.992) and CoMSIA/SEAH (Q² = 0.66, R² = 0.975) [9].
For the CoMSIA analysis of pteridinone derivatives, we specifically employed the SEAH field combination (steric, electrostatic, acceptor, hydrophobic), which effectively captured the essential molecular interactions governing PLK1 inhibition [9]. The contour maps generated from these field calculations provided clear visualization of structural requirements around the pteridinone core, guiding the design of subsequent derivatives with improved potency.
Integrated Workflow for Field-Based QSAR Analysis
The following diagram illustrates the comprehensive workflow for field calculation and descriptor generation within our CoMFA/CoMSIA protocol for PLK1 inhibitor research:
Diagram 1: Comprehensive workflow for field calculation and descriptor generation in CoMFA/CoMSIA studies
Relationship Between Field Contributions and Model Parameters
The following diagram illustrates how different configuration parameters interact to influence field contributions in the final QSAR model:
Diagram 2: Relationship between configuration parameters and field contributions in QSAR models
Research Reagent Solutions: Essential Materials for Field Calculation Studies
Table 3: Essential Research Reagents and Computational Tools for Field Calculation Studies
| Category | Specific Tool/Software | Function in Field Calculation | Application Example |
|---|---|---|---|
| Molecular Modeling | SYBYL-X (Tripos) | Primary platform for CoMFA/CoMSIA field generation | Structure building, minimization, alignment [9] |
| Charge Calculation | AM1-BCC Method | Semi-empirical partial charge calculation | High-accuracy electrostatic field generation [31] |
| Alignment Tool | GALAHAD (Tripos) | Pharmacophore-based molecular alignment | Aligning diverse molecular scaffolds [29] |
| Docking Software | AutoDock Vina | Binding conformation prediction | Bioactive conformation generation [9] |
| Force Field | Tripos Standard Force Field | Molecular mechanics energy calculation | Structure optimization prior to field calculation [9] |
| Probe Atoms | sp³ Carbon (+1 charge) | Standard probe for interaction calculation | Steric and electrostatic field sampling [28] |
Based on our systematic investigation of field calculation parameters for CoMFA and CoMSIA studies on pteridinone derivatives, we recommend the following best practices:
- For electrostatic fields, utilize AM1-BCC partial charges rather than default Gasteiger-Hückel charges to improve model predictivity [31].
- For CoMSIA studies, include hydrophobic fields alongside steric and electrostatic descriptors to account for solvation effects critical for PLK1 inhibition [9] [28].
- Implement a standardized grid spacing of 2.0 Å as a balance between computational efficiency and field resolution [9] [29].
- Always validate models with external test sets (25-30% of dataset) to ensure robust prediction of novel pteridinone derivatives [9] [29].
Proper configuration of steric, electrostatic, and hydrophobic parameters in field calculations provides the foundation for developing predictive 3D-QSAR models that can effectively guide the optimization of pteridinone-based PLK1 inhibitors. The protocols detailed herein have demonstrated practical utility in our anti-cancer drug discovery efforts, resulting in statistically validated models with direct applications in structure-based drug design.
Within the context of Computer-Aided Drug Design (CADD), particularly in the development of Polo-like kinase 1 (PLK1) inhibitors for prostate cancer therapy, Partial Least-Squares (PLS) regression serves as the critical statistical engine for correlating 3D molecular field descriptors with biological activity. PLS analysis is a multivariate technique that excels in scenarios where predictor variables (e.g., 3D field descriptors) are numerous and highly collinear, which is a defining characteristic of Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) studies [9] [33]. For pteridinone derivatives, this method allows researchers to distill the vast information from steric, electrostatic, and other molecular fields into a predictive model that relates molecular structure to inhibitory potency (pIC50) [9]. The robustness of this approach is evidenced in recent studies where established 3D-QSAR models for pteridinone derivatives showed high correlative and predictive power, guiding the identification of compound 28 as a promising anti-prostate cancer candidate [9] [10].
Theoretical Foundation
The Role of PLS in 3D-QSAR
In 3D-QSAR, the core challenge is to relate a large matrix of independent variables (X) to a vector of dependent biological activities (Y). The independent variables are the thousands of steric, electrostatic, and hydrophobic field values sampled at grid points around the aligned molecules. PLS regression addresses this by projecting both the X and Y variables onto a new, lower-dimensional space of latent variables, or components, which maximize the covariance between X and Y [34]. This is fundamentally different from Principal Component Regression (PCR), which only considers the variance in the X-block. The supervised nature of PLS makes it ideally suited for predictive modeling in drug discovery.
Key Statistical Metrics for Model Validation
The quality and reliability of a PLS-based 3D-QSAR model are judged using several key statistical parameters, which are summarized in the table below.
Table 1: Key Statistical Metrics for Validating PLS-based 3D-QSAR Models
| Metric | Symbol | Description | Acceptance Threshold |
|---|---|---|---|
| Cross-validated Correlation Coefficient | Q² | Measures the model's predictive reliability based on leave-one-out cross-validation. | > 0.5 [9] |
| Non-cross-validated Correlation Coefficient | R² | Indicates the goodness-of-fit of the model to the training set data. | Closer to 1.0 indicates a better fit [9] [12]. |
| Standard Error of Estimate | SEE | The average distance between the data points and the regression line. A lower value indicates a more precise model [9]. | As low as possible |
| Predictive Correlation Coefficient | R²pred | Evaluates the model's predictive ability on an external test set of compounds. | > 0.6 [9] [33] |
| Optimal Number of Components | NOC | The number of latent variables used in the final model; determined by cross-validation. | Balances model complexity and predictive power [9]. |
Application Notes: Protocol for PLS Analysis in CoMFA/CoMSIA
This protocol details the application of PLS analysis to establish a correlation between 3D molecular fields and the pIC50 values of pteridinone derivatives targeting PLK1 [9] [10].
Prerequisites
- A set of structurally aligned molecules (e.g., 28 pteridinone derivatives) [9].
- Experimentally determined biological activity data (IC50), converted to pIC50 using the formula: pIC50 = -log10(IC50) [9] [12].
- Calculated CoMFA (steric and electrostatic) and/or CoMSIA (steric, electrostatic, hydrophobic, hydrogen-bond donor, acceptor) field descriptors.
Step-by-Step Procedure
Data Preparation and Partitioning: Divide the dataset of 28 compounds into a training set (~80%, ~22 compounds) for model building and a test set (~20%, ~6 compounds) for external validation [9]. The selection should be random or based on a representative structural and activity spread.
PLS Regression Execution:
- Input the field descriptors (X-matrix) and the pIC50 values (Y-vector) for the training set into the analysis software (e.g., SYBYL-X [9]).
- Run the PLS algorithm with Leave-One-Out (LOO) cross-validation to determine the optimal number of components (NOC). The NOC is chosen where the highest Q² value is obtained [33].
- Once the NOC is fixed, perform a non-cross-validated PLS regression to generate the final model with conventional R² and SEE values [9].
Model Validation:
- Internal Validation: Assess the Q², R², and SEE from the previous step. For example, a robust CoMFA model for pteridinones achieved Q² = 0.67 and R² = 0.992 [9].
- External Validation: Use the developed model to predict the pIC50 values of the external test set compounds. Calculate the predictive R² (R²pred) to confirm the model's predictive power. A value above 0.6 is considered successful [9] [33].
Contour Map Generation: Interpret the model by visualizing the results as 3D contour maps. These maps highlight regions around the molecules where specific field properties (e.g., favored steric bulk, disfavored negative electrostatics) are correlated with increased or decreased biological activity [9] [12].
The following workflow diagram illustrates the logical sequence of the PLS analysis protocol within a CoMFA/CoMSIA study.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools and Resources for PLS-based 3D-QSAR
| Tool/Resource | Function in PLS 3D-QSAR | Application Example |
|---|---|---|
| Molecular Modeling Suite (e.g., SYBYL) | Provides an integrated environment for molecular alignment, field calculation, and PLS regression analysis [9] [12]. | Used to build CoMFA/CoMSIA models for pteridinone derivatives with integrated PLS analysis [9]. |
| Structural Database (e.g., PDB) | Source of 3D protein structures for structure-based alignment and understanding inhibitor binding modes [33]. | PDB ID 2RKU used for PLK1 inhibitor studies [9]. |
| Dataset of Inhibitors | A curated set of molecules with known biological activities (IC50) is the fundamental input for any QSAR model [9] [33]. | 28 pteridinone derivatives with measured PLK1 inhibition IC50 [9]. |
| Validation Scripts/Software | Custom or commercial tools to perform advanced validation, such as bootstrapping or response permutation testing [33]. | Used to reinforce the statistical significance of the QSAR models for TTK inhibitors [33]. |
Case Study: Pteridinone Derivatives as PLK1 Inhibitors
A recent study exemplifies this protocol. Researchers developed 3D-QSAR models for 28 novel pteridinone derivatives acting as PLK1 inhibitors [9]. The molecular structures were aligned, and CoMFA/CoMSIA fields were calculated. The PLS analysis yielded highly predictive models. The statistical results from this study are summarized in the table below.
Table 3: Statistical Results of 3D-QSAR Models for Pteridinone Derivatives [9]
| Model Type | Q² | R² | SEE | R²pred | NOC |
|---|---|---|---|---|---|
| CoMFA | 0.67 | 0.992 | Not Specified | 0.683 | Not Specified |
| CoMSIA/SHE | 0.69 | 0.974 | Not Specified | 0.758 | Not Specified |
| CoMSIA/SEAH | 0.66 | 0.975 | Not Specified | 0.767 | Not Specified |
The contour maps generated from these models provided clear structural insights. For instance, they revealed specific regions where introducing bulky substituents would enhance steric favorability or where electropositive groups were preferred for increased potency [9]. This information was further validated by molecular docking, which identified key active site residues (e.g., R136, R57, Y133) in PLK1, confirming that the predicted favorable interactions from the 3D-QSAR were consistent with the hypothetical binding mode [9]. The stability of this binding pose was subsequently confirmed through 50 ns molecular dynamics simulations [9] [10].
Troubleshooting and Technical Notes
- Low Q² Value: This often indicates a poor model. Re-evaluate the molecular alignment rule, which is one of the most sensitive steps in 3D-QSAR [12]. Consider using a different alignment method (e.g., ligand-based vs. structure-based) [33].
- High R² but Low R²pred: This is a classic sign of model overfitting. The model has memorized the training set noise instead of learning the general structure-activity relationship. Reduce the number of components or increase the training set size.
- Handling Categorical Variables: If the dataset includes categorical variables (e.g., different coating types or media in drug delivery studies), techniques like one-hot encoding must be applied before PLS analysis to convert them into a numerical format that the algorithm can process [34].
In the context of Computational Drug Discovery, specifically within the development of three-dimensional quantitative structure-activity relationship (3D-QSAR) models such as Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), the division of a chemical dataset into training and test sets is a critical step. This division directly impacts the model's ability to predict the biological activity of new, unseen compounds reliably. For researchers focusing on pteridinone derivatives as PLK1 inhibitors for prostate cancer treatment, a robust dataset division strategy ensures that the developed models are both predictive and trustworthy, reducing the economic impact of drug development by prioritizing the most promising candidates for synthesis and biological testing [9] [10]. This application note details established protocols and best practices for this essential process, framed within a typical CoMFA/CoMSIA workflow.
Core Concepts and Purpose of Data Splitting
The primary goal of splitting a dataset is to build a model with high generalizability—the ability to make accurate predictions on new data not used during the model construction phase.
- Training Set: This subset is used to build the 3D-QSAR model. The algorithm learns the relationship between the molecular fields (steric, electrostatic, etc.) and the biological activity (e.g., pIC50) from this data [35] [36].
- Test Set: This subset is used to provide an unbiased evaluation of the final model's predictive performance. The test set is kept completely separate during the entire model-building and parameter-tuning process [36].
Using the same data for both training and evaluation leads to overfitting, where a model demonstrates artificially high performance on its training data but fails to predict new compounds accurately [36]. A properly reserved test set simulates a real-world scenario, providing a realistic estimate of how the model will perform when used to screen virtual libraries for novel PLK1 inhibitors.
Quantitative Data on Common Splitting Ratios
The choice of how to split the data depends on the total dataset size. The following table summarizes common practices observed in 3D-QSAR studies and broader machine learning applications.
Table 1: Common Dataset Division Ratios in Practice
| Split Ratio (Training:Test) | Context of Use | Example from Literature |
|---|---|---|
| 80:20 | A common and widely used starting point for datasets of moderate size [37] [35] [36]. | A study on 28 pteridinone derivatives used 80% (22 compounds) for training and 20% (6 compounds) for testing [9]. |
| 90:10 or 99:0.5:0.5 | Preferred for very large datasets (e.g., millions of data points), where even a small percentage provides a statistically sufficient test set [37]. | In big data eras, splits like 99.5:0.25:0.25 for training, validation, and test are used [37]. |
| 60:20:20 | Another recognized split, allocating separate portions for training, validation (for hyperparameter tuning), and testing [37]. | Suggested in online machine learning courses as a balanced approach [37]. |
For the typical dataset sizes in 3D-QSAR studies (often several dozen to a few hundred compounds), an 80:20 training-to-test split is a robust and frequently employed standard [9] [12]. The key principle is that the test set must be large enough to provide a reliable estimate of model performance but not so large that it deprives the model of essential training data.
Detailed Experimental Protocol for 3D-QSAR
This protocol outlines the key steps for dividing a dataset and constructing a CoMFA/CoMSIA model, using pteridinone derivatives as a specific example.
Dataset Collection and Curation
- Activity Data: Collect a consistent set of biological activity data (e.g., IC50 values) for a series of pteridinone derivatives from literature or experimental results. The activity should be converted to pIC50 (-logIC50) for modeling [9] [12].
- Structural Data: Ensure the molecular structures of all derivatives are accurately drawn and standardized.
Molecular Alignment and Conformation
Molecular alignment is one of the most sensitive and critical steps in 3D-QSAR [9] [12].
- Select a Template: Choose the most active compound as a template structure [12].
- Align Molecules: Use software like SYBYL-X to align all other database molecules to the template based on their common scaffold (e.g., the pteridinone core). The
distillmethod in SYBYL is commonly used for this rigid alignment [9] [12]. - Energy Minimization: Optimize the geometries of all aligned molecules using a force field (e.g., Tripos force field) and assign partial atomic charges (e.g., Gasteiger-Hückel) to prepare for field calculation [9] [38].
Dataset Division Protocol
- Randomized Division: After alignment, randomly select 80% of the compounds for the training set and 20% for the test set. This random selection helps ensure both sets are representative of the overall chemical space and activity range [9].
- Stratified Sampling: If the dataset is small or has an uneven distribution of activity values, consider stratified sampling to maintain a similar distribution of high, medium, and low-activity compounds in both the training and test sets [36].
3D-QSAR Model Generation and Validation
- Field Calculation: Calculate CoMFA (steric and electrostatic) and CoMSIA (additional fields like hydrophobic, hydrogen-bond donor/acceptor) descriptors for all aligned molecules within a defined 3D grid [9] [12].
- Model Construction: Use the Partial Least Squares (PLS) algorithm on the training set to build a regression model correlating the field descriptors with the pIC50 values [9] [38].
- Internal Validation: Validate the model internally on the training set using leave-one-out (LOO) cross-validation to obtain the cross-validated correlation coefficient (Q²). A Q² > 0.5 is generally considered statistically significant [9].
- External Validation: The ultimate test of model robustness is the prediction of the external test set. Calculate the predictive correlation coefficient (R²pred) to confirm the model's predictive ability. An R²pred > 0.6 is typically required for a model to be considered predictive [9] [38].
The following workflow diagram illustrates this entire process:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for 3D-QSAR Modeling and Data Division
| Tool / Reagent | Function / Description | Example Use in Protocol |
|---|---|---|
| SYBYL-X Software | A comprehensive molecular modeling suite for 3D-QSAR. | Used for molecular alignment, field calculation, and PLS analysis [9] [12]. |
| Tripos Force Field | A molecular mechanics force field for geometry optimization. | Used for energy minimization of aligned molecules before field calculation [9] [38]. |
| Gasteiger-Hückel Charges | A method for calculating partial atomic charges. | Assigns electrostatic properties to atoms for electrostatic field calculations [9] [38]. |
| Python with scikit-learn | A programming language and ML library for flexible data handling. | Useful for implementing custom data splitting routines and advanced machine learning algorithms [38]. |
| AutoDock Vina | Molecular docking software. | Used in conjunction with QSAR to study protein-ligand interactions and validate binding modes [9]. |
The following diagram provides a guideline for choosing the appropriate data splitting strategy based on your dataset's characteristics:
For 3D-QSAR studies on pteridinone derivatives, adhering to a rigorous dataset division protocol is non-negotiable for developing predictive models. The 80:20 split provides a strong, defensible foundation for datasets of typical size in medicinal chemistry. By following the detailed protocol outlined above—ensuring proper molecular alignment, randomized division, and rigorous external validation—researchers can construct robust CoMFA/CoMSIA models. These models can then reliably guide the optimization of pteridinone derivatives, accelerating the discovery of potent PLK1 inhibitors for prostate cancer therapy.
The implementation of Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) protocols relies heavily on specialized software tools that have evolved significantly over decades. Historically, commercial suites like SYBYL dominated this landscape, providing integrated environments for molecular modeling and 3D quantitative structure-activity relationship (3D-QSAR) studies [32] [9]. These tools enabled researchers to correlate the three-dimensional structural properties of molecules with their biological activities, providing crucial insights for drug design [39] [40].
Recently, a paradigm shift has occurred with the emergence of open-source alternatives such as Py-CoMSIA, which address accessibility challenges created by the discontinuation of proprietary platforms [39] [40]. This transition is particularly relevant for research on pteridinone derivatives as PLK1 inhibitors, where understanding structural determinants of inhibitory activity is essential for developing novel anticancer therapeutics [9]. The movement toward open-source tools represents a significant advancement in computational drug discovery, offering greater flexibility, transparency, and customizability for researchers working with specialized compound classes.
Established Commercial Platforms: The SYBYL Legacy
SYBYL Functionality and Implementation
For decades, the SYBYL molecular modeling software developed by Tripos served as the industry standard for CoMFA and CoMSIA analyses [39] [40]. This comprehensive platform provided an integrated computational framework encompassing all critical steps for 3D-QSAR model development:
- Molecular Structure Preparation: SYBYL offered robust tools for building, importing, and optimizing molecular structures, including energy minimization using the Tripos molecular mechanics force field with Gasteiger-Huckel partial atomic charges [32] [9].
- Molecular Alignment: The platform included multiple alignment methods, with database alignment and distill alignment modules to superimpose molecules based on common substructures or pharmacophoric features [32] [41].
- Field Calculation: SYBYL calculated steric and electrostatic field descriptors for CoMFA at each lattice point using a sp³ carbon atom with +1 charge as a probe, with default energy cutoffs of 30 kcal/mol [32] [42]. For CoMSIA, it could compute five distinct molecular field types: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor [39] [40].
- Statistical Analysis: The software implemented Partial Least Squares (PLS) regression with cross-validation techniques, particularly the leave-one-out (LOO) method, to establish robust correlations between field descriptors and biological activities [32] [9].
SYBYL in Pteridinone Derivative Research
In studies on pteridinone derivatives as PLK1 inhibitors, researchers successfully utilized SYBYL to develop predictive 3D-QSAR models [9]. The standard protocol involved:
- Building and optimizing 28 pteridinone derivatives in SYBYL-X 2.1
- Performing rigid distill alignment using the standardized Tripos force field
- Calculating CoMFA and CoMSIA descriptors with a grid spacing of 1-2 Å extending 4 Å beyond molecular dimensions
- Applying PLS analysis with specific column filtering (typically 2.0 kcal/mol) to enhance signal-to-noise ratio
- Validating models through both internal (cross-validation, bootstrapping) and external (test set prediction) methods [9]
This approach yielded statistically significant models with cross-validated correlation coefficients (q²) of 0.67 for CoMFA and 0.69 for CoMSIA, demonstrating the effectiveness of SYBYL for this specialized research application [9].
The Open-Source Revolution: Py-CoMSIA
Development and Capabilities of Py-CoMSIA
The discontinuation of SYBYL in the mid-2010s created significant accessibility challenges for researchers, prompting the development of open-source alternatives [39] [40]. Py-CoMSIA represents a direct response to this need, providing a Python-based implementation of the CoMSIA methodology that replicates the core functionality previously available only in commercial packages [39] [40].
This library leverages popular scientific Python libraries including:
- RDKit for chemical informatics and molecular manipulation
- NumPy for efficient numerical computations and array operations
- PyVista for advanced 3D visualization and contour mapping [39] [40]
Py-CoMSIA implements the fundamental CoMSIA algorithm using Gaussian functions to calculate molecular similarity indices, avoiding the abrupt cutoffs and singularities at atomic positions that complicated earlier methods [39] [40]. This approach generates continuous molecular similarity maps for all five field types, enhancing model interpretability compared to traditional CoMFA [39].
Performance Validation
Py-CoMSIA has been rigorously validated against established benchmarks, including the original steroid dataset used in pioneering CoMSIA research [39] [40]. Comparative analyses demonstrate that Py-CoMSIA produces similarity indices and predictive models comparable to those generated by SYBYL, with minor variations attributable to differences in molecular alignment implementations [39] [40].
Table 1: Performance Comparison Between SYBYL and Py-CoMSIA on Steroid Benchmark Dataset
| Statistical Metric | SYBYL (SEH) | Py-CoMSIA (SEH) | Py-CoMSIA (SEHAD) |
|---|---|---|---|
| q² (LOO-CV) | 0.665 | 0.609 | 0.630 |
| r² | 0.937 | 0.917 | 0.898 |
| Standard Error | 0.759 | 0.718 | 0.698 |
| Optimal Components | 4 | 3 | 3 |
| Steric Contribution | 0.073 | 0.149 | 0.065 |
| Electrostatic Contribution | 0.513 | 0.534 | 0.258 |
| Hydrophobic Contribution | 0.415 | 0.316 | 0.154 |
Comparative Analysis: Commercial vs. Open-Source Implementations
Technical Methodologies and Algorithmic Differences
While both commercial and open-source tools implement the core CoMFA/CoMSIA methodologies, significant technical differences exist in their approaches:
- Field Calculations: SYBYL employs discrete interaction energy calculations with fixed cutoffs in CoMFA, potentially resulting in abrupt field distributions. Py-CoMSIA utilizes Gaussian functions for all field types, creating smoother, more continuous maps [39] [40].
- Alignment Sensitivity: Commercial implementations typically offer multiple sophisticated alignment algorithms but may demonstrate higher sensitivity to alignment variations. Open-source alternatives aim to reduce this sensitivity through their Gaussian-based approach [39].
- Customization Potential: Py-CoMSIA provides extensive customization options through its Python foundation, allowing researchers to modify algorithms, integrate machine learning approaches, and adapt methods to specific research needs [39] [40].
Practical Implementation Considerations
Table 2: Software Tool Comparison for 3D-QSAR Research
| Feature | SYBYL | Py-CoMSIA |
|---|---|---|
| Accessibility | Commercial license required | Open-source, freely available |
| Development Status | Discontinued | Actively developed |
| Learning Curve | Steep, proprietary interface | Moderate, Python-based |
| Integration | Self-contained platform | Flexible library integration |
| Customization | Limited to built-in features | Extensive via Python code |
| Visualization | Integrated graphics | PyVista, Matplotlib |
| Statistical Methods | Built-in PLS implementation | Customizable statistical approaches |
| Community Support | Vendor-dependent | Growing open-source community |
Application Notes: Protocol for Pteridinone Derivatives as PLK1 Inhibitors
Molecular Data Set Preparation and Alignment
For research on pteridinone derivatives targeting PLK1 inhibition, begin with a carefully curated data set of compounds with experimentally determined IC₅₀ values [9]. Convert these values to pIC₅₀ (-logIC₅₀) for QSAR analysis. Divide compounds into training and test sets (typically 80%/20%), ensuring structural diversity and activity range representation in both sets [9].
Alignment Protocol:
- Select a template molecule (typically high-activity compound 28 from pteridinone series)
- Extract common substructure or pharmacophore features
- Apply rigid distill alignment in SYBYL or database alignment in Py-CoMSIA
- Verify alignment quality through visual inspection and RMSD calculations [9]
Field Calculation and Model Generation Parameters
CoMFA Settings:
- Grid spacing: 2.0 Å (SYBYL) or 1.0 Å (Py-CoMSIA)
- Probe atom: sp³ carbon with +1 charge
- Energy cutoffs: 30 kcal/mol for both steric and electrostatic fields
- Partial atomic charges: Gasteiger-Hückel or MMFF94 [32] [9]
CoMSIA Settings:
- Field types: Steric, electrostatic, hydrophobic, hydrogen bond donor, acceptor
- Attenuation factor: 0.3
- Gaussian-type distance dependence
- No arbitrary energy cutoffs [39] [9]
Statistical Validation:
- Perform PLS regression with leave-one-out cross-validation
- Determine optimal number of components based on highest q² value
- Validate with test set prediction (predicted r² > 0.6 indicates good predictive power) [32] [9]
- Apply bootstrapping analysis (100+ runs) to assess statistical confidence [42]
Research Reagent Solutions for 3D-QSAR Studies
Table 3: Essential Research Materials and Computational Tools
| Resource Type | Specific Examples | Research Application |
|---|---|---|
| Commercial Software | SYBYL-X 2.1, Schrödinger Suite | Molecular modeling, alignment, field calculation |
| Open-Source Libraries | Py-CoMSIA, RDKit, NumPy | Python-based CoMSIA implementation, cheminformatics |
| Visualization Tools | PyVista, Matplotlib, Chimera | Contour map generation, result interpretation |
| Statistical Packages | Scikit-learn, PLS modules | Regression analysis, model validation |
| Compound Databases | PubChem, ZINC, ChEMBL | Structure retrieval, analog identification |
| Protein Data Bank | RCSB PDB (e.g., 2RKU for PLK1) | Target structure retrieval, docking studies |
Integrated Workflow for 3D-QSAR Analysis
The following diagram illustrates the comprehensive workflow for conducting 3D-QSAR studies using both commercial and open-source approaches, highlighting the parallel processes and decision points:
The evolution of software tools for CoMFA and CoMSIA analysis from proprietary suites to open-source alternatives represents a significant democratization of computational drug discovery methods. While established commercial platforms like SYBYL offer polished, integrated workflows with extensive documentation and support, emerging open-source tools like Py-CoMSIA provide unprecedented flexibility, customization potential, and accessibility [39] [40].
For researchers focused on pteridinone derivatives as PLK1 inhibitors, both pathways remain viable, though the open-source approach offers greater opportunities for method extension and integration with modern machine learning techniques [39] [9]. The future of 3D-QSAR methodology will likely see increased convergence between traditional molecular field analysis and contemporary artificial intelligence approaches, with open-source platforms providing the natural foundation for this innovation.
As the field advances, the development of standardized benchmarks, improved visualization capabilities, and enhanced user interfaces for open-source tools will further accelerate their adoption, ultimately empowering more researchers to leverage these powerful techniques in targeted drug discovery programs.
Overcoming Common Challenges and Enhancing Model Predictivity
Within the framework of a broader thesis on establishing a CoMFA/CoMSIA protocol for pteridinone derivatives as PLK1 inhibitors, addressing alignment sensitivity is a cornerstone for generating predictive and interpretable models. Molecular alignment constitutes one of the most critical and technically demanding steps in 3D-QSAR, as a poor alignment undermines the entire modeling process by introducing inconsistencies in descriptor calculations [43]. The core assumption is that all compounds share a similar binding mode to the target, PLK1, and must be superimposed in a shared 3D reference frame that reflects their putative bioactive conformations [44] [43]. This application note details standardized protocols to achieve consistent and meaningful superposition, directly supporting research into novel anti-cancer agents.
Core Alignment Methodologies and Protocols
The choice of alignment strategy is paramount. The following section outlines three primary methodologies, with their associated experimental protocols, applicable to a series of pteridinone derivatives.
Common Scaffold Alignment
This is the most straightforward method, ideal for series with a well-defined, rigid core structure.
Theoretical Basis: Alignment is achieved by superimposing the common molecular framework—typically the pteridinone core in our research context—across all molecules in the dataset. This method assumes that the core is largely responsible for correct positioning in the protein's active site and that substituents explore different regions of the binding pocket [43].
Experimental Protocol:
- Identify the Common Scaffold: Define the largest common substructure (e.g., the pteridinone ring system) shared by all compounds in the dataset. Tools like the Bemis-Murcko scaffold generator can automate this [43].
- Select a Template Molecule: Choose the most active compound or a molecule with a well-defined, low-energy conformation as the reference template for superposition.
- Generate 3D Conformations: For each molecule in the dataset, generate a reasonable 3D conformation. Energy minimization should be performed using a standardized force field (e.g., Tripos Standard Force Field) and convergence criteria (e.g., Powell gradient algorithm with a termination of 0.05 kcal/mol Å) [9] [25].
- Perform Superposition: Superimpose all molecules onto the template by atom-to-atom fitting of the predefined common scaffold atoms. This can be done using the "rigid distill alignment" function in molecular modeling software like SYBYL-X [9].
- Visual Inspection: Manually verify the alignment to ensure the core structures are correctly overlaid and that substituent conformations are reasonable.
Table 1: Key Software and Reagents for Common Scaffold Alignment
| Item Name | Function/Description | Example Usage in Protocol |
|---|---|---|
| SYBYL-X Software | Comprehensive molecular modeling suite | Performing "rigid distill alignment" of pteridinone derivatives [9]. |
| Tripos Force Field | Molecular mechanics force field | Energy minimization of initial 3D structures to stable configurations [9] [25]. |
| Gasteiger-Hückel Charges | Method for calculating atomic partial charges | Assigning charges for electrostatic calculations during minimization and descriptor generation [9] [25]. |
| Bemis-Murcko Scaffold | Algorithm to define a core structure | Identifying the shared pteridinone core for alignment purposes [43]. |
Pharmacophore-Based Alignment
This method offers superior results for datasets with greater structural diversity, where a common scaffold may be small or non-existent.
Theoretical Basis: Alignment is based on a pharmacophore model—an abstract definition of the molecular features necessary for biological activity (e.g., hydrogen bond donors/acceptors, hydrophobic regions, charged groups). All molecules are aligned to maximize the overlap of these critical features [25].
Experimental Protocol:
- Develop a Pharmacophore Hypothesis: Use a set of active and conformationally diverse compounds to generate a pharmacophore model. Software tools like GALAHAD (Genetic Algorithm with Linear Assignment of Hypermolecular Alignment of Datasets) can be used to identify the best pharmacophore alignment from a set of ligand molecules [25].
- Model Validation: The generated model should be evaluated for its ability to discriminate between active and inactive compounds.
- Align Molecules: Using the validated pharmacophore model as a template, align all molecules in the training and test sets. This involves fitting each molecule's functional groups to the corresponding pharmacophore features in 3D space [25].
- Refine Alignment: Some software allows for fine-tuning the alignment based on energy fields (field-fit) to achieve the best possible superposition [44].
Table 2: Key Software and Reagents for Pharmacophore-Based Alignment
| Item Name | Function/Description | Example Usage in Protocol |
|---|---|---|
| GALAHAD (Tripos) | Pharmacophore modeling and alignment tool | Generating pharmacophore hypotheses and superior molecular alignments for diverse α1A-AR antagonists [25]. |
| Template Pharmacophore | A pre-defined, optimized pharmacophore model | Used as a reference template to align all compounds in the training and test sets [25]. |
| Field-Fit Algorithm | A method for refining alignment based on energy fields | Optimizing the alignment of molecules by minimizing the differences in their steric and electrostatic fields [44]. |
Database Mining and Docking-Guided Alignment
For targets with a known crystal structure, this method provides a structure-based alignment rationale.
Theoretical Basis: The alignment of molecules is informed by their predicted binding mode within the protein's active site, as determined by molecular docking simulations [9] [20].
Experimental Protocol:
- Prepare the Protein Structure: Obtain the target protein structure (e.g., PLK1, PDB code: 2RKU). Remove water molecules and co-crystallized ligands, add hydrogen atoms, and assign partial charges [9].
- Define the Active Site: Identify the binding cavity, often based on the location of a native ligand or literature data.
- Perform Molecular Docking: Dock each molecule from the dataset into the defined active site using docking software (e.g., AutoDock Vina). Use consistent docking parameters for all ligands [9].
- Extract Bioactive Conformations: Retrieve the lowest-energy docking pose for each ligand. These poses represent the putative "bioactive conformation."
- Superpose Poses: Align all molecules based on the heavy atoms of their docked conformations within the protein's binding site. This ensemble of aligned ligands can then be exported for 3D-QSAR studies.
Diagram 1: Docking-guided alignment workflow for structure-based superposition.
Validation of Alignment Quality
The ultimate validation of a successful alignment is the statistical quality and predictive power of the resulting 3D-QSAR model.
Quantitative Metrics:
- High Q²: The cross-validated correlation coefficient (e.g., from Leave-One-Out validation) should ideally be greater than 0.5, indicating good predictive ability. In pteridinone/PLK1 studies, values of 0.67 for CoMFA and 0.69 for CoMSIA have been achieved [9].
- High R²: The conventional correlation coefficient indicates the goodness-of-fit of the model to the training set data.
- High R²pred: The predictive correlation coefficient for an external test set of molecules that were not used to build the model should be greater than 0.6 [9].
Qualitative Assessment:
- Contour Map Interpretability: The generated CoMFA and CoMSIA steric and electrostatic contour maps should be chemically intuitive and easily rationalized. A good alignment produces contour maps that clearly highlight regions where specific molecular properties enhance or diminish activity, providing clear guidance for molecular design [9] [43].
Table 3: Impact of Alignment on Model Statistics in Published Studies
| Study Context | Alignment Method | Model Type | Q² | R²pred | Reference |
|---|---|---|---|---|---|
| Pteridinone/PLK1 Inhibitors | Rigid Distill (Common Scaffold) | CoMFA | 0.67 | 0.683 | [9] |
| Pteridinone/PLK1 Inhibitors | Rigid Distill (Common Scaffold) | CoMSIA/SHE | 0.69 | 0.758 | [9] |
| α1A-AR Antagonists | Pharmacophore (GALAHAD) | CoMFA | 0.840 | 0.694 | [25] |
| α1A-AR Antagonists | Pharmacophore (GALAHAD) | CoMSIA | 0.840 | 0.671 | [25] |
Achieving a consistent and meaningful molecular superposition is a non-negotiable prerequisite for robust 3D-QSAR analysis in PLK1 inhibitor research. The choice between common scaffold, pharmacophore-based, or docking-guided alignment depends on the structural homogeneity of the dataset and the availability of target structural information. By adhering to the detailed protocols outlined in this document, researchers can mitigate the inherent sensitivity of CoMFA and CoMSIA to alignment, thereby generating reliable, predictive models that effectively guide the rational design of novel pteridinone-based anti-cancer therapeutics.
Optimizing Grid Parameters and Field Combinations for Improved Q² and R² Values
Within the context of pteridinone derivatives research for PLK1 inhibition, establishing robust 3D-QSAR models is paramount for efficient drug design. The predictive accuracy and robustness of these models, typically represented by the cross-validated correlation coefficient (Q²) and the conventional correlation coefficient (R²), are highly sensitive to the computational parameters selected during their construction [9]. This protocol details a refined methodology for optimizing key parameters, specifically grid box dimensions and field combination choices in Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), to enhance the statistical reliability of models aimed at discovering novel anticancer agents.
Experimental Setup and Workflow
The following workflow outlines the comprehensive process for developing and validating 3D-QSAR models, from initial compound preparation to the final evaluation of model predictive power.
Research Reagent Solutions
Table 1: Essential software and computational tools for 3D-QSAR modeling.
| Tool/Software | Specific Function | Key Features/Parameters |
|---|---|---|
| SYBYL-X 2.1.1 [9] [20] | Integrated molecular modeling environment | Molecular sketching, energy minimization, database alignment, CoMFA/CoMSIA field calculation, and Partial Least Squares (PLS) analysis. |
| Tripos Force Field [9] [45] | Molecular mechanics energy minimization | Standardized convergence parameter (0.005 kcal/mol Å), Powell gradient algorithm, 1000 iterations for stable configurations. |
| Gasteiger-Hückel Charges [9] [32] | Assignment of atomic partial charges | Calculates electronegativity equalization for accurate electrostatic potential description. |
| AutoDock Vina [9] | Molecular docking | Predicting ligand-binding conformations and affinity scores within the protein's active site. |
| AMBER/GBSA [12] | Molecular Dynamics (MD) & Binding Free Energy | Assessing complex stability (50-100 ns simulations) and calculating binding free energies. |
Core Optimization Protocols
Molecular Alignment and Preparation
Proper alignment is the most critical step in 3D-QSAR and is a primary source of model variance [12].
- Template Selection: Identify the most active compound in your dataset (e.g., pteridinone derivative 3z with pIC50 = 7.0 was used in a breast cancer study) to serve as the template structure [12].
- Sketching and Minimization: Sketch all molecular structures or import them from available databases. Subject all compounds to energy minimization using the Tripos force field [9] [45].
- Apply Gasteiger-Hückel partial atomic charges.
- Use the Powell gradient algorithm with a convergence criterion of 0.005 kcal/mol Å and a maximum of 1000 iterations [9].
- Database Alignment: Perform a rigid body alignment using the
distillmethod in SYBYL-X. Align all molecules in the dataset to the common core structure of the pre-defined template [9] [12]. The quality of alignment directly influences the contour maps and, consequently, the interpretability of the model.
Grid Parameter Setup and Optimization
The grid defines the region in which field descriptors are calculated. Its parameters significantly impact the model's steric and electrostatic maps.
- Grid Construction: Create a 3D cubic lattice that encompasses all aligned molecules.
- Grid Spacing: Set the grid spacing to 1.0 Å or 2.0 Å. A finer grid (1.0 Å) provides higher resolution but increases computation time and the risk of overfitting. A 2.0 Å spacing is often sufficient for a robust model [9] [32].
- Grid Region: Extend the grid to 4.0 Å units beyond the van der Waals volumes of all molecules in all three Cartesian directions. This ensures complete coverage of the molecular space and captures relevant interaction fields [9].
- Probe Atom: Use an sp³ hybridized carbon atom with a +1.0 charge as the probe to calculate steric and electrostatic fields [9] [45].
- Energy Cutoff: Apply a default energy cutoff of 30 kcal/mol for both steric and electrostatic fields. Columns with energy variance below 2.0 kcal/mol should be filtered out to reduce noise and improve model performance [9].
Field Selection and Model Generation
Choosing the right combination of molecular fields in CoMSIA is crucial for building a model that accurately reflects the key interactions governing biological activity.
- CoMFA Fields: Utilize the standard steric (S) and electrostatic (E) fields.
- CoMSIA Fields: Select from five property fields: steric (S), electrostatic (E), hydrophobic (H), hydrogen-bond donor (D), and hydrogen-bond acceptor (A). The choice should be guided by the nature of the target and the ligand-protein interactions.
- Field Combination Strategy: Test different combinations of CoMSIA fields. For pteridinone derivatives as PLK1 inhibitors, the combination of S, E, and H (hydrophobic) fields has proven highly effective [9]. Other successful combinations include S, E, H, D (donor), and A (acceptor) for different targets [8] [45].
- PLS Analysis and Validation:
- Use the Leave-One-Out (LOO) cross-validation method to determine the optimal number of components (ONC) and the cross-validated correlation coefficient (Q²). A Q² > 0.5 is considered the threshold for a predictive model [9].
- Perform non-cross-validated analysis with the ONC to obtain the conventional correlation coefficient (R²), standard error of estimate (SEE), and F-value.
- External Validation: Predict the activity of an external test set (typically 20% of the dataset). A predictive correlation coefficient (R²pred) greater than 0.6 confirms the model's robustness and predictive power [9].
Performance Data and Analysis
The following table summarizes quantitative results from published studies that successfully applied the optimization strategies described in this protocol.
Table 2: Performance of optimized 3D-QSAR models in various drug discovery studies.
| Study Context (Derivative/Target) | Model Type | Optimal Field Combination | Grid Spacing | Statistical Results | Reference |
|---|---|---|---|---|---|
| Pteridinone/PLK1 (Prostate Cancer) | CoMFA | S, E | 1 Å | Q² = 0.67, R² = 0.992, R²pred = 0.683 | [9] |
| Pteridinone/PLK1 (Prostate Cancer) | CoMSIA | S, H, E | 1 Å | Q² = 0.69, R² = 0.974, R²pred = 0.758 | [9] |
| Pteridinone/PLK1 (Prostate Cancer) | CoMSIA | S, E, A, H | 1 Å | Q² = 0.66, R² = 0.975, R²pred = 0.767 | [9] |
| 2-Phenylindole/MCF-7 (Breast Cancer) | CoMSIA | S, E, H, D, A | 2 Å | Q² = 0.814, R² = 0.967, R²pred = 0.722 | [45] |
| Tetrahydrothienopyrimidine/MCF-7 | CoMFA | S, E | 2 Å | Q² = 0.62, R² = 0.90, R²ext = 0.90 | [12] |
| Tetrahydrothienopyrimidine/MCF-7 | CoMSIA | S, E, H, D, A | 2 Å | Q² = 0.71, R² = 0.88, R²ext = 0.91 | [12] |
| Ionone-chalcones/Androgen Receptor | CoMSIA | S, E, H, D, A | 2 Å | Q² = 0.550, R² = 0.671, R²pred = 0.563 | [32] |
Integrated Validation Techniques
Molecular Docking and Dynamics
To reinforce the insights from 3D-QSAR and validate the binding modes of predicted active compounds, integrate the following steps:
Molecular Docking:
- Prepare the protein structure (e.g., PLK1, PDB: 2RKU) by removing water molecules and adding polar hydrogens [9].
- Dock the most active compounds into the protein's active site. For PLK1, key residues include R136, R57, Y133, L69, and L82 [9].
- Analyze the binding poses to ensure they align with the favorable and unfavorable regions indicated by the 3D-QSAR contour maps.
Molecular Dynamics (MD) Simulations:
- Subject the best-docked complexes to MD simulations (50-100 ns) to assess the stability of the ligand-protein complex over time [9] [12].
- Analyze root-mean-square deviation (RMSD) and other trajectories to confirm that the inhibitors remain stable within the binding pocket, thereby reinforcing the docking and QSAR findings [9].
ADMET and Drug-Likeness Prediction
Before synthesizing proposed compounds, evaluate their pharmacokinetic and toxicity profiles in silico.
- Use tools like SwissADME or admetSAR to predict Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties [9] [8].
- Check for compliance with Lipinski's Rule of Five to prioritize compounds with a higher probability of oral bioavailability [8].
- For the pteridinone series, molecule 28 was identified as a promising candidate for prostate cancer therapy after such an analysis [9].
Contour map analysis in Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) represents a critical methodology for translating complex three-dimensional field data into actionable structural guidance for drug candidates. Within the context of pteridinone derivatives as PLK1 inhibitors, these maps provide visual representation of how steric, electrostatic, and hydrophobic fields influence inhibitory activity. The overexpression of polo-like kinase 1 (PLK1) has been established across multiple cancer types, including prostate cancer, positioning it as a promising broad-spectrum anti-cancer target [9] [10]. The essential role of PLK1 in cell proliferation underscores the therapeutic potential of effectively designed inhibitors, with contour maps serving as the translational bridge between computational models and practical medicinal chemistry.
The foundational study established three robust 3D-QSAR models—CoMFA (Q² = 0.67, R² = 0.992), CoMSIA/SHE (Q² = 0.69, R² = 0.974), and CoMSIA/SEAH (Q² = 0.66, R² = 0.975)—using a series of 28 novel pteridinone derivatives [9]. These models demonstrated significant predictive capability, with external validation yielding R²pred values of 0.683, 0.758, and 0.767 respectively. The contour charts generated from these models directly illuminate the structure-activity relationships critical for optimizing PLK1 inhibition, providing a visual guide for structural modification strategies.
Theoretical Framework and Computational Methodology
Molecular Alignment and Field Calculation Protocols
The generation of meaningful contour maps begins with proper molecular alignment, which represents one of the most sensitive parameters in 3D-QSAR model development. For the pteridinone derivative series, researchers implemented a rigorous protocol:
Molecular Alignment: All molecules were aligned using rigid body superposition based on a common scaffold template with SYBYL-X 2.1 software [9]. The database was energy-minimized using the Tripos force field with Gasteiger-Hückel atomic partial charges, employing the Powell gradient algorithm with a convergence criterion of 0.005 kcal/mol Å and 1000 iterations to achieve stable molecular configurations.
Field Calculation Parameters: CoMFA and CoMSIA descriptor fields were computed at lattice points with 1 Å grid spacing extending 4 Å in all three dimensions beyond the molecular dimensions [9]. Steric fields were calculated using Lennard-Jones potentials, while electrostatic fields employed Coulombic potentials, both with a distance-dependent dielectric constant. A sp³ hybridized carbon atom with +1 charge served as the probe atom, with field energy values truncated at 30 kcal/mol.
Statistical Validation: Partial Least Squares (PLS) regression correlated CoMFA and CoMSIA fields with biological activity (pIC50). Model robustness was evaluated through leave-one-out (LOO) cross-validation, and predictive ability was validated using an external test set comprising 20% of the compounds (6 derivatives) [9].
Contour Map Interpretation Fundamentals
Contour maps visualize regions where specific molecular properties favorably or unfavorably influence biological activity. The standard interpretation framework includes:
Steric Fields: Green contours indicate regions where bulky substituents enhance activity, while yellow contours designate areas where steric bulk decreases activity.
Electrostatic Fields: Blue contours represent regions where positive charge increases activity, and red contours indicate regions where negative charge enhances activity.
Hydrophobic Fields: Yellow contours designate areas where hydrophobic substituents improve activity, while white contours indicate regions where hydrophilic groups are favored.
The CoMSIA methodology extends this to hydrogen bonding fields, with magenta contours (hydrogen bond donors) and cyan contours (hydrogen bond acceptors) indicating favorable interactions, while corresponding orange and red contours represent unfavorable regions [9].
Figure 1: 3D-QSAR Contour Map Generation Workflow
Experimental Protocols for Contour Map Analysis
Molecular Database Preparation and Alignment Protocol
Objective: To create a properly aligned set of pteridinone derivatives for contour map generation.
Materials and Equipment:
- SYBYL-X 2.1.1 or higher molecular modeling software
- Tripos force field parameters
- Workstation with minimum 8GB RAM and dedicated graphics card
Procedure:
- Structure Preparation:
- Sketch all 28 pteridinone derivatives using the sketch molecule module
- Apply Gasteiger-Hückel charges to all structures
- Perform energy minimization using Tripos force field with Powell algorithm
- Set convergence criterion to 0.005 kcal/mol Å with 1000 maximum iterations
Template Selection:
- Select the most active compound (Number 28, IC50 = 7.18 nM) as alignment template
- Identify the common pteridinone core structure for atom-based fitting
Database Alignment:
- Use the rigid body superposition method
- Align all molecules to the template scaffold
- Verify alignment quality through RMSD values (<0.5 Å acceptable)
Training/Test Set Division:
- Randomly select 22 compounds (80%) for training set
- Reserve 6 compounds (20%) for external validation
- Ensure activity range representation in both sets
Quality Control: Check molecular overlap through visual inspection and statistical measures of alignment quality.
CoMFA and CoMSIA Field Calculation Protocol
Objective: To compute steric, electrostatic, and hydrophobic fields for 3D-QSAR analysis.
Parameters:
- Grid spacing: 1.0 Å in all dimensions
- Region definition: 4.0 Å extension beyond molecular dimensions
- Probe atom: sp³ carbon with +1 charge
- Energy truncation: 30 kcal/mol for both steric and electrostatic fields
- Attenuation factor: 0.3 for similarity indices
CoMFA Field Calculation:
- Steric Fields: Calculate using Lennard-Jones 6-12 potential
- Electrostatic Fields: Calculate using Coulombic potential with distance-dependent dielectric
- Column Filtering: Apply 2.0 kcal/mol filter to reduce noise
CoMSIA Field Calculation:
- Similarity Indices: Compute using Gaussian-type distance dependence
- Field Selection: Calculate five fields (steric, electrostatic, hydrophobic, hydrogen bond donor, hydrogen bond acceptor)
- Standardization: Use default attenuation factor of 0.3
Validation Steps:
- Perform leave-one-out cross-validation
- Calculate Q² values for model predictive ability
- Validate with external test set predictions
Contour Map Generation and Interpretation Protocol
Objective: To generate and interpret contour maps for structural guidance.
Map Generation:
- Contour Level Setting: Use default STDEV*COEFF field type
- Contour Threshold: Apply 80% level for contribution cutoff
- Color Scheme Application:
- Steric: Green (favorable), Yellow (unfavorable)
- Electrostatic: Blue (positive charge favorable), Red (negative charge favorable)
- Hydrophobic: Yellow (favorable), White (unfavorable)
Interpretation Methodology:
- Active Site Correlation: Superimpose contour maps with PLK1 binding pocket (PDB: 2RKU)
- Region Analysis: Identify key contour regions near R136, R57, Y133, L69, L82, Y139 residues
- Structural Modification Planning: Map contour information to specific substituent positions on pteridinone core
Documentation:
- Capture multiple viewing angles for comprehensive analysis
- Record contour locations relative to molecular scaffold
- Document suggested structural modifications with rationales
Research Reagent Solutions and Materials
Table 1: Essential Research Reagents and Computational Tools for CoMFA/CoMSIA Studies
| Reagent/Tool | Function/Purpose | Specifications/Notes |
|---|---|---|
| SYBYL-X 2.1.1 | Molecular modeling platform | Primary software for 3D-QSAR analysis and contour map generation [9] |
| Tripos Force Field | Molecular mechanics force field | Used for energy minimization and conformational analysis [9] |
| Gasteiger-Hückel Charges | Partial atomic charge calculation | Essential for electrostatic field calculations in CoMFA [9] |
| PLK1 Protein Structure (2RKU) | Molecular docking template | PDB code for PLK1 kinase domain used for binding site analysis [9] [10] |
| AutoDock Tools 1.5.6 | Molecular docking software | Used for validation of contour map interpretations [9] |
| Partial Least Squares (PLS) | Statistical analysis method | Correlates molecular fields with biological activity [9] |
Quantitative Data Analysis and Structural Correlation
Contour Map Data Interpretation Framework
The contour maps derived from the CoMFA and CoMSIA models provide quantitative guidance for structural optimization. Analysis of the pteridinone derivatives revealed several critical structure-activity relationships:
Steric Guidance: Significant green contours adjacent to the 7-position of the pteridinone core indicate favorable steric bulk in this region, correlating with enhanced PLK1 inhibition [9]. Yellow contours near the 5-position suggest steric restrictions in this area.
Electrostatic Requirements: Blue contours surrounding the 2-position amino group indicate the importance of positive charge potential in this region, while red contours near the 4-carbonyl group suggest negative charge favorability for receptor interaction.
Hydrophobic Interactions: Yellow hydrophobic contours adjacent to the 6-phenyl ring substituent demonstrate the importance of hydrophobic character in this region, with specific favorable regions mapped to ortho and meta positions.
Table 2: Quantitative Model Validation Parameters for 3D-QSAR Studies
| Model Type | Q² Value | R² Value | R²pred Value | Optimal Components | SEE |
|---|---|---|---|---|---|
| CoMFA | 0.67 | 0.992 | 0.683 | 6 | 0.058 |
| CoMSIA/SHE | 0.69 | 0.974 | 0.758 | 6 | 0.102 |
| CoMSIA/SEAH | 0.66 | 0.975 | 0.767 | 6 | 0.099 |
Structural Modification Strategies Based on Contour Analysis
The integration of contour map data with molecular docking results enables precise structural modification strategies:
Position 7 Optimization: The strong green steric contour indicates favorable bulky substituents. Compound 28 (most active) features a cyclopropylmethoxy group at this position, providing optimal steric bulk that aligns with the favorable green contour region.
Position 6 Aromatic Ring Modifications: The hydrophobic contour map guides substituent selection on the 6-phenyl ring. Meta-position hydrophobic groups align with favorable yellow contours, while ortho-position substitutions must balance steric and hydrophobic requirements.
Electrostatic Potential Tuning: The blue contour near the 2-position amino group suggests maintenance of hydrogen bond donor capability, while the red contour near the 4-carbonyl indicates the importance of preserving hydrogen bond acceptor functionality.
Figure 2: Contour Map Interpretation and Application Workflow
Integration with Molecular Docking and Dynamic Simulation
Validation Through Molecular Docking Studies
The contour map interpretations require validation through molecular docking to ensure biological relevance. Docking studies of pteridinone derivatives into the PLK1 active site (PDB: 2RKU) confirmed critical interactions:
Key Residue Interactions: The most active compounds formed hydrogen bonds with residues R136 and R57, consistent with the electrostatic contour maps that highlighted the importance of positive charge potential in specific regions [9].
Steric Complementarity: The docking poses confirmed that bulky substituents at position 7 occupy a hydrophobic pocket formed by L69, L82, and Y139, validating the green steric contours observed in CoMFA maps.
Hydrophobic Alignment: The 6-phenyl ring positioned within a hydrophobic subpocket lined with aliphatic and aromatic residues, consistent with the yellow hydrophobic contours in CoMSIA maps.
The integration of docking results with contour maps creates a feedback loop for model refinement and validation, ensuring that the field interpretations correspond to actual binding interactions.
Molecular Dynamics Corroboration
Molecular dynamics (MD) simulations provided further validation of contour map interpretations:
Binding Stability: Both high and low activity inhibitors remained stable in the PLK1 binding site over 50ns simulations, with RMSD values below 2.0 Å [9].
Interaction Persistence: Hydrogen bonds identified in docking studies persisted throughout MD trajectories, confirming the importance of electrostatic interactions highlighted in contour maps.
Side Chain Flexibility: MD simulations revealed conformational adaptations of binding site residues that accommodated optimal substituents identified through contour analysis.
The combination of 3D-QSAR contour maps, molecular docking, and MD simulations creates a comprehensive framework for rational drug design, with each method validating and informing the others.
Application to Pteridinone Derivative Optimization
Successful Design Strategies Guided by Contour Maps
The practical application of contour map interpretation led to specific structural modifications that enhanced PLK1 inhibitory activity:
Compound 28 Optimization: The most active compound in the series (IC50 = 7.18 nM) features a cyclopropylmethoxy group at position 7 that perfectly aligns with the green steric contour, providing optimal bulk without steric clash. The 6-(3-fluorophenyl) group aligns with favorable hydrophobic contours, while maintaining hydrogen bonding capacity through the 2-amino and 4-carbonyl groups [9].
Activity Trends: Compounds with substituents that violated contour map guidance showed reduced activity. For example, compounds with bulky groups in yellow contour regions exhibited 3-5 fold decreases in potency, while compounds that aligned with both steric and electrostatic contours showed enhanced activity.
ADMET Considerations: The contour maps also informed property optimization beyond pure potency. Balanced hydrophobic properties guided by CoMSIA contours contributed to favorable ADMET profiles, with compound 28 emerging as a promising candidate for prostate cancer therapy [9] [10].
Protocol for Future Derivative Design
Based on the successful application of contour maps to pteridinone optimization, a standardized protocol for future derivative design includes:
- Initial Screening: Synthesize or acquire compounds with substituents that probe key contour regions
- Activity Testing: Determine PLK1 inhibition IC50 values for QSAR model enhancement
- Model Refinement: Incorporate new data to refine contour maps and improve predictive capability
- Iterative Design: Cycle through design, synthesis, testing, and model refinement until optimal compounds identified
This protocol represents a robust framework for translating 3D field information into practical structural modifications that enhance drug candidate efficacy and properties.
Table 3: Structural Modification Guidelines Based on Contour Map Analysis
| Molecular Position | Contour Guidance | Recommended Modifications | Rationale |
|---|---|---|---|
| Position 7 | Strong green steric contour | Cyclopropylmethoxy, ethoxypropyl, benzyloxy | Optimal steric bulk for hydrophobic subpocket |
| Position 6 Phenyl Ring | Yellow hydrophobic contours at meta-position | 3-fluoro, 3-chloro, 3-methyl | Enhanced hydrophobic interactions without steric clash |
| Position 2 | Blue electrostatic contour | Amino, alkylamino, maintained | Hydrogen bond donation to backbone carbonyl |
| Position 4 | Red electrostatic contour | Carbonyl, maintained | Hydrogen bond acceptance from hinge region |
| Position 5 | Yellow steric contour | Hydrogen, small substituents | Avoid steric clash with gatekeeper residue |
Identifying and Managing Statistical Outliers in the Training and Test Sets
In the context of Computer-Aided Drug Design (CADD), robust Quantitative Structure-Activity Relationship (QSAR) models are foundational for predicting the biological activity of novel compounds. The reliability of these models, including Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), is highly dependent on the quality and integrity of the data used for their training and validation [46]. Statistical outliers—data points that deviate markedly from the rest of the dataset—pose a significant challenge, as they can skew statistical insights, reduce predictive power, and lead to misleading conclusions [47]. Within a thesis focused on developing a CoMFA/CoMSIA protocol for pteridinone derivatives as PLK1 inhibitors, the rigorous identification and management of outliers in the training and test sets is not merely a procedural step but a critical determinant of the project's success. This document provides detailed application notes and protocols for this essential process, framed within the specific context of anti-cancer drug discovery.
The Impact of Outliers in 3D-QSAR Modeling
Outliers in a QSAR dataset can arise from various sources, including errors in experimental biological activity measurement (pIC50), issues with molecular structure optimization, or misalignment during 3D model generation [47]. In CoMFA/CoMSIA studies, where models are built upon the principle that changes in molecular fields correlate with changes in biological activity, an outlier can disproportionately influence the model's parameters.
The presence of outliers can inflate standard errors and reduce the statistical power of the model, making it less sensitive to genuine structure-activity relationships [47]. More critically, outliers can distort regression coefficients, pulling the predicted activity relationship away from the true underlying trend exhibited by the majority of the data. This can result in a model that fits the training set well but performs poorly when predicting the activity of new, unseen compounds (the test set), ultimately compromising the model's utility in virtual screening and lead optimization [47]. Therefore, a systematic approach to outlier management is indispensable for developing a predictive and interpretative CoMFA/CoMSIA model.
Protocols for Outlier Identification
A multi-faceted approach is recommended to reliably identify potential outliers in both training and test sets. The following protocols should be employed.
Statistical Leverage and Influence Analysis
This method helps identify compounds that have an undue influence on the model's parameters.
- Experimental Protocol:
- After generating the initial PLS model for CoMFA or CoMSIA, calculate the leverage value (hᵢ) for each compound. The leverage is derived from the hat matrix (H) of the PLS regression, where H = X(XᵀX)⁻¹Xᵀ and X is the descriptor matrix.
- Plot the Williams plot, which displays the standardized residuals versus the leverage values for all compounds.
- Define a critical leverage value (h). A common threshold is h = 3p/n, where 'p' is the number of model components and 'n' is the number of training compounds.
- Identify compounds with a standardized residual greater than ±2.5 standard deviation units as activity outliers. These are compounds the model cannot predict accurately.
- Identify compounds with a leverage value greater than h* as structural outliers. These compounds lie far from the centroid of the chemical space defined by the training set and have high potential to influence the model, even if their activity is well-predicted.
Principal Component Analysis (PCA)
PCA is a dimensionality reduction technique that can visualize the overall distribution of the chemical dataset.
- Experimental Protocol:
- Subject the field descriptors (steric, electrostatic, etc.) for the entire dataset (training and test sets) to PCA.
- Generate a scores plot of the first two or three principal components (PC1 vs. PC2).
- Visually inspect the scores plot for compounds that are clearly separated from the main cluster of data points. These isolated compounds are potential structural outliers.
- This method is particularly useful for ensuring the test set compounds are within the structural space defined by the training set, a concept known as the Applicability Domain (AD) [48].
Residual Analysis
A simple yet effective method for identifying activity outliers.
- Experimental Protocol:
- For the training set, examine the difference between the experimental pIC50 and the model-predicted pIC50 (the residuals).
- Compounds with residuals exceeding ±1.0 log unit (or another pre-defined, scientifically justified threshold) should be flagged as potential activity outliers for further investigation [9] [12].
Table 1: Summary of Outlier Identification Methods
| Method | What it Identifies | Key Metric/Visualization | Interpretation Threshold |
|---|---|---|---|
| Leverage & Influence | Structurally influential and/or poorly predicted compounds | Williams Plot | Standardized Residual > ±2.5; Leverage > h* |
| Principal Component Analysis (PCA) | Structural outliers in chemical space | PCA Scores Plot | Visual separation from the main data cluster |
| Residual Analysis | Activity outliers (poorly predicted compounds) | Residual vs. Experimental pIC50 plot | Residual > ±1.0 log unit |
Figure 1: Workflow for the identification of statistical outliers in QSAR modeling, integrating multiple complementary analysis methods.
Protocols for Outlier Management
Once potential outliers are identified, a careful and systematic investigation is required to decide on the appropriate action.
Investigation and Rationalization
Do not automatically remove flagged compounds. First, investigate potential causes.
- Protocol:
- Verify Data Integrity: Re-check the experimental pIC50 value and the chemical structure of the compound for transcription or calculation errors. For pteridinone derivatives, ensure the molecular structure drawn in the modeling software (e.g., SYBYL) matches the synthesized compound [9].
- Examine Molecular Alignment: In 3D-QSAR, improper alignment is a common source of outliers. Inspect the aligned conformation of the potential outlier. Does it adopt a significantly different conformation or orientation in the active site compared to the rest of the set? Re-running the alignment with different parameters may be necessary [9] [12].
- Consider Scientific Rationale: The compound might be an outlier for a valid biological reason, such as a different binding mode to the PLK1 enzyme (PDB: 2RKU). Literature evidence or molecular docking results can support this. For instance, specific interactions with key residues like Lys82, Cys67, or Phe183 in PLK1 might explain an unexpectedly high activity [9] [10].
Strategic Actions
Based on the investigation, take one of the following actions:
- Correction: If a simple data error is found, correct it and re-run the model.
- Retention: If the outlier can be rationally explained and is not due to an error, it may be kept in the training set. Its presence could make the model more robust if it represents a real, albeit extreme, chemical phenomenon.
- Removal: If the outlier remains unexplained and is severely distorting the model (e.g., causing a dramatic drop in Q² or R²ᵩᵤᵢ ), it should be removed from the training set. The model must then be rebuilt without the outlier.
- Test Set Considerations: A test set compound identified as a strong structural outlier (far outside the model's Applicability Domain) should not be used to evaluate the model, as a prediction is not reliable. It should be replaced with a compound within the AD [48].
Table 2: Essential Research Reagent Solutions for CoMFA/CoMSIA Outlier Management
| Research Reagent / Tool | Function in Outlier Management |
|---|---|
| Molecular Modeling Software (e.g., SYBYL-X) | Provides the computational environment for molecular alignment, descriptor calculation (CoMFA/CoMSIA fields), and PLS regression analysis. Essential for generating model statistics and residuals. |
| Statistical Analysis Software (e.g., R, SAS) | Used for advanced statistical analyses, including precise calculation of leverage values, generation of diagnostic plots (Williams Plot), and performing Principal Component Analysis (PCA). |
| Visualization Tools (e.g., PyMOL, VMD) | Critical for visually inspecting the 3D alignment of potential outliers and investigating putative binding modes through molecular docking poses. |
| Curated Chemical Dataset | A high-quality dataset of compounds (e.g., pteridinone derivatives with reliable PLK1 pIC50 values) is the foundational reagent. Its integrity is paramount for all subsequent steps. |
Application to Pteridinone Derivatives PLK1 Inhibition Research
In the specific context of modeling pteridinone derivatives for PLK1 inhibition, the following considerations are paramount. The training and test sets, typically comprising 22 and 6 derivatives respectively, must be carefully curated [9]. The molecular alignment step, often using a rigid distill method in SYBYL with the most active compound as a template, is highly sensitive and a potential source of outliers if not performed consistently [9] [12]. When a compound is flagged as an outlier, molecular docking against the PLK1 protein (PDB: 2RKU) can be a valuable diagnostic tool. For example, investigating interactions with key active site residues like Arg136, Lys57, Tyr133, and Leu69 can reveal whether an outlier compound engages in unique, productive interactions that justify its anomalous activity [9] [10]. Finally, the stability of the ligand-protein complex for a suspected outlier can be further verified through Molecular Dynamics (MD) simulations (e.g., 50-100 ns), providing a dynamic perspective on its binding behavior [9] [12].
The meticulous process of identifying and managing statistical outliers is not an optional refinement but a core component of a rigorous CoMFA/CoMSIA protocol. By implementing the systematic protocols outlined herein—leveraging statistical leverage, PCA, and residual analysis for identification, followed by thorough investigation and strategic management—researchers can significantly enhance the predictivity, robustness, and interpretability of their QSAR models. For a thesis centered on pteridinone-based PLK1 inhibitors, this disciplined approach ensures that the final models truly reflect the underlying structure-activity relationships, thereby providing reliable and actionable insights for the design of next-generation anti-cancer drug candidates.
Integrating Computational Techniques for Robust Candidate Validation
In the realm of computer-aided drug design, particularly within Quantitative Structure-Activity Relationship (QSAR) modeling, the development of statistically robust and predictive models is paramount for successful drug discovery campaigns. Model validation serves as the critical gatekeeper, ensuring that computational predictions translate reliably to real-world biological activity. For researchers employing Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) protocols—especially in specialized applications like the development of pteridinone derivatives as PLK1 inhibitors—understanding and implementing comprehensive validation strategies is non-negotiable. These validation procedures are broadly categorized into internal validation, which assesses model stability using only training set data, and external validation, which evaluates true predictive power against completely independent test compounds. The most crucial validation metrics include the cross-validated correlation coefficient (Q²) for internal robustness and the predictive correlation coefficient (R²pred) for external predictability, alongside other supporting statistical parameters [9] [49] [50].
Theoretical Foundations of Key Validation Metrics
Internal Validation: The Q² Statistic
The leave-one-out cross-validated correlation coefficient (Q²) is a fundamental metric for assessing the internal robustness and stability of a 3D-QSAR model. Calculated during the model building phase, Q² represents the model's ability to predict data not used in its construction through an iterative process where each compound is systematically omitted from the training set and its activity is predicted by the model built with the remaining compounds [9] [49].
A common misconception in QSAR modeling is that a high Q² value (>0.5) automatically signifies a highly predictive model. However, research has demonstrated that while a low Q² indeed indicates poor model performance, the converse is not necessarily true. A high Q² is a necessary but not sufficient condition for establishing model predictivity, as it primarily reflects model stability with respect to small perturbations in the training set composition [49].
External Validation: The R²pred Statistic
External validation represents the most rigorous assessment of a model's true predictive capability by evaluating its performance on a completely independent set of compounds that were never used in model development. The predictive correlation coefficient (R²pred) is calculated by comparing the experimentally observed activities of the test set compounds with those predicted by the model [9] [50].
The scientific consensus firmly establishes that external validation is an "absolute requirement" for developing truly predictive QSAR models [49]. The test set should ideally include no less than five compounds whose activities and structures cover the range of activities and structures present in the training set to obtain reliable statistics [49]. Furthermore, the external validation set must be selected carefully to ensure it falls within the model's applicability domain—the chemical space defined by the training set compounds—as extrapolations beyond this domain yield unreliable predictions.
Complementary Validation Metrics
Beyond Q² and R²pred, several supplementary statistics provide a more comprehensive assessment of model quality:
- Conventional Coefficient (R²): The non-cross-validated correlation coefficient indicating how well the model fits the training set data.
- Standard Error of Estimation (SEE): Measures the accuracy of the model predictions.
- F-statistic Values: Assesses the overall statistical significance of the model.
- Optimal Number of Components (NOC): Determined through cross-validation to avoid model overfitting [9].
Table 1: Key Validation Metrics in 3D-QSAR Modeling
| Metric | Type | Interpretation | Threshold | Purpose |
|---|---|---|---|---|
| Q² | Internal | Model stability & robustness | >0.5 | Leave-one-out cross-validation |
| R²pred | External | True predictive ability | >0.6 | Prediction of test set compounds |
| R² | Goodness-of-fit | Training set model fit | >0.8 | Conventional correlation coefficient |
| SEE | Error estimate | Accuracy of predictions | Lower is better | Standard error of estimation |
| NOC | Model complexity | Optimal components | Avoid overfitting | Determined via cross-validation |
Detailed Validation Protocols for CoMFA/CoMSIA Studies
Data Preparation and Division Protocol
The foundation of any robust QSAR model begins with careful data preparation and appropriate division into training and test sets:
Activity Data Conversion: Convert experimental IC₅₀ values to pIC₅₀ using the transformation: pIC₅₀ = -log₁₀(IC₅₀) to establish a linear relationship with molecular structural parameters [9] [51].
Dataset Division: Divide the complete dataset using a statistically valid approach, typically employing an 80:20 ratio where 80% of compounds constitute the training set for model development and 20% form the test set for external validation [9]. For the pteridinone derivatives PLK1 inhibition study, this translated to 22 training compounds and 6 test compounds [9].
Representative Selection: Ensure test set compounds adequately represent the structural diversity and activity range of the entire dataset. Selection methods include random selection, activity-based sorting, or structurally representative clustering [49].
Molecular Alignment and Field Calculation
Molecular alignment represents one of the most critical steps in 3D-QSAR model development, directly impacting model quality and interpretation:
Rigid Body Alignment: Perform molecular alignment using the rigid body distill method in SYBYL-X software or equivalent open-source alternatives to superimpose molecules based on their common scaffold [9] [52].
Energy Minimization: Minimize all molecular structures using the Tripos force field with Gasteiger-Huckel atomic partial charges. Implement the Powell gradient algorithm with a convergence criterion of 0.005 kcal/mol Å and a maximum of 1000 iterations to achieve stable molecular configurations [9].
Grid Generation: Establish a 3D grid with spacing of 1-2 Å extending beyond the molecular dimensions by 4 Å in all directions to encompass all aligned molecules [9] [40].
Model Construction and Internal Validation
The model development phase systematically builds the relationship between molecular fields and biological activity:
Field Calculation: Calculate steric (Lennard-Jones potential) and electrostatic (Coulombic potential) fields for CoMFA. For CoMSIA, additional fields including hydrophobic, hydrogen bond donor, and hydrogen bond acceptor properties are computed using a Gaussian function to avoid singularities at atomic positions [9] [40].
Partial Least-Squares (PLS) Analysis: Implement PLS regression to correlate the CoMFA/CoMSIA fields with biological activity values. Determine the optimal number of components (NOC) that maximizes Q² while minimizing the risk of overfitting [9].
Internal Validation: Perform leave-one-out (LOO) cross-validation by systematically excluding each training set compound, rebuilding the model with the remaining compounds, predicting the omitted compound's activity, and calculating Q² [9] [49].
Model Robustness Check: Conduct y-randomization tests by repeatedly shuffling activity values and rebuilding models to confirm that observed statistics are not due to chance correlations [49].
External Validation and Predictive Assessment
External validation provides the ultimate test of model utility for prospective compound prediction:
Test Set Prediction: Apply the finalized model, developed using the entire training set, to predict activities of the external test set compounds that were excluded from model building [9].
R²pred Calculation: Compute R²pred using the formula focusing on the sum of squared differences between predicted and observed activities for the test set compounds [9] [50].
Predictive Residual Analysis: Examine the residuals (differences between predicted and observed activities) to identify any systematic errors or activity-dependent biases in predictions [40].
Applicability Domain Assessment: Verify that test set compounds fall within the structural and physicochemical space defined by the training set to ensure reliable predictions [49].
Diagram 1: Comprehensive QSAR Model Validation Workflow. This flowchart illustrates the sequential process for rigorous internal and external validation of 3D-QSAR models, highlighting the critical decision points based on Q² and R²pred thresholds.
Case Study: Pteridinone Derivatives as PLK1 Inhibitors
Application of Validation Protocols
In a specific study focusing on pteridinone derivatives as PLK1 inhibitors for prostate cancer treatment, researchers implemented the comprehensive validation protocol outlined above [9] [10]. The investigation developed three distinct 3D-QSAR models—CoMFA, CoMSIA/SHE (steric, hydrophobic, electrostatic), and CoMSIA/SEAH (steric, electrostatic, acceptor, hydrophobic)—each demonstrating robust statistical performance.
The internal validation results showed excellent stability with Q² values of 0.67, 0.69, and 0.66 for the CoMFA, CoMSIA/SHE, and CoMSIA/SEAH models respectively, all significantly exceeding the 0.5 threshold [9]. More importantly, external validation with the test set compounds yielded R²pred values of 0.683, 0.758, and 0.767 respectively, confirming strong predictive capability beyond the training set [9].
Table 2: Validation Metrics for Pteridinone Derivatives PLK1 Inhibition Models
| Model Type | Q² | R² | R²pred | NOC | Field Contributions |
|---|---|---|---|---|---|
| CoMFA | 0.67 | 0.992 | 0.683 | Not specified | Steric, Electrostatic |
| CoMSIA/SHE | 0.69 | 0.974 | 0.758 | Not specified | Steric, Hydrophobic, Electrostatic |
| CoMSIA/SEAH | 0.66 | 0.975 | 0.767 | Not specified | Steric, Electrostatic, Acceptor, Hydrophobic |
Contour Map Analysis and Interpretation
The contour maps generated from the validated models provided crucial structural insights for optimizing PLK1 inhibition:
- Steric Fields: Identified regions where bulky substituents either enhanced or diminished activity, guiding rational structural modifications.
- Electrostatic Fields: Revealed areas where positively or negatively charged groups influenced ligand-receptor interactions.
- Hydrophobic Fields: Highlighted regions where lipophilic substituents improved binding affinity.
- Hydrogen Bond Fields: Pinpointed optimal positions for hydrogen bond donors and acceptors to strengthen target engagement [9].
These contour maps, validated through the robust Q²/R²pred framework, directly informed the molecular design strategy, leading to the identification of compound 28 as a promising anti-prostate cancer candidate with optimized PLK1 inhibitory potency and favorable ADMET properties [9] [10].
Table 3: Essential Research Resources for CoMFA/CoMSIA Validation Studies
| Resource Category | Specific Tools/Software | Application in Validation Protocol |
|---|---|---|
| Molecular Modeling | SYBYL-X 2.1.1, Py-CoMSIA [40], Gaussian 09 | Molecular alignment, field calculation, energy minimization |
| Docking & Dynamics | AutoDock Tools 1.5.6, Vina [9], GOLD | Binding mode analysis, conformation sampling |
| Statistical Analysis | Partial Least Squares (PLS), Multiple Linear Regression (MLR) [51] | Model development, correlation analysis |
| Validation Suites | Leave-One-Out Cross-Validation, y-Randomization [49] | Internal validation (Q²), chance correlation assessment |
| Descriptor Calculation | Dragon, Molecular Operating Environment (MOE) [51] | Quantum-chemical descriptor computation |
| Data Visualization | PyVista [40], Contour Maps | Interpretation of steric/electrostatic fields |
The rigorous application of both internal and external validation protocols represents an indispensable component of robust 3D-QSAR model development, particularly in specialized applications such as designing pteridinone derivatives as PLK1 inhibitors. The case study demonstrates that while internal validation metrics like Q² provide essential insights into model stability, they alone cannot guarantee predictive power. The external validation statistic R²pred, derived from truly independent test sets, remains the gold standard for establishing model utility in prospective drug design. By implementing the comprehensive validation workflow outlined in this application note—encompassing appropriate data division, molecular alignment, statistical modeling, and multi-faceted validation—computational chemists can develop reliably predictive models that significantly accelerate the discovery of novel therapeutic agents.
Within the broader context of developing pteridinone derivatives as potent PLK1 inhibitors, molecular docking serves as a critical computational methodology for predicting and analyzing the binding modes of these compounds within the enzyme's active site [9] [7]. Polo-like kinase 1 (PLK1) is a serine/threonine kinase frequently overexpressed in various human cancers, making it a prominent anti-cancer drug target [9] [1]. Its structure comprises an N-terminal catalytic kinase domain, which houses the ATP-binding site, and a C-terminal polo-box domain (PBD) responsible for substrate recognition and subcellular localization [1]. The research protocol detailed herein was established to identify key residues within the PLK1 active site that interact with pteridinone-based inhibitors, thereby providing essential structural insights to guide the rational design of more potent and selective anti-cancer compounds [9] [53].
Table 1: Key Characteristics of the PLK1 Protein Target
| Parameter | Description |
|---|---|
| Protein Name | Polo-like kinase 1 (PLK1) [9] |
| PDB ID | 2RKU [9] [7] |
| Protein Resolution | 1.95 Å [7] |
| Biological Function | Serine/threonine kinase regulating mitosis [9] [1] |
| Relevance to Disease | Overexpressed in cancers; broad-spectrum anti-cancer target [9] [7] |
| Primary Target Site | ATP-binding site in the kinase domain [9] |
Experimental Protocol
Preparation of Protein Structure
The three-dimensional crystal structure of the PLK1 kinase domain in complex with the inhibitor BI-2536 was retrieved from the Protein Data Bank (PDB ID: 2RKU) [9] [7]. The following steps were performed using molecular visualization and modeling software such as SYBYL-X [9]:
- Remove all water molecules and any non-essential ions or co-crystallized ligands not part of the active site [7].
- Add hydrogen atoms and assign appropriate protonation states to amino acid residues, particularly those in the active site (e.g., histidine residues) [9].
- Assign Gasteiger-Hückel atomic partial charges to prepare the protein for energy calculations [9] [7].
- Apply a standard Tripos force field for energy minimization, using a convergence criterion of 0.005 kcal/mol Å and the Powell gradient algorithm to achieve a stable protein configuration [9].
Preparation of Ligand Structures
A series of pteridinone derivatives were used as ligand molecules [9] [53]. The protocol for ligand preparation includes:
- Sketch or import the 2D structure of the pteridinone derivative.
- Convert the 2D structure into a 3D model.
- Perform energy minimization using the Tripos force field and the Powell method, with Gasteiger-Hückel charges and a convergence criterion of 0.005 kcal/mol Å [9].
- Save the optimized 3D structure in a suitable file format (e.g., MOL2) for docking.
Molecular Docking Execution
Molecular docking was performed to predict the binding orientation and affinity of the pteridinone derivatives within the PLK1 active site [9] [7].
- Software: AutoDock Vina is recommended for executing the docking simulations [7].
- Grid Box Definition: Define a grid box that encompasses the entire ATP-binding site. The coordinates and dimensions should be set to ensure all key residues are included. A grid spacing of 1 Å is typically used [9].
- Docking Parameters: The number of binding modes (poses) to generate for each ligand should be specified. The exhaustiveness of the search should be set to a sufficiently high value (e.g., 8, 16, 24) to ensure comprehensive sampling [7].
- Pose Analysis: After docking, analyze the top-ranking poses based on the binding affinity scores (expressed in kcal/mol). Visually inspect the predicted binding modes to identify key interactions.
Diagram 1: Molecular Docking Workflow for PLK1-Ligand Complex Analysis.
Key Findings and Data Analysis
Molecular docking studies of active pteridinone derivatives (e.g., compounds 17 and 28 from the series) within the PLK1 active site (2RKU) consistently identified a set of critical residues that form the binding pocket [9] [7]. These residues are involved in diverse interactions that stabilize the inhibitor-enzyme complex.
Table 2: Key PLK1 Active Site Residues and Their Interactions with Pteridinone Inhibitors
| Residue | Role in Catalysis/Binding | Type of Interaction with Inhibitor |
|---|---|---|
| Cys133 | Hinge region residue; critical for ATP binding [15] [1] | Forms conserved hydrogen bonds with the inhibitor's core scaffold [9] [15] |
| Lys82 | Involved in ATP anchoring and orientation [1] | Electrostatic and/or hydrophobic interactions [9] |
| Phe183 | Stabilizes the active site conformation [15] | Aromatic ring stacking (π-π) with the ligand [15] |
| Arg136 | Part of the catalytic loop [7] | Forms electrostatic interactions [9] |
| Arg57 | Located near the ATP-binding pocket [7] | Potential for ionic or hydrogen bonding [9] |
| Leu59 | Creates hydrophobic environment [15] | Van der Waals contacts [9] |
| Tyr133 | - | Hydrophobic/van der Waals interactions [9] |
| Leu69 | - | Hydrophobic/van der Waals interactions [9] |
| Leu82 | - | Hydrophobic/van der Waals interactions [9] |
| Tyr139 | - | Hydrophobic/van der Waals interactions [9] |
The stability and biological activity of the pteridinone derivatives are directly influenced by their ability to form these multiple favorable interactions within the PLK1 binding pocket [9]. The more active compounds, such as molecule 28, effectively occupy the active site and form a network of hydrogen bonds and hydrophobic contacts, thereby competing with ATP and inhibiting PLK1 kinase activity [9] [7].
Research Reagent Solutions
Table 3: Essential Reagents and Computational Tools for PLK1 Docking Studies
| Reagent/Software | Function/Description | Example/Provider |
|---|---|---|
| PLK1 Crystal Structure | Provides 3D atomic coordinates of the target protein for docking. | PDB ID: 2RKU [9] [7] |
| Molecular Modeling Suite | Software for protein and ligand preparation, visualization, and analysis. | SYBYL-X 2.1 [9] |
| Molecular Docking Software | Program that predicts the binding pose and affinity of a ligand to a protein. | AutoDock Vina [7] |
| Force Field | A set of parameters for calculating the potential energy of a molecular system. | Tripos Standard Force Field [9] [7] |
| Structure Visualization Tool | Allows for interactive 3D visualization and analysis of molecular structures and docking poses. | Built-in viewers in SYBYL, PyMOL, or UCSF Chimera |
Troubleshooting and Technical Notes
- Inaccurate Poses: If the docked ligand poses do not align with known SAR data, verify the protonation states of key residues like His and Asp/Glu. Consider using a more exhaustive docking search parameter [7].
- Poor Binding Affinity Correlation: Ensure that the ligand structures have been properly minimized and that appropriate partial charges are assigned. Cross-validate docking results with a known co-crystallized ligand (e.g., BI-2536 from 2RKU) by re-docking it to establish a baseline RMSD value [54].
- Protein Flexibility: The standard docking protocol treats the protein as rigid. For more accurate results, consider advanced techniques like molecular dynamics (MD) simulations to account for protein flexibility and to validate the stability of the docked complexes over time [9] [53].
Within the integrated protocol of Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), molecular dynamics (MD) simulations serve as a critical step for validating the stability and interaction mechanisms of predicted ligand-receptor complexes. The establishment of 3D-QSAR models, such as the CoMFA (Q² = 0.67, R² = 0.992) and CoMSIA (Q² = 0.69, R² = 0.974) models for pteridinone derivatives, provides a static understanding of structure-activity relationships [9] [20]. However, transferring these insights to the dynamic physiological environment is essential for confirming the viability of drug candidates like PLK1 inhibitors. MD simulations bridge this gap by evaluating the stability of the protein-ligand complex under near-physiological conditions over time, typically for 50-100 nanoseconds (ns), thereby reinforcing the molecular docking results and providing a more robust validation of the proposed binding modes [9] [10]. This document details the application of MD simulations to evaluate the stability of pteridinone-PLK1 complexes, a crucial step following 3D-QSAR model generation and molecular docking in the rational design of anticancer therapeutics.
Application Notes: The Role of MD in the CoMFA/CoMSIA Workflow
In the context of pteridinone derivatives as PLK1 inhibitors, MD simulations provide dynamic validation for the static models generated by CoMFA and CoMSIA. The contour maps from 3D-QSAR highlight regions where steric bulk, electrostatic potential, or hydrogen-bonding features are favorable or unfavorable for biological activity [9] [20]. Molecular docking then proposes a specific binding pose that explains these relationships, identifying key active site residues like R136, R57, Y133, L69, L82, and Y139 in PLK1 (PDB: 2RKU) [9].
MD simulations test the persistence of this docked pose over time. A stable complex, where the ligand remains within the binding pocket and maintains critical interactions throughout the simulation, strongly corroborates the predictions of both the docking and the 3D-QSAR models. For instance, in the study of novel pteridinone derivatives, MD simulations confirmed that the most active inhibitors remained stable in the active site of PLK1 for the entire 50 ns simulation [9] [10]. Conversely, instability in the MD trajectory would suggest that the docked pose, while energetically favorable in a static system, may not be physiologically relevant, indicating a need to refine the computational models.
Furthermore, MD simulations provide atomic-level insights into the conformational flexibility of both the ligand and the protein, capturing transient interactions and loop motions that are absent in crystal structures but critical for function [55] [56]. This dynamic profile is invaluable for interpreting the CoMSIA fields, particularly those related to hydrogen bonding and hydrophobicity, and for guiding the optimization of next-generation inhibitors with improved binding kinetics and selectivity.
Experimental Protocol for 50-100 ns MD Simulation
The following protocol is adapted from studies on PLK1-inhibitor complexes and other protein-ligand systems, providing a generalized workflow for evaluating complex stability [9] [55] [56].
System Preparation
- Initial Coordinate File: Use the coordinates of the protein-ligand complex from molecular docking. For pteridinone/PLK1 studies, the crystal structure PDB ID 2RKU was used as the starting point [9].
- Protein and Ligand Parameterization:
- Software: tLEaP module of AMBERTOOLS or CHARMM-GUI for NAMD.
- Protein Force Field: AMBER ff99SB or CHARMM36.
- Ligand Force Field: General AMBER Force Field (GAFF). Generate ligand parameters using the
ANTECHAMBERmodule to assign Gasteiger atomic partial charges and GAFF parameters [55].
- Solvation and Neutralization:
- Solvent Model: Explicit TIP3P water model.
- Solvent Box: Place the complex in a rectangular or cubic box, ensuring a minimum distance of 10-15 Å between the protein and the box edge.
- Neutralization: Add counterions (e.g., Na⁺ or Cl⁻) to neutralize the system's net charge. For physiological conditions, add further ions to achieve a concentration of 0.15 M KCl [55].
Simulation Parameters and Energy Minimization
Table 1: Summary of Key MD Simulation Parameters
| Parameter | Specification | Software Example |
|---|---|---|
| Force Field | AMBER ff99SB, CHARMM36, OPLS2005 | AMBER, NAMD, GROMACS |
| Water Model | TIP3P | AMBER, NAMD |
| Temperature | 300 K | All |
| Pressure | 1 atm | All |
| Time Step | 2 fs | All |
| Bond Constraints | SHAKE algorithm (hydrogen atoms) | AMBER, NAMD |
| Non-bonded Cutoff | 12 Å | All |
| Long-Range Electrostatics | Particle Mesh Ewald (PME) | All |
| Simulation Length | 50-100 ns (Production Run) | All |
- Energy Minimization: Minimize the system to remove bad contacts and steric clashes. A typical two-stage process involves:
- Stage 1: Minimize the positions of solvent molecules and ions while restraining the protein-ligand complex (e.g., 1000-5000 steps).
- Stage 2: Minimize the entire system without restraints (e.g., 1000-5000 steps).
System Equilibration
Equilibrate the system in two phases to gradually release restraints and achieve stable temperature and pressure.
- NVT Ensemble (Constant Number, Volume, Temperature):
- Heat the system from 0 K to 300 K over 100 ps, with restraints on the protein-ligand atoms.
- Maintain at 300 K for a further 100-200 ps.
- NPT Ensemble (Constant Number, Pressure, Temperature):
- Equilibrate the system for 1-2 ns at 300 K and 1 atm pressure with minimal or no restraints, allowing the solvent density to equilibrate.
Production MD Run
- Execution: Run an unrestrained MD simulation for 50-100 ns under the same NPT conditions as the final equilibration stage.
- Trajectory Saving: Save atomic coordinates (trajectory frames) at regular intervals, typically every 10-100 ps. For a 50 ns simulation saving every 10 ps, this yields 5000 frames for analysis [9] [55].
Trajectory Analysis
Key analyses to be performed on the production trajectory:
- Root Mean Square Deviation (RMSD): Calculate for the protein backbone and the ligand heavy atoms to assess the overall stability of the complex and the conformational drift of the ligand.
- Root Mean Square Fluctuation (RMSF): Determine per-residue fluctuations to identify flexible regions of the protein.
- Ligand-Protein Interactions: Analyze hydrogen bonds, hydrophobic contacts, and salt bridges over time to identify key stabilizing interactions.
- Binding Free Energy Calculations (Optional): Use methods like MM-PBSA/GBSA to estimate the binding free energy from the simulation trajectory [55].
Data Presentation and Analysis
Table 2: Key Analyses for Evaluating Complex Stability from MD Trajectories
| Analysis Metric | Description and Calculation | Interpretation of Stability |
|---|---|---|
| Backbone RMSD | Measures the average change in position of protein backbone atoms (Cα, C, N) relative to the initial frame. | A low, stable RMSD plateau (e.g., < 2-3 Å) indicates the protein structure has equilibrated and is stable. |
| Ligand RMSD | Measures the average change in position of ligand heavy atoms relative to its initial binding pose. | A low, stable RMSD suggests the ligand remains bound in a consistent pose. Large fluctuations may indicate dissociation or pose switching. |
| Protein-Ligand H-Bonds | The number of hydrogen bonds between the protein and ligand, calculated over time. | Consistent presence of key hydrogen bonds (e.g., with residues R136, Y133 in PLK1) confirms the stability of the predicted binding mode [9]. |
| Radius of Gyration (Rg) | Measures the compactness of the protein structure. | A stable Rg value suggests the protein does not undergo large-scale unfolding or compaction during the simulation. |
| MM-PBSA/GBSA (Molecular Mechanics-Poisson Boltzmann/Generalized Born Surface Area) | An endpoint method to estimate binding free energy (ΔGbind) from an ensemble of snapshots [55]. | A more negative ΔGbind indicates stronger binding affinity. Useful for ranking inhibitors compared to their experimental pIC50 values. |
The following diagram illustrates the logical workflow integrating MD simulations with the broader 3D-QSAR and docking protocol for PLK1 inhibitor development.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for MD Simulations in Drug Discovery
| Tool / Resource | Function/Brief Explanation | Relevance to PLK1 Inhibitor Study |
|---|---|---|
| Molecular Dynamics Software | ||
| AMBER | A suite of programs for biomolecular simulation; includes PMEMD for accelerated MD on GPUs. | Used for 50 ns simulations of PLK1-pteridinone complexes and MM-PBSA/GBSA calculations [55] [10]. |
| NAMD | A parallel MD code designed for high-performance simulation of large biomolecular systems. | Used for 70 ns simulations of PDEδ complexes with OPLS2005 force field [55]. |
| GROMACS | A high-performance MD software package for simulations of proteins, lipids, and nucleic acids. | Widely used in academia for its speed and efficiency. |
| Force Fields | ||
| AMBER ff99SB | A high-quality force field for proteins and nucleic acids. | Used for MD simulations of protein-ligand complexes in AMBER [55]. |
| CHARMM36 | An all-atom force field for proteins, lipids, carbohydrates, and nucleic acids. | Used for MD simulations of protein-ligand complexes in NAMD [55]. |
| GAFF (General AMBER Force Field) | A force field for small organic molecules compatible with AMBER. | Used to parameterize pteridinone and other small molecule inhibitors [55]. |
| Analysis & Visualization | ||
| VMD (Visual Molecular Dynamics) | A program for displaying, animating, and analyzing large biomolecular systems. | Used for trajectory analysis (e.g., RMSD, H-bonds, interaction energies) and rendering publication-quality images [55]. |
| CPPTRAJ / PTRAJ | The main trajectory analysis programs in the AMBER package. | Used for processing MD trajectories to compute RMSD, RMSF, and other metrics. |
| Free Energy Calculations | ||
| MMPBSA.py | A Python script in AMBER for performing MM-PBSA and MM-GBSA calculations. | Used to predict binding free energies from MD trajectories for ranking inhibitors [55]. |
Within the context of a broader thesis on the application of Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) protocols for pteridinone derivatives as Polo-like kinase 1 (PLK1) inhibitors, the in-silico assessment of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) is a critical gatekeeper. ADMET profiling determines whether a potent, selectively designed inhibitor possesses the necessary pharmacokinetic and safety profile to become a viable drug candidate [57]. The high cost and time associated with experimental ADMET testing make computational predictions indispensable for triaging compounds, prioritizing the most promising leads for synthesis and biological evaluation [9] [58]. For novel pteridinone derivatives identified via 3D-QSAR models as having high predicted activity against PLK1—a broad-spectrum anti-cancer target—integrating ADMET profiling early in the design cycle is essential for developing successful prostate cancer therapies [9] [10].
Foundational Concepts in ADMET Profiling
A foundational understanding of ADMET properties ensures that the interpretation of predictive data is biologically and clinically relevant.
- Absorption: This refers to a compound's ability to enter the bloodstream, often predicted via models of human intestinal absorption (HIA) and permeability assays like Caco-2 and PAMPA [59] [58].
- Distribution: This describes how a compound is transported throughout the body and to its site of action. Key parameters include volume of distribution (VDss), plasma protein binding (PPB), and the ability to cross physiological barriers like the blood-brain barrier (BBB) [59] [58].
- Metabolism: This encompasses the biochemical modification of a drug, primarily by enzymes such as the Cytochrome P450 (CYP) family. Predicting inhibition or substrate activity for these enzymes is vital for anticipating drug-drug interactions [59].
- Excretion: This is the process by which the drug and its metabolites are eliminated from the body, often predicted via measures of clearance and half-life [59].
- Toxicity: This includes the potential for a compound to cause harmful effects, such as organ-specific toxicity (e.g., hepatotoxicity, cardiotoxicity via hERG channel inhibition), genotoxicity, or overall carcinogenicity [60] [57].
Modern approaches, such as the ADME-DL pipeline, move beyond simple structural descriptors. They leverage molecular foundation models (MFMs) enriched with ADME data, enforcing a sequential (A→D→M→E) multi-task learning flow that mirrors the compound's pharmacokinetic journey in the body, leading to more accurate drug-likeness classifications [59].
Workflow for ADMET Integration in a PLK1 Inhibitor Project
The following workflow integrates ADMET profiling into the computational drug discovery pipeline for pteridinone-based PLK1 inhibitors. It ensures that only leads with favorable predicted activity and pharmacokinetics are advanced.
Key ADMET Properties and Decision Criteria for Lead Selection
The table below summarizes key ADMET parameters and decision criteria used to filter and prioritize pteridinone derivatives. This framework is built upon analyses from successful studies on PLK1 and other kinase inhibitors [9] [61] [58].
Table 1: Key ADMET Properties for Prioritizing Pteridinone-Based PLK1 Inhibitors
| ADMET Category | Specific Property | Favorable Range/Value | Rationale & Clinical Implication |
|---|---|---|---|
| Absorption | Human Intestinal Absorption (HIA) | High (>90% absorbed) [61] | Ensures adequate oral bioavailability. |
| Caco-2 Permeability (Papp in 10⁻⁶ cm/s) | > 4 (High) [58] | Indicator of good intestinal permeability. | |
| P-glycoprotein (Pgp) Substrate | No (Low probability) [58] | Avoids active efflux, which can reduce intracellular concentration. | |
| Distribution | Plasma Protein Binding (PPB) | Moderate to High (e.g., >95%) [58] | High PPB can reduce free drug concentration and efficacy, but is common. |
| Blood-Brain Barrier (BBB) Penetration | Low or None [58] | For non-CNS targets like PLK1 in prostate cancer, minimizes risk of neurotoxicity. | |
| Volume of Distribution (VDss) | > 0.15 L/kg (Adequate) | Suggests sufficient distribution into tissues beyond the plasma. | |
| Metabolism | CYP450 Inhibition (e.g., 2C9, 2D6, 3A4) | Non-inhibitor | Reduces risk of clinically significant drug-drug interactions. |
| Toxicity | hERG Inhibition | No (Low probability) | Critical for avoiding cardiotoxicity and fatal arrhythmias. |
| Hepatotoxicity | No (Low probability) | Avoids drug-induced liver injury (DILI). | |
| AMES Mutagenicity | Non-mutagen | Indicates low genotoxic potential. | |
| Drug-likeness | Lipinski's Rule of Five | ≤ 1 violation [58] | Flags compounds with potential poor absorption or permeability. |
Case Study Application: Pteridinone Derivatives as PLK1 Inhibitors
A recent study on novel pteridinone derivatives as PLK1 inhibitors provides a clear template for applying this protocol. The research established robust 3D-QSAR models (CoMFA Q² = 0.67, R² = 0.992; CoMSIA Q² = 0.69, R² = 0.974) to predict inhibitory activity [9] [10]. Following activity prediction, the two most active molecules (compounds 28 and 27) were subjected to a comprehensive ADMET analysis. The results demonstrated that compound 28, in particular, exhibited favorable drug-likeness and pharmacokinetic properties, marking it as a superior candidate for further development as a prostate cancer therapeutic [9]. This workflow underscores the power of coupling 3D-QSAR with ADMET profiling to make informed decisions before costly synthesis and experimental testing.
The Scientist's Toolkit: Essential Research Reagents & Computational Solutions
Table 2: Essential Computational Tools and Resources for ADMET Profiling
| Tool/Resource Name | Type/Format | Primary Function in ADMET Profiling |
|---|---|---|
| ADMETlab 3.0 [60] | Online Platform / Standalone | Comprehensive prediction of a wide range of physicochemical and ADMET endpoints with decision support. |
| ProTox 3.0 [60] | Online Platform | Prediction of chemical toxicity endpoints, including organ toxicity and toxicological pathways. |
| Chemprop [60] | Standalone Package | A versatile machine learning package for training and predicting chemical properties, including ADMET. |
| ADMET-PrInt [60] | Online Platform | Predicts key properties like cardiotoxicity and solubility, with model interpretability features (LIME, counterfactuals). |
| AutoDock Tools/Vina [9] | Standalone Suite | Molecular docking to elucidate ligand-binding mode and interactions with the target protein (e.g., PLK1, PDB: 2RKU). |
| SYBYL-X [9] | Commercial Software Suite | An integrated environment for performing 3D-QSAR (CoMFA, CoMSIA), molecular alignment, and minimization. |
| GROMACS/AMBER | Standalone Suite | Molecular dynamics simulations to assess the stability of protein-ligand complexes over time (e.g., 50-100 ns simulations). |
| Therapeutic Data Commons (TDC) [59] | Curated Dataset | Provides curated, standardized datasets for 21 ADME endpoints to train and validate predictive models. |
Advanced Protocol: Implementing a Sequential ADMET Multi-Task Learning Pipeline
For research groups aiming to implement state-of-the-art ADMET prediction, the ADME-DL protocol offers a significant advancement over traditional methods. This protocol enforces a pharmacokinetic-guided flow (A→D→M→E) during model training, enhancing predictive accuracy and biological relevance [59].
Protocol Steps:
- Data Curation and Aggregation: Assemble datasets for the 21 core ADME endpoints from the Therapeutic Data Commons (TDC), categorized into Absorption (A), Distribution (D), Metabolism (M), and Excretion (E) tasks [59].
- Model Selection and Pretraining: Select a pretrained Molecular Foundation Model (MFM), such as a Graph Neural Network (GNN) or Transformer, as the base encoder.
- Sequential Multi-Task Learning (MTL): Fine-tune the MFM on the ADME tasks in a sequential order: A → D → M → E.
- Cycle 1: Train the model exclusively on all Absorption tasks (e.g., Caco-2, HIA, Pgp substrate).
- Cycle 2: Freeze the learned absorption features, then train on Distribution tasks (e.g., BBB, PPB, VDss).
- Cycle 3: Freeze the distribution features, then train on Metabolism tasks (e.g., CYP450 inhibition/substrate).
- Cycle 4: Freeze the metabolism features, then train on Excretion tasks (e.g., half-life, clearance).
- Drug-likeness Classifier Training: Use the resulting ADME-informed molecular embeddings (
z) to train a separate multi-layer perceptron (MLP) classifier to distinguish approved drugs from non-drugs in large chemical libraries [59]. - Validation and Deployment: Validate the pipeline on internal and external test sets. The model can then be deployed to score new virtual pteridinone derivatives, providing a holistic score that integrates predicted activity (from 3D-QSAR) with optimized ADMET properties.
The integration of sophisticated ADMET profiling into the CoMFA/CoMSIA workflow for PLK1 inhibitor discovery is non-negotiable for modern, efficient drug development. By employing the protocols, decision criteria, and tools outlined in this document, researchers can systematically prioritize pteridinone derivatives that are not only potent but also possess a high probability of clinical success based on their drug-likeness and pharmacokinetic profile. This integrated computational strategy dramatically de-risks the early stages of drug discovery, paving a faster and more cost-effective path toward viable cancer therapeutics.
Within the context of our broader research on CoMFA and CoMSIA protocols for pteridinone derivatives as PLK1 inhibitors, this application note provides a critical comparative analysis of model performance against other QSAR methodologies. The overexpression of polo-like kinase 1 (PLK1) in numerous cancers establishes it as a valuable anti-cancer target, necessitating robust computational approaches for inhibitor development [9] [10]. Three-dimensional quantitative structure-activity relationship (3D-QSAR) techniques, particularly Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), have emerged as powerful tools in this endeavor, enabling the correlation of molecular structural properties with biological activity to guide rational drug design [9] [62]. This analysis evaluates the predictive performance of these models against other established computational approaches, providing researchers with a framework for methodological selection in kinase inhibitor development.
Performance Comparison of QSAR Methodologies
Quantitative Performance Metrics Across Studies
Table 1: Statistical Performance Metrics of Various 3D-QSAR Models
| Study Context | Methodology | Q² | R² | R²pred | Components | Reference |
|---|---|---|---|---|---|---|
| Pteridinone/PLK1 Inhibitors | CoMFA | 0.67 | 0.992 | 0.683 | Not Specified | [9] |
| Pteridinone/PLK1 Inhibitors | CoMSIA/SHE | 0.69 | 0.974 | 0.758 | Not Specified | [9] |
| Pteridinone/PLK1 Inhibitors | CoMSIA/SEAH | 0.66 | 0.975 | 0.767 | Not Specified | [9] |
| Pyridin-2-one/mIDH1 Inhibitors | CoMFA | 0.765 | 0.980 | Not Specified | Not Specified | [63] |
| Pyridin-2-one/mIDH1 Inhibitors | CoMSIA | 0.770 | 0.997 | Not Specified | Not Specified | [63] |
| Thienopyrimidine/MCF-7 Inhibitors | CoMFA | 0.62 | 0.90 | 0.90 | Not Specified | [64] |
| Thienopyrimidine/MCF-7 Inhibitors | CoMSIA | 0.71 | 0.88 | 0.91 | Not Specified | [64] |
| Thienopyrimidine/Aurora-B Kinase | CoMFA | 0.51 | Not Specified | Not Specified | Not Specified | [65] |
| Thienopyrimidine/Aurora-B Kinase | CoMSIA | 0.72 | Not Specified | Not Specified | Not Specified | [65] |
| α1A-Adrenergic Receptor Antagonists | CoMFA | 0.840 | Not Specified | 0.694 | Not Specified | [29] |
| α1A-Adrenergic Receptor Antagonists | CoMSIA | 0.840 | Not Specified | 0.671 | Not Specified | [29] |
Comparative Analysis of Model Performance
The statistical metrics presented in Table 1 demonstrate that CoMFA and CoMSIA methodologies consistently generate robust models with high predictive capability across diverse biological targets. Our PLK1 inhibitor models show exceptional explanatory power with R² values exceeding 0.97, indicating excellent correlation between observed and predicted activities [9]. The cross-validated coefficients (Q²) ranging from 0.66-0.69 for the PLK1 inhibitors fall within acceptable parameters for predictive models (Q² > 0.5), though they are modest compared to the α1A-adrenergic receptor antagonist models which achieved Q² values of 0.840 [29]. The external predictive ability (R²pred) of our CoMSIA models for PLK1 inhibition (0.758-0.767) exceeds the threshold of 0.6, confirming their robustness for forecasting the activity of novel compounds [9].
When compared to other kinase-targeted studies, our PLK1 inhibitor models demonstrate comparable or superior performance to Aurora-B kinase inhibitors (Q² = 0.51-0.72) [65] and MCF-7 breast cancer cell line inhibitors (Q² = 0.62-0.71) [64]. The integration of CoMSIA's additional field types (hydrogen bond donor/acceptor, hydrophobic) in our PLK1 research provided enhanced insights into the structural determinants of biological activity beyond the standard steric and electrostatic fields of CoMFA [9].
Integrated QSAR Modeling Protocol
Experimental Workflow for Comparative QSAR Analysis
The following diagram illustrates the comprehensive workflow for conducting comparative QSAR analysis, from initial data preparation through final model validation:
Detailed Methodological Protocols
Molecular Database Preparation and Alignment
For our PLK1 inhibitor study, a dataset of 28 pteridinone derivatives with experimental IC50 values was compiled from literature [9]. The biological activities were converted to pIC50 values using the formula pIC50 = -log10(IC50) to create a normally distributed dependent variable [9]. The dataset was divided into training (80%, 22 compounds) and test sets (20%, 6 compounds) using structural diversity and activity range as stratification criteria [9].
Molecular alignment was performed using the distill method in SYBYL-X 2.1.2 software, with the most active compound serving as the template structure [9]. All molecules were energy-minimized using the Tripos force field with Gasteiger-Hückel atomic partial charges, employing the Powell gradient algorithm with a convergence criterion of 0.005 kcal/mol Å and 1000 iterations [9] [64]. This alignment approach ensures that molecules are superimposed based on their common structural features, which is critical for meaningful field calculation in CoMFA and CoMSIA studies.
CoMFA and CoMSIA Field Calculations and Model Development
Following molecular alignment, CoMFA and CoMSIA fields were calculated using a 3D cubic lattice with grid spacing of 2.0 Å in all Cartesian directions, extending 4.0 Å beyond the molecular dimensions [9] [29]. For CoMFA, steric (Lennard-Jones) and electrostatic (Coulombic) fields were computed using an sp³ carbon probe atom with +1.0 charge, with energy values truncated at 30 kcal/mol [29]. For CoMSIA, five similarity fields were evaluated: steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor, using a Gaussian-type function with attenuation factor α = 0.3 [29].
Partial least-squares (PLS) regression was employed to correlate the CoMFA/CoMSIA field descriptors with biological activity values [9]. The optimal number of components was determined by leave-one-out (LOO) cross-validation, followed by non-cross-validated analysis using this component number [29]. Column filtering was set to 2.0 kcal/mol to reduce noise and accelerate analysis [9]. Model robustness was evaluated through external prediction using the test set molecules, with predictive correlation coefficient (R²pred) values > 0.6 considered acceptable [9].
Advanced Computational Validation Techniques
To complement the 3D-QSAR models, molecular docking studies were performed using AutoDock Tools 1.5.6 to explore binding interactions between PLK1 inhibitors and the target protein (PDB: 2RKU) [9]. Key active site residues identified included R136, R57, Y133, L69, L82, and Y139 [9]. Molecular dynamics simulations were conducted for 50-100 ns using AMBER or NAMD to assess complex stability and binding free energies through MM/GBSA calculations [9] [64]. Finally, ADMET properties were predicted to evaluate drug-likeness and pharmacokinetic parameters of promising candidates [9] [64].
Research Reagent Solutions and Computational Tools
Table 2: Essential Research Reagents and Computational Tools for QSAR Studies
| Category | Specific Tool/Reagent | Function/Application | Example Use Case |
|---|---|---|---|
| Molecular Modeling Software | SYBYL-X 2.1.2 [9] | Molecular alignment, field calculation, and PLS analysis | CoMFA/CoMSIA model development for pteridinone derivatives |
| Docking Software | AutoDock Tools 1.5.6 [9] | Protein-ligand docking simulations | Identification of key PLK1 binding site interactions |
| Dynamics Software | NAMD 2.13 [66] | Molecular dynamics simulations | Assessment of protein-ligand complex stability over time |
| Force Fields | Tripos Force Field [29] | Molecular mechanics calculations | Energy minimization of molecular structures |
| Partial Charge Method | Gasteiger-Hückel [29] | Atomic partial charge calculation | Charge assignment for electrostatic field calculations |
| Database Resources | Protein Data Bank (PDB) [62] | Protein structure retrieval | Source of PLK1 structure (2RKU) for docking studies |
| Chemical Databases | ZINC Database [66] | Compound libraries for virtual screening | Identification of potential RdRp inhibitors |
Comparative Performance Visualization
The following diagram illustrates the relative performance of different QSAR methodologies across key statistical metrics, providing an at-a-glance comparison of model quality:
This comparative analysis demonstrates that CoMFA and CoMSIA methodologies generate highly predictive models for PLK1 inhibitor development, with statistical performance comparable or superior to other established QSAR approaches across diverse biological targets. The integration of these 3D-QSAR techniques with molecular docking, dynamics simulations, and ADMET profiling creates a robust computational framework for rational drug design [9]. The consistently high performance of CoMSIA across multiple studies suggests its particular utility in kinase inhibitor development, likely due to its ability to capture hydrogen bonding and hydrophobic interactions critical for ATP-binding site recognition [9] [62]. As the field advances, the integration of machine learning approaches with traditional 3D-QSAR methods shows promise for further enhancing predictive accuracy and accelerating the discovery of novel kinase inhibitors [62].
Conclusion
The integrated application of CoMFA and CoMSIA provides a powerful, rational framework for designing potent pteridinone-based PLK1 inhibitors. This protocol demonstrates that robust 3D-QSAR models, validated through molecular docking and dynamics, can successfully identify key structural features governing inhibitory activity—such as the critical roles of residues R136, R57, and Y133 in the PLK1 active site. The workflow's ability to predict compounds with favorable ADMET properties, as evidenced by candidate molecules like compound 28, significantly de-risks the early drug discovery pipeline. Future directions should focus on the experimental synthesis and biological testing of the in silico-predicted leads, the exploration of PBD-targeting inhibitors for enhanced selectivity, and the application of this validated protocol to other kinase targets. This comprehensive computational approach holds strong promise for accelerating the development of novel, targeted therapies for prostate cancer and other PLK1-dependent malignancies.
References
- 1. Polo-like Kinase 1 (PLK1) Inhibitors Targeting Anticancer ... [https://www.mdpi.com/2813-3757/3/4/23]
- 2. Structural regulation of PLK1 activity: implications for cell ... [https://www.nature.com/articles/s41417-025-00907-7]
- 3. PLK1 in cancer therapy: a comprehensive review ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1602752/full]
- 4. Pan-cancer analysis of polo-like kinase family genes ... [https://www.sciencedirect.com/science/article/pii/S2405844024054045]
- 5. Overexpression of oncogenic polo-like kinase 1 disrupts the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12181926/]
- 6. The plk1 Gene Regulatory Network Modeling Identifies Three ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12113133/]
- 7. 3D-QSAR Studies, Molecular Docking, Molecular Dynamic ... [https://www.mdpi.com/2075-1729/13/1/127]
- 8. Design and evaluation of novel triazole derivatives as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11922854/]
- 9. 3D-QSAR Studies, Molecular Docking, Molecular Dynamic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9865323/]
- 10. 3D-QSAR Studies, Molecular Docking, Molecular Dynamic ... [https://pubmed.ncbi.nlm.nih.gov/36676076/]
- 11. 3D-QSAR Studies, Molecular Docking ... [https://doaj.org/article/b0e5d9f2b0ec4e18a55e3971648121ab]
- 12. 3D computer modeling of inhibitors targeting the MCF-7 ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2024.1384832/full]
- 13. Discovery of a novel PLK1 inhibitor with high inhibitory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11892073/]
- 14. 3D-QSAR : Principles and Methods [https://drugdesign.org/chapters/3d-qsar/]
- 15. Combined Pharmacophore Modeling, Docking, and 3D- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3257097/]
- 16. 3D-QSAR CoMFA/CoMSIA, molecular docking and ... [https://pubmed.ncbi.nlm.nih.gov/36718094/]
- 17. CoMFA, CoMSIA, Topomer CoMFA, HQSAR, molecular ... [https://www.sciencedirect.com/science/article/abs/pii/S147692711830536X]
- 18. Docking-based CoMFA and CoMSIA studies of non- ... [https://pubmed.ncbi.nlm.nih.gov/15595461/]
- 19. CoMFA, CoMSIA, molecular docking and MOLCAD studies ... [https://www.sciencedirect.com/science/article/pii/S002228602031108X]
- 20. Design and Synthesis of a Novel PLK1 Inhibitor Scaffold ... [https://www.mdpi.com/1422-0067/22/8/3865]
- 21. CoMFA and CoMSIA analysis [https://bio-protocol.org/exchange/minidetail?id=2515250&type=30]
- 22. CoMFA and CoMSIA analysis of ACE-inhibitory ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523414006217]
- 23. Plk1-Targeted Small Molecule Inhibitors: Molecular Basis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3887635/]
- 24. Discovery and optimization of dihydropteridone derivatives ... [https://www.sciencedirect.com/science/article/abs/pii/S0968089624000233]
- 25. Comparative Molecular Field Analysis (CoMFA) and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3211024/]
- 26. 3D-QSAR CoMFA/CoMSIA models based on theoretical ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523409003080]
- 27. 3D QSAR and HQSAR analysis of protein kinase B (PKB/Akt ... [https://arabjchem.org/3d-qsar-and-hqsar-analysis-of-protein-kinase-b-pkb-akt-inhibitors-using-various-alignment-methods/]
- 28. Comparative Molecular Similarity Indices Analysis [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/comparative-molecular-similarity-indices-analysis]
- 29. Comparative Molecular Field Analysis (CoMFA) and ... [https://www.mdpi.com/1422-0067/12/10/7022]
- 30. 3.4. CoMSIA Studies [https://bio-protocol.org/exchange/minidetail?id=10197942&type=30]
- 31. A comparison of different electrostatic potentials on ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523410000309]
- 32. CoMFA, CoMSIA, HQSAR and Molecular Docking Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4852576/]
- 33. Combined 3D-QSAR, molecular docking and dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9666879/]
- 34. Implementing partial least squares and machine learning ... [https://www.nature.com/articles/s41598-025-06227-y]
- 35. Splitting training data into Train, Validation, and Test Sets [https://help.pecan.ai/en/articles/6454518-splitting-training-data-into-train-validation-and-test-sets]
- 36. Training, Validation, Test Split for Machine Learning Datasets [https://encord.com/blog/train-val-test-split/]
- 37. Is there a rule-of-thumb for how to divide a dataset into ... [https://stackoverflow.com/questions/13610074/is-there-a-rule-of-thumb-for-how-to-divide-a-dataset-into-training-and-validatio]
- 38. Integration of machine learning in 3D-QSAR CoMSIA models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10654693/]
- 39. Py-CoMSIA: An Open-Source Implementation of ... [https://www.mdpi.com/1424-8247/18/3/440]
- 40. Py-CoMSIA: An Open-Source Implementation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11945924/]
- 41. CoMFA, CoMSIA, kNN MFA and docking studies of 1,2,4 ... [https://arabjchem.org/comfa-comsia-knn-mfa-and-docking-studies-of-124-oxadiazole-derivatives-as-potent-caspase-3-activators/]
- 42. CoMFA and CoMSIA 3D-QSAR analysis of DMDP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2533057/]
- 43. Beginner's Guide to 3D-QSAR in Drug Design [https://neovarsity.org/blogs/beginners-guide-3d-qsar-drug-design]
- 44. Comparative Molecular Field Analysis - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/comparative-molecular-field-analysis]
- 45. Insights from 3D-QSAR, molecular docking, and dynamics for ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0326245]
- 46. A (Comprehensive) Review of the Application ... [https://www.mdpi.com/2076-3417/15/3/1206]
- 47. Missing Data & Outliers in Biocomputational Analysis [https://sapient.bio/resources/biocomputational-analysis-addressing-outliers/]
- 48. A Statistical Exploration of QSAR Models in Cancer Risk ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12030765/]
- 49. Beware of q2! [https://www.sciencedirect.com/science/article/abs/pii/S1093326301001231]
- 50. Comparison of various methods for validity evaluation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9396839/]
- 51. QSAR modeling, molecular docking and molecular dynamic ... [https://arabjchem.org/qsar-modeling-molecular-docking-and-molecular-dynamic-simulation-of-phosphorus-substituted-quinoline-derivatives-as-topoisomerase-i-inhibitors/]
- 52. HQSAR, CoMFA, CoMSIA docking studies and... | Research Square [https://www.researchsquare.com/article/rs-1465758/v1]
- 53. Structural investigation of tetrahydropteridin analogues as ... [https://www.sciencedirect.com/science/article/abs/pii/S0167732221009612]
- 54. Binding Studies and Lead Generation of Pteridin-7(8H) [https://pmc.ncbi.nlm.nih.gov/articles/PMC9319409/]
- 55. Molecular Simulations of Solved Co-crystallized X-Ray ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4576317/]
- 56. Crystal Structures and Molecular Dynamics Simulations of ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0083091]
- 57. ADMET Predictions - Computational Chemistry Glossary [https://deeporigin.com/glossary/admet-predictions]
- 58. Prediction of ADMET profile and anti-inflammatory potential ... [https://www.nature.com/articles/s41598-025-86809-y]
- 59. ADME-drug-likeness: enriching molecular foundation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12261486/]
- 60. ADMET predictions [https://www.vls3d.com/ADMET.html]
- 61. In-silico investigations of novel tacrine derivatives potency ... [https://www.sciencedirect.com/science/article/pii/S2468227623005021]
- 62. Tracking protein kinase targeting advances: integrating QSAR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11980485/]
- 63. 3D-QSAR, Scaffold Hopping, Virtual Screening, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11242256/]
- 64. 3D computer modeling of inhibitors targeting the MCF-7 breast ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11181028/]
- 65. 3D-QSAR (CoMFA, CoMFA-RG, CoMSIA) and molecular ... [https://www.sciencedirect.com/science/article/abs/pii/S0928098715300063]
- 66. Virtual Screening and Molecular Docking Studies for ... [https://www.mdpi.com/2073-4352/11/5/471]